

The successful preparation of a mutant KH domain representing the first KH domain of PCBP1 and its crystallization in complex with a C-rich DNA are reported. This structure is anticipated to provide high-resolution information that will allow better understanding of the basis of cytosine specificity by PCBPs.
Keywords: polycytosine-binding proteins, KH domains, cytosine specificity
Abstract
Polycytosine-binding proteins (PCBPs) are triple KH-domain proteins that play an important role in the regulation of translation of eukaryotic mRNA. They are also utilized by viral RNA and have been shown to interact with ssDNA. Underlying their function is the specific recognition of C-rich nucleotides by their KH domains. However, the structural basis of this recognition is only partially understood. Here, the preparation of a His-tagged KH domain is described, representing the first domain of PCBP1 that incorporates a C54S mutation as well as the addition of a C-terminal tryptophan. This construct has facilitated the preparation of highly diffracting crystals in complex with C-rich DNA (sequence ACCCCA). Crystals of the KH1–DNA complex were grown using the hanging-drop vapour-diffusion method in 0.1 M phosphate–citrate pH 4.2, 40%(v/v) PEG 300. X-ray diffraction data were collected to 1.77 Å resolution and the diffraction was consistent with space group P21, with unit-cell parameters a = 38.59, b = 111.88, c = 43.42 Å, α = γ = 90.0, β = 93.37°. The structure of the KH1–DNA complex will further our insight into the basis of cytosine specificity by PCBPs.
1. Introduction
During the lifetime of a eukaryotic mRNA, from its synthesis in the cell nucleus and export to the cell cytoplasm for translation to its ultimate degradation, the mRNA interacts with and is modified by a large number of combinatorial factors consisting of proteins and small noncoding RNAs (Moore, 2005 ▶; Soller, 2006 ▶). These act directly upon the mRNA to regulate the level of expression and the nature of the gene product. Polycytosine-binding proteins (PCBPs) are one such class of RNA-binding proteins involved in the regulation of protein translation. These proteins are ubiquitously expressed and can be divided into two subsets: heterogeneous nuclear ribonucleoprotein K (hnRNP K) and isoforms of PCBP (also known as α-complex protein, αCP or hnRNP E), which include PCBP1–4. They are characterized by their high and specific affinity for C-rich oligonucleotide sequences (Ostareck-Lederer et al., 1998 ▶; Makeyev & Liebhaber, 2002 ▶).
PCBPs are involved in the post-transcriptional regulation of mRNA (Makeyev & Liebhaber, 2002 ▶). PCBPs recognize and bind to C-rich regions in the 3′ UTR (Kiledjian et al., 1995 ▶; Waggoner & Liebhaber, 2003 ▶) and have been found to interact with a variety of mRNAs, including androgen receptor (Yeap et al., 2002 ▶), α-globin (Waggoner & Liebhaber, 2003 ▶), tyrosine hydroxylase (Holcik & Liebhaber, 197), erythropoietin (Czyzyk-Krzeska & Bendixen, 1999 ▶) and lipoxygenase mRNA (Holcik & Liebhaber, 1997 ▶; Ostareck et al., 2001 ▶). They have also been found to bind to some viral mRNAs, including human papillomavirus (Collier et al., 1998 ▶) and the 5′ UTR of poliovirus (Gamarnik & Andino, 1997 ▶). In general, binding by PCBPs has the effect of increasing mRNA stability (Holcik & Liebhaber, 1997 ▶; Waggoner & Liebhaber, 2003 ▶; Hollams et al., 2002 ▶; Murray et al., 2001 ▶). However, in the case of lipoxygenase and human papillomavirus the effect is different. Here, binding leads to the translational silencing of the mRNA, which is thought to be a consequence of the inhibition of 60s ribosomal subunit recruitment (Ostareck et al., 2001 ▶; Collier et al., 1998 ▶). In the case of viral RNA, PCBPs have been found to be utililized to coordinate a switch from RNA translation to replication in the propagation of the virus (Gamarnik & Andino, 1998 ▶). Besides binding to mRNA, in a more recent study PCBPs have also been discovered to bind to single-stranded DNA and to regulate the transcription of mouse µ-opioid receptor gene expression (Malik et al., 2006 ▶; Kim et al., 2005 ▶; Ko & Loh, 2005 ▶).
PCBPs consist of RNA-binding modules which belong to the family of KH (hnRNP K homology) domains. The first and the third KH domains (referred to in the following as KH1 and KH3) have been shown to individually bind to C-rich RNA, whereas this has not yet been demonstrated for the second KH domain (KH2; Dejgaard & Leffers, 1996 ▶). KH1 and KH2 are closely spaced along the protein sequence, whereas KH3 is separated by a linker that varies considerably between PCBP isoforms. As well as oligonucleotide binding, the PCBP KH domains have been shown to be capable of dimer formation; in fact, in the absence of oligonucleotide KH1 and KH2 have been shown to form a back-to-back heterodimer that does not preclude oligonucleotide binding (Du et al., 2008 ▶). The linker between KH2 and KH3 is also known to contain a nuclear localization signal involved in its shuttling role between the nucleus and the cytoplasm (Chkheidze & Liebhaber, 2003 ▶).
The basis of KH-domain recognition of C-rich oligonucleotides has been the subject of considerable study (Du et al., 2005 ▶, 2007 ▶; Fenn et al., 2007 ▶). It is known that the PCBP KH oligonucleotide-binding site accommodates four bases and that the central two positions are clearly configured to accommodate the cytosine base. Specific hydrogen bonds to the unique cytosine-base functional groups have been well documented. In contrast, these same studies provided no consistent explanation for the preference for cytosine binding at the first and fourth nucleotide-binding sites. In an effort to clarify our understanding of the basis of PCBP KH-domain specificity for polycytosine tracts, we have embarked on the production of a highly diffracting crystal of the first KH domain (KH1) of PCBP1 in complex with a C-rich nucleotide sequence. In this paper, we detail how this has been achieved using a mutant form of PCBP KH1 in which a cysteine shown previously to form intermolecular disulfide bonds in crystallo is replaced by serine, and a tryptophan is added to assist in the spectrophotometric detection of the purified product. We describe the formation of the complex of the KH1 domain with a unique olignucleotide comprising exclusively cytosine for protein recognition (ACCCCA) and the successful production of high-quality crystals.
2. Methods and results
2.1. DNA manipulation
The original construct encoding full-length human PCBP1 was kindly provided by Peter Leedman. The sequence corresponds to that deposited in GenBank (accession No. Q15365). A construct for the expression of the first KH domain (corresponding to amino-acid residues 14–90) was created by cloning using a 5′ primer (gtctgtCATATGCTCACCATTCGGCTTCTTATG) incorporating an NdeI restriction site and a 3′ primer (GAAGATATCAACAGCTCCTGGTAACTCGAGacagac) incorporating an XhoI restriction site into a pET-15b plasmid. The 3′ primer was also designed to incorporate a tryptophan residue at the C-terminus to facilitate detection by absorbance at 280 nm. PCR was performed using standard methods. After digestion with XhoI and NdeI, the PCR fragment was ligated into a likewise digested pET-15b plasmid downstream of a hexahistidine DNA sequence.
In a previous crystallization effort using wild-type KH1, a low-resolution crystal structure of a protein–oligonucleotide complex was obtained (PDB entry 1ztg; M. Sidiqi, J. A. Wilce, A. Barker, J. Schmidgerger, P. J. Leedman & M. C. J. Wilce, unpublished work). A disulfide bridge was found to be formed between two cysteines at residue 54 within the crystal. Since this disulfide bridge could have limited the optimization of the crystal form, a mutant of KH1 was created by mutating this cysteine to a serine to improve the potential crystal quality. The mutation was made using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies, Stratagene, USA). After sequencing had confirmed the successful cloning and mutagenesis, the resulting pET-15b-KH1.W.C54S plasmid was transformed into Escherichia coli BL21 (DE3) for overexpression of the protein.
The final construct consisted of residues 14–90 of the PCBP1 sequence with a C54S mutation, a C-terminal tryptophan and 21 residues present at the N-terminus including the uncleaved histidine tag (sequence MGSSHHHHHHSSGLVPRGSHM) and will henceforth be referred to as KH1.W.C54S.
2.2. Recombinant protein expression and purification
E. coli BL21 (DE3) cells harbouring pET15b-KH1.W.C54S were grown at 310 K in lysogeny broth (LB) medium with 100 µg ml−1 ampicillin to an OD600 of 0.6–0.8. Expression was induced with 0.5 mM IPTG for 4 h at 303 K. Cells were harvested by centrifugation at 5000 rev min−1 for 30 min at 277 K and were resuspended in buffer A (10 mM HEPES, 300 mM NaCl pH 8.0, 5 mM imidazole) with 0.2 mM PMSF and a ground tablet of EDTA-free protease-inhibitor cocktail (Roche). Cells were lysed using a homogenisator (Thomas) and centrifuged at 15 000 rev min−1 to remove the cellular debris.
The first step of the purification of His-tagged KH1.W.C54S was performed using nickel immobilized metal-affinity chromatography (IMAC). 4 ml 50% nickel-resin slurry was added to the supernatant and incubated at 277 K for 2 h. The resin was then applied onto a gravity column and washed with two column volumes of buffer A followed by two column volumes of buffer A containing 150 mM imidazole. KH1.W.C54S was eluted with buffer A containing 250 mM imidazole.
KH1.W.C54S was then further purified using a HiLoad 16/60 SP-Sepharose HP cation-exchange column (GE Healthcare) and fractionated using a 0–1.5 M NaCl gradient at a rate of 1 ml min−1. Interestingly, KH1.W.C54S eluted as two peaks: an early peak at 600 mM NaCl and a late peak at 750 mM NaCl (Fig. 1 ▶). The fractions from the two KH1.W.C54S peaks were then separately applied onto a HiLoad 16/60 Superdex 75 HP size-exclusion column (GE Healthcare) equilibrated with buffer B (10 mM HEPES, 150 mM NaCl, 1 mM EDTA). The results of the size-exclusion chromatography revealed that both the early and the late peaks corresponded to the same-size molecule (Fig. 2 ▶). The two fractions were also indistinguishable by mass-spectrometric analysis, with the masses of the early and late peaks found to be 10 345.26 and 10 345.28, respectively (the expected mass is 10 338.36). KH1.W.C54S from each peak was then concentrated separately using a Vivaspin 3 kDa molecular-weight cutoff concentrator (Sartorius Stedim Biotech, Victoria, Australia) to 11 mg ml−1 for the early peak and 14 mg ml−1 for the late peak and stored at 277 K in buffer B.
Figure 1.

A chromatogram of KH1.W.C54S cation exchange shows two peaks, A and B, eluting at around 600 and 750 mM NaCl, respectively. The smaller dashed chromatograms show that upon separate re-injection samples from the two peaks do not interconvert. SDS–PAGE analysis of the early and late fractions marked by different dashed lines is also shown.
Figure 2.

Size-exclusion chromatogram of early fractions (dashed line) and late fractions (continuous line) from the cation-exchange purification step of KH1.W.C54S.
2.3. Crystallization
The protein from each peak was used in cocrystallization trials with a 6-mer DNA sequence ACCCCA (purchased from Geneworks Australia) using the JCSG+ screen (Qiagen). After the DNA had been solubilized in water and its concentration measured using the OD260, it was freeze-dried. The DNA was resolubilized in buffer B containing 11 mg ml−1 (early peak) or 14 mg ml−1 (late peak) KH1.W.C54S at a 1:1.2 protein:DNA ratio to form the protein–DNA complex.
Crystallization trials were set up using 500 µl of each reagent from the JCSG+ screen dispensed into Linbro tissue-culture plates. 1 µl protein–DNA complex was mixed with 1 µl reservoir solution on Hampton siliconized 22 mm glass slides and the mixture was equilibrated over the reservoir at room temperature using the hanging-drop vapour-diffusion method. Crystals of the protein–DNA complex were observed after approximately two weeks from the late KH1.W.C54S peak in 0.1 M phosphate–citrate pH 4.2, 40%(v/v) PEG 300. The crystals possessed an irregular form and had dimensions of approximately 0.5 × 0.2 × 0.1 mm (Fig. 3 ▶).
Figure 3.

Crystals of the KH1.W.C54S–ACCCCA complex of approximately 0.5 × 0.2 × 0.1 mm in size.
The crystal was prised apart with a microprobe (Hampton Research) and a fragment of the crystal was picked up using a silicon loop (Hampton Research) and streaked through cryoprotectant solution consisting of the reservoir solution with 25% glycerol. Initial diffraction screening was carried out in-house on a Rigaku MicroMax-007 HF rotating-anode generator with an R-AXIS IV++ image-plate detector. X-ray data were then collected at a wavelength of 0.9801 Å on the high-throughput protein crystallography beamline (MX2) at the Australian Synchrotron (McPhillips et al., 2002 ▶) using an ADSC Quantum 315r detector. 252 diffraction images were recorded with an oscillation angle of 0.5° and an exposure time of 1 s (Fig. 4 ▶). Diffraction data were integrated with XDS (Kabsch, 2010 ▶) and intensities were scaled using SCALA (Evans, 2006 ▶; Winn et al., 2011 ▶).
Figure 4.

Diffraction of the KH1.W.C54S–ACCCA crystal to 1.77 Å resolution. The resolution at the edge of the diffraction image is 1.50 Å.
3. Results and discussion
The first KH domain of PCBP1 (residues 14–90) was expressed as a His-tagged protein with the cysteine at position 54 mutated to serine to avoid possible intermolecular disulfide formation as well as with a C-terminal tryptophan to facilitate absorbance detection at 280 nm for purification and concentration-determination purposes. Previous studies of the PCBP1 KH1 domain in our laboratory utilized a GST-tagged construct. However, it was noted that the PCBP1 KH1 domain exhibited poor solubility upon the removal of the GST tag that limited its yield. We therefore recloned the construct to produce the protein with a purification tag that would not require removal prior to crystallization trials. Whilst an unstructured hexa-His sequence could potentially hinder crystal formation, the advantages of improved solubility and yield outweighed this possible impediment.
The His-tagged KH1.W.C54S protein was expressed in E. coli and purified using nickel IMAC and cation-exchange chromatography followed by size-exclusion chromatography. Two peaks were eluted from the cation-exchange column that both corresponded to the KH1.W.C54S protein according to SDS–PAGE and mass spectrometry. Furthermore, each of these peaks eluted at the same position upon size-exclusion chromatography. These peaks did not represent forms of the protein that were in exchange with one another, as they both eluted at the same positions (i.e. either early or late) upon re-injection onto the cation-exchange column. Thus, although these peaks appeared to represent the same protein, the eluted fractions were not combined but were treated in parallel. The purified samples were complexed with a 6-mer oligonucleotide ACCCCA in a 1:1.2 molar ratio and subjected to hanging-drop crystallization trials. Crystals appeared after approximately two weeks and were stable at room temperature. The crystals were found to diffract well and efforts to optimize the crystallization conditions did not give rise to better quality crystals. The crystals were frozen and data sets were collected both in-house and using synchrotron radiation, the latter to 1.77 Å resolution. The diffraction data and systematic absences are consistent with space group P21, with unit-cell parameters a = 38.59, b = 111.88, c = 43.42 Å, α = 90.00, β = 93.37, γ = 90.00°. Based on the Matthews coefficient, the unit-cell volume is consistent with the presence of four protein–DNA complexes in the asymmetric unit. Diffraction data statistics are summarized in Table 1 ▶. Starting phases were found by molecular-replacement techniques using Phaser (McCoy et al., 2007 ▶) with the coordinate set of KH1 previously determined in our laboratory (PDB entry 1ztg). The electron density clearly revealed the presence of the oligonucleotide bound to the KH1 protein.
Table 1. Crystallographic data-collection statistics for the KH1.W.C54S–DNA complex.
Values in parentheses are for the highest resolution shell.
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 38.59, b = 114.88, c = 43.42, α = 90.00, β = 93.37, γ = 90.00 |
| Resolution (Å) | 30.00–1.77 (1.87–1.77) |
| No. of unique reflections | 35997 (5268) |
| Multiplicity | 3.2 (3.1) |
| Completeness (%) | 99.7 (100) |
| 〈I/σ(I)〉 | 7.9 (2.2) |
| Rmerge† (%) | 9.0 (60.2) |
| Rp.i.m.‡ (%) | 6.0 (41.0) |
R
merge = 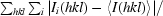
 , where I
i(hkl) is the intensity of individual reflections.
, where I
i(hkl) is the intensity of individual reflections.
R p.i.m. is the multiplicity-weighted R merge.
The high-resolution structure of this KH1.W.C54S–ACCCCA complex will represent the first structure of a KH domain in complex with a C-rich tetrad oligonucleotide and will provide sufficient resolution to ascertain whether specific interactions at nucleotide-binding sites 1 and 4 underlie specificity for cytosines. This in turn will help to clarify the mode of interaction of PCBPs in their recognition of host as well as viral RNA in the regulation of its translation.
Acknowledgments
We wish to thank the staff of the Protein Crystallography Beamline (MX2) at the Australian Synchrotron, Victoria, Australia, where the diffraction data were collected. This work was supported by a grant from the Australian Research Council awarded to MCJW and JAW and a National Health and Medical Research Senior Research Fellowship awarded to MCJW.
References
- Chkheidze, A. N. & Liebhaber, S. A. (2003). Mol. Cell. Biol. 23, 8405–8415. [DOI] [PMC free article] [PubMed]
- Collier, B., Goobar-Larsson, L., Sokolowski, M. & Schwartz, S. (1998). J. Biol. Chem. 3, 22648–22656. [DOI] [PubMed]
- Czyzyk-Krzeska, M. & Bendixen, A. (1999). RNA, 93, 2111–2120. [PubMed]
- Dejgaard, K. & Leffers, H. (1996). Eur. J. Biochem. 241, 425–431. [DOI] [PubMed]
- Du, Z., Fenn, S., Tjhen, R. & James, T. L. (2008). J. Biol. Chem. 283, 28757–28766. [DOI] [PMC free article] [PubMed]
- Du, Z., Lee, J. K., Fenn, S., Tjhen, R., Stroud, R. M. & James, T. L. (2007). RNA, 13, 1043–1051. [DOI] [PMC free article] [PubMed]
- Du, Z., Lee, J. K., Tjhen, R., Li, S., Pan, H., Stroud, R. M. & James, T. L. (2005). J. Biol. Chem. 280, 38823–38830. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fenn, S., Du, Z., Lee, J. K., Tjhen, R., Stroud, R. M. & James, T. L. (2007). Nucleic Acids Res. 35, 2651–2660. [DOI] [PMC free article] [PubMed]
- Gamarnik, A. & Andino, R. (1997). RNA, 3, 2111–2120. [PMC free article] [PubMed]
- Gamarnik, A. V. & Andino, R. (1998). Genes Dev. 12, 2293–2304. [DOI] [PMC free article] [PubMed]
- Holcik, M. & Liebhaber, S. A. (1997). Proc. Natl Acad. Sci. USA, 94, 2410–2414. [DOI] [PMC free article] [PubMed]
- Hollams, E. M., Giles, K. M., Thomson, A. M. & Leedman, P. J. (2002). Neurochem. Res. 27, 957–980. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kiledjian, M., Wang, X. & Liebhaber, S. A. (1995). EMBO J. 14, 4357–4364. [DOI] [PMC free article] [PubMed]
- Kim, S.-S., Pandey, K. K., Choi, H. S., Kim, S.-Y., Law, P.-Y., Wei, L.-N. & Loh, H. H. (2005). Mol. Pharmacol. 68, 729–736. [DOI] [PubMed]
- Ko, J. L. & Loh, H. H. (2005). J. Neurochem. 93, 749–761. [DOI] [PubMed]
- Makeyev, A. V. & Liebhaber, S. A. (2002). RNA, 8, 265–278. [DOI] [PMC free article] [PubMed]
- Malik, A. K., Flock, K. E., Godavarthi, C. L., Loh, H. H. & Ko, J. L. (2006). Brain Res. 1112, 33–45. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad. 9, 401–406. [DOI] [PubMed]
- Moore, M. J. (2005). Science, 309, 1514–1518. [DOI] [PubMed]
- Murray, K. E., Roberts, A. W. & Barton, D. J. (2001). RNA, 7, 1126–1141. [DOI] [PMC free article] [PubMed]
- Ostareck, D. H., Ostareck-Lederer, A., Shatsky, I. N. & Hentze, M. W. (2001). Cell, 104, 281–290. [DOI] [PubMed]
- Ostareck-Lederer, A., Ostareck, D. H. & Hentze, M. W. (1998). Trends Biochem. Sci. 23, 409–411. [DOI] [PubMed]
- Soller, M. (2006). Cell. Mol. Life Sci. 63, 796–819. [DOI] [PMC free article] [PubMed]
- Waggoner, S. & Liebhaber, S. (2003). Exp. Biol. Med. (Maywood), 228, 387–395. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yeap, B. B., Voon, D. C., Vivian, J. P., McCulloch, R. K., Thomson, A. M., Giles, K. M., Czyzyk-Krzeska, M. F., Furneaux, H., Wilce, M. C., Wilce, J. A. & Leedman, P. J. (2002). J. Biol. Chem. 277, 27183–27192. [DOI] [PubMed]