

To further understand the structural basis of MmeI (a type IIL restriction enzyme) DNA-binding specificity, the enzyme has been crystallized in complex with its DNA substrate.
Keywords: type II restriction enzymes, MmeI, DNA binding
Abstract
Type IIL restriction enzymes have rejuvenated the search for user-specified DNA binding and cutting. By aligning and contrasting the highly comparable amino-acid sequences yet diverse recognition specificities across the family of enzymes, amino acids involved in DNA binding have been identified and mutated to produce alternative binding specificities. To date, the specificity of MmeI (a type IIL restriction enzyme) has successfully been altered at positions 3, 4 and 6 of the asymmetric TCCRAC (where R is a purine) DNA-recognition sequence. To further understand the structural basis of MmeI DNA-binding specificity, the enzyme has been crystallized in complex with its DNA substrate. The crystal belonged to space group P1, with unit-cell parameters a = 61.73, b = 94.96, c = 161.24 Å, α = 72.79, β = 89.12, γ = 71.68°, and diffracted to 2.6 Å resolution when exposed to synchrotron radiation. The structure promises to reveal the basis of MmeI DNA-binding specificity and will complement efforts to create enzymes with novel specificities.
1. Introduction
The ability to design new DNA-binding and DNA-cleaving proteins with the discrimination of the type II restriction enzymes would open new avenues of research for biologists (Jeltsch et al., 1996 ▶). Targeting a specific genomic sequence would permit researchers to test the function of individual genes by accurately cleaving or blocking specific sequences, as well as opening up new avenues for whole genome analysis and fingerprinting. One approach towards designing a binding motif has been to modify the natural, well documented and highly accurate type II restriction enzymes (Osuna et al., 1991; Aggarwal, 1995 ▶; Pingoud et al., 2005 ▶; Gormley et al., 2005 ▶) such as BamHI (Newman et al., 1994 ▶, 1995 ▶; Dorner et al., 1999 ▶), EcoRI (Rosenberg et al., 1987 ▶; Heitman & Model, 1990a ▶,b ▶; Osuna et al., 1991 ▶), EcoRV (Lanio et al., 2000 ▶; Jeltsch et al., 1996 ▶; Thielking et al., 1991 ▶; Winkler et al., 1993 ▶), BglII (Lukacs et al., 2000 ▶; Townson et al., 2005 ▶) and BstYI (Townson et al., 2004 ▶, 2005 ▶; Samuelson & Xu, 2002 ▶). However, even when guided by three-dimensional structures, mutating the DNA-contacting residues has generally failed to produce viable enzymes with altered specificity (Alves & Vennekohl, 2004 ▶).
Type IIL restriction enzymes (REs) provide a new framework for designing DNA-binding specificity (Morgan & Luyten, 2009 ▶). The type IIL RE subtype links host-protective modification and endonuclease functions to a common DNA-recognition module, which allows the rapid evolution of DNA-recognition specificity (Morgan et al., 2009 ▶). Thus, we observe families of type IIL REs that contain a large collection of REs with highly similar amino-acid sequences yet divergent recognition sequences (Morgan et al., 2009 ▶). The sequence similarities of type IIL REs are shown by their method of discovery: cloning MmeI (from Methylophilus methylotrophus) and comparing its sequence to genomic databases of known bacterial and archaeal sequences. As such, comparing the analogous protein sequences and divergent recognition sequences of 21 type IIL REs suggested the amino acids that make contact with the bound DNA and which substitutions might change the specificity (Morgan & Luyten, 2009 ▶). By interchanging the contacting amino acids between homologs, we have designed a subset of type IIL REs that can bind alternative recognition sites with high efficiency (Morgan & Luyten, 2009 ▶). However, these studies are greatly hampered by the lack of structural information on type IIL REs, limiting the repertoire of enzymes that can be rationally engineered from sequence analysis.
To better understand the structural basis of DNA recognition and cleavage by type IIL REs, we have crystallized MmeI in complex with DNA. MmeI is a large enzyme (919 amino acids) that encompasses DNA-recognition, methyltransferase and endonuclease activities in the same polypeptide (Boyd et al., 1986 ▶; Tucholski et al., 1998 ▶; Nakonieczna et al., 2009 ▶; Morgan et al., 2008 ▶). The enzyme recognizes the asymmetric DNA sequence TCCRAC (where R is a purine) and cleaves the DNA two helical turns away from the recognition sequence: TCCRAC20/18. We have grown crystals of the complete MmeI enzyme in complex with its DNA substrate. The structure will be helpful in understanding the amino acids contacting DNA as well as providing a basis for the long reach between the recognition sequence and the DNA-cleavage point. We have also grown cocrystals of selenomethionine (SeMet) MmeI and efforts are under way to solve the structure using the multiwavelength anomalous diffraction (MAD) method.
2. Expression and purification
2.1. Native MmeI cell growth and purification
The MmeI gene was placed under plac promoter control in the pRRS vector in Escherichia coli ER2683 cells, which were grown in LB medium containing 100 µg ml−1 ampicillin. 537 g of cells were suspended in three volumes of buffer A (20 mM Tris–HCl pH 8, 50 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 5% glycerol) and passed through a microfluidizer at 124 MPa. The lysate was clarified by centrifugation, yielding 1900 ml crude extract at a pH of 7.27. All purification steps were performed at 277 K.
The crude extract solution, which contained approximately 5 700 000 units or 190 mg MmeI, was applied onto a 397 ml Heparin Hyper-D column (Pall BioSepra, Ann Arbor, Michigan, USA) equilibrated in buffer A. The column was washed with 800 ml buffer A; a 4 l gradient of NaCl from 0.050 to 1 M in buffer A was then applied and 25 ml fractions were collected between 0.15 and 0.6 M NaCl. Fractions were assayed for MmeI endonuclease activity, which was found to elute between 0.35 and 0.45 M NaCl.
Heparin Hyper-D column fractions containing peak MmeI activity were pooled, diluted 1:7 with buffer A without NaCl and applied onto a 132 ml Source TM15Q column (GE Healthcare, Piscataway, New Jersey, USA). The column was washed with two volumes of buffer A; MmeI activity was found in the flowthrough and wash, having not bound to the column.
The 3.125 l Source TM15Q flowthrough and wash pool was diluted with 1 l buffer B (20 mM potassium phosphate pH 6.8, 50 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 5% glycerol) and applied onto a 120 ml Source TM15S column (GE Healthcare, Piscataway, New Jersey, USA). The column was washed with two volumes of buffer B; a 20-column-volume linear gradient from 0.05 to 0.5 M NaCl in buffer B was then applied and 20 ml fractions were collected. MmeI eluted between 0.13 and 0.15 M NaCl.
Fractions containing peak MmeI activity were pooled, diluted 1:3 with buffer B and applied onto a 56 ml Heparin-TSK column (Tosoh Bioscience, Tokyo, Japan). The column was washed with two column volumes of buffer B and a 20-column-volume linear gradient from 0.05 to 0.5 M NaCl was then applied. MmeI eluted between 0.21 and 0.27 M NaCl. Fractions containing peak MmeI activity were dialyzed into MmeI storage buffer (10 mM Tris–HCl pH 7.4, 300 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 50% glycerol).
The concentrated MmeI solution (30 ml) was applied onto a Superdex 75 XK 50/100 column (GE Healthcare, Piscataway, New Jersey, USA). A single large UV peak was obtained which contained the MmeI activity. Active fractions were dialyzed overnight against storage buffer. This preparation yielded 42.5 ml highly pure MmeI enzyme solution at 64 000 units ml−1, or 2.17 mg ml−1, which was used for crystallographic analysis (Fig. 1 ▶).
Figure 1.

Purified wild-type MmeI in twofold serial dilutions from 16 to 1 µg on a 10–20% Tris–glycine gel (1 mm × 12 wells; Invitrogen, catalog No. EC61352) with an NEB 10–250 kDa protein ladder (NEB, catalog No. P7703S). The gel was run at 125 V constant voltage and stained using Coomassie Blue. The molecular weights of the two markers bracketing MmeI are 80 and 100 kDa.
Native MmeI was buffer-exchanged into 10 mM Tris–HCl pH 7.4, 300 mM NaCl, 1 mM DTT, 0.1 mM EDTA and 5% glycerol and concentrated to 135 µM prior to being aliquoted and stored. Prior to crystallization, 10 mM CaCl2, 100 mM NaCl, 1 mM sinefungin, 70 µM DNA and buffer were added to the RE solution, diluting the final protein concentration for use in hanging-drop formation to 30 µM.
2.2. SeMet MmeI cell growth and purification
The MmeI gene was placed under T7 promoter control in the vector pSapv6 and transformed into the methionine-auxotroph strain T7 Express Crystal C3022 cells (NEB). A single colony was grown overnight at 303 K in 250 ml minimal medium supplemented with 1.0 mM l-methionine containing 30 µg ml−1 chloramphenicol, using 3% glycerol as a carbon source. 80 ml of the seed culture was added to 9 l minimal medium as above in a high-density fermentor in which the pH was controlled at 6.8 and dissolved oxygen was maintained at 20%. The carbon-source feed stock contained 1.0 mM selenomethionine. Cells were grown at 303 K to an OD600 of 12.8; selenomethionine was then added to 10 mM and the MmeI gene was induced by IPTG at 0.4 mM. The culture was grown at 303 K for 7 h to an OD600 of 25.0 and produced 497 g cells (wet weight). These were assayed and found to contain 25 000 units of MmeI per gram. SeMet MmeI was purified using the same purification protocol as used for native MmeI, yielding 162 mg MmeI protein. The incorporation level of SeMet in this preparation of MmeI was found to be 94.6% by amino-acid analysis. The SeMet protein had the same specific endonuclease activity as the wild-type MmeI.
3. Results and discussion
We sought to cocrystallize native MmeI with DNAs of different lengths and terminal ends encompassing the MmeI recognition sequence. Crystals were grown using 2 µl hanging drops over 1 ml reservoirs at 293 K. The initial cocrystals were obtained with a 30-mer from solutions consisting of 10%(w/v) polyethylene glycol (PEG) 8K, 0.1 M HEPES pH 7.5, 0.2 M calcium acetate. The largest cocrystals were subsequently obtained with a blunt-end double-stranded 29-mer with sequence 5′-TATCCGACATAACGCTAGTCACTAGCTTC-3′/3′-ATAGGCTGTATTGCGATCAGTGATCGAAG-5′). The DNA oligos were synthesized on a 1 µM scale and PAGE-purified (Integrated DNA Technologies, Coralville, Iowa, USA). These cocrystals were highly mosaic and diffracted to 4 Å resolution when exposed to synchrotron radiation. The resolution and mosaicity were improved by dehydrating the crystals (in reservoir solution with 30% glycerol) and by changing the mother liquor for crystal growth and dehydration to 20% PEG 4K, 0.1 M HEPES pH 7.5 and 0.1 M ammonium sulfate. The resolution was further improved to 2.6 Å (see Fig. 2 ▶ a) by replacing several thymines outside the recognition site with 5-bromouracil (5′-TATCCGACAUAACGCUAGUCACUAGCUUC-3′/3′-ATAGGCTGUATUGCGAUCAGUGAUCGAAG-5′, where U is 5-bromouracil). The brominated oligos were synthesized at New England Biolabs and PAGE-purified for crystallization. For cryoprotection, the crystals were soaked for 5 min in solutions of mother liquor plus increasing concentrations of glycerol (final concentration of 30% glycerol). Cocrystals with SeMet MmeI (14 methionines per molecule) were obtained under similar conditions as those used for the native enzyme, although the crystals were typically smaller. Interestingly, the program DIBER, which computes the largest local average value of the diffraction intensity at 3.4 Å resolution, reported a 94% probability of the crystals containing a protein–DNA complex, as opposed to protein or DNA only (Chojnowski & Bochtler, 2010 ▶; McCoy et al., 2007 ▶).
Figure 2.

(a) Typical crystals of MmeI in complex with 5-bromouracil-substituted DNA. The red scale bar is 100 µm in length. (b) The best X-ray diffraction pattern of the MmeI–brominated DNA complex recorded on APS NE-CAT 24ID-C with 1° oscillation. The resolution at the edge of the detector is 2.45 Å.
Diffraction data from the native MmeI–29-mer cocrystals were collected to 2.6 Å resolution on beamline 24ID-C at the Advanced Photon Source (APS), Argonne National Laboratory (Fig. 2 ▶ b) at a wavelength of 0.91938 Å. Diffraction statistics are given in Table 1 ▶. The HKL-2000 package (Otwinowski & Minor, 1997 ▶) was used to determine that the crystals belonged to space group P1, with unit-cell parameters a = 61.73, b = 94.96, c = 161.24 Å, α = 72.79, β = 89.12, γ = 71.68°. From solvent calculations the crystals may contain two or three molecules per asymmetric unit. Assuming two molecules per asymmetric unit gives a solvent content of 68% and a Matthews coefficient of 3.84 Å3 Da−1, whereas three molecules per asymmetric unit gives a more probable solvent content of 48.1% and a Matthews coefficient of 2.37 Å3 Da−1 (Matthews, 1968 ▶; Adams et al., 2010 ▶). However, a self-rotation search performed with POLARRFN in the CCP4 package (Winn et al., 2011 ▶) gives the highest peak at κ = 180° and not at κ = 120° (using a radius of integration of 60 Å). The peak at κ = 180° occurs at spherical polar angles ω = 90°, ϕ = −85°, suggesting a noncrystallographic twofold axis lying roughly along the c* × a axis of the crystals (Fig. 3 ▶ a). Moreover, the top Phaser rotation-function solutions as reported by DIBER using a shell of data around 3.4 Å and a B-DNA model are also roughly along the c* × a axis (Fig. 3 ▶ b), suggesting this to be roughly the direction of the DNA in the crystals (Chojnowski & Bochtler, 2010 ▶; Adams et al., 2010 ▶). Data were collected over 360° with a crystal-to-detector distance of 400 mm and 1° oscillation per frame. The SeMet MmeI–DNA crystals diffracted to 3.4 Å resolution using synchrotron radiation. We have measured MAD data from these crystals and efforts are under way to solve the structure by the MAD method using the Se and Br atoms.
Table 1. Diffraction statistics for MmeI in complex with 5-bromouracil-substituted DNA collected on APS NE-CAT 24ID-C.
Values in parentheses are for the highest resolution shell.
| Beamline | APS 24ID-C |
| Wavelength (Å) | 0.91938 |
| Space group | P1 |
| Unit-cell parameters (Å, °) | a = 61.73, b = 94.96, c = 161.24, α = 72.79, β = 89.12, γ = 71.68 |
| Resolution range (Å) | 50–2.6 (2.69–2.60) |
| Completeness (%) | 89.3 (75.2) |
| Rmerge† (%) | 12.8 (51.1) |
| 〈I/σ(I)〉 | 11.6 (2.0) |
| Mosaicity range (°) | 0.55–1.04 |
| Type of detector | ADSC Q315 [315 × 315 mm] |
| Temperature of crystal during data collection (K) | 100 |
| Total reflections | 321516 |
| Unique reflections | 91018 |
| Criteria for observed reflections | −3σ |
| Multiplicity | 3.5 |
R
merge = 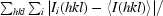
 , where I(hkl) is the intensity of reflection hkl.
, where I(hkl) is the intensity of reflection hkl.
Figure 3.

(a) A stereographic projection of the κ = 180° section of the self-rotation function obtained with the program POLARFFN in the CCP4 package. In the plot, ω varies from 0 to 90° in the radial direction, while ϕ varies from 0 to 360° as measured in an anticlockwise direction from the a axis. The a axis points to the right in the plane of the paper, while the c* × a axis points up in the plane of the paper. Data between resolution limits of 9 and 4.0 Å were used in the calculation. A Patterson integration radius of 60 Å was also used in the calculation. (b) A plot from the program DIBER showing the top ten solutions of the Phaser rotation function (marked ×) on top of the local intensity averages in the thin 3.4 Å resolution shell on a stereographic net.
We chose to study MmeI because of its highly similar amino-acid sequence to other type IIL REs with divergent recognition sequences. The structure promises to reveal the MmeI residues that underlie the recognition of its asymmetric recognition sequence TCCRAC and provide a basis for the unusually long reach between the recognition sequence and the DNA-cleavage point. The structure will complement our bioinformatic analysis of type IIL RE sequences, expanding the range of amino acids that can be rationally engineered to create enzymes with novel substrate specificities.
Acknowledgments
This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aggarwal, A. K. (1995). Curr. Opin. Struct. Biol. 5, 11–19. [DOI] [PubMed]
- Alves, J. & Vennekohl, P. (2004). Restriction Endonucleases, edited by A. Pingoud, pp. 393–411. Berlin, Heidelberg: Springer-Verlag.
- Boyd, A. C., Charles, I. G., Keyte, J. W. & Brammar, W. J. (1986). Nucleic Acids Res. 14, 5255–5274. [DOI] [PMC free article] [PubMed]
- Chojnowski, G. & Bochtler, M. (2010). Acta Cryst. D66, 643–653. [DOI] [PubMed]
- Dorner, L. F., Bitinaite, J., Whitaker, R. D. & Schildkraut, I. (1999). J. Mol. Biol. 285, 1515–1523. [DOI] [PubMed]
- Gormley, N. A., Watson, M. A. & Halford, S. E. (2005). Encyclopedia of Life Sciences doi:10.1038/npg.els.0003897.
- Heitman, J. & Model, P. (1990a). EMBO J. 9, 3369–3378. [DOI] [PMC free article] [PubMed]
- Heitman, J. & Model, P. (1990b). Proteins, 7, 185–197. [DOI] [PubMed]
- Jeltsch, A., Wenz, C., Wende, W., Selent, U. & Pingoud, A. (1996). Trends Biotechnol. 14, 235–238. [DOI] [PubMed]
- Lanio, T., Jeltsch, A. & Pingoud, A. (2000). Protein Eng. 13, 275–281. [DOI] [PubMed]
- Lukacs, C. M., Kucera, R., Schildkraut, I. & Aggarwal, A. K. (2000). Nature Struct. Biol. 7, 134–140. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Morgan, R. D., Bhatia, T. K., Lovasco, L. & Davis, T. B. (2008). Nucleic Acids Res. 36, 6558–6570. [DOI] [PMC free article] [PubMed]
- Morgan, R. D., Dwinell, E. A., Bhatia, T. K., Lang, E. M. & Luyten, Y. A. (2009). Nucleic Acids Res. 37, 5208–5221. [DOI] [PMC free article] [PubMed]
- Morgan, R. D. & Luyten, Y. A. (2009). Nucleic Acids Res. 37, 5222–5233. [DOI] [PMC free article] [PubMed]
- Nakonieczna, J., Kaczorowski, T., Obarska-Kosinska, A. & Bujnicki, J. M. (2009). Appl. Environ. Microbiol. 75, 212–223. [DOI] [PMC free article] [PubMed]
- Newman, M., Strzelecka, T., Dorner, L. F., Schildkraut, I. & Aggarwal, A. K. (1994). Structure, 2, 439–452. [DOI] [PubMed]
- Newman, M., Strzelecka, T., Dorner, L. F., Schildkraut, I. & Aggarwal, A. K. (1995). Science, 269, 656–663. [DOI] [PubMed]
- Osuna, J., Flores, H. & Soberón, X. (1991). Gene, 106, 7–12. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pingoud, A., Fuxreiter, M., Pingoud, V. & Wende, W. (2005). Cell. Mol. Life Sci. 62, 685–707. [DOI] [PubMed]
- Rosenberg, J., Wang, B., Frederic, C., Reich, N., Greene, P., Grable, J. & McClarin, J. (1987). Protein Engineering, edited by P. McPhie, pp. 237–250. New York: Alan R. Liss.
- Samuelson, J. C. & Xu, S. (2002). J. Mol. Biol. 319, 673–683. [DOI] [PubMed]
- Thielking, V., Selent, U., Köhler, E., Wolfes, H., Pieper, U., Geiger, R., Urbanke, C., Winkler, F. K. & Pingoud, A. (1991). Biochemistry, 30, 6416–6422. [DOI] [PubMed]
- Townson, S. A., Samuelson, J. C., Vanamee, É, S., Edwards, T. A., Escalante, C. R., Xu, S. & Aggarwal, A. K. (2004). J. Mol. Biol. 338, 725–733. [DOI] [PubMed]
- Townson, S. A., Samuelson, J. C., Xu, S. & Aggarwal, A. K. (2005). Structure, 13, 791–801. [DOI] [PubMed]
- Tucholski, J., Zmijewski, J. W. & Podhajska, A. J. (1998). Gene, 223, 293–302. [DOI] [PubMed]
- Winkler, F. K., Banner, D. W., Oefner, C., Tsernoglou, D., Brown, R. S., Heathman, S. P., Bryan, R. K., Martin, P. D., Petratos, K. & Wilson, K. S. (1993). EMBO J. 12, 1781–1795. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.