

Abstract
Methyl-transverse relaxation optimized spectroscopy is rapidly becoming the preferred NMR technique for probing structure and dynamics of very large proteins up to ~1 MDa in molecular size. Data interpretation, however, necessitates assignment of methyl groups which still presents a very challenging and time-consuming process. Here we demonstrate that, in combination with a known 3D structure, paramagnetic relaxation enhancement (PRE), induced by nitroxide spin-labels incorporated at only a few surface-exposed engineered cysteines, provides fast, straightforward and robust access to methyl group resonance assignments, including stereoassignments for the methyl groups of leucine and valine. Neither prior assignments, including backbone assignments, for the protein, nor experiments that transfer magnetization between methyl groups and the protein backbone, are required. PRE-derived assignments are refined by 4D methyl–methyl nuclear Overhauser enhancement data, eliminating ambiguities and errors that may arise due to the high sensitivity of PREs to the potential presence of sparsely-populated transient states.
Keywords: Paramagnetic relaxation enhancement, Methyl-TROSY, 1H–13C HMQC, Paramagnetic spin-labeling, Methyl group labeling
Introdution
In combination with transverse relaxation optimized spectroscopy (TROSY) techniques (Pervushin et al. 1997; Tugarinov et al. 2003), selective isotope-labeling methodologies have significantly increased the molecular weight range accessible to solution NMR. In particular, selective incorporation of 1H and 13C into the methyl groups of Ala, Ile, Leu, Met and Val in a perdeuterated background (Tugarinov et al. 2006; Gelis et al. 2007; Ayala et al. 2009) provides excellent site-specific probes for the investigation of protein structure and dynamics in systems up to ~1 MDa molecular size (Religa et al. 2010; Sprangers and Kay 2007). The main advantage of using methyl groups in NMR studies of large macromolecules arises from their favorable relaxation properties (Tugarinov et al. 2003), the three-fold degeneracy of 1H methyl (1HM) chemical shifts, and their insensitivity to solvent exchange. Buried methyl groups report on side chain packing and protein folding (Tugarinov et al. 2005), while surface-exposed methyl groups can serve as probes for protein–protein (Gross et al. 2003) and protein–ligand (Hajduk et al. 2000) interactions. In addition, Ala methyl groups are not subject to multiple rotameric conformations and are therefore excellent targets for residual dipolar coupling measurements (Godoy-Ruiz et al. 2010) that provide important restraints on interdomain orientations (Schwieters et al. 2010; Tjandra and Bax 1997).
Conventional methyl group assignment strategies rely on a set of multidimensional experiments that transfer methyl magnetization to assigned backbone resonances (Gardner et al. 1996; Tugarinov and Kay 2003). This approach has been successfully applied to well-behaved proteins up to ~80 kDa (Tugarinov and Kay 2003). However, obtaining the prerequisite backbone assignments in the presence of extensive chemical shift overlap and/or exchange line-broadening can be a challenging and time consuming endeavor (if not impossible), even if the methyl-TROSY correlation spectrum is well resolved. In such cases strategies that eliminate the need for backbone assignments are essential.
A different class of methods makes use of the known 3D structure of the protein under investigation (obtained either directly by crystallography or indirectly by homology modeling based on a related crystal structure) combined with tailored experimental techniques to derive methyl group resonance assignments. This is the approach that has been used to date for all NMR studies of very large systems that cannot be dissected into small isolated domains from which resonance assignments can be directly transferred to the intact system (Sprangers and Kay 2007; Gelis et al. 2007; Godoy-Ruiz et al. 2010; Velyvis et al. 2007). If the protein contains a lanthanide binding site, resonance assignments can be obtained by minimizing the difference between experimental and predicted pseudocontact shifts (PCS) (John et al. 2007). The PCS-based assignment strategy, however, requires prior knowledge of the magnetic susceptibility tensor, which in turn requires known assignments for a subset of cross-peaks or, alternatively, combined analysis of PCS, PRE and RDC data derived from 1H–15N correlation spectroscopy (Pintacuda et al. 2004). More commonly, methyl–methyl nuclear Overhauser enhancement (NOE) experiments have been used in combination with site-directed mutagenesis (Sprangers and Kay 2007; Gelis et al. 2007) or chemical shift prediction programs (Xu et al. 2009) to assign methyl groups in proteins. Due to the short-range nature of the NOE, however, these methods work best for ordered regions with a high density of methyl groups; when the methyl groups are sparse, such as on the protein surface, this approach either fails or requires the generation of a large number of site-directed mutants.
Here, we show that analysis of paramagnetic relaxation enhancements (PRE) induced by nitroxide spin-labels, in combination with an available 3D structure, provides a powerful and robust method for methyl group assignment. Since PREs can be observed out to distances of ~25 Å from a nitroxide spin label (and up to ~35 Å for other paramagnetic centers such as Mn2+), the number of cross-peaks that can be potentially assigned from a single mutation is dramatically increased compared to assignment by mutagenesis/NOE techniques, and further permits assignment of isolated methyl groups for which there are no detectable NOEs.
Briefly, the assignment strategy consists in producing one or more NMR samples of the target protein with a nitroxide spin label introduced at a single position, measuring the 1HM-Γ2 PRE rates induced by the spin-label, and then searching for the assignment that results in the best match between predicted and experimental PREs. The obtained resonance assignments are then refined by comparison of experimentally observed methyl–methyl NOE patterns to the protein structure. The approach is tested on the 27 kDa N-terminal domain of E. coli Enzyme I (EIN) using R1p as the nitroxide spin-label (Fig. 1). When introduced at a surface α-helical site, the conformational space available to the R1p side chain is highly restricted compared to commonly used nitroxide spin-labels, thereby greatly simplifying the interpretation of PRE data (Fawzi et al. 2011). Since R1p is conjugated to the protein via an engineered surface exposed Cys residue involving a disulfide bond, all native Cys residues must be removed by mutagenesis to ensure that spin-labeling occurs at the single engineered site. In cases where such modifications result in either significantly reduced stability or inability of the protein to fold correctly, alternative spin-labeling strategies involving unnatural amino acids (Fleissner et al. 2009) can be employed.
Fig 1.
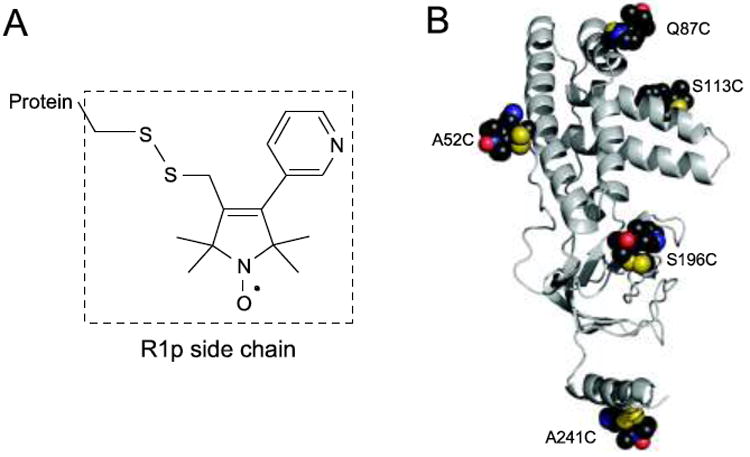
a Chemical structure of the R1p paramagnetic side chain. b 3D structure of EIN showing the positions of the five spin-labeling sites
Materials and methods
Sample preparation
A list of the EIN samples used is provided in Table 1. EIN was expressed and purified as described previously (Garrett et al. 1997). AILMV, AILV and ILV labeled samples, as well as single residue labeled samples, were prepared using appropriate isotope-enrichment of M9 minimal medium according to previously published protocols (Godoy-Ruiz et al. 2010; Gelis et al. 2007; Tugarinov et al. 2006; Ayala et al. 2009). EIN mutant genes (A52C, Q87C, S113C, S196C and A241C) were constructed using the Quik-Change (Stratagene) kit. The reducing agent dithiothreitol (DTT) was present at all times during purification of the EIN mutant proteins to maintain the engineered surface cysteines in the reduced state. DTT was removed by passage through a desalting column immediately prior to incubating the mutant EIN proteins for 1 h at room temperature with a fivefold excess of the R1p precursor (Fawzi et al. 2011). Excess R1p precursor was subsequently removed by an additional desalting step. The reaction was tested for completion by liquid chromatography/mass spectrometry. Diamagnetic samples for PRE measurements were prepared by adding 2 mM ascorbic acid to the corresponding paramagnetic samples.
Table 1.
List of NMR samples used in the present study
| Sample | Labeling | Buffer | Construct | NMR experiment |
|---|---|---|---|---|
| A1–A5a | AILMV | 99.99% 2H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~0.2 mM EIN-R1p | PRE experiment |
| B | AILV | 99.99% 2H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~1 mM WT EIN | 1H–13C HMQC-NOESY-HMQC |
| C | Uniformly 15N + ILV (linearized spin systems) | 5% 2H2O/95% H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~1 mM WT EIN | HC(CO)NH-TOCSY |
| D | A | 99.99% 2H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~0.2 mM WT EIN | 1H–13C HMQC |
| E | I | 99.99% 2H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~0.2 mM WT EIN | 1H–13C HMQC |
| F | M | 99.99% 2H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~0.2 mM WT EIN | 1H–13C HMQC |
| G | LV | 99.99% 2H2O, 100 mM NaCl, 0.1× EDTA-free complete protease inhibitor (Roche), and 20 mM Tris-pH 7.4 | ~0.2 mM WT EIN | 1H–13C HMQC |
All samples are in a uniform 2H, 12C and 14N background unless otherwise specified. Methyl-bearing side chains with 1H–13C labeled methyl groups are listed in the second column
The five samples correspond to R1p conjugated to surface engineered cysteines at A52C, Q87C, S113C, S196C and A241C
NMR spectroscopy
All NMR data were recorded at 37°C on a Bruker Avance 600 MHz spectrometer equipped with a triple resonance z-gradient cryoprobe. Transverse 1HM-Γ2 PRE rates were obtained from the differences in transverse relaxation rates (1HM-R2) between the paramagnetic and corresponding diamagnetic samples (Clore and Iwahara 2009) using the pulse scheme shown in Fig. 2. Six relaxation delays (T = 6, 15, 25, 36, 50 and 68 ms) were acquired in an interleaved manner, and the observed decays in cross-peak intensity, as a function of the delay T, were fit to a two-parameter single exponential function to obtain 1HM-R2. In the diamagnetic spectra, chemical shift changes up to ~0.2 ppm were observed both in the 1H and 13C dimensions as a consequence of the mutations and subsequent incorporation of the spin-label. However, these cross-peaks are completely broadened out in the paramagnetic spectra. For these cross-peaks (as well as for all the other broadened out peaks) the experimental PRE rate was set to the highest measured PRE rate for that particular mutant plus 10 s−1.
Fig. 2.
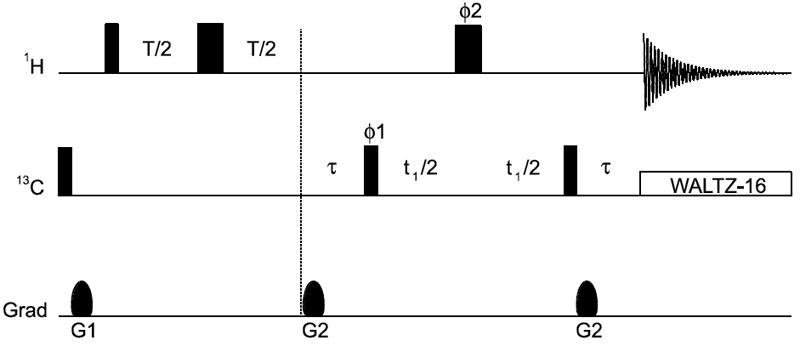
1H–13C HMQC-based pulse scheme used for measuring 1HM-R2 rates. Narrow and wide rectangular pulses are applied with flip angles of 90° and 180°, respectively, along the x-axis unless indicated otherwise. T is the relaxation delay. The delay τ is set to 4 ms. Quadrature detection in t1 is obtained using States-TPPI (Marion et al. 1989) of phase ϕ1. The phase cycling employed is: ϕ1 = (x, −x); ϕ2 = 2(x), 2(y), 2(−x), 2(−y); receiver phase = x, −x, −x, x. The duration and strength of the gradients are as follows: G1, 1 ms, 15 G/cm; G2, 0.5 ms, 20 G/cm
Methyl-methyl NOE data were obtained by acquiring a 4D 1H–13C HMQC-NOE-HMQC spectrum (Vuister et al. 1993) on sample B (Table 1) using a data matrix of 20*(t1) × 22*(t2) × 20*(t3) × 1024(t4)* complex points with acquisition times of 8, 27, 8 and 122 ms, sweep widths of 16.2, 1.4, 18.7 and 1.0 ppm, and carrier positions of 18.7, 1.0, 18.7 and 1.0 ppm in the F1(13C), F2(1H), F3(13C) and F4(1H) dimensions, respectively (total duration 90 h). A 4D HC(CO)NH-TOCSY experiment (Gardner et al. 1996) (total duration 120 h) was also acquired on sample C (Table 1).
PRE Q-factor
The PRE Q-factor is a measure of the agreement between observed and calculated values of Γ2 and is given by (Iwahara et al. 2004):
| (1) |
where and are the observed and calculated transverse PRE rates, respectively, for residue i. Lower Q-factors correspond to better agreement between experimental and calculated PRE data.
Algorithm for matching assignments to the experimental PRE data
The search for the best assignment satisfying the experimental PRE was accomplished by a Metropolis Monte-Carlo optimization algorithm in which randomly chosen pairs of (initially arbitrary) assignments are iteratively swapped to minimize the PRE energy (EPRE) given by:
| (2) |
where is the observed transverse PRE rate for cross-peak i, and the corresponding calculated rate for residue i, in mutant N; α is a constant with value unity and units of kBT*s2, where kB is the Boltzman constant and T* the (arbitrary) reduced temperature.
For Leu and Val residues, a chemical shift potential (ECS) was also included in the target function:
| (3) |
| (4) |
where k is a force constant set to 106 kBT*ppm−2, is the observed chemical shift for cross-peak i, and δ̅C and σC are the average 13C chemical-shift and corresponding standard deviation, respectively, for the methyl groups of Leu and Val in the Biological Magnetic Resonance Data Bank (Ulrich et al. 2008). (For Leu, δ̅C = 24.4 ppm and σC = 1.71 ppm; for Val, δ̅C = 21.4 ppm and σC = 1.56 ppm). During the optimization process, the probability P of accepting swaps is evaluated using the Boltzmann criteria (Metropolis et al. 1953):
| (5) |
where ΔE is the energy difference between the trial and current states, and β is increased geometrically (i.e. temperature is decreased) from 10−3 to 0.5 (kBT*)−1 over ~6 million minimization steps. Finally, statistics on the resulting assignments are collected by running an additional 1 million steps at constant β. The calculation converges in a few seconds on a standard desktop computer.
The software used for the PRE-based methyl group assignment is available for download at http://spin.niddk.nih.gov/clore.
Fitting nitroxide positions to the experimental PRE data
The position, or ensemble of positions, of the nitroxide spin label that best fits the PRE observables was computed by comparing predicted and observed 1HM-Γ2 PREs using the coordinates of the structure of EIN (Garrett et al. 1997). 1HM-Γ2 PREs were back-calculated in Xplor-NIH (Schwieters et al. 2003) with either a single conformer or a three-conformer ensemble for the spin label together with the Solomon-Bloembergen Model Free (SBMF) representation (Iwahara et al. 2004). The label and immediately adjacent side chain coordinate positions were optimized in torsion angle space by simulated annealing to minimize the difference between observed and calculated 1HM-Γ2 PREs, as previously described (Iwahara et al. 2004). In addition to the PRE pseudo-potential, the target function includes stereochemical restraints, a quartic van der Waals repulsion term to prevent atomic overlap (note atomic overlap between spin labels in the three-conformer ensemble is allowed since the ensemble represents a distribution of states), and a multidimensional torsion angle database potential of mean force (Clore and Kuszewski 2002). The PRE correlation time τc value was allowed to vary between 7 and 15 ns during the course of simulated annealing as described previously (Iwahara et al. 2004). An optimized τc value of 11 ns was obtained for all the calculations.
Ensemble refinement of transient intra- and inter-molecular interactions sampled by the C-terminal helix
Calculations were performed in Xplor-NIH using an ensemble representation for the C-terminal fragment (residues 230–249) of EIN together with the Solomon-Bloembergen (SB) representation (Iwahara et al. 2004). The ensemble size was set to 5 and 10 for the intra- and the inter-molecular calculations, respectively. When simulating inter-molecular effects, ten copies of the isolated C-terminal fragment were simulated together with one copy of full length EIN that was kept fixed during refinement; in addition, only the experimental PREs for residues 54–144 were included in the inter-molecular calculations. The ensemble simulated annealing refinements were carried out according to previously published protocols (Tang et al. 2006).
Results and discussion
1HM-Γ2 PRE rates were measured on five single Cys mutants of perdeuterated EIN in which the methyl groups of Ala, Ile (δ1), Leu, Met and Val were protonated and 13C labeled as described in “Materials and methods”. The five mutations (A52C, Q87C, S113C, S196C and A241C) were designed to optimally sample the protein structure (Fig. 1). Initial sets of predicted PREs were back-calculated from the coordinates of the EIN structure (PDB code: 1EZA) (Garrett et al. 1997) using the Solomon-Bloembergen equation (Solomon and Bloembergen 1956) (with τc = 10 ns). For this calculation, the starting conformation of the R1p sidechain bearing the paramagnetic center was modeled on the basis of the X-ray structure of an R1p analogue introduced into T4 lysozyme (PDB code: 1ZUR; Fleissner and Hubbell, unpublished results).
Resonance assignment was achieved using a three-step protocol that simultaneously searches for the best assignment using the algorithm described in “Materials and methods” and refines the nitroxide position and value of τc. To reduce the number of ambiguous results, the methyl resonances were grouped into four different classes based on their residue types (Ala, Ile, Met and Leu/Val) (Fig. 3), and the search steps were performed separately for each class.
Fig. 3.
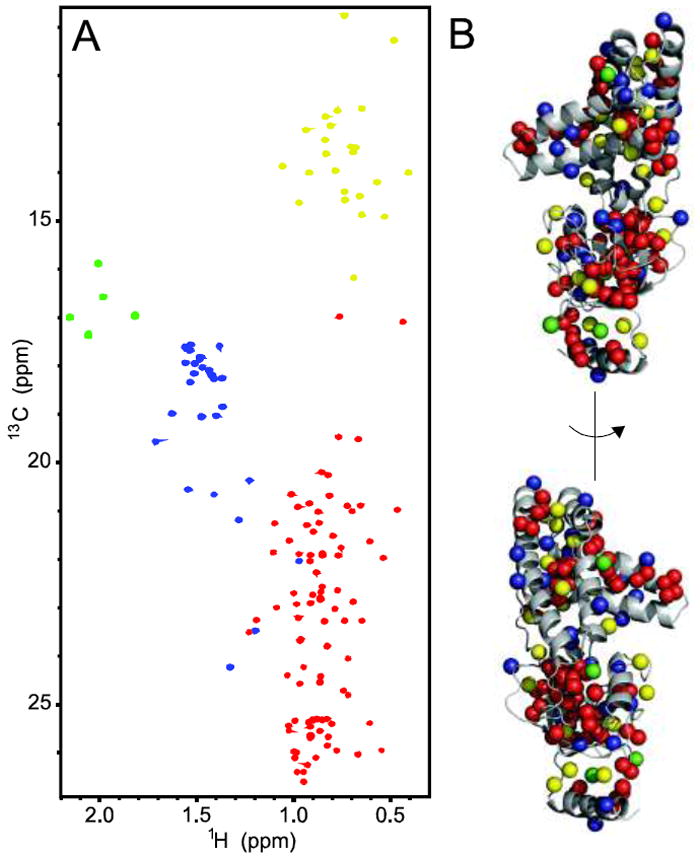
a 1H–13C HMQC spectrum of U-[2H,12C], Ala-[13CH3], Ile-δ1-[13CH3], Met-[13CH3], Val-Leu-[13CH3, 12CD3] EIN. b Two views of the 3D structure of EIN with methyl groups displayed as spheres. The color coding of the methyl groups used in both panels is as follows: Ala blue; Ile yellow; Met green; Leu/Val red. Classification of the methyl cross-peaks based on residue type was obtained by running methyl-TROSY experiments on samples D–G (Table 1)
In step 1, initial assignments were obtained for Ala, Ile and Met (Supplementary Table S1). Subsequently, the experimental PREs for methyl groups that were assigned with certainty and are located at least 5 Å away from any other methyl group of the same residue type, were selected to refine the nitroxide positions and τc value by simulated annealing in Xplor-NIH (Schwieters et al. 2003) as described by Iwahara et al. (2004). In step 2 the resulting optimized parameters were used to create a new set of predicted PREs that was used for a second assignment run. At this stage all the Ala, Ile and Met methyl groups assigned with certainty were selected for further refinement of the nitroxide positions and τc value in Xplor-NIH. Finally, in step 3 assignments were obtained for all the methyl groups of EIN. These resonance assignments were then verified by comparison of the experimentally observed methyl–methyl NOE patterns in a 4D 13C/13C-separated HMQC-NOE-HMQC spectrum (Vuister et al. 1993) to the 3D structure of EIN.
The combined PRE/NOE approach afforded complete assignments of Ala, Ile (δ1) and Met methyl groups (Table S1). In the case of Val/Leu residues, sequence- and stereo-specific assignments were obtained for 79 out of 88 cross-peaks (Supplementary Table S1). For the nine unassigned cross-peaks, the search for the best match between predicted and experimental PREs did not result in a unique answer and analysis of the methyl–methyl NOEs was not sufficient to resolve the ambiguities. PRE and NOE data are in complete disagreement for only 20 out of 140 cross-peaks (Supplementary Table S1). Small fluctuations in methyl-bearing side chain and spin label positions are unlikely to be the origin of these discrepancies. Indeed, although a better fit of the nitroxide positions to the experimental PRE data can be obtained by accounting for side chain and spin label flexibility (Fig. 4), no dramatic changes in the PRE-based assignments are observed and the 20 cross-peak assignment discrepancies remain unresolved.
Fig. 4.
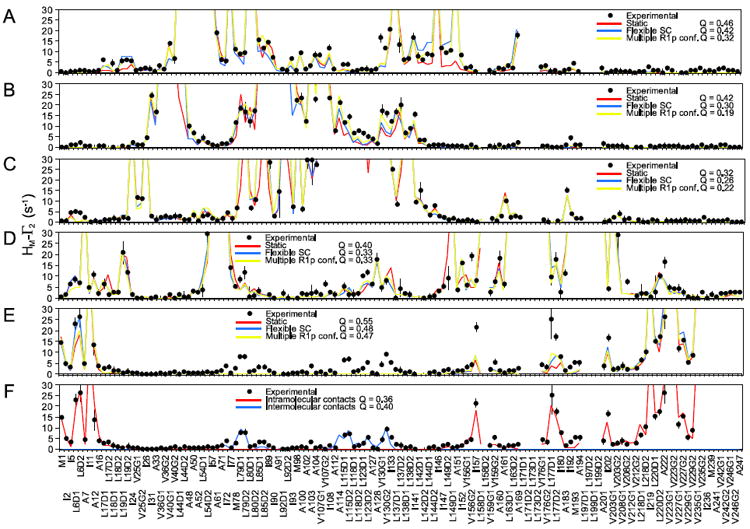
Fitting the experimental PRE data for the a A52C, b Q87C, c S113C, d S196C, and e A241C spin-labeled mutants of EIN. The experimental PRE profiles are displayed as closed black circles (with error bars equal to 1 standard deviation). PREs too large (>30 s−1) to be accurately measured are plotted at the top of the charts. PRE profiles back-calculated from the structure of EIN are shown as solid lines: red, side chains fixed during the fitting; blue, side chains allowed to move; yellow, side chains allowed to move and R1p treated as a three-conformer ensemble. For every fit the corresponding PRE Q-factor is also reported. f Fitting of the experimental PRE data for the A241C mutant that accounts for the effect of transient intra- and inter-molecular interactions (see “Materials and methods”). A listing of the experimental PRE rates is provided in Supplementary Table S2
A more careful analysis reveals that the 20 problematic methyl groups cluster in two specific regions of the protein structure (Fig. 5). The largest cluster is located close to the A241C mutation situated in the C-terminal helix of EIN. This mutation yields poor agreement between experimental and back-calculated PREs (Fig. 4e), which is partially restored only if the C-terminal helix is refined as an ensemble of five equally weighted conformers that are allowed to move as a rigid body during the calculation (Figs. 4f, 5a). Discrepancies, however, between experimental and back-calculated PRE data persist in the HPr binding site, where the other group of incorrect assignments clusters (Fig. 5). The large PREs observed in this region cannot be explained by intramolecular contacts with the C-terminal helix which is located ~50 Å away. These PREs can therefore only be accounted for by considering the existence of weak, highly transient intermolecular complexes formed by interaction of the C-terminal helix of one molecule of EIN with the HPr binding site of a second EIN molecule (Figs. 4f, 5b). This finding is not too surprising since transient, non-specific interactions involving non-physiological ligands have been observed in the active sites of several well-characterized proteins (Bernini et al. 2009). By incorporating information about transient states and intermolecular interactions sampled by the C-terminal helix into the EIN structural model, a new set of predicted PREs is obtained for the A241C mutant that accounts for most of the discrepancies between the PRE and NOE data (Supplementary Table S1), confirming that transient, sparsely-populated intra- and inter-molecular interactions are indeed the main source of error in the PRE-based resonance assignment. These effects, however, are completely undetected by NOE data (Fig. 6) that are much less sensitive to the existence of sparsely-populated states (Clore 2011), thus providing a powerful validation tool for PRE-based resonance assignment. It is is worth noting that a few small discrepancies between experimental and back-calculated PREs are also observed for the A52C (Fig. 4a) and S196C (Fig. 4e) mutants. The fact that these anomalous data are observed for different methyl groups in the two mutants suggests that they arise for protein–protein interactions rather than from specific interations of the nitroxide with the protein. However, such small discrepancies are limited to a few methyl groups and do not affect the overall assignment. Thus, the transient intermolecular interactions sampled by these mutants were not investigated further.
Fig. 5.
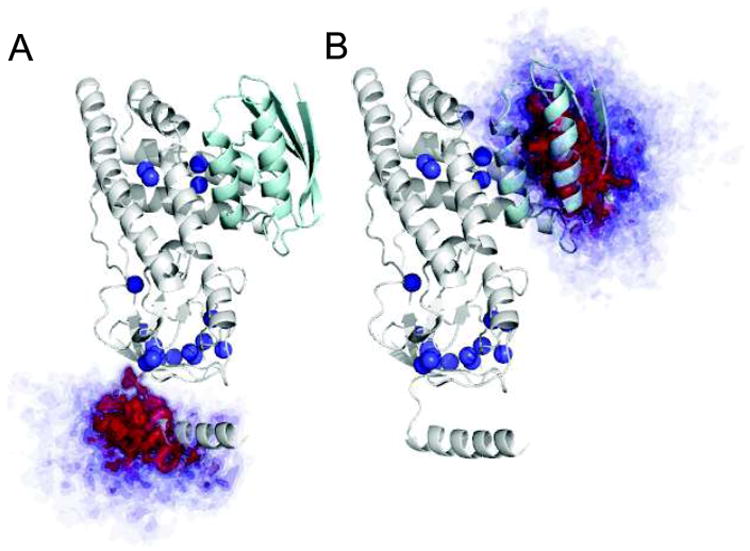
Atomic probability density maps (Schwieters and Clore 2002) showing the conformational space sampled by the C-terminal fragment of EIN in PRE-driven ensemble refinements. The atomic probability maps are derived from ten independent simulated annealing calculations, and plotted at a threshold ranging from 10 (transparent blue) to 50% (opaque red) of maximum. These plots clearly show that the C-terminal fragment of EIN a samples a quite extensive region of conformational space and b is also involved in transient inter-molecular contacts localized exclusively in the HPr binding site. Methyl groups for which discrepancies between PRE and NOE data are observed are displayed as blue spheres. The structure of EIN complexed to HPr (Garrett et al. 1999) is shown as ribbon with EIN in gray and HPr in cyan
Fig. 6.
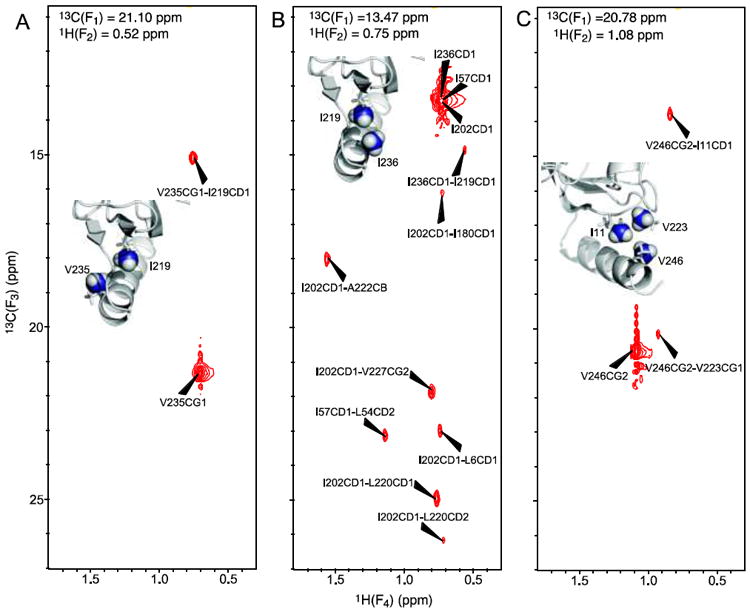
Selection of 13C(F3)-1H(F4) planes of the 4D methyl–methyl HMQC-NOE-HMQC spectrum showing that the methyl–methyl NOEs for the C-terminal helix are fully consistent with the experimental structure of EIN. The planes display methyl–methyl NOEs originating from a V235, b I236, and c V246. In each panel, the 13C(F1)/1H(F2) chemical shifts are indicated
Concluding remarks
We have presented a new method that makes use of combined PRE and NOE data to obtain reliable, stereo- and sequence-specific 1H/13C assignments of methyl groups in proteins of known three-dimensional structure. Most importantly, no previous knowledge of either backbone or side chain assignments is needed, allowing resonance assignment even in situations where connectivities to the backbone are difficult to establish or the backbone assignments are incomplete.
Due to the insensitivity of the NOE to the existence of sparsely-populated transient states (Clore 2011), NOE data provide a powerful refinement and verification tool for assignments obtained by PRE. Thus, when discrepancies between PRE and NOE data are present, the correct assignment must fully account for the observed NOEs, as the discrepancies may well arise from the impact of sparsely-populated states on the PRE. If the discrepancy cannot be readily rationalized, the conservative approach would involve discarding cases where the PRE and NOE data conflict. It is also important to note that NOE data either alone or combined with chemical shift predictions are not sufficient to obtain comprehensive and reliable methyl resonance assignments of the EIN methyl-TROSY spectrum. Indeed, in the case of EIN, the recently published algorithm of Xu et al (2009) that combines methyl chemical shift prediction with methyl–methyl NOE data afforded correct assignments for only 32 out of 140 methyl cross-peaks (Supplementary Table S1). This is due to several factors including the limitations in structure-based chemical shift calculations, the intrinsically low resolution of the 4D 13C/13C-separated HMQC-NOE-HMQC spectrum that hampers, in the absence of additional information, correct discrimination of NOEs originating from diagonal peaks with very similar chemical shifts, and the short range distance nature of the NOE.
Although we demonstrated that the PRE/NOE-based methyl group assignment method works well for the relatively small 27 kDa N-terminal domain of enzyme I, the NMR experiments used in this work are still highly effective for proteins with molecular weight in excess of 300 kDa (Sprangers and Kay 2007; Religa et al. 2010; Gelis et al. 2007). Further, accurate conversion of 1HM-Γ2 PRE data into distance restraints has been recently shown to be feasible for large protein complexes of similar size (Gelis et al. 2007; Religa et al. 2010), so that the method is well suited for methyl group assignment in extremely large systems not accessible to conventional assignment techniques.
From the present work, one can roughly estimate that approximately one spin-label per ~6 kDa is required to obtain near complete and reliable assignments of methyl resonances. This estimate, however, is strongly dependent on protein shape, distribution of methyl groups and the nature of the paramagnetic center. In particular, for larger systems the use of stronger paramagnetic groups (such as Mn2+ or Gd3+) may help reduce the number of paramagnetic labels required for assignments. Further, PRE effects on methyl groups located at the center of a spherical globular domain of molecular weight in excess of ~ 150 kDa may be too weak to measure accurately. In such cases the method would be limited to the assignment of methyl groups within ~30–35 Å of the protein surface.
Supplementary Material
Acknowledgments
We thank Drs. Mark Fleissner, Kálmán Hideg, Tamás Kálai and Wayne Hubbell for generously providing the reagent for generating the R1p paramagnetic side chain. This work was supported by funds from the Intramural Program of the NIH, NIDDK, and the Intramural AIDS Targeted Antiviral Program of the Office of the Director of the NIH (to G.M.C.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10858-011-9559-4) contains supplementary material, which is available to authorized users.
References
- Ayala I, Sounier R, Use N, Gans P, Boisbouvier J. An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J Biomol NMR. 2009;43:111–119. doi: 10.1007/s10858-008-9294-7. [DOI] [PubMed] [Google Scholar]
- Bernini A, Venditti V, Spiga O, Niccolai N. Probing protein surface accessibility with solvent and paramagnetic molecules. Prog Nucl Magn Reson Spectrosc. 2009;54:278–289. [Google Scholar]
- Clore GM. Exploring sparsely populated states of macromolecules by diamagnetic and paramagnetic NMR relaxation. Protein Sci. 2011;20:229–246. doi: 10.1002/pro.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Kuszewski J. χ1 rotamer populations and angles of mobile surface side chains are accurately predicted by a torsion angle database potential of mean force. J Am Chem Soc. 2002;124:2866–2867. doi: 10.1021/ja017712p. [DOI] [PubMed] [Google Scholar]
- Fawzi NL, Fleissner MR, Anthis NJ, Kálai T, Hideg K, Hubbell WL, Clore GM. A rigid, small, cysteine-reactive nitroxide spin label simplifies the quantitative analysis of PRE data. J Biomol NMR. 2011 doi: 10.1007/s10858-011-9545-x. 10:1007/s10858-011-9545-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleissner MR, Brustad EM, Kalai T, Altenbach C, Cascio D, Peters FB, Hideg K, Peuker S, Schultz PG, Hubbell WL. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc Natl Acad Sci USA. 2009;106:21637–21642. doi: 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner KH, Konrat R, Rosen MK, Kay LE. An (H)C(CO)NH-TOCSY pulse scheme for sequential assignment of protonated methyl groups in otherwise deuterated (15)N, (13)C-labeled proteins. J Biomol NMR. 1996;8:351–356. doi: 10.1007/BF00410333. [DOI] [PubMed] [Google Scholar]
- Garrett DS, Seok YJ, Liao DI, Peterkofsky A, Gronenborn AM, Clore GM. Solution structure of the 30 kDa N-terminal domain of enzyme I of the Escherichia coli phosphoenolpyruvate: sugar phosphotransferase system by multidimensional NMR. Biochemistry. 1997;36:2517–2530. doi: 10.1021/bi962924y. [DOI] [PubMed] [Google Scholar]
- Garrett DS, Seok YJ, Peterkofsky A, Gronenborn AM, Clore GM. Solution structure of the 40,000 Mr phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr. Nat Struct Biol. 1999;6:166–173. doi: 10.1038/5854. [DOI] [PubMed] [Google Scholar]
- Gelis I, Bonvin AM, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy-Ruiz R, Guo C, Tugarinov V. Alanine methyl groups as NMR probes of molecular structure and dynamics in high-molecular-weight proteins. J Am Chem Soc. 2010;132:18340–18350. doi: 10.1021/ja1083656. [DOI] [PubMed] [Google Scholar]
- Gross JD, Gelev VM, Wagner G. A sensitive and robust method for obtaining intermolecular NOEs between side chains in large protein complexes. J Biomol NMR. 2003;25:235–242. doi: 10.1023/a:1022890112109. [DOI] [PubMed] [Google Scholar]
- Hajduk P, Augeri D, Mack J, Mendoza R, Yang J, Betz S, Fesik S. NMR-based screening of proteins containing 13C-labeled methyl groups. J Am Chem Soc. 2000;122:7898–7904. [Google Scholar]
- Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against 1H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- John M, Schmitz C, Park AY, Dixon NE, Huber T, Otting G. Sequence-specific and stereospecific assignment of methyl groups using paramagnetic lanthanides. J Am Chem Soc. 2007;129:13749–13757. doi: 10.1021/ja0744753. [DOI] [PubMed] [Google Scholar]
- Marion D, Ikura M, Tschudin R, Bax A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J Magn Reson. 1989;85:393–399. [Google Scholar]
- Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equations of state calculations by fast computing machines. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintacuda G, Keniry MA, Huber T, Park AY, Dixon NE, Otting G. Fast structure-based assignment of 15N HSQC spectra of selectively 15N-labeled paramagnetic proteins. J Am Chem Soc. 2004;126:2963–2970. doi: 10.1021/ja039339m. [DOI] [PubMed] [Google Scholar]
- Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science. 2010;328:98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Clore GM. Reweighted atomic densities to represent ensembles of NMR structures. J Biomol NMR. 2002;23:221–225. doi: 10.1023/a:1019875223132. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Suh JY, Grishaev A, Ghirlando R, Takayama Y, Clore GM. Solution structure of the 128 kDa enzyme I dimer from Escherichia coli and its 146 kDa complex with HPr using residual dipolar couplings and small- and wide-angle X-ray scattering. J Am Chem Soc. 2010;132:13026–13045. doi: 10.1021/ja105485b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon I, Bloembergen N. Nuclear magnetic interactions in the HF molecule. J Chem Phys. 1956;25:261–266. [Google Scholar]
- Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- Tang C, Iwahara J, Clore GM. Visualization of transient encounter complexes in protein–protein association. Nature. 2006;444:383–386. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced 1H-13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Choy WY, Orekhov VY, Kay LE. Solution NMR-derived global fold of a monomeric 82-kDa enzyme. Proc Natl Acad Sci USA. 2005;102:622–627. doi: 10.1073/pnas.0407792102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velyvis A, Yang YR, Schachman HK, Kay LE. A solution NMR study showing that active site ligands and nucleotides directly perturb the allosteric equilibrium in aspartate transcarbamoylase. Proc Natl Acad Sci USA. 2007;104:8815–8820. doi: 10.1073/pnas.0703347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuister GM, Clore GM, Gronenborn AM, Powers R, Garrett DS, Tschudin R, Bax A. Increased resolution and improved spectral quality in four-dimentional 13C/13C separated HMQC-NOESY-HMQC spectra using pulsed field gradients. J Magn Reson Ser B. 1993;101:210–213. [Google Scholar]
- Xu Y, Liu M, Simpson PJ, Isaacson R, Cota E, Marchant J, Yang D, Zhang X, Freemont P, Matthews S. Automated assignment in selectively methyl-labeled proteins. J Am Chem Soc. 2009;131:9480–9481. doi: 10.1021/ja9020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.