

A high-resolution X-ray crystallographic study of the effects of solvent deuteration on the crystallization of proteinase K shows negligibly small degradations of the crystals owing to solvent deuteration and small structural differences between nondeuterated and deuterated crystals of proteinase K.
Keywords: deuteration, polyethylene glycol, proteinase K
Abstract
Deuteration of macromolecules is an important technique in neutron protein crystallography. Solvent deuteration of protein crystals is carried out by replacing water (H2O) with heavy water (D2O) prior to neutron diffraction experiments in order to diminish background noise. The effects of solvent deuteration on the crystallization of proteinase K (PK) with polyethylene glycol as a precipitant were investigated using high-resolution X-ray crystallography. In previous studies, eight NO3 − anions were included in the PK crystal unit cell grown in NaNO3 solution. In this study, however, the PK crystal structure did not contain NO3 − anions; consequently, distortions of amino acids arising from the presence of NO3 − anions were avoided in the present crystal structures. High-resolution (1.1 Å) X-ray diffraction studies showed that the degradation of PK crystals induced by solvent deuteration was so small that this degradation would be negligible for the purpose of neutron protein crystallography experiments at medium resolution. Comparison of the nonhydrogen structures of nondeuterated and deuterated crystal structures demonstrated very small structural differences. Moreover, a positive correlation between the root-mean-squared differences and B factors indicated that no systematic difference existed.
1. Introduction
Replacement of hydrogen with deuterium in crystals is an important technique in neutron crystallography. H atoms produce a much larger incoherent neutron scattering effect than other atoms such as deuterium. The replacement of hydrogen with deuterium diminishes background noise significantly, thereby providing data with better signal-to-noise ratios. Ideally, H atoms in both the protein and the solvent should be replaced with deuterium. Although perdeuteration of the protein is becoming more commonplace, it is still rather difficult to accomplish. More commonly, only a deuterated solvent is used (Blakeley et al., 2008 ▶; Niimura et al., 2006 ▶). Solvent deuteration is carried out by preparing all reagent solutions in heavy water (D2O). Previous studies have reported that the effects of deuteration on protein structures are usually small when investigating the three-dimensional structures of proteins (Meilleur et al., 2005 ▶; Artero et al., 2005 ▶; Di Costanzo et al., 2007 ▶; Blum et al., 2010 ▶). However, some studies have reported that deuteration of the solvent may sometimes have an effect on the crystallization conditions of proteins and/or their crystal structures. Haloalkane dehalogenase, which was crystallized using ammonium sulfate as a precipitant and investigated at 1.5 Å resolution, exhibited unexpected structural changes in the deuterated crystal (Liu et al., 2007 ▶). In our previous study of the crystallization of dissimilatory sulfite reductase D using ammonium sulfate, the optimal crystallization conditions in nondeuterated solution differed from those in deuterated solution (Chatake et al., 2003 ▶). These changes may be the result of a shift in the pH/pD on deuteration of the crystallization solution. In particular, the pH of a high-salt solution such as ammonium sulfate is lowered considerably on deuteration. The crystallization of proteins in deuterated solutions containing salts is therefore adjusted in pD in order to maintain good crystallization conditions and avoid potential structural changes. On the other hand, polyethylene glycol (PEG) does not significantly change in pH on deuteration. We thus expected that when PEG was used as a precipitant and the pH/pD was carefully adjusted during crystallization, the deuterated protein crystals would be identical to the nondeuterated crystals, except that D/H exchange would be obtained, using the same crystallization conditions.
Proteinase K (PK) was used in our present study. PK is a powerful protease which has high activity in various environments (Sweeney & Walker, 1993 ▶; Ebeling et al., 1974 ▶). It exhibits 80% of its maximum protease activity over a wide pH range (4.0–12.0) and at high temperature (333 K) in a buffer solution containing Ca2+ cations. It is possible to collect high-resolution X-ray diffraction data from a single crystal of PK obtained from Tritirachium album (Betzel et al., 2001 ▶). Crystallization of PK was successfully carried out using NaNO3 as a precipitant. Crystallization of PK in deuterated solution was also carried out and a neutron diffraction experiment was successfully conducted using LADI-III at ILL (Gardberg et al., 2009 ▶). Crystallization was accomplished under high-salt conditions; thus, several NO3 − anions were found to be contained within the PK crystals and the presence of NO3 − ions distorted some amino-acid side chains. On the other hand, PK crystals could be obtained from solutions comprising cacodylate buffer, calcium acetate (CaAc2) and polyethylene glycol 8000 (PEG 8000). In the present study, the crystallization of PK using PEG 8000 was performed at various degrees of solvent deuteration and X-ray crystallographic analysis at 1.1 Å resolution confirmed that deuteration had no effect on crystal quality or crystal structure in this case.
2. Materials and methods
PK was purchased from Merck Co. and used without further purification. Crystals were obtained using the sitting-drop vapour-diffusion technique. Partially deuterated protein solutions were prepared by mixing nondeuterated and deuterated protein solutions. 10 mg PK powder was dissolved in 0.5 ml H2O or 0.5 ml D2O. Subsequently, 20 mg ml−1 PK in 100% H2O and 20 mg ml−1 PK in 100% D2O were mixed in 1:3 and 1:1 ratios to obtain partially deuterated 20 mg ml−1 PK solutions (75% H2O/25% D2O and 50% H2O/50% D2O, respectively). Reservoir solutions were prepared in a similar manner. Initially, 100% H2O and 100% D2O reservoir solutions containing 100 mM sodium cacodylate, 100 mM CaAc2 and 9.0% PEG 8000 were prepared. The pH (pD) values of the two reservoirs were adjusted to 6.8 prior to diluting the solution to the final concentration. Concentrated hydrochloric acid was diluted to 1 M with H2O and D2O and this 1 M HCl (in H2O or D2O) was added until a pH meter reading of pH/pD 6.8 was obtained. Partially deuterated reservoir solutions were prepared by mixing the two reservoir solutions. Reservoir solution was poured into a well of a crystal plate (700 µl per well) and a 20 µl droplet was placed on a sitting bridge. The 20 µl droplets were prepared by mixing 10 µl PK solution and 10 µl reservoir solution. The pH (pD) of each droplet was measured using a pH meter, confirming that the pH (pD) values of the four types of droplets were maintained at 6.7. This small decrease in pH (pD) was derived from the unbuffered PK solution. The crystal plate was then sealed with sealing tape and stored in an incubator at 298 K. To investigate the effects of solvent deuteration on the PK crystal properties, PK crystallizations were carried out in four H2O/D2O solutions (100% H2O, 75% H2O/25% D2O, 50% H2O/50% D2O and 100% D2O).
X-ray diffraction experiments were carried out using synchrotron radiation on beamline 5A at the Photon Factory (PF) and beamline 38B1 at SPring-8 (SP8; Okazaki et al., 2008 ▶). The crystals used for X-ray experiments were similar in size (0.5 × 0.5 × 0.5 mm). Crystals obtained from 100% H2O (PK000), 75% H2O/25% D2O (PK025), 50% H2O/50% D2O (PK050) and 100% D2O (PK100) solutions were used for X-ray data collection. The crystals were soaked for 5 s in cryoprotectant solution and were then flash-cooled in N2 gas at 100 K prior to carrying out X-ray diffraction experiments. The cryoprotectant solution was also deuterated. The deuteration of the cryoprotectant solution was matched to that of the crystallization solution for each PK crystal. The cryoprotectant solutions for PK000 and PK100 consisted of 100 mM sodium cacodylate (pH/pD 6.8), 100 mM CaAc2, 9.0% PEG 8000 and 50% glycerol in H2O and D2O, respectively. The cryoprotectant solutions for PK025 and PK050 were prepared by mixing the 100% H2O and 100% D2O cryoprotectant solutions in the same manner as for preparing crystallization solutions. All of the crystals diffracted very well and the effective resolutions of all of the crystals were superior to the diffractometer limit (1.05 Å). Aluminium attenuators were used to control saturated diffractions in lower resolution shells. For each data set, 210 diffraction images were collected using the oscillation method with an interval of 1.0° and an exposure time of 10 s per frame. The diffraction patterns were indexed and integrated to 1.05 Å resolution using the MOSFLM software (Leslie, 1992 ▶) and the intensities were merged using SCALA from the CCP4 software suite (Winn et al., 2011 ▶).
Two crystal structures, PK000 and PK100, were determined. Initial phases were determined by the molecular-replacement method with the AMoRe program (Navaza, 1994 ▶), employing coordinates from the PK structure solved at 0.98 Å resolution by X-ray crystallographic analysis of a crystal obtained from H2O solution using NaNO3 as a precipitant (PDB entry 1ic6; Betzel et al., 2001 ▶). Atomic parameters were refined with the PHENIX program (Adams et al., 2010 ▶) at 1.1 Å resolution and the molecular structures were constructed and modified on a graphics workstation to fit to the 2|F o| − |F c| and |F o| − |F c| electron-density maps using the Coot program (Emsley et al., 2010 ▶). Table 1 ▶ summarizes the data-collection parameters, data processing and data statistics.
Table 1. X-ray experimental statistics and PK structure determinations.
Values in parentheses are for the outermost shell.
| Name | PK000 | PK025 | PK050 | PK100 |
|---|---|---|---|---|
| Crystallization | ||||
| D2O (%) | 0 | 25 | 50 | 100 |
| H2O (%) | 100 | 75 | 50 | 0 |
| X-ray experiment | ||||
| Source | PF 5A | SP8 38B1 | SP8 38B1 | PF 5A |
| Wavelength () | 1.0† | 0.7† | 0.7† | 1.0† |
| Oscillation method | ||||
| Step () | 1.0 | 1.0 | 1.0 | 1.0 |
| Range () | 210.0 | 210.0 | 210.0 | 210.0 |
| Space group | P43212 | P43212 | P43212 | P43212 |
| Unit-cell parameters () | ||||
| a = b | 67.9 | 67.8 | 67.7 | 68.1 |
| c | 102.0 | 101.7 | 101.9 | 102.5 |
| d min () | 1.05 | 1.05 | 1.05 | 1.05 |
| R merge (%) | 4.3 (16.2) | 6.3 (20.7) | 7.0 (23.6) | 8.7 (31.2) |
| Completeness (%) | 99.7 | 99.9 | 100.0 | 99.7 |
| Multiplicity | 12.7 | 14.3 | 14.8 | 14.0 |
| I/(I) | 34.9 | 32.4 | 30.0 | 23.6 |
| B factor‡ (2) | 3.69 | 3.99 | 4.04 | 4.86 |
| Structure determination | ||||
| Resolution () | 501.1 | 251.1 | ||
| R factor (%) | 11.8 (9.1) | 14.7 (14.1) | ||
| R free (%) | 13.2 (10.6) | 16.2 (17.6) | ||
| R.m.s.d. bonds () | 0.009 | 0.008 | ||
| R.m.s.d. angles () | 1.425 | 1.301 | ||
| Solvent molecules | ||||
| H2O | 380 | 376 | ||
| Ca2+ | 2 | 2 | ||
| Glycerol | 1 | 1 | ||
| PDB code | 3aj8 | 3aj9 | ||
Using an aluminium attenuator.
These values were estimated from the Wilson plot.
3. Results and discussion
3.1. Effect of deuteration on crystalline parameters
The four types of crystals obtained from differently deuterated solutions looked alike. The nucleation rates, growth rates and final sizes of the crystals were all similar. The crystals could not be distinguished using optical microscopy. However, some differences were observed using X-ray diffraction experiments. The PK100 (100% D2O) unit cell was larger (ΔV = +1.5%) than the unit cells of the other crystals. Additionally, R iso indicated the presence of structural differences between PK000 and PK100 (Fig. 1 ▶). The R iso values for PK100 (19.5% against PK000, 20.0% against PK025 and 23.4% against PK100) were obviously different to the R iso values between PK000, PK025 and PK050 (9.2% on average), which were comparable with the crystal R merge values. These R iso values suggested that the crystal structures of PK000, PK025 and PK050 are the same and that PK100 has a different crystal structure from the other three crystals. R merge also increased gradually from PK000 to PK100 in increments of 2.0% (PK000–PK025), 0.7% (PK025–PK050) and 1.7% (PK050–PK100); however, the increase in R merge from PK000 to PK100 (2.7%) was smaller than the increase in R iso from PK050 to PK100 (15.3%), suggesting that although the high R iso of PK100 was mainly derived from the differences in crystal structure between PK100 and the other crystals, the increase in R merge contributes to the increase in R iso for PK100. These values suggested that the four crystals are isomorphous; meanwhile, comparison of the crystal structure of PK100 with the other structures was necessary to investigate the identities of the crystal structures because of the relatively high R iso values for PK100 compared with those for PK000, PK025 and PK050.
Figure 1.

R
iso for the four crystals, where R
iso between crystal1 and crystal2 is defined as R
iso = 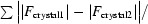
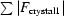 .
.
As shown in Fig. 2 ▶, the R merge and Wilson B factor of the data gradually increased with increasing D2O proportion, suggesting that the presence of D2O solution induced degradation of the PK crystals. Complete deuteration increased the R merge and Wilson B factor of the data by 4.4% and 1.17 Å2, respectively. The 〈I/σ(I)〉 value for PK100 (100% D2O) was smaller than that for PK000 (100% H2O). These three values indicate that deuteration of the crystallization solution was the cause of the observed degradation. As a result, the R factor and R free values for PK100 were larger than those for PK000 by 2.9% and 3.1%, respectively. However, this degradation was so small that it was undetectable at medium resolution and was thus negligible for neutron crystallographic analysis. One of the possible causes of degradation is that the glycerol in the cryoprotectant solution for all PK crystals was not deuterated. The concentration of glycerol in the cryoprotectant solution was very high (50%); therefore, degradation may occur by H/D exchange while soaking the deuterated crystals in this solution. The present degradation would be a combination of the effects of deuteration of the crystallization solution and H/D exchange during soaking.
Figure 2.

Comparison of PK crystal characteristics. The bars correspond to Wilson plot B factors. The line indicates R merge.
3.2. Crystal structure of PK crystallized from PEG 8000 solution
This study is the first structural determination of PK crystals obtained from PEG 8000 solution. We did not observe any nitrate anions, which were present in previous crystallographic studies. Two Ca2+ cations and one glycerol molecule from the cryoprotectant were present in the PK000 and PK100 unit cells. All non-H atoms of PK were precisely determined at ultrahigh resolution (1.1 Å). Moreover, densities corresponding to large numbers of H atoms were observed in |F o| − |F c| Fourier maps, although no H atoms were included in the present model. We found alternative water-molecule positions using these analyses, as shown in Fig. 3 ▶. 19 alternative positions of water molecules were determined for PK000 and 21 alternative positions were found for PK100. The overall structures were similar to the 0.98 Å resolution structure obtained by crystallization using NaNO3 as a precipitant (PKNO3; Betzel et al., 2001 ▶). The average root-mean-square differences (r.m.s.d.s) of atoms in the main chain and the side chains were 0.14 and 0.27 Å, respectively. However, the conformations of the side chains of several residues differed greatly from those reported for PKNO3 because distortions arising from the presence of NO3 − anions were avoided. The r.m.s.d. values for the three side chains Tyr61, Arg80 and Arg250 were greater than 2.0 Å. Each of these three residues was present at the PK surface and the conformational differences from the literature report were a consequence of the absence of NO3 − anions. In particular, Tyr61 was strongly affected by NO3 − anions, as shown in Fig. 4 ▶. All 19 residues which had side-chain r.m.s.d. values of greater than 1.0 Å were also present at the surface. No ions were detected within the crystal structure of PK000 except for two Ca2+ cations which were necessary for the activity of the protein, showing one conformation of PK under low-salt conditions. Comparison of this structure with the previous PK structure under high-salt conditions suggests that the core structure of PK is very stable and that the salt ions only affect local structural changes which were not critical for its activity. This variability is reasonable, as PK maintains its high protease activity under various conditions.
Figure 3.

2|F o| − |F c| Fourier maps around alternate positions of water molecules in PK000 (a) and PK100 (b).
Figure 4.

Three-dimensional structure of Tyr61 in PK000, superimposed on PKNO3.
3.3. Comparison between PK000 and PK100
The crystal structures of PK000 and PK100 are very similar to each other. The averaged r.m.s.d. values for the main chain and side chains are 0.05 and 0.07 Å, respectively. The largest side-chain r.m.s.d. was 0.28 Å (Arg250), which is about a quarter of a C—H bond length. Moreover, this value was only 2.8 and 2.3 times the magnitudes of the coordinate errors of PK000 (0.10 Å) and PK100 (0.12 Å), respectively, as estimated using the maximum-likelihood method. There were seven amino-acid residues for which the side-chain r.m.s.d. values were greater than 0.2 Å. All seven amino acids were located on the surface on the protein and five of the seven directly interacted with symmetry-related neighbouring protein molecules. It is plausible that these slight differences could be promoted by small changes in crystal packing owing to the expansion of the unit cell (ΔV = +1.5%). Of 380 PK000 H2O molecules, 373 (98%) were conserved as D2O molecules in PK100. The average r.m.s.d. value of water molecules between PK000 and PK100 was 0.33 Å and only 12 H2O molecules (3%) were greater than 1 Å away from the corresponding D2O molecules. About four-fifths of the water molecules located in alternative positions in PK000 conserved their alternative positions in PK100, as shown in Fig. 3 ▶. The B-factor values of amino-acid residues and water molecules were positively correlated with their r.m.s.d. values, indicating that there were no striking structural changes and supporting the high isomorphism of the deuterated and nondeuterated crystal structures of PK, thus supporting the above results.
4. Conclusions
In the present study, we attempted to obtain deuterated crystals of PK using PEG 8000 with the same crystal structure, the same crystal quality and under the same crystallization conditions as nondeuterated crystals. Four types of PK crystals were obtained at the same pH/pD from nondeuterated, 25% deuterated, 50% deuterated and fully deuterated crystallization solutions. These crystals could not be distinguished using optical microscopy and the degradation of the crystals with increasing deuteration was negligibly small. In the previous studies, PK crystals were obtained from NaNO3 solution and some NO3 − ions were found in their crystal structures. On the other hand, only two Ca2+ cations, which were essential for PK function, were found in the present crystal. Comparison between the previous high-salt structure and the present low-salt structure suggested that they were in principle isomorphous, except for local structural changes induced by NO3 − ions. Comparison between PK000 and PK100 at 1.1 Å resolution indicated extremely high isomorphism. At least for non-H atoms, the two structures could be considered to be identical in crystallographic studies. Such an identity would be convenient for attempting neutron experiments on PK under low-salt conditions. On the other hand, crystallization using PEG has a disadvantage for neutron diffraction studies. PEG itself has many non-exchangeable H atoms which contribute to a high background in neutron diffraction images. It is desirable to deuterate PEG for neutron experiments in order to solve this problem. It is concluded that the present high-resolution X-ray crystallographic analyses detected small structural changes which were induced by solvent deuteration in the crystallization of PK using PEG 8000 as a precipitant; however, they are negligibly small for structural studies at medium resolution such as neutron crystallographic analyses.
Supplementary Material
PDB reference: proteinase K, 3aj8
PDB reference: 3aj9
Acknowledgments
This work was part of a collaborative research project (21P9-2, 19S18). This work was supported in part by Grants-in-Aid for Scientific Research from the Chiba Institute of Sciences and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 18790030 to TC). The synchrotron-radiation experiments were performed on the BL38B1 beamline at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; Proposal No. 2007B1503).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Artero, J.-B., Härtlein, M., McSweeney, S. & Timmins, P. (2005). Acta Cryst. D61, 1541–1549. [DOI] [PubMed]
- Betzel, C., Gourinath, S., Kumar, P., Kaur, P., Perbandt, M., Eschenburg, S. & Singh, T. P. (2001). Biochemistry, 40, 3080–3088. [DOI] [PubMed]
- Blakeley, M. P., Langan, P., Niimura, N. & Podjarny, A. (2008). Curr. Opin. Struct. Biol. 18, 593–600. [DOI] [PMC free article] [PubMed]
- Blum, M.-M., Tomanicek, S. J., John, H., Hanson, B. L., Rüterjans, H., Schoenborn, B. P., Langan, P. & Chen, J. C.-H. (2010). Acta Cryst. F66, 379–385. [DOI] [PMC free article] [PubMed]
- Chatake, T., Mizuno, N., Voordouw, G., Higuchi, Y., Arai, S., Tanaka, I. & Niimura, N. (2003). Acta Cryst. D59, 2306–2309. [DOI] [PubMed]
- Di Costanzo, L., Moulin, M., Haertlein, M., Meilleur, F. & Christianson, D. W. (2007). Arch. Biochem. Biophys. 465, 82–89. [DOI] [PMC free article] [PubMed]
- Ebeling, W., Hennrich, N., Klockow, M., Metz, H., Orth, H. D. & Lang, H. (1974). Eur. J. Biochem. 47, 91–97. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gardberg, A. S., Blakeley, M. P. & Myles, D. A. A. (2009). Acta Cryst. F65, 184–187. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr. 26
- Liu, X., Hanson, B. L., Langan, P. & Viola, R. E. (2007). Acta Cryst. D63, 1000–1008. [DOI] [PubMed]
- Meilleur, F., Dauvergne, M.-T., Schlichting, I. & Myles, D. A. A. (2005). Acta Cryst. D61, 539–544. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Niimura, N., Arai, S., Kurihara, K., Chatake, T., Tanaka, I. & Bau, R. (2006). Cell. Mol. Life Sci. 63, 285–300. [DOI] [PMC free article] [PubMed]
- Okazaki, N., Hasegawa, K., Ueno, G., Murakami, H., Kumasaka, T. & Yamamoto, M. (2008). J. Synchrotron Rad. 15, 288–291. [DOI] [PMC free article] [PubMed]
- Sweeney, P. J. & Walker, J. M. (1993). Methods Mol. Biol. 16, 305–311. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: proteinase K, 3aj8
PDB reference: 3aj9