

The structural characterization of cellobiose phosphorylase from C. thermocellum reveals the residues involved in phosphate coordination and provides insight into substrate binding and discrimination.
Keywords: cellobiose, phosphate, phosphorylases, Clostridium thermocellum, glycoside hydrolase family 94
Abstract
Clostridium thermocellum is a cellulosome-producing bacterium that is able to efficiently degrade and utilize cellulose as a sole carbon source. Cellobiose phosphorylase (CBP) plays a critical role in cellulose degradation by catalyzing the reversible phosphate-dependent hydrolysis of cellobiose, the major product of cellulose degradation, into α-d-glucose 1-phosphate and d-glucose. CBP from C. thermocellum is a modular enzyme composed of four domains [N-terminal domain, helical linker, (α/α)6-barrel domain and C-terminal domain] and is a member of glycoside hydrolase family 94. The 2.4 Å resolution X-ray crystal structure of C. thermocellum CBP reveals the residues involved in coordinating the catalytic phosphate as well as the residues that are likely to be involved in substrate binding and discrimination.
1. Introduction
Clostridium thermocellum, a thermophilic, anaerobic, Gram-positive bacterium, displays one of the highest rates of cellulose degradation for bacteria and shows promise as a catalyst for the production of biofuels from lignocellulosic biomass (Demain et al., 2005 ▶; Johnson et al., 1982 ▶). The extracellular degradation of cellulose results in the formation of soluble oligosaccharides, including cellotetraose, cellotriose and cellobiose, which are subsequently imported into the cell in an ATP-dependent manner (Strobel et al., 1995 ▶). Inside the cell, cellobiose phosphorylase (CBP) catalyzes the reversible inorganic phosphate-dependent phosphorolysis of the β-1,4-glucosidic bond of cellobiose, yielding α-d-glucose 1-phosphate and d-glucose with inversion of the anomeric carbon (Fig. 1 ▶). The resulting α-d-glucose 1-phosphate is metabolized to glucose 6-phosphate, which serves as the entry point to the Embden–Meyerhof fermentation pathway (Lynd et al., 2002 ▶). Recent genome-wide microarray studies of C. thermocellum showed that all of the required enzymes for this conversion are highly upregulated during growth on cellulose and cellobiose (Riederer et al., 2011 ▶). Owing to the energetic benefit of the ATP-independent formation of glucose 1-phosphate in the CBP reaction (Zhang & Lynd, 2005 ▶), C. thermocellum can readily grow on cellobiose and other oligosaccharides as a sole carbon source and will preferentially utilize these even in the presence of glucose (Lynd et al., 2002 ▶; Mitchell, 1998 ▶).
Figure 1.

Schematic of the proposed mechanism of the reaction catalyzed by ctCBP.
CBPs are found in a wide range of cellulolytic bacteria and were originally classified as members of glycosyl transferase (GT) family 36 owing to sequence similarity to other GT36 enzymes, a lack of hydrolytic activity and their ability to utilize α-d-glucose 1-phosphate to form disaccharides (Coutinho et al., 2003 ▶). The structures of CBP from Cellvibrio gilvus (cgCBP) and chitobiose phosphorylase from Vibrio proteolyticus (vpChBP), which share 63 and 33% identity with CBP from C. thermocellum (ctCBP), respectively, revealed that CBPs and ChBPs were structurally similar to glucoamylases and maltose phosphorylases from glycoside hydrolase (GH) families 15 and 65, respectively (Hidaka et al., 2004 ▶; Stam et al., 2005 ▶). Based on the vpChBP structures, CBPs, together with the homologous cellodextrin phosphorylases and cyclic 1,2-glucan synthases, were reclassified into the novel GH family GH94.
Here, we present the X-ray crystal structure of the GH94 ctCBP at a resolution of 2.4 Å. The structure presented here reveals the residues involved in coordinating the catalytic phosphate as well as those that are likely to be involved in binding cellobiose. The structure of ctCBP also provides insight into the residues that are responsible for discrimination between soluble oligosaccharides of different chain lengths and compositions, which could thus be used as a guide to generate variants with altered substrate specificity.
2. Materials and methods
2.1. Protein purification
DNA encoding ctCBP was cloned into the pEC plasmid, which contained a tobacco etch protease (TEV) cleavable His8–maltose-binding protein tag. The ctCBP construct was expressed in Escherichia coli BL21 cells as described previously (Sreenath et al., 2005 ▶). Harvested cells were lysed by sonication and ctCBP was purified from the cellular supernatant using immobilized nickel-affinity chromatography. The affinity/solubility tag was cleaved utilizing TEV protease, which was subsequently captured by subtractive nickel-affinity chromatography. Fractions that contained ctCBP, as determined by SDS–PAGE, were pooled and passed over a gel-filtration column that was equilibrated with 5 mM HEPES buffered at pH 7.0 containing 50 mM NaCl and 3 mM NaN3. After gel filtration, ctCBP was concentrated to 5 mg ml−1 and utilized for structural determination.
2.2. Crystallization, diffraction data collection and structure determination
Rectangular-shaped ctCBP crystals were observed in the UW192 screen (Center for Eukaryotic Structural Genomics) after 3 d at 293 K. Crystals utilized for data collection were grown by hanging-drop vapor diffusion by mixing 1 µl of the ctCBP protein solution, as described above, with 1 µl reservoir solution [100 mM Tris buffer pH 7.5 containing 157 mM (NH4)2HPO4 and 2.1 M (NH4)H2PO4]. ctCBP crystals were cryoprotected by the addition of 15% ethylene glycol to the final well solutions as described above and cooled directly in liquid N2.
X-ray diffraction data for ctCBP were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-G beamline at the Advanced Photon Source, Argonne National Laboratory. The data were indexed, integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶). The ctCBP structure was solved by molecular replacement with Phaser (McCoy et al., 2007 ▶) using cgCBP (PDB entry 2cqs; Hidaka et al., 2006 ▶) as an initial model. The structure was completed with alternating rounds of manual model building in Coot (Emsley & Cowtan, 2004 ▶) and refinement in PHENIX (Adams et al., 2009 ▶). All refinement steps were monitored using an R free value based on selection of 4.9% of the independent reflections. The final model was refined to a resolution of 2.4 Å with an R cryst of 0.151 and an R free of 0.208. Model quality was assessed using MolProbity (Chen et al., 2010 ▶). All pertinent information on data collection, refinement and model statistics is summarized in Table 1 ▶. Figures were generated using PyMOL (DeLano, 2002 ▶).
Table 1. Crystal parameters, data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Crystal parameters | |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 83.26, b = 122.06, c = 181.996 |
| Data-collection statistics | |
| Wavelength (Å) | 0.97857 |
| Resolution range (Å) | 34.33–2.4 (2.46–2.4) |
| No. of reflections (measured/unique) | 459689/70340 |
| Completeness (%) | 99.5 (97.6) |
| Rmerge† | 0.127 (0.42) |
| Multiplicity | 6.2 (5.3) |
| Mean I/σ(I) | 13.4 (3.93) |
| Refinement and model statistics | |
| Resolution range (Å) | 34.33–2.4 |
| No. of reflections (work/test) | 70340/3512 |
| Rcryst‡ | 0.151 (0.197) |
| Rfree§ | 0.208 (0.245) |
| R.m.s.d. bonds (Å) | 0.006 |
| R.m.s.d. angles (°) | 0.933 |
| B factors (Å2) | |
| Protein | 34.08 |
| Solvent | 35.92 |
| Phosphate | 33.56 |
| Tris | 42.59 |
| No. of protein atoms | 13163 |
| No. of protein waters | 692 |
| No. of auxiliary molecules | 2 phosphate and 2 Tris |
| Ramachandran plot (%) | |
| Favorable region | 96.1 |
| Additional allowed region | 3.9 |
| PDB entry | 3qde |
R
merge = 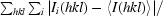
 , where I
i(hkl) is the intensity of an individual measurement of the reflection and 〈I(hkl)〉 is the mean intensity of the reflection.
, where I
i(hkl) is the intensity of an individual measurement of the reflection and 〈I(hkl)〉 is the mean intensity of the reflection.
R
cryst = 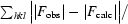
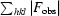 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes.
R free was calculated as R cryst using a randomly selected 4.9% of the unique reflections that were omitted from the structure refinement.
3. Results and discussion
3.1. Overall structure and structure quality
The structure of CBP from C. thermocellum (ctCBP) had well defined electron density for residues 1–811. Two subunits of ctCBP in complex with phosphate were observed per asymmetric unit and the structure belonged to space group P212121. ctCBP is a modular protein, is a member of GH family 94 and is composed of four distinct domains: an N-terminal domain (residues 1–279), a helical linker (residues 280–314), an (α/α)6-barrel domain (residues 321–734) and a C-terminal β-sandwich domain (residues 315–320 and 735–811) (Fig. 2 ▶ a). The N-terminal domain is composed of 18 antiparallel β-strands that form two β-sheets which stack against each other. Similar domains are observed in β-galactosidase (Jacobson et al., 1994 ▶) from GH2 and 4-α-glucantransferase (Imamura et al., 2003 ▶) from GH57. The N-terminal domain is connected to the (α/α)6-barrel domain via a helical linker that is composed of two α-helices, which form a 90° bend. The (α/α)6-barrel domain is formed by two concentric rings of six α-helices and contains several highly conserved residues, a bound phosphate ion and the catalytic residue (Asp483) near the center of the (α/α)6 barrel (Fig. 3 ▶ a). Catalytic domains from several GH families (8, 15, 37, 48, 63 and 65; Henrissat & Bairoch, 1996 ▶) are known to adopt a similar fold. The C-terminal domain adopts a two-layered jelly-roll fold that is structurally similar to the starch-binding domain of glucoamylase from Aspergillus niger (Sorimachi et al., 1996 ▶), but the exact function of the C-terminal domain is unclear.
Figure 2.

(a) The overall fold of a ctCBP monomer shown in cartoon rendering with the N-terminal domain in blue, the helical linker in green, the active-site-containing (α/α)6-barrel domain in red and the C-terminal domain in brown. (b) The dimeric form of ctCBP shown in carton rendering. Bound phosphates are shown as spheres and the helical extensions (residues 158–169) from the N-terminal domain that contribute to the formation of the active-site pocket are shown in red.
Figure 3.

(a) Residues that interact with the bound phosphate in the (α/α)6-barrel domain are shown as sticks. Dashed black lines represent hydrogen bonds. The 2F o − F c electron-density map shown is contoured at 1.5σ. (b) Comparison of the sugar-binding sites of ctCBP in red, vpChBP in brown and cgCBP in green. A bound Tris molecule is shown with C atoms in white, N atoms in green and O atoms in red. Glucose-binding subsites are labeled. The major differences between the three structures are located at Arg355 and Tyr640. In vpChBP Arg355 adopts an altered position and the Tyr is replaced by a Val.
The overall fold of ctCBP is identical to those of other GH94 members, such as cgCBP (Hidaka et al., 2006 ▶; r.m.s.d. 0.9 Å) and vpChBP (Hidaka et al., 2004 ▶; r.m.s.d. 1.5 Å). In addition to cgCBP and vpChBP, a number of ctCBP structural homologues from additional GH families were identified by DALI (Holm & Rosenstrom, 2010 ▶). Despite low sequence identity, ctCBP is structurally homologous to glucoamylase from Thermoanaerobacterium thermosaccharolyticum (Aleshin et al., 2003 ▶; GH15; r.m.s.d. 3.6 Å), glucodextranase from Arthrobacter globiformis (Mizuno et al., 2004 ▶; GH15; r.m.s.d. 3.7 Å) and maltose phosphorylase from Lactobacillus brevis (Egloff et al., 2001 ▶; GH65; r.m.s.d. 4.1 Å). Based on their structural similarities, it is possible that ctCBP and the enzymes mentioned above evolved from a common ancestor.
The two subunits in the asymmetric unit form a dimer which is held together through a series of hydrophobic interactions and hydrogen bonds. The dimer interface is primarily formed by the (α/α)6-barrel domain and the N-terminal domain (Fig. 2 ▶ b). Upon dimer formation, 3590 Å2 of solvent-accessible surface area is buried as calculated by the PISA server (Krissinel & Henrick, 2007 ▶). The ctCBP dimer formation is similar to those of other structurally characterized CBPs (Hidaka et al., 2004 ▶, 2006 ▶) and is likely to represent a biologically relevant conformation.
3.2. Active-site pocket of ctCBP
Two short helical extensions (residues 158–169) from the N-terminal domain of the adjacent subunit extend into the cleft which runs along the face of the (α/α)6-barrel domain, forming an active-site pocket (Fig. 2 ▶ b). An additional loop (residues 488–507) lays on top of the active-site pocket, forming a restrictive active-site pocket that is large enough to bind a disaccharide but not large enough to bind an oligosaccharide, thus providing discrimination based on the length of the oligosaccharide. In comparison, the active-site pocket of vpChBP, which binds the bulkier substrate chitobiose, is larger and more solvent-exposed. The interactions between the N-terminal domain and the (α/α)6-barrel domain in ctCBP determine the size of the active-site pocket and act as a size filter for substrates.
3.3. Phosphate-binding site
The side chains of His653, Gln699 and Thr718 and the backbone N atom of Gly719 coordinate the phosphate bound in the interior of the (α/α)6-barrel domain (Fig. 3 ▶ a). A Tris molecule dervied from the crystallization buffer is positioned between the phosphate and Asp483 and is close enough to form two hydrogen bonds to the phosphate (Fig. 3 ▶ a). A glycerol or N-acetylglucosamine (GlcNAc) molecule occupies a similar position in the phosphate-bound cgCBP structure or vpChBP structure, respectively (Hidaka et al., 2004 ▶, 2006 ▶). The Tris molecule occupies a position analogous to the bound GlcNAc in the vpChBP structure, indicating that it occupies a glucose-binding subsite. This would place a glucose moiety in position for nucleophilic attack by the bound phosphate (Fig. 1 ▶).
3.4. Glucose-binding subsites
Based on the structures of vpChBP in complex with GlcNAc and of cgCBP in complex with a molecule of glucose, and the fortuitous positioning of Tris in the ctCBP structure, the two glucose-binding subsites (−1 and +1) of ctCBP can be proposed (Hidaka et al., 2004 ▶, 2006 ▶). The Tris molecule at the −1 glucose-binding subsite (sugar-donor site) superposes with the C4, C6, O5 and O6 atoms of GlcNAc bound in the −1 GlcNAc-binding subsite of vpChBP (Fig. 3 ▶ b). The residues (Arg355, Asp361 and Gln699) surrounding the −1 glucose-binding subsite of ctCBP are highly conserved and adopt similar conformations in both the vpChBP and ctCBP structures (Fig. 3 ▶ b). The only significant difference between the ctCBP and vpChBP structures at the −1 subsite is the position of the side chain of Arg355 (Arg343 of vpChBP). The rearrangement of Arg355 is presumably owing to the lack of the N-acetyl group which is located at C2 of chitin and allows the side chain to move further into the active-site pocket. Trp481, which is conserved in cgCBP and vpChBP, forms the back of the −1 subsite, effectively limiting the size of the functional groups on C4 and C5 of a bound sugar moiety.
Residues near the +1 glucose-binding subsite are structurally conserved, with the exception of Tyr640 (Fig. 3 ▶ b). Glu637, Lys636 and Gln168 from the N-terminal domain of the adjacent subunit adopt similar positions to their sugar-bound homologues (Fig. 3 ▶ b). Again, the major difference between the +1 glucose-binding subsite of ctCBP and the +1 GlcNAc-binding subsite of vpChBP is localized at the residue near the C2 position. Tyr640 of ctCBP, which would clash with the N-acetyl group of chitin, is replaced by Val631 in vpChBP and is likely to play a role in discrimination between oligosaccharides of different compositions. Asp483 is located between the −1 and +1 glucose-binding subsites, placing it in position to donate a proton to the leaving glucose molecule.
4. Conclusion
CBPs play an important role in providing an energetic advantage to C. thermocellum and other organisms during growth on cellulose (Zhang & Lynd, 2005 ▶). The ctCBP structure reveals the amino acids responsible for binding the catalytic phosphate and composing the cellobiose-binding site. Significant differences surrounding the C2 position of subsites −1 and +1 are observed when ctCBP is compared with the chitobiose-binding vpChBP. It appears that modulation of substrate specificity can be obtained by a limited number of amino-acid substitutions. These active-site alterations would be applicable to CBPs from other bacterial species and perhaps additional structurally homologous GH94-family members.
Supplementary Material
PDB reference: cellobiose phosphorylase, 3qde
Acknowledgments
This work was funded in part by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). The authors would like to thank the Center for Eukaryotic Structural Genomics for the use of various equipment and reagents.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aleshin, A. E., Feng, P. H., Honzatko, R. B. & Reilly, P. J. (2003). J. Mol. Biol. 327, 61–73. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Coutinho, P. M., Deleury, E., Davies, G. J. & Henrissat, B. (2003). J. Mol. Biol. 328, 307–317. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Demain, A. L., Newcomb, M. & Wu, J. H. D. (2005). Microbiol. Mol. Biol. Rev. 69, 124–154. [DOI] [PMC free article] [PubMed]
- Egloff, M. P., Uppenberg, J., Haalck, L. & van Tilbeurgh, H. (2001). Structure, 9, 689–697. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Henrissat, B. & Bairoch, A. (1996). Biochem. J. 316, 695–696. [DOI] [PMC free article] [PubMed]
- Hidaka, M., Honda, Y., Kitaoka, M., Nirasawa, S., Hayashi, K., Wakagi, T., Shoun, H. & Fushinobu, S. (2004). Structure, 12, 937–947. [DOI] [PubMed]
- Hidaka, M., Kitaoka, M., Hayashi, K., Wakagi, T., Shoun, H. & Fushinobu, S. (2006). Biochem. J. 398, 37–43. [DOI] [PMC free article] [PubMed]
- Holm, L. & Rosenstrom, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Imamura, H., Fushinobu, S., Yamamoto, M., Kumasaka, T., Jeon, B.-S., Wakagi, T. & Matsuzawa, H. (2003). J. Biol. Chem. 278, 19378–19386. [DOI] [PubMed]
- Jacobson, R. H., Zhang, X.-J., DuBose, R. F. & Matthews, B. W. (1994). Nature (London), 369, 761–766. [DOI] [PubMed]
- Johnson, E. A., Sakajoh, M., Halliwell, G., Madia, A. & Demain, A. L. (1982). Appl. Environ. Microbiol. 43, 1125–1132. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lynd, L. R., Weimer, P. J., van Zyl, W. H. & Pretorius, I. S. (2002). Microbiol. Mol. Biol. Rev. 66, 506–577. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mitchell, W. J. (1998). Adv. Microb. Physiol. 39, 31–130. [DOI] [PubMed]
- Mizuno, M., Tonozuka, T., Suzuki, S., Uotsu-Tomita, R., Kamitori, S., Nishikawa, A. & Sakano, Y. (2004). J. Biol. Chem. 279, 10575–10583. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Riederer, A., Takasuka, T. E., Makino, S., Stevenson, D. M., Bukhman, Y. V., Elsen, N. L. & Fox, B. G. (2011). Appl. Environ. Microbiol. 77, 1243–1253. [DOI] [PMC free article] [PubMed]
- Sorimachi, K., Jacks, A. J., Le Gal-Coëffet, M.-F., Williamson, G., Archer, D. B. & Williamson, M. P. (1996). J. Mol. Biol. 259, 970–987. [DOI] [PubMed]
- Sreenath, H. K., Bingman, C. A., Buchan, B. W., Seder, K. D., Burns, B. T., Geetha, H. V., Jeon, W. B., Vojtik, F. C., Aceti, D. J., Frederick, R. O., Phillips, G. N. & Fox, B. G. (2005). Protein Expr. Purif. 40, 256–267. [DOI] [PubMed]
- Stam, M. R., Blanc, E., Coutinho, P. M. & Henrissat, B. (2005). Carbohydr. Res. 340, 2728–2734. [DOI] [PubMed]
- Strobel, H. J., Caldwell, F. C. & Dawson, K. A. (1995). Appl. Environ. Microbiol. 61, 4012–4015. [DOI] [PMC free article] [PubMed]
- Zhang, Y.-H. P. & Lynd, L. R. (2005). J. Bacteriol. 187, 99–106. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: cellobiose phosphorylase, 3qde