

The extracellular part of the human epithelial cell-adhesion molecule (EpCAM; CD326) was cloned, mutated to prevent N-linked glycosylation, expressed in insect cells, purified and crystallized. Diffraction data were collected to 1.95 Å resolution.
Keywords: epithelial cell-adhesion molecule, EpCAM, CD326
Abstract
The epithelial cell-adhesion molecule (EpCAM; CD326) is a transmembrane glycoprotein involved in epithelial cell–cell adhesion, cell proliferation and differentiation. Its elevated expression level in various carcinomas is exploited by several antitumour therapies that are at various stages of clinical development. The 35 kDa polypeptide chain of EpCAM is divided into a large extracellular part, a transmembrane helix and a short cytoplasmic tail. The modular extracellular part of human EpCAM was cloned and mutated to prevent N-linked glycosylation. After expression in insect cells and purification using standard chromatographic techniques, the extracellular part was crystallized. The crystals belonged to space group C2, with unit-cell parameters a = 86.83, b = 50.16, c = 66.56 Å, β = 127.9°. The crystal diffracted to 1.95 Å resolution and contained one molecule in the asymmetric unit.
1. Introduction
Cell–cell adhesion is a fundamental mechanism that is tightly linked to cell proliferation and differentiation. The intercellular adhesion–communication bridge is typically mediated by cell-adhesion molecules (CAMs) belonging to three distinct superfamilies: cadherins, claudins and immunoglobulin-like CAMs (Cavallaro & Dejana, 2011 ▶). The epithelial cell-adhesion molecule (EpCAM), which was first discovered as a colon carcinoma marker (Herlyn et al., 1979 ▶), does not belong to any of these major and widely expressed groups of CAMs. Rather, its expression is limited to epithelial cells at various stages of differentiation, in which it mediates homophilic calcium-independent cell–cell adhesive interactions (Litvinov et al., 1994 ▶). The highest expression levels are observed on the surface of undifferentiated embryonic cells, which classifies EpCAM as a human embryonic stem-cell marker, with cellular differentiation marker name CD326 (Baeuerle & Gires, 2007 ▶; Lu et al., 2010 ▶). This location determines the role of EpCAM in promoting cell proliferation via its cytoplasmic tail (EpIC), which is liberated by a specific proteolytic cleavage. The complex formed by EpIC, four-and-a-half LIM domains protein 2 (FHL2), β-catenin and lymphoid enhancer-binding factor (Lef-1) is then transported to the nucleus, where it activates gene transcription at Lef-1 consensus sites (Maetzel et al., 2009 ▶). Enhanced cell proliferation is also a hallmark of cancer cells of epithelial origin (carcinomas), which express EpCAM at elevated levels compared with normal differentiated cells (Göttlinger et al., 1986 ▶). In addition, on carcinoma cells EpCAM forms a complex with junction protein claudin-7, tumour-associated scaffolding protein tetraspanin CO-029 and extracellular matrix receptor CD44v6 which is thought to promote tumourigenesis (Kuhn et al., 2007 ▶). As a cancer-associated molecule, EpCAM is the target of several antibody-based therapies which are at various stages of clinical development (Münz et al., 2010 ▶). The existing therapies rely only on the differential expression pattern of EpCAM; structure–function relationships have not yet been exploited owing to the complete lack of structural data.
Amino-acid sequence analysis revealed that the EpCAM molecule can be divided into three regions: a large N-terminal extracellular part (242 residues), a single transmembrane helix (23 residues) and a small C-terminal cytosolic tail (26 residues) (Strnad et al., 1989 ▶). The extracellular part can be further divided into three distinct regions: a unique small N-terminal cysteine-rich domain, a central thyroglobulin type 1 (TY) domain and an exclusive C-terminal region. The TY domain is a protein module which is found in many structurally and functionally unrelated proteins, where it can serve as an inhibitor/regulator of the activity of papain-like cysteine peptidases (Lenarčič & Bevec, 1998 ▶) or as a binding site for heparin, as in the case of the insulin growth-factor-like binding protein (IGFBP) family (Kiefer et al., 1992 ▶). The TY domain is also the only region of the EpCAM molecule for which structural insights have been provided: three-dimensional structures have been determined of the TY domain of human invariant chain p41 bound to cathepsin L (Gunčar et al., 1999 ▶) as well as of the TY domains of IGFBP6, IGFBP2 and IGFBP1 (Headey et al., 2004 ▶; Kuang et al., 2006 ▶; Sala et al., 2005 ▶). The TY domain of EpCAM further diversifies the roles of TY modules since it mediates lateral interactions of EpCAM molecules. Two dimers from adjacent cells then form a tetramer via the small N-terminal domains and the adhesion unit is anchored to the actin cytoskeleton via interaction of the C-terminal cytosolic tail with α-actinin (Balzar et al., 2001 ▶).
The structure of the extracellular part of EpCAM (EpEC) would offer important insights into the function and adhesion mechanism of this unique member of the CAMs. These could serve as important starting points for the refinement of already existing therapies. In this report, we describe the cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of the complete extracellular part of human EpCAM mutated to prevent N-linked glycosylation (ngEpEC).
2. Materials and methods
2.1. Cloning and expression
The DNA fragment encoding the extracellular part of EpCAM with native signal peptide (residues 1–265) was amplified by PCR using a full-length EpCAM clone as a template (cDNA clone library clone ID IRAKp961G0321Q; Source BioScience imaGenes) with the simultaneous introduction of a C-terminal His6 tag followed by a stop codon using appropriate primer overhangs. Three point mutations (N74Q, N111Q and N198Q) were introduced by the method of two-sided splicing by overlap extension to prevent N-linked glycosylation of the polypeptide chain (Horton et al., 1989 ▶). The resulting mutated DNA fragment was digested and ligated into the pFastBac1 plasmid vector. Recombinant bacmids were prepared by homologous recombination in Escherichia coli DH10Bac cells according to the Bac-to-Bac system (Invitrogen). Bacmids containing the EpCAM EC insert were identified by PCR and transfected into Spodoptera frugiperda Sf9 insect cells to generate recombinant baculoviruses. After several rounds of amplification the viral titre was determined by plaque assay.
The optimal parameters for expression [S. frugiperda Sf9 or Trichoplusia ni High Five (Invitrogen) insect cells, cell density, multiplicity of infection (MOI) and time from infection to harvest] were determined by small-scale expression experiments using suspension cultures in Erlenmeyer flasks. Insect-XPRESS Protein-free medium with l-glutamine (Lonza) was used in all experiments.
Scaled-up expression was performed using the determined optimal parameters in suspension cultures (Sf9 cells; 2.5 × 106 cells ml−1 at time of infection; MOI = 5). 36 h after infection the medium was harvested by centrifugation and concentrated to 1/10 of the initial volume by ultrafiltration in Amicon stirred cells (Millipore) with 3 kDa cutoff filter discs (Pall). The concentrate was extensively dialysed against 50 mM sodium phosphate buffer pH 7.4, 200 mM NaCl, 10%(v/v) glycerol in dialysis tubing with 3.5 kDa cutoff (Spectrum), clarified by centrifugation and loaded onto an Ni2+-charged IMAC column using gravity flow (GE Healthcare) to remove most of the contaminants. Unbound proteins were washed from the column with IMACBW buffer (50 mM sodium phosphate pH 7.4, 500 mM NaCl, 25 mM imidazole) and elution was performed using the same buffer supplemented with 500 mM imidazole (IMACE buffer). The pooled eluate was dialysed against IMACBW buffer to lower the imidazole concentration and again loaded onto an Ni2+-charged IMAC column on an FPLC system (GE Healthcare). After washing with the same buffer, the bound protein was eluted with a linear gradient of IMACE buffer. The pooled eluate was dialysed against IEXBW buffer (20 mM Na HEPES pH 8.0), loaded onto a Mono Q column (GE Healthcare) and eluted with a linear gradient of IEXE buffer (IEXBW containing 500 mM NaCl). The pooled eluate containing pure ngEpEC was loaded onto a Superdex 200 10/300 GL size-exclusion column (GE Healthcare) equilibrated with 20 mM Na HEPES pH 7.4, 100 mM NaCl (Fig. 1 ▶). The single protein peak eluting at 15.2 ml was concentrated to a final concentration of 15.5 mg ml−1 using Amicon centrifugal filter units with 3 kDa cutoff membranes (Millipore). For analytical purposes, size-exclusion chromatography was also performed using a buffer with low pH in which the dimeric form is not stable (20 mM sodium acetate pH 4.7, 100 mM NaCl; Fig. 1 ▶). The size-exclusion chromatography column was calibrated with samples of proteins with known molecular masses and the molecular mass of ngEpEC was calculated from the linear log(MW)–V e relationship. The homogeneity of the protein sample was determined by SDS–PAGE analysis (12.5% gel) under reducing conditions. Correct processing of the signal peptide was confirmed by N-terminal sequence analysis using Edman degradation, in which the expected sequence of the mature protein (QEECVCE) matched the experimentally determined sequence (QEExVxE, where x denotes missing cysteine residues which are destroyed during derivatization).
Figure 1.

Size-exclusion chromatography and SDS–PAGE analysis of ngEpEC. Chromatogram from a Superdex 200 10/300 column at pH 7.4 (solid line) and 4.7 (dashed line). The elution volumes of proteins used for column calibration are shown at the top of chromatogram. Inset, SDS–PAGE analysis (reducing conditions) of both peaks shown in the chromatogram.
2.2. Crystallization
Crystallization conditions were initially screened at 293 K by the sitting-drop vapour-diffusion method using commercially available screens from Sigma–Aldrich (Basic and Extension Crystallography Kits based on sparse-matrix screens; Jancarik & Kim, 1991 ▶). 1 µl protein solution and 1 µl reservoir solution were mixed and allowed to equilibrate against 100 µl reservoir solution. Crystals grew within two weeks to dimensions of 20 × 20 × 200 µm in a condition consisting of 0.2 M magnesium chloride, 0.1 M Tris–HCl pH 8.5, 30%(w/v) PEG 4000 (Fig. 2 ▶ a). A set of optimization experiments over a range of pH values (8–9.5) and PEG concentrations (25–35%) did not yield better diffracting crystals (Figs. 2 ▶ b and 2 ▶ c).
Figure 2.

Crystals of ngEpEC grown in (a) 0.2 M magnesium chloride, 0.1 M Tris–HCl pH 8.5, 30%(w/v) PEG 4000, (b) 0.2 M magnesium chloride, 0.1 M Tris–HCl pH 8.5, 33%(w/v) PEG 4000, (c) 0.2 M magnesium chloride, 0.1 M Tris–HCl pH 8.5, 30%(w/v) PEG 4000, 1.8 mM n-decyl β-d-maltopyranoside. (d) X-ray diffraction image from an ngEpEC crystal [the smaller crystal shown in (a)]. The resolution of the detector edge is 1.95 Å.
2.3. Data collection and processing
For data collection, the crystal was directly flash-frozen in a stream of nitrogen gas (100 K) without any additional cryoprotectant. A 1.95 Å resolution native diffraction data set was collected on beamline XRD1 at the Elettra Synchrotron Light Laboratory, Italy (Fig. 2 ▶ d). Data were recorded with 30 s exposure and 1° rotation per image as a total of 180 images using an SX-165 CCD detector (MAR Research). Reflections (excluding ice rings) were indexed and processed using MOSFLM (Leslie, 1992 ▶). The unit-cell content was estimated from calculation of the Matthews coefficient (Matthews, 1968 ▶). Data-collection and processing statistics are summarized in Table 1 ▶.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.10697 |
| Resolution range (Å) | 27.59–1.95 (2.06–1.95) |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 86.83, b = 50.16, c = 66.56, β = 127.9 |
| No. of observed reflections | 59531 (6754) |
| No. of unique reflections | 16390 (2180) |
| Average mosaicity (°) | 0.61 |
| Multiplicity | 3.6 (3.1) |
| Mean I/σ(I) | 13.7 (5.1) |
| Completeness (%) | 98.8 (91.6) |
| Rmerge† | 0.051 (0.154) |
R
merge = 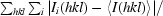
 .
.
3. Results and discussion
The preparation of small amounts of recombinant human EpCAM has been described previously (Strassburg et al., 1992 ▶). In the extracellular part of EpCAM expressed in T. ni insect cells two of the three Asn residues (Asn74 and Asn111) which are part of the N-linked glycosylation consensus motif (NxS/NxT) are heterogeneously glycosylated (Chong & Speicher, 2001 ▶). EpCAM isolated from human tissue also has glycans attached to the third Asn residue (Asn198) and the extent of glycosylation is linked to the cell state (Pauli et al., 2003 ▶). To achieve the highest homogeneity of the sample for crystallization, we prepared a mutant extracellular part of EpCAM in which all three Asn residues known to be involved in N-linked glycosylation were mutated to Gln residues. The mutant nonglycosylated EpEC (ngEpEC) was purified from recombinant baculovirus-infected insect-cell culture supernatant using affinity, ion-exchange and size-exclusion chromatographic steps, with a final yield of 12 mg pure ngEpEC per litre of culture. The size-exclusion chromatograms of nonglycosylated mutant (Fig. 1 ▶) and glycosylated wild-type EpEC (results not shown) were essentially the same, indicating the same tendency towards the formation of dimers. The oligomeric state of ngEpEC was analysed using size-exclusion chromatography at pH 7.4 and 4.7 (Fig. 1 ▶). The lower pH enabled us to obtain the monomeric form owing to the instability of the dimer. The apparent molecular mass of His6-tagged ngEpEC is 46 kDa at pH 7.4 and 28 kDa at pH 4.7 as calculated from the elution volumes from a calibrated size-exclusion column (using the linear relationship between the elution volume and the logarithm of the molecular mass). The molecular mass of 28 kDa clearly corresponds to the monomer since it is the same as the molecular mass of ngEpEC calculated from the amino-acid sequence (28.3 kDa). The 46 kDa species could correspond to the ngEpEC dimer. The difference between the molecular masses of the dimer as calculated from the elution volume (46 kDa) and from the amino-acid sequence (56.6 kDa) could be attributed to changes in the spatial arrangement of the individual domains which affect the overall molecular shape of the dimer. The pH-dependent transition from the dimeric to the monomeric state can be reversed simply by raising the pH (data not shown). The absence of heterogeneous Asn-attached glycans resulted in highly homogenous protein. The crystals diffracted to 1.95 Å resolution and belonged to space group C2. Crystals obtained using variations of these conditions did not diffract better. A summary of the crystal parameters and the statistics of the diffraction data are presented in Table 1 ▶. The Matthews coefficient was determined to be 2.02 Å3 Da−1 with a solvent content of 39.3%, corresponding to the presence of one molecule in the asymmetric unit (Matthews, 1968 ▶). Molecular replacement using known three-dimensional structures of TY domains is not feasible since the TY domain of EpCAM represents only 25% of the whole EpEC. For phasing, we intend to prepare a selenomethionine variant of EpEC since it contains four methionine residues. This work is currently in progress.
Acknowledgments
This work was supported by the Slovenian Research Agency. We thank Dr Katja Galeša for help with data collection, Dr Gregor Gunčar and Professor Kristina Djinović Carugo for helpful discussions and Professor Antonio Baici for critical reading of the manuscript. We gratefully acknowledge the Elettra Synchrotron Light Laboratory (Italy) for providing the beamtime and Drs Maurizio Polentarutti (Elettra, Italy) and Doriano Lamba (CNR, Italy) for help with data-collection setup.
References
- Baeuerle, P. A. & Gires, O. (2007). Br. J. Cancer, 96, 417–423. [DOI] [PMC free article] [PubMed]
- Balzar, M., Briaire-de Bruijn, I. H., Rees-Bakker, H. A., Prins, F. A., Helfrich, W., de Leij, L., Riethmüller, G., Alberti, S., Warnaar, S. O., Fleuren, G. J. & Litvinov, S. V. (2001). Mol. Cell. Biol. 21, 2570–2580. [DOI] [PMC free article] [PubMed]
- Cavallaro, U. & Dejana, E. (2011). Nature Rev. Mol. Cell Biol. 12, 189–197. [DOI] [PubMed]
- Chong, J. M. & Speicher, D. W. (2001). J. Biol. Chem. 276, 5804–5813. [DOI] [PubMed]
- Göttlinger, H., Johnson, J. & Riethmuller, G. (1986). Hybridoma, 5, S29–S37. [PubMed]
- Gunčar, G., Pungerčič, G., Klemenčič, I., Turk, V. & Turk, D. (1999). EMBO J. 18, 793–803. [DOI] [PMC free article] [PubMed]
- Headey, S. J., Keizer, D. W., Yao, S., Brasier, G., Kantharidis, P., Bach, L. A. & Norton, R. S. (2004). Mol. Endocrinol. 18, 2740–2750. [DOI] [PubMed]
- Herlyn, M., Steplewski, Z., Herlyn, D. & Koprowski, H. (1979). Proc. Natl Acad. Sci. USA, 76, 1438–1442. [DOI] [PMC free article] [PubMed]
- Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J. K. & Pease, L. R. (1989). Gene, 77, 61–68. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kiefer, M. C., Schmid, C., Waldvogel, M., Schläpfer, I., Futo, E., Masiarz, F. R., Green, K., Barr, P. J. & Zapf, J. (1992). J. Biol. Chem. 267, 12692–12699. [PubMed]
- Kuang, Z., Yao, S., Keizer, D. W., Wang, C. C., Bach, L. A., Forbes, B. E., Wallace, J. C. & Norton, R. S. (2006). J. Mol. Biol. 364, 690–704. [DOI] [PubMed]
- Kuhn, S., Koch, M., Nübel, T., Ladwein, M., Antolovic, D., Klingbeil, P., Hildebrand, D., Moldenhauer, G., Langbein, L., Franke, W. W., Weitz, J. & Zöller, M. (2007). Mol. Cancer Res. 5, 553–567. [DOI] [PubMed]
- Lenarčič, B. & Bevec, T. (1998). Biol. Chem. 379, 105–111. [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr. 26
- Litvinov, S. V., Velders, M. P., Bakker, H. A., Fleuren, G. J. & Warnaar, S. O. (1994). J. Cell Biol. 125, 437–446. [DOI] [PMC free article] [PubMed]
- Lu, T.-Y., Lu, R.-M., Liao, M.-Y., Yu, J., Chung, C.-H., Kao, C.-F. & Wu, H.-C. (2010). J. Biol. Chem. 285, 8719–8732. [DOI] [PMC free article] [PubMed]
- Maetzel, D., Denzel, S., Mack, B., Canis, M., Went, P., Benk, M., Kieu, C., Papior, P., Baeuerle, P. A., Munz, M. & Gires, O. (2009). Nature Cell Biol. 11, 162–171. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Münz, M., Murr, A., Kvesic, M., Rau, D., Mangold, S., Pflanz, S., Lumsden, J., Volkland, J., Fagerberg, J., Riethmüller, G., Rüttinger, D., Kufer, P., Baeuerle, P. A. & Raum, T. (2010). Cancer Cell Int. 10, 44. [DOI] [PMC free article] [PubMed]
- Pauli, C., Münz, M., Kieu, C., Mack, B., Breinl, P., Wollenberg, B., Lang, S., Zeidler, R. & Gires, O. (2003). Cancer Lett. 193, 25–32. [DOI] [PubMed]
- Sala, A., Capaldi, S., Campagnoli, M., Faggion, B., Labò, S., Perduca, M., Romano, A., Carrizo, M. E., Valli, M., Visai, L., Minchiotti, L., Galliano, M. & Monaco, H. L. (2005). J. Biol. Chem. 280, 29812–29819. [DOI] [PubMed]
- Strassburg, C. P., Kasai, Y., Seng, B. A., Miniou, P., Zaloudik, J., Herlyn, D., Koprowski, H. & Linnenbach, A. J. (1992). Cancer Res. 52, 815–821. [PubMed]
- Strnad, J., Hamilton, A. E., Beavers, L. S., Gamboa, G. C., Apelgren, L. D., Taber, L. D., Sportsman, J. R., Bumol, T. F., Sharp, J. D. & Gadski, R. A. (1989). Cancer Res. 49, 314–317. [PubMed]