

A 17 kDa capsid protease domain from Aura virus was purified, crystallized together with its complex with dioxane and characterized by the X-ray diffraction method.
Keywords: Aura virus, capsid protease
Abstract
The C-terminal protease domain of capsid protein from Aura virus expressed in a bacterial expression system has been purified to homogeneity and crystallized. Crystals suitable for X-ray diffraction analysis were obtained by the vapour-diffusion method using 0.1 M bis-tris and polyethylene glycol monomethyl ether 2000. Crystals of the C-terminal protease domain of capsid protein in complex with dioxane were also produced and crystal data were obtained. Both crystals belonged to space group C2, with unit-cell parameters a = 79.6, b = 35.2, c = 49.5 Å. High-resolution data sets were collected to a resolution of 1.81 Å for the native protein and 1.98 Å for the complex. Preliminary crystallographic studies suggested the presence of a single molecule in the crystallographic asymmetric unit, with a solvent content of 38.5%.
1. Introduction
The Alphavirus genus, belonging to the Togaviridae family, contains a positive-sense single-stranded RNA genome of ∼11.8 kb. These viruses are responsible for causing encephalitis, fever, arthritis and rash in mammals. Some members of the Alphavirus genus, including Chikungunya virus, Venezuelan equine encephalitis, Eastern equine encephalitis and Western equine encephalitis virus, are lethal human pathogens and are considered to be possible bioterrorism threats (Reichert et al., 2009 ▶; Jose et al., 2009 ▶). At present, there is no human vaccine or medical treatment available for alphavirus infection.
Alphaviruses are icosahedral enveloped viruses of approximately 700 Å in diameter (Jose et al., 2009 ▶). The nucleocapsid core is composed of 240 copies of the capsid protein which are arranged in a T = 4 icosahedral lattice. The nucleocapsid core is surrounded by the phospholipid bilayer, in which trimeric spikes of E1 and E2 heterodimers are embedded (Cheng et al., 1995 ▶; Mukhopadhyay et al., 2002 ▶). The genome of alphaviruses is comprised of two domains: the 5′-terminal two-thirds encodes nonstructural proteins, while the 3′-terminal third encodes structural proteins. The structural proteins are translated from the subgenomic 26S RNA as a polyprotein. Capsid protein, a serine proteinase present at the N-terminus of the polyprotein, cleaves itself from the structural polyprotein precursor by cis-autoproteolysis (Aliperti & Schlesinger, 1978 ▶; Tong et al., 1993 ▶). The C-terminal tryptophan residue of the alphavirus capsid remains bound in the active site after cis-cleavage and thus the proteolytic activity of capsid protease is turned off (Choi et al., 1991 ▶). However, in vitro trans-cleavage activity of the C-terminally truncated Semliki Forest virus (SFV) capsid protease using synthetic substrates has recently been demonstrated (Morillas et al., 2008 ▶). After cis-cleavage, the released alphavirus capsid protein interacts with the genomic RNA, encapsidates it and forms the nucleocapsid (NC) assembly. The capsid protein in the nucleocapsid core also interacts with the cytoplasmic tail of viral glycoproteins and directs the budding of virus particles (Owen & Kuhn, 1997 ▶; Suomalainen et al., 1992 ▶; Kail et al., 1991 ▶).
The alphavirus capsid, a multifunctional protein, consists of an intrinsically unstructured N-terminal domain (∼113 residues) and a structured C-terminal protease domain (∼150 residues) containing a chymotrypsin-like serine protease fold (Choi et al., 1991 ▶; Hong et al., 2006 ▶; Perera et al., 2001 ▶). Intrinsically disordered regions in proteins are engaged in important molecular interactions with proteins, nucleic acids or membranes that are crucial for functional activity of the protein (Uversky, 2002 ▶). The disordered N-terminal domain of the alphavirus capsid interacts with the genomic RNA and also participates in protein–protein interactions that lead to capsid dimerization. These interactions eventually result in nucleocapsid core formation (Owen & Kuhn, 1996 ▶; Perera et al., 2001 ▶). Similarly, the N-terminal domain (∼98 residues) of another alphavirus protein nsP4 is predicted to be disordered and also participates in protein–protein interaction (Tomar et al., 2006 ▶; Rupp et al., 2011 ▶). As intrinsically disordered regions perform important structural or biological functions, disordered regions in proteins, including the alphavirus capsid, are therefore considered to be potential drug targets (Uversky et al., 2008 ▶; Metallo, 2010 ▶).
The autoprotease activity of alphavirus capsid protein resides in the C-terminal domain and is carried out by the His-Ser-Asp catalytic triad (Tong et al., 1993 ▶; Choi et al., 1991 ▶). The crystal structures of capsid proteins from Sindbis virus and Semliki Forest virus show the post-cleavage state with the C-terminal Trp residue residing in the active site, which prevents trans-proteolytic activity (Tong et al., 1993 ▶; Choi et al., 1997 ▶). This Trp residue present at the C-terminus is fully conserved among all alphaviruses (Faragher et al., 1988 ▶; Kinney et al., 1986 ▶; Levinson et al., 1990 ▶). Additionally, the capsid protein contains a hydrophobic pocket which participates in protein–protein interaction with the glycoprotein shell that facilitates the virus budding process (Skoging et al., 1996 ▶; Owen & Kuhn, 1997 ▶; Suomalainen et al., 1992 ▶). In the crystal structure of Sindbis virus capsid protein, this hydrophobic pocket was occupied by a solution-derived dioxane (Lee et al., 1998 ▶). Based on this crystal structure data, dioxane-based antiviral compounds have been designed that inhibit Sindbis virus replication (Kim et al., 2007 ▶). Thus, the multifunctional capsid protein is an excellent target for developing antiviral inhibitors to combat alphaviral diseases.
Aura virus is a member of the alphavirus family and was first isolated from Aedes serratus in Brazil and northern Argentina (Rümenapf, Strauss et al., 1995 ▶). Unlike other alphaviruses, the capsid protein of Aura virus encapsidates both the 4.2 kb subgenomic RNA and the genomic RNA in virion particles. This is a unique feature of Aura viruses which is responsible for their less precise virus assembly in comparison to other alphaviruses. Additionally, the specific infectivity of Aura virions has been found to be lower than other members of the alphavirus family (Rümenapf, Brown et al., 1995 ▶). Although the members of the alphavirus family share a high protein sequence identity of greater than 36% overall, they interestingly exhibit differences in RNA packaging, virus assembly, inhibition of host cellular processes and specific infectivity. Three-dimensional structures of the viral proteins associated with packaging and assembly from different members of the family will assist in understanding the underlying mechanism that is responsible for the variation in these processes. Hence, it is a requisite to crystallize proteins involved in virus assembly and the life cycle from different alphaviruses.
Here, we report the heterologous expression, purification, crystallization and preliminary X-ray diffraction analysis of Aura virus capsid protease (AVCP) and its complex with dioxane.
2. Materials and methods
2.1. Construction of expression plasmids
Aura virus genomic cDNA (Rümenapf et al., 1994 ▶) was used as the template for polymerase chain reaction (PCR) amplification of the DNA fragments encoding the full-length capsid protein and the carboxy-terminal capsid protease domain (residues 110–267). The oligonucleotide primers 5′-ACGAACATATGAACTCTGTCTTTTACAATCCGTT-3′ (forward), 5′-AAGCACTCGAGTTACCACTCTACAGTATCTTCGTGGG-3′ (reverse) and 5′-CTGGAATTCATATGGCCCTGAAATTTGAAGCCGAC-3′ (forward), 5′-CTAGATACTCGAGCTACCACTCTACAGTATCTTCGTGG-3′ (reverse) containing NdeI and XhoI sites were synthesized for the amplification of DNA fragments encoding the full length and the capsid protease domain, respectively. The primers were designed on the basis of GenBank accession No. NP_819015. The NdeI and XhoI restriction sites introduced into the primers allowed the cloning of these DNA fragments into the corresponding sites of the pET28c vector in which a thrombin protease cleavage site had been substituted by a TEV (tobacco etch virus) protease cleavage site (a kind gift from Dr S. Karthikeyan, Institute of Microbial Technology, Chandigarh). The resulting recombinant proteins had an additional 21 amino-acid residues at the N-terminus including the His6 tag and TEV protease cleavage site (MGSSHHHHHHSSENLYFQGHM). The DNA fragments were amplified by PCR using these primers and purified using a PCR purification kit (Qiagen, USA) according to the manufacturer’s instructions to remove unincorporated nucleotides and primers. The purified PCR fragment and pET28c vector were restriction-digested with NdeI and XhoI restriction enzymes. The digested DNA fragments were separated by 1% agarose gel electrophoresis, purified using a DNA gel extraction kit (Qiagen, USA) and ligated with T4 DNA ligase. Escherichia coli DH5α competent cells were transformed by the heat-shock method (Inoue et al., 1990 ▶) using the ligation mixture. Some of the colonies obtained by plating the transformed cells on Luria–Bertani (LB) agar plates containing 50 µg ml−1 kanamycin were picked and grown overnight. Plasmids were isolated using a MiniPrep plasmid-isolation kit (Qiagen, USA) and screened for the presence of the constructs by PCR and restriction-enzyme digestion. The integrity of the resulting plasmids was confirmed by sequencing in both directions using T7 promoter and T7 terminator primers.
2.2. Expression of capsid protein
For the production of N-terminally His-tagged recombinant AVCP, Rosetta (DE3) cells were transformed with plasmid containing the AVCP-encoding DNA fragment. Bacterial cultures of the transformed cells were grown in LB broth supplemented with 50 µg ml−1 kanamycin and 35 µg ml−1 chloramphenicol at 310 K to an optical density of 0.6 at 600 nm (OD600). Expression was then induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the culture was grown for 4 h at 310 K after induction. Finally, the cells were harvested by centrifugation at 8000g and 277 K and the pellets were stored at 253 K until further use.
2.3. Purification of AVCP
The cell pellet from 1 l culture was resuspended in binding buffer (50 mM Tris–HCl pH 7.6, 15 mM imidazole, 100 mM NaCl) and lysed using a cell disruptor (Constant Systems Ltd, Daventry, England). The cell lysate was centrifuged at 14 000g for 45 min at 277 K. The clarified supernatant was loaded and incubated for half an hour on a gravity-flow column containing Ni–NTA beads (Qiagen, USA) pre-equilibrated with binding buffer at 277 K. Recombinant capsid protein was eluted with 250 mM imidazole in 50 mM Tris–HCl pH 7.6, 100 mM NaCl. The fractions containing purified protein were pooled and dialyzed overnight at 277 K against dialysis buffer consisting of 50 mM Tris–HCl pH 7.6, 20 mM NaCl, 3 mM EDTA. TEV protease was added to the protein sample in a 1:25 ratio for His-tag cleavage. EDTA was removed from the protein sample by dialysis in the same buffer but without EDTA. A reverse Ni–NTA column was run to remove the uncleaved protein, cleaved His tag and His-tagged TEV protease. His-tag-cleaved AVCP present in the flowthrough was collected and concentrated to ∼6 mg ml−1. The protein was further purified by gel-filtration chromatography using a pre-equilibrated HiLoad Superdex 75 16/60 column (GE Healthcare) and ÄKTApurifier system (GE Healthcare) that was operated at 277 K with a flow rate of 0.5 ml min−1. The size-exclusion column was calibrated with an LMW Calibration Kit containing bovine serum albumin (66 kDa), ovalbumin (45 kDa), trypsin (23 kDa) and lysozyme (14 kDa) for determination of the void volume, construction of the standard curve and estimation of the molecular weight of the purified protein. The gel-filtration eluate was collected in 2 ml fractions and the purity of the fractions was analyzed by Coomassie Blue-stained SDS–PAGE. The fractions containing pure protein sample were then pooled and concentrated to ∼10 mg ml−1 using an Amicon Ultra-15 concentrator with a cutoff value of 3 kDa (Millipore, Bedford, Massachusetts, USA). The concentration of the purified protein was determined by UV absorbance spectroscopy at 280 nm using a calculated extinction coefficient of 22 460 M −1 cm−1.
2.4. Crystallization
The purified protein was crystallized using the sitting-drop vapour-diffusion method. The protein concentration for crystallization was ∼10 mg ml−1 in 50 mM Tris–HCl pH 7.6, 20 mM NaCl. Crystal screens from Hampton Research were used for optimization of the crystal-growth conditions. The protein and the reservoir buffer were used in a 1:1 ratio and were equilibrated against 50 µl reservoir buffer. Crystals were grown in 100 mM bis-tris pH 6.5, 28%(w/v) polyethylene glycol monomethyl ether 2000 at 293 K. Crystals of the AVCP–dioxane complex were prepared by soaking the native protein crystals in 0.1 M bis-tris pH 6.5, 25%(w/v) polyethylene glycol 3350 containing 10 mM dioxane at 293 K for a few seconds.
2.5. Data collection
For high-resolution data collection, the composition of the cryoprotectant was optimized by testing various cryoprotectant agents. The AVCP crystals were first briefly soaked in 15%(w/v) glycerol for 1–2 s; this was followed by crystal annealing and the crystals were then re-soaked in 12.5%(w/v) ethylene glycol prior to X-ray data collection. A similar procedure was followed for data collection from the AVCP–dioxane complex crystals. X-ray data were collected at the home source with a MAR 345 imaging-plate system using Cu Kα radiation generated by a Bruker–Nonius Microstar H rotating-anode generator operated at 45 kV and 60 mA. The data were collected under cryogenic conditions (100 K) at a wavelength of 1.5418 Å. The diffraction data were processed with the HKL-2000 package (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
The full-length Aura virus capsid protein-encoding gene was cloned into pET28c vector for bacterial expression. However, the full-length recombinant capsid protein did not show any expression; this may be the consequence of the presence of an unstructured amino-terminal RNA-binding domain in Aura virus capsid protein. Therefore, the N-terminal domain (residues 1–109) of Aura virus capsid protein was deleted. Another construct containing the carboxy-terminal protease domain (residues 110–267) was designed and cloned into pET28c vector for bacterial expression. The recombinant plasmids were sequenced and the sequencing results showed that the Val120, Gly121, Val190, Val212, Val240, Pro241 and Gly242 residues of the GenBank sequence were substituted by Ala120, Val121, Gly190, Ala212, Gly240, Ala241 and Arg242 in the AVCP construct. AVCP showed expression in the soluble form and the recombinant protein was purified using the IMAC (immobilized metal-affinity chromatography) method. The N-terminal His tag was cleaved by TEV protease and the protein was re-purified using a reverse Ni–NTA column. The flowthrough of the reverse Ni–NTA column containing AVCP without His tag was collected, concentrated and loaded onto the gel-filtration column. Size-exclusion chromatography, which was the last step of purification, provided a homogenous preparation of purified AVCP (Fig. 1 ▶). Using a standard curve based on molecular-weight markers, the molecular weight of the major elution peak containing AVCP protein was calculated and was estimated to be approximately 17 kDa. This suggests that AVCP exists in a monomeric form. The estimated yield of pure protein was ∼13 mg per litre of culture. The purified protein was concentrated to ∼10 mg ml−1 and used for crystallization.
Figure 1.

Gel-filtration profile and SDS–PAGE analysis of AVCP. Lane 1, molecular-weight markers (kDa); lane 2, pellet containing insoluble protein fraction; lane 3, supernatant containing soluble protein fraction; lane 4, purified His-tagged AVCP; lane 5, purified AVCP without His tag.
Crystals of AVCP were obtained in 15–20 d at 293 K using 100 mM bis-tris buffer pH 6.5 and 28%(w/v) polyethylene glycol monomethyl ether 2000 as the precipitant (Fig. 2 ▶). The crystals of native AVCP diffracted to 1.81 Å resolution with one molecule per asymmetric unit. The crystals belonged to the monoclinic space group C2, with 90.2% completeness and an R merge of 5.7%. For AVCP–dioxane complex formation, the native crystals were soaked in mother liquor containing dioxane. The AVCP–dioxane complex crystals also belonged to the monoclinic space group C2 with the same unit-cell parameters. Diffraction data were collected to a resolution of 1.98 Å with 86.2% completeness and an R merge of 4.8% (Fig. 3 ▶). The data-collection statistics for the native and complex crystals are summarized in Table 1 ▶. As capsid protein is the first and the key enzyme required for structural polyprotein processing, it is a potential antiviral drug target. The crystal structure of the capsid protease from Aura virus will not only assist in structure-based drug design but will also highlight the differences between alphavirus members that govern viral RNA-packaging and virus-assembly processes.
Figure 2.

Crystals of Aura virus capsid protease. The longest dimensions of a typical crystal were ∼50–100 µm.
Figure 3.

Diffraction of AVCP crystals using in-house radiation at the Macromolecular Crystallographic Facility (MCU), IIC. (a) Native AVCP crystal diffraction; the resolution at the edge of the plate is 1.81 Å. (b) AVCP–dioxane complex crystal diffraction; the resolution at the edge of the plate is 1.98 Å.
Table 1. Data-collection statistics for AVCP and its complex with dioxane.
Values in parentheses are for the last resolution shell.
| Capsid protein | Complex with dioxane | |
|---|---|---|
| Space group | C2 | C2 |
| Unit-cell parameters (Å) | a = 79.6, b = 35.2, c = 49.5 | a = 79.6, b = 35.2, c = 49.5 |
| Resolution range (Å) | 50.0–1.81 (1.85–1.81) | 50.0–1.98 (2.02–1.98) |
| Completeness (%) | 90.2 (70.7) | 86.2 (45.7) |
| Rmerge† (%) | 5.7 (25.5) | 4.8 (28.2) |
| Mean I/σ(I) | 22.6 (2.9) | 20.3 (1.8) |
| No. of observed reflections | 45053 | 24420 |
| No. of unique reflections | 11410 (436) | 7989 (160) |
| Molecules per asymmetric unit | 1 | 1 |
| Matthews coefficient (Å3 Da−1) | 2.0 | 1.99 |
| Solvent content (%) | 38.5 | 38.5 |
| Multiplicity | 3.9 (2.1) | 3.1 (1.9) |
R
merge = 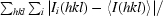
 , where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Acknowledgments
The authors are grateful to and thank the Macromolecular Crystallographic Facility (MCU) at IIC, IIT Roorkee for data collection. The authors thank Dr S. Karthikeyan for his kind gift. ST thanks the Defence Research and Development Organization (DRDO), Government of India for funding. MA thanks the Council of Scientific and Industrial Research (CSIR), SD thanks the MHRD and SP thanks the DBT, Government of India for financial support.
References
- Aliperti, G. & Schlesinger, M. J. (1978). Virology, 90, 366–369. [DOI] [PubMed]
- Cheng, R. H., Kuhn, R. J., Olson, N. H., Rossmann, M. G., Choi, H.-K., Smith, T. J. & Baker, T. S. (1995). Cell, 80, 621–630. [DOI] [PMC free article] [PubMed]
- Choi, H.-K., Lu, G., Lee, S., Wengler, G. & Rossmann, M. G. (1997). Proteins, 27, 345–359. [DOI] [PubMed]
- Choi, H.-K., Tong, L., Minor, W., Dumas, P., Boege, U., Rossmann, M. G. & Wengler, G. (1991). Nature (London), 354, 37–43. [DOI] [PubMed]
- Faragher, S. G., Meek, A. D., Rice, C. M. & Dalgarno, L. (1988). Virology, 163, 509–526. [DOI] [PubMed]
- Hong, E. M., Perera, R. & Kuhn, R. J. (2006). J. Virol. 80, 8848–8855. [DOI] [PMC free article] [PubMed]
- Inoue, H., Nojima, H. & Okayama, H. (1990). Gene, 96, 23–28. [DOI] [PubMed]
- Jose, J., Snyder, J. E. & Kuhn, R. J. (2009). Future Microbiol. 4, 837–856. [DOI] [PMC free article] [PubMed]
- Kail, M., Hollinshead, M., Ansorge, W., Pepperkok, R., Frank, R., Griffiths, G. & Vaux, D. (1991). EMBO J. 10, 2343–2351. [DOI] [PMC free article] [PubMed]
- Kim, H. Y., Kuhn, R. J., Patkar, C., Warrier, R. & Cushman, M. (2007). Bioorg. Med. Chem. 15, 2667–2679. [DOI] [PMC free article] [PubMed]
- Kinney, R. M., Johnson, B. J., Brown, V. L. & Trent, D. W. (1986). Virology, 152, 400–413. [DOI] [PubMed]
- Lee, S., Kuhn, R. J. & Rossmann, M. G. (1998). Proteins, 33, 311–317. [PubMed]
- Levinson, R. S., Strauss, J. H. & Strauss, E. G. (1990). Virology, 175, 110–123. [DOI] [PubMed]
- Metallo, S. J. (2010). Curr. Opin. Chem. Biol. 14, 481–488. [DOI] [PMC free article] [PubMed]
- Morillas, M., Eberl, H., Allain, F. H., Glockshuber, R. & Kuennemann, E. (2008). J. Mol. Biol. 376, 721–735. [DOI] [PubMed]
- Mukhopadhyay, S., Chipman, P. R., Hong, E. M., Kuhn, R. J. & Rossmann, M. G. (2002). J. Virol. 76, 11128–11132. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Owen, K. E. & Kuhn, R. J. (1996). J. Virol. 70, 2757–2763. [DOI] [PMC free article] [PubMed]
- Owen, K. E. & Kuhn, R. J. (1997). Virology, 230, 187–196. [DOI] [PubMed]
- Perera, R., Owen, K. E., Tellinghuisen, T. L., Gorbalenya, A. E. & Kuhn, R. J. (2001). J. Virol. 75, 1–10. [DOI] [PMC free article] [PubMed]
- Reichert, E., Clase, A., Bacetty, A. & Larsen, J. (2009). Biosecur. Bioterror. 4, 413–427. [DOI] [PubMed]
- Rümenapf, T., Brown, D. T., Strauss, E. G. & Strauss, J. H. (1995). J. Virol. 69, 1741–1746. [DOI] [PMC free article] [PubMed]
- Rümenapf, T., Strauss, E. G. & Strauss, J. H. (1994). J. Virol. 69, 56–62. [DOI] [PMC free article] [PubMed]
- Rümenapf, T., Strauss, E. G. & Strauss, J. H. (1995). Virology, 208, 621–633. [DOI] [PubMed]
- Rupp, J. C., Jundt, N. & Hardy, R. W. (2011). J. Virol. 85, 3449–3460. [DOI] [PMC free article] [PubMed]
- Skoging, U., Vihinen, M., Nilsson, L. & Liljeström, P. (1996). Structure, 15, 519–529. [DOI] [PubMed]
- Suomalainen, M., Liljeström, P. & Garoff, H. (1992). J. Virol. 66, 4737–4747. [DOI] [PMC free article] [PubMed]
- Tomar, S., Hardy, R. W., Smith, J. L. & Kuhn, R. J. (2006). J. Virol. 80, 9962–9969. [DOI] [PMC free article] [PubMed]
- Tong, L., Wengler, G. & Rossmann, M. G. (1993). J. Mol. Biol. 230, 228–247. [DOI] [PubMed]
- Uversky, V. N. (2002). Protein Sci. 11, 739–756. [DOI] [PMC free article] [PubMed]
- Uversky, V. N., Oldfield, C. J. & Dunker, A. K. (2008). Annu. Rev. Biophys. 37, 215–246. [DOI] [PubMed]