

The cloning, purification, crystallization and preliminary X-ray diffraction analysis of a novel staphylococcal phage dUTPase is reported. This protein contains a specific polypeptide insertion that is potentially responsible for modulation of expression of superantigenicity island genes.
Keywords: ϕ11 helper phage, dUTPases
Abstract
Staphylococcus aureus superantigen-carrying pathogenicity islands (SaPIs) play a determinant role in spreading virulence genes among bacterial populations that constitute a major health hazard. Repressor (Stl) proteins are responsible for the transcriptional regulation of pathogenicity island genes. Recently, a derepressing interaction between the repressor Stl SaPIbov1 and dUTPase from the ϕ11 helper phage has been suggested [Tormo-Más et al. (2010 ▶), Nature (London), 465, 779–782]. Towards elucidation of the molecular mechanism of this interaction, this study reports the expression, purification and X-ray analysis of ϕ11 dUTPase, which contains a phage-specific polypeptide segment that is not present in other dUTPases. Crystals were obtained using the hanging-drop vapour-diffusion method at room temperature. Data were collected to 2.98 Å resolution from one type of crystal. The crystal of ϕ11 dUTPase belonged to the cubic space group I23, with unit-cell parameters a = 98.16 Å, α = β = γ = 90.00°.
1. Introduction
Staphylococcus aureus is a major human bacterial pathogen responsible for frequent infections causing severe diseases. It constitutes a serious healthcare problem especially owing to the rapid appearance of resistant strains, most notably methicillin-resistant Staphylococcus aureus (MRSA; van Belkum, 2011 ▶). Bacterial virulence in S. aureus has multiple major factors, including an intriguing network of communication between pathogenicity islands and helper phages (Chen & Novick, 2009 ▶). Recently, it has been proposed that transcriptional regulation of superantigen-carrying pathogenicity islands (SaPIs) relies on helper-phage proteins with multiple functions (Tormo-Más et al., 2010 ▶). Importantly, binding of the transcription-related repressor factor Stls encoded within the SaPI genomic regions to their specific promoter elements has been suggested to be modulated by interaction with moonlighting proteins. In one such interaction, the binding of the Stl repressor of the pathogenicity island SaPIbov1 to the ϕ11 helper phage dUTPase protein suppressed the repressor function of SaPIbov1. This interaction has been suggested to rely on a protein segment of ϕ11 dUTPase that is not involved in catalytic activity (Tormo-Más et al., 2010 ▶; Vértessy & Tóth, 2009 ▶).
To obtain insight into the molecular details of this intriguing interaction, we aim to examine complex formation between the Stl repressor SaPIbov1 and ϕ11 dUTPase by determination of the three-dimensional structure of the interacting proteins. As a first step in this process, we report the cloning, purification and crystallization of ϕ11 dUTPase.
2. Materials and methods
2.1. Cloning
The cDNA of the dUTPase protein (GenBank ID AAL82253.1) from the ϕ11 helper phage was synthesized as a codon-optimized (EnCor Biotechnology Inc.) construct. The codon-optimized construct was cloned into the vector pETDuet-1 from Novagen with NdeI and XhoI restriction sites using the services of Eurofins MWG Operon. No affinity tag was attached to the protein sequence. The recombinant plasmid DUET-ϕDUT was verified by DNA sequencing on both strands using DuetUP2 TTGTACACGGCCGCATAATC and T7 terminator GCTAGTTATTGCTCAGCGG primers.
2.2. Protein expression and purification
The plasmid DUET-ϕDUT was transformed into Escherichia coli strain BL21 Rosetta (DE3). The cells were cultured at 310 K in LB medium. The cultures were induced using 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at the logarithmic growth phase. After induction, the cell cultures were grown for a further 4 h followed by centrifugation at 277 K. All subsequent procedures were carried out on ice, except where noted otherwise.
The cell pellet was resuspended in lysis buffer [10 mM HEPES pH 7.5, 10 mM KCl, 10 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 µg ml−1 DNase I, 10 µg ml−1 RNase and one tablet of EDTA-free Complete ULTRA protease-inhibitor preparation (Roche, Switzerland) per 50 ml solution], sonicated and then centrifuged at 16 000g. The supernatant solution was applied onto a Q-Sepharose (GE Healthcare) anion-exchange column in 10 mM HEPES, 10 mM KCl, 10 mM β-mercaptoethanol, 0.1 mM PMSF pH 8.0 (buffer A). Elution was followed at a wavelength of 280 nm. The column was washed with buffer A until no further protein elution was observed. ϕ11 dUTPase protein was eluted using a 45 ml linear gradient of buffer A and buffer B (10 mM HEPES, 10 mM KCl, 1 M NaCl, 10 mM β-mercaptoethanol, 0.1 mM PMSF pH 8.0). Elution of ϕ11 dUTPase was observed at 0.35 M NaCl.
Ion-exchange chromatography was followed by gel filtration on a Superdex 75 column (GE Healthcare) using an ÄKTApurifier instrument in buffer A. Elution of ϕ11 dUTPase was observed at an elution volume corresponding to a native molecular mass of 51.8 kDa. Considering that the molecular mass of the protein calculated from the primary sequence was 18.35 kDa, the gel-filtration data indicate that ϕ11 dUTPase most probably adopts the trimeric oligomer structure characteristic of dUTPases.
Protein fractions were analyzed by SDS–PAGE, which indicated that the protein purity was >90% after the second chromatography step. The protein concentration was determined using an A 0.1% 280nm of 0.786 estimated from the amino-acid composition. The protein solution was concentrated to 10 mg ml−1.
2.3. Crystallization
Protein samples were used for crystallization immediately after purification. Initial crystallization trials were performed using the JCSG-plus screen (Molecular Dimensions) and the vapour-diffusion method at room temperature. Hanging-drop plates were set up using 1 µl protein solution and an equal amount of reservoir solution. The protein solution consisted of 5 mg ml−1 ϕ11 dUTPase, 2.3 mM α,β-imido-dUTP (a slowly hydrolysable dUTP substrate analogue) and 5 mM MgCl2 (the metal cofactor). Crystals could be observed in several conditions from the first screen. Crystals of up to 0.2 mm in size were grown using a well solution consisting of 0.1 M ammonium acetate, 0.1 M bis-tris pH 5.5, 17%(w/v) PEG 10 000 (condition A; Fig. 1 ▶ a). Smaller crystals were grown using a well solution consisting of 0.2 M ammonium nitrate pH 6.3, 20%(w/v) PEG 3350 (condition B; Fig. 1 ▶ b). Cryoprotection of the reservoir solution was tested in a liquid-nitrogen stream at 100 K (Oxford Cryosystem); samples were flash-cooled and prepared for X-ray testing.
Figure 1.

Crystals of native ϕ11 dUTPase. (a) Crystals grown from condition A, pre-tested on the home source and used for data collection. (b) Crystals from condition B and pre-tested on the home source. Scale bars are shown.
2.4. X-ray diffraction, data collection and processing
Pre-experimental home-source testing was performed on a Rigaku rotating-anode instrument (RU-200 generator, confocal optics, R-AXIS IV++ detector, Cu Kα radiation) and on a SuperNova sealed-tube system equipped with an Eos CCD detector (Agilent).
X-ray data were collected on ESRF beamline ID14-1 at 0.9334 Å wavelength and 100 K. Diffraction data were collected to a resolution of 2.98 Å (Fig. 2 ▶). Molecular replacement was employed using the structure of Mycobacterium tuberculosis dUTPase (PDB entry 3hza; Pecsi et al., 2010 ▶), which shows 32% sequence identity to ϕ11 dUTPase. Crystallographic data were processed using iMOSFLM (Battye et al., 2011 ▶) and SCALA (Evans, 2006 ▶) from the CCP4 software package (Winn et al., 2011 ▶).
Figure 2.

A diffraction image collected on a synchrotron beamline. The black circle corresponds to the resolution limit of 2.98 Å.
3. Results and discussion
The ϕ11 dUTPase was successfully expressed using the E. coli expression host and the T7–pET vector system in accordance with our previous results on dUTPase proteins from other sources (Varga et al., 2007 ▶, 2008 ▶; Németh-Pongrácz et al., 2007 ▶; Kovári et al., 2004 ▶; Barabás et al., 2004 ▶; Mustafi et al., 2003 ▶). Purification using ion-exchange and size-exclusion chromatography steps resulted in protein preparations that were suitable for crystallization. Denaturing SDS–PAGE analysis indicated that the purified protein has an apparent molecular mass of 18 kDa, corresponding to the monomer mass of the ϕ11 dUTPase, which includes a phage-specific polypeptide segment of approximately 40 residues (Tormo-Más et al., 2010 ▶). The oligomerization status of ϕ11 dUTPase in solution was assessed by analytical gel filtration and indicated a trimeric organization, as observed for most dUTPases (Persson et al., 2001 ▶; Cedergren-Zeppezauer et al., 1992 ▶; Vértessy & Tóth, 2009 ▶; Fiser & Vértessy, 2000 ▶).
Using the JCSG-plus screen, many conditions provided crystals; however, only two conditions led to diffracting protein crystal specimens (Fig. 1 ▶). The crystals were pre-tested on the home source. A full data set was collected on the ESRF ID14-1 beamline from a crystal segment broken away from a specimen similar to that shown in Fig. 1 ▶(a) and the results are summarized in Table 1 ▶. No evidence of twinning was found. X-ray data analysis showed that the asymmetric unit contains one molecule. Matthews coefficient and solvent-content estimations were performed using the CCP4 software (Winn et al., 2011 ▶). The calculated Matthews coefficient (Matthews, 1968 ▶) and solvent content are 2.19 Å3 Da−1 and 43.85%, respectively. Considering the high homology of the ϕ11 and M. tuberculosis dUTPase proteins (32% sequence identity), we plan to solve the phase problem by molecular replacement using the structure of the monomer of the M. tuberculosis dUTPase (PDB entry 3hza) as the search model.
Table 1. X-ray data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | I23 |
| Resolution (Å) | 40.07–2.98 (3.14–2.98) |
| Unit-cell parameters (Å, °) | a = 98.16, α = β = γ = 90.00 |
| Total reflections | 18394 (2571) |
| Unique reflections | 3337 (490) |
| Completeness (%) | 99.7 (99.6) |
| Rmerge† | 0.096 (0.474) |
| 〈I/σ(I)〉 | 11.2 (3.5) |
R
merge = 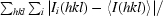
 , where I
i(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity over symmetry-related observations of reflection hkl.
, where I
i(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity over symmetry-related observations of reflection hkl.
Acknowledgments
This work was supported by OTKA K68229, OTKA-A08 CK78646, OTKA NK-84008, OTKA K72973, OTKA NK67800, Howard Hughes Medical Institutes No. 55000342, Alexander von Humboldt-Stiftung, Germany, the New Hungary Development Plan (Project ID TAMOP-4.2.1/B-09/1/KMR-2010-0002, -0003) and the Baross program of the New Hungary Development Plan (Project ID 3DSTRUCT, OMFB-00266/2010 REG-KM-09-1-2009-0050). We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron-radiation facilities and we thank Dr Stéphanie Monaco for assistance in using beamline ID14-1.
References
- Barabás, O., Pongrácz, V., Kovári, J., Wilmanns, M. & Vértessy, B. G. (2004). J. Biol. Chem. 279, 42907–42915. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Belkum, A. van (2011). Adv. Exp. Med. Biol. 697, 273–288. [DOI] [PubMed]
- Cedergren-Zeppezauer, E. S., Larsson, G., Nyman, P. O., Dauter, Z. & Wilson, K. S. (1992). Nature (London), 355, 740–743. [DOI] [PubMed]
- Chen, J. & Novick, R. P. (2009). Science, 323, 139–141. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fiser, A. & Vértessy, B. G. (2000). Biochem. Biophys. Res. Commun. 279, 534–542. [DOI] [PubMed]
- Kovári, J., Barabás, O., Takács, E., Békési, A., Dubrovay, Z., Pongrácz, V., Zagyva, I., Imre, T., Szabó, P. & Vértessy, B. G. (2004). J. Biol. Chem. 279, 17932–17944. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mustafi, D., Bekesi, A., Vertessy, B. G. & Makinen, M. W. (2003). Proc. Natl Acad. Sci. USA, 100, 5670–5675. [DOI] [PMC free article] [PubMed]
- Németh-Pongrácz, V., Barabás, O., Fuxreiter, M., Simon, I., Pichová, I., Rumlová, M., Zábranská, H., Svergun, D., Petoukhov, M., Harmat, V., Klement, E., Hunyadi-Gulyás, E., Medzihradszky, K. F., Kónya, E. & Vértessy, B. G. (2007). Nucleic Acids Res. 35, 495–505. [DOI] [PMC free article] [PubMed]
- Pecsi, I., Leveles, I., Harmat, V., Vertessy, B. G. & Toth, J. (2010). Nucleic Acids Res. 38, 7179–7186. [DOI] [PMC free article] [PubMed]
- Persson, R., Cedergren-Zeppezauer, E. S. & Wilson, K. S. (2001). Curr. Protein Pept. Sci. 2, 287–300. [DOI] [PubMed]
- Tormo-Más, M. A., Mir, I., Shrestha, A., Tallent, S. M., Campoy, S., Lasa, I., Barbé, J., Novick, R. P., Christie, G. E. & Penadés, J. R. (2010). Nature (London), 465, 779–782. [DOI] [PMC free article] [PubMed]
- Varga, B., Barabás, O., Kovári, J., Tóth, J., Hunyadi-Gulyás, E., Klement, E., Medzihradszky, K. F., Tölgyesi, F., Fidy, J. & Vértessy, B. G. (2007). FEBS Lett. 581, 4783–4788. [DOI] [PubMed]
- Varga, B., Barabás, O., Takács, E., Nagy, N., Nagy, P. & Vértessy, B. G. (2008). Biochem. Biophys. Res. Commun. 373, 8–13. [DOI] [PubMed]
- Vértessy, B. G. & Tóth, J. (2009). Acc. Chem. Res. 42, 97–106. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.