

Two orthologous putative tRNA methyltransferases from P. furiosus and T. thermophilus have been expressed, purified and crystallized. X-ray diffraction data were collected to 2.2 and 2.05 Å, respectively.
Keywords: tRNA, methyltransferases, ttc1157, pf1002
Abstract
Methyltransferases form a major class of tRNA-modifying enzymes that are needed for the proper functioning of tRNA. Here, the expression, purification and crystallization of two related putative tRNA methyltransferases from two kingdoms of life are reported. The protein encoded by the gene pf1002 from the archaeon Pyrococcus furiosus was crystallized in the monoclinic space group P21. A complete data set was collected to 2.2 Å resolution. The protein encoded by the gene ttc1157 from the eubacterium Thermus thermophilus was crystallized in the trigonal space group P3221. A complete data set was collected to 2.05 Å resolution.
1. Introduction
During their synthesis, tRNA molecules undergo a series of maturation steps that convert a precursor nucleotide chain into the final functional tRNA (Hopper & Phizicky, 2003 ▶). In this process, tRNAs undergo a series of post-transcriptional nucleoside modifications that are needed for (i) the proper folding and stability of tRNA, (ii) correct anticodon–codon recognition at the decoding centre of the small ribosomal subunit and (iii) recognition by the correct aminoacyltransferase. Presently, more than 100 such RNA modifications are known, with methylations, catalysed by RNA methyltransferases, being the most common (Motorin & Helm, 2010 ▶).
RNA methyltransferases are classified into four superfamilies. The largest superfamily consists of the Rossmann-fold methyltransferases (RFMs), which show a fold very similar to the classical (di-)nucleotide-binding Rossmann fold. The SPOUT superfamily, named after the first two proteins identified with this fold (spoU and TrmD), is characterized by a fold showing resemblance to the RFMs but with a deep topological knot forming the cofactor-binding site. The radical SAM methyltransferases catalyse methyl transfer using a radical reaction and adopt a fold resembling a TIM barrel. Finally, the smallest superfamily is the FAD/NAD(p) methyltransferase family (Urbonavicius et al., 2005 ▶). With the exception of the members of the latter family, all of these enzymes use S-adenosylmethionine (SAM) as a methyl donor (Motorin & Helm, 2011 ▶). Some methyltransferases are capable of recognizing their target and catalysing the modification reaction independently of other domains. However, a second domain can often be observed in the protein adjacent to the methyltransferase domain, which is involved in binding the RNA substrate. The most common RNA-binding domains are PUA (named after pseudouridine synthase and archaeosine transglycosylase), TRAM (named after Trm2p and MiaB) and THUMP (named after 4-thiouridine, methyltransferases and pseudouridine synthases) (Czerwoniec et al., 2009 ▶).
The THUMP domain was initially proposed from sequence-profile searches to be an ancient RNA-binding domain (Aravind & Koonin, 2001 ▶) and is found in RNA deaminases, methyltransferases, pseudouridine synthases and 4-thiouridine synthases, amongst others. The minimal core of the THUMP domain, as originally proposed by Aravind and Koonin, consists of a five-stranded mixed β-sheet packed against two α-helices (Aravind & Koonin, 2001 ▶; Waterman et al., 2006 ▶). Often, this core THUMP domain is N-terminally fused to a so-called NFLD domain (N-terminal ferredoxin-like domain) consisting of a four-stranded antiparallel β-sheet packed against two helices (Waterman et al., 2006 ▶).
To the best of our knowledge, only two examples of proteins containing a THUMP domain linked to a methyltransferase domain have been described in the literature: the tRNA:m2 2G10 methyltransferase from Pyrococcus abyssi (Pab1283; Gabant et al., 2006 ▶; Armengaud et al., 2004 ▶) and Trm14 from Methanocaldococcus jannaschii (Menezes et al., 2011 ▶). Pab1283 catalyses the formation of either N 2-monomethylguanosine (m2G) or N 2-dimethylguanosine (m2 2G) at position 10 of certain tRNA molecules, whereas Trm14 catalyses the formation of m2G at position 6. Although it has been proposed that the THUMP domain is involved in the modification of nucleotides in the three-dimensional core of the tRNA molecules, very little is yet known on the exact role and binding mode of the THUMP domain in this respect.
Here, we report the expression, purification, crystallization and initial X-ray crystallographic analysis of two orthologous proteins from two kingdoms of life: the archaeal protein encoded by the gene pf1002 from P. furiosus, hereafter referred to as Pf1002, and the eubacterial homologue encoded by the gene ttc1157 from Thermus thermophilus, hereafter referred to as TTC1157. Sequence alignment shows that these proteins are similar to known tRNA methyltransferases and suggests that these proteins consist of an N-terminal THUMP domain coupled to a C-terminal methyltransferase domain. Indeed, analysis of Pf1002 and TTC1157 using the Phyre server (Kelley & Sternberg, 2009 ▶) predicts that their N-terminal domains have high similarity (E values of 1.6 × 10−6 for Pf1002 and 12.2 × 10−10 for TTC1157) to the THUMP domain of the 4-thiouridine synthase ThiI (PDB entry 2c5s; Waterman et al., 2006 ▶), while their C-terminal domains have high similarity (E values of 1.1 × 10−13 for Pf1002 and 9.1 × 10−10 for TTC1157) to the RFM domain of the 16S rRNA methyltransferase RmsC (PDB entry 2pjd; Sunita et al., 2007 ▶). Hence, both proteins are speculated to be involved in the methylation of tRNA. The structures of these proteins, combined with biochemical studies, will be used to reveal their role in tRNA modification. Furthermore, these structures might provide further insights into the role of the THUMP domain in RNA binding and modification.
2. Experimental
2.1. Protein expression and purification
The gene pf1002 was amplified by PCR from P. furiosus genomic DNA using the forward and reverse oligonucleotides PF1002-1 (5′-GAC CAT ATG AAG TTT TTG CTC ACA ACA GCC-3′), bearing an NdeI site (bold), and PF1002-2 (5′-GAC CTC GAG TTT CAC TAC ATA CAA GTG AAC-3′), bearing an XhoI site (bold). The 1098 bp fragment was cloned into pJet1.2/blunt vector and subsequently digested by NdeI/XhoI for subcloning into the pET30 expression vector, allowing the expression of a C-terminally His-tagged protein.
The His-tagged Pf1002 protein was expressed in Escherichia coli Rosetta (DE3). The cells were grown at 310 K to an OD600 of 0.5 in LB medium. After induction with 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) the cells were grown at 288 K and were harvested the next day by centrifugation. The cells were resuspended in buffer A (50 mM Tris–HCl, 500 mM NaCl pH 8) and lysed by sonication. The His-tagged protein was purified using a nickel Sepharose column. Bound protein was eluted using a linear imidazole gradient and the fractions were analysed on SDS–PAGE (Laemmli, 1970 ▶).
The gene ttc1157 was amplified by PCR from T. thermophilus genomic DNA using the forward and reverse oligonucleotides TTC1157-1 (5′-G ATC CAT ATG TGG CTT GAG GCC ACC ACC CAC-3′), bearing an NdeI site (bold), and TTC1157-2 (5′-G ATC GTC GAC CTA GAG CTT CTC TAG GAC GAA GAC C-3′), bearing a SalI site (bold). The 1008 bp fragment was cloned into pJet1.2/blunt vector and subsequently digested by NdeI/SalI for subcloning into pET28, allowing the expression of an N-terminally His-tagged protein.
The His-tagged TTC1157 protein was expressed in E. coli BL21 (DE3). Cells were grown and the protein was purified according to the method described for Pf1002, except for the use of 250 mM NaCl in all buffers.
2.2. Crystallization and dynamic light scattering
Dynamic light-scattering (DLS) experiments were performed on a DynaPro Plate Reader (Wyatt Technology) using a protein solution containing either 1 mg ml−1 Pf1002 or TTC1157.
Initial crystallization conditions were established using sitting-drop vapour diffusion at 293 K in a 96-well screen format using a Phoenix crystallization robot (Art Robbins Instruments). Screening kits were purchased from Molecular Dimensions (Morpheus, Stura Footprint, MacroSol, JCSG+) and Jena Bioscience (JB Classic 1–8, JB Cryo 5–8, JB Pentaerythritol, JB Basic 1–4). For the initial screening 100 nl precipitant solution was mixed with 100 nl protein solution. In the case of Pf1002, protein solutions at 10 and 5 mg ml−1 were used in a buffer consisting of 50 mM Tris–HCl pH 8, 10 mM MgCl2, 500 mM NaCl, 280 mM imidazole, 1 mM DTT. For TTC1157, protein solutions at 1 and 0.5 mg ml−1 were used in a buffer consisting of 50 mM Tris–HCl pH 8, 250 mM NaCl and 350 mM imidazole. After refinement of initial hits, the proteins were crystallized at 293 K using hanging-drop vapour diffusion. In the case of Pf1002, 1 µl of a 19 mg ml−1 protein solution was mixed with 1 µl crystallization solution consisting of 100 mM Tris acetate pH 8, 32% PEG 4000, 15% glycerol. In the case of TTC1157, 1 µl protein solution at 2 mg ml−1 was mixed with 1 µl crystallization solution consisting of 100 mM citrate/phosphate pH 3.5, 15% PEG 6000, 200 mM NaCl, 100 mM sodium citrate.
2.3. X-ray diffraction and data processing
The crystals of Pf1002 were flash-cooled in liquid nitrogen using the crystallization reservoir solution as a cryoprotectant. X-ray data were collected at 100 K using a MAR225 CCD detector on beamline ID23-2 at the ESRF (Grenoble, France). Data were collected over a total oscillation range of 180° using oscillation steps of 1° per image and lateral translation of the crystal after 90°. The crystals of TTC1157 were flash-cooled in liquid nitrogen using the mother liquor containing 20% glycerol as a cryoprotectant. X-ray data were collected at 100 K using a MAR225 CCD detector on beamline ID23-2 at the ESRF (Grenoble, France). Data were collected over a total oscillation range of 360° with an oscillation range of 0.5° per image, using the helical shift function provided at the beamline to scan the full length of the crystal in order to circumvent radiation damage. The raw data were processed and scaled using the XDS suite (Kabsch, 2010 ▶).
Attempts to solve the structure using molecular replacement were performed using Phaser (McCoy et al., 2007 ▶).
3. Results
Pf1002 from P. furiosus was overexpressed as a soluble His-tagged protein in the cytoplasm of E. coli Rosetta (DE3). Purification of the protein was performed using a single Ni–NTA affinity chromatography step. Typical yields of 2 mg protein per litre of E. coli culture were obtained. SDS–PAGE confirmed the expected molecular weight of 42 kDa (Fig. 1 ▶ a), whereas gel-filtration experiments on a Superdex 200 16/60 column suggested the protein to be monomeric (data not shown). Finally, DLS experiments showed a polydispersity of the protein in solution of 5.7%, meaning that the sample is monodisperse under the given buffer conditions.
Figure 1.

SDS–PAGE of the purified proteins Pf1002 (a) and TTC1157 (b) used for crystallization. Molecular-weight markers are labelled in kDa.
The best crystals of Pf1002 were generated using a crystallization solution consisting of 100 mM Tris acetate pH 8, 32% PEG 4000, 15% glycerol. Using this condition, crystals grew to dimensions of about 0.10 × 0.025 × 0.005 mm within 3 d (Fig. 2 ▶ a). A data set was collected on beamline ID23-2 at the ESRF (Grenoble, France) to a resolution of 2.0 Å (Fig. 3 ▶). The data were processed with XDS using a resolution cutoff of 2.2 Å. A total of 162 685 reflections were merged into 46 452 unique reflections, resulting in an R meas of 12.7% and a completeness of 99.7%. The data-collection statistics are summarized in Table 1 ▶. The protein crystallized in space group P21 and the asymmetric unit was estimated to contain two molecules of Pf1002, corresponding to a Matthews coefficient of 2.66 Å3 Da−1 (Matthews, 1968 ▶) and a solvent content of 53.8%.
Figure 2.

Crystals of P. furiosus Pf1002 (a) and T. thermophilus TTC1157 (b).
Figure 3.

X-ray diffraction image from a P. furiosus Pf1002 crystal. The edge of the detector corresponds to a resolution of 2.0 Å.
Table 1. Data-collection statistics for Pf1002 and TTC1157.
Values in parentheses are for the highest resolution shell.
| Pf1002 | TTC1157 | |
|---|---|---|
| X-ray source | ID23-2, ESRF | ID23-2, ESRF |
| X-ray wavelength (Å) | 0.8726 | 0.8726 |
| Temperature (K) | 100 | 100 |
| Space group | P21 | P3221 |
| Unit-cell parameters (Å, °) | a = 85.52, b = 45.33, c = 122.23, β = 91.23 | a = b = 65.86, c = 144.55 |
| Crystal mosaicity (°) | 0.257 | 0.282 |
| Resolution range (Å) | 50–2.2 (2.3–2.2) | 50–2.05 (2.16–2.05) |
| Total/unique reflections | 162685/46452 (19871/5748) | 502157/23534 (74454/3366) |
| Rmerge† (%) | 10.8 (57.7) | 10.1 (52.5) |
| Rmeas‡ (%) | 12.7 (68.4) | 10.4 (53.7) |
| Data completeness (%) | 99.7 (99.7) | 100 (100) |
| Average I/σ(I) | 9.3 (2.7) | 22.4 (8.1) |
| Multiplicity | 3.5 (3.5) | 21.3 (22.1) |
R
merge = 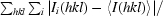
 .
.
R
meas = 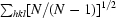
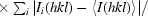 .
.
TTC1157 from T. thermophilus was overexpressed as a soluble His-tagged fusion protein in the cytoplasmic fraction of the E. coli BL21 (DE3) expression strain. After an Ni–NTA affinity-chromatography step pure protein was obtained, with a typical yield of 8 mg protein per litre of E. coli culture. SDS–PAGE confirmed the expected molecular weight of 39 kDa (Fig. 1 ▶ b) and size-exclusion chromatography experiments on a Superdex 75 16/60 column showed the protein to be monomeric in solution (data not shown). DLS experiments showed a polydispersity of the protein in solution of 6.0%, meaning that the sample is monodisperse under the given buffer conditions.
The best crystals of TTC1157 grew using a crystallization buffer consisting of 100 mM citrate–phosphate pH 3.5, 15% PEG 6000, 200 mM NaCl, 100 mM sodium citrate. The crystals grew to dimensions of 0.04 × 0.04 × 0.04 mm (Fig. 2 ▶ b) within 2 d. A data set was collected on beamline ID23-2 at the ESRF (Grenoble, France) to 1.7 Å resolution (Fig. 4 ▶) and was processed to a resolution of 2.05 Å. A total of 502 157 reflections were measured and merged into 23 534 unique reflections, resulting in an R meas of 10.4% and a completeness of 100%. The data-collection statistics are summarized in Table 1 ▶. The protein crystallized in space group P3221 and the asymmetric unit is estimated to contain one molecule of TTC1157, corresponding to a Matthews coefficient of 2.45 Å3 Da−1 (Matthews, 1968 ▶) and a solvent content of 49.7%.
Figure 4.

X-ray diffraction image of a T. thermophilus TTC1157 crystal. The edge of the detector corresponds to a resolution of 1.7 Å.
Initial attempts were made to solve the crystal structures of Pf1002 and TTC1157 by molecular replacement using Phaser (McCoy et al., 2007 ▶). Different search models were generated starting from PDB entry 3ldu (30% sequence identity over 240 amino acids to Pf1002; 24% sequence identity over 170 amino acids to TTC1157; K. Tan, R. Wu, K. Buck & A. Joachimiak, unpublished work). In the best cases, maximal Z scores of 5.1 (Pf1002) and 4.5 (TTC1157) were obtained for the molecular-replacement solutions. These solutions could not be refined to an R free below 40% and the resulting electron-density maps were not interpretable.
Attempts to solve the structure of Pf1002 using the MAD method with selenomethionine-labelled protein are in progress. Since TTC1157 does not contain any methionines apart from the N-terminal methionine (nor does it contain any cysteines), the same approach is not applicable to this protein in a straightforward way. Therefore, we plan to solve this structure using molecular-replacement procedures after successfully solving the Pf1002 structure.
Acknowledgments
We would like to thank the staff of beamline ID23-2 at the ESRF, France for assistance during data collection. Financial support was provided by FWO grant G025909N. WV is the recipient of an FWO postdoctoral grant. MF is the recipient of an IWT predoctoral grant.
References
- Aravind, L. & Koonin, E. V. (2001). Trends Biochem. Sci. 26, 215–217. [DOI] [PubMed]
- Armengaud, J., Urbonavicius, J., Fernandez, B., Chaussinand, G., Bujnicki, J. M. & Grosjean, H. (2004). J. Biol. Chem. 279, 37142–37152. [DOI] [PubMed]
- Czerwoniec, A., Kasprzak, J. M., Kaminska, K. H., Rother, K., Purta, E. & Bujnicki, J. M. (2009). DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution, edited by H. Grosjean, pp. 289–302. Austin, Texas: Landes Bioscience.
- Gabant, G., Auxilien, S., Tuszynska, I., Locard, M., Gajda, M. J., Chaussinand, G., Fernandez, B., Dedieu, A., Grosjean, H., Golinelli-Pimpaneau, B., Bujnicki, J. M. & Armengaud, J. (2006). Nucleic Acids Res. 34, 2483–2494. [DOI] [PMC free article] [PubMed]
- Hopper, A. K. & Phizicky, E. M. (2003). Genes Dev. 17, 162–180. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kelley, L. A. & Sternberg, M. J. (2009). Nature Protoc. 4, 363–371. [DOI] [PubMed]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Menezes, S., Gaston, K. W., Krivos, K. L., Apolinario, E. E., Reich, N. O., Sowers, K. R., Limbach, P. A. & Perona, J. J. (2011). Nucleic Acids Res. 39, 7641–7655. [DOI] [PMC free article] [PubMed]
- Motorin, Y. & Helm, M. (2010). Biochemistry, 49, 4934–4944. [DOI] [PubMed]
- Motorin, Y. & Helm, M. (2011). Wiley Interdiscip. Rev. RNA, 2, 611–631. [DOI] [PubMed]
- Sunita, S., Purta, E., Durawa, M., Tkaczuk, K. L., Swaathi, J., Bujnicki, J. M. & Sivaraman, J. (2007). Nucleic Acids Res. 35, 4264–4274. [DOI] [PMC free article] [PubMed]
- Urbonavicius, J., Skouloubris, S., Myllykallio, H. & Grosjean, H. (2005). Nucleic Acids Res. 33, 3955–3964. [DOI] [PMC free article] [PubMed]
- Waterman, D. G., Ortiz-Lombardía, M., Fogg, M. J., Koonin, E. V. & Antson, A. A. (2006). J. Mol. Biol. 356, 97–110. [DOI] [PubMed]