

Abstract
The underlying inflammatory component of chronic kidney disease may predispose blood vessels to intimal hyperplasia (IH), which is the primary cause of dialysis access failure. We hypothesize that vascular pathology andmarkers of IH formation are antecedent to arteriovenous (AV) fistula creation. Blood, cephalic, and basilic vein segments were collected from predialysis chronic kidney disease (CKD) patients with no previous AV access and patients with end-stage renal disease (ESRD). Immunohistochemistry was performed with antibodies against mast cell chymase, transforming growth factor-beta (TGF-β) and interleukin-6 (IL-6), which cause IH. Plasma chymase was measured by ELISA. IH was present in 91% of CKD and 75% of ESRD vein segments. Chymase was abundant in vessels with IH, with the greatest expression in intima and medial layers, and virtually absent in the controls. Chymase colocalized with TGF-β1 and IL-6. Plasma chymase concentration was elevated up to 33-fold in patients with CKD versus controls and was associated with increased chymase in vessels with IH. We show that chymase expression in vessels with IH corresponds with plasma chymase concentrations. As chymase inhibition attenuates IH in animal models, and we find chymase is highly expressed in IH lesions of patients with CKD and ESRD, we speculate that chymase inhibition could have therapeutic value in humans.
Intimal hyperplasia (IH) in dialysis arteriovenous (AV) access is accelerated (1,2), and patency following angioplasty or stenting is particularly poor among patients with end-stage renal disease (ESRD) (3–6), compared with long-term patency of other vascular beds (7–11). Consequently, patients with ESRD may require an access intervention every 3–6 months (12) to reestablish and maintain patency, and over time can exhaust sites for all AV access options in the upper extremities.
Currently, there is no effective therapy to treat IH in patients with ESRD. Given the significant contribution of angiotensin II to IH (13–15), interruption of the renin-angiotensin system with angiotensin converting enzyme (ACE) inhibition has been investigated, but does not attenuate IH in humans (16–19). The lack of benefit may be because of the presence of the ACE-independent angiotensin II-forming enzyme, chymase, which is present in blood vessels, and contained in the secretory granules of mast cells. With inflammatory stimulus, chymase is released into the vascular interstitium and forms Ang II independent of ACE.
Chymase is proposed to play a causative role in the formation of venous neointimal hyperplasia, via stimulation of two primary contributors to IH formation, angiotensin II and transforming growth factor-beta 1 (TGF-β1) (13,14,20–22). Chymase causes Ang II formation, which has been shown to increase the expression of pro-TGF-β and MMP-9 (23,24). In addition to this indirect effect, chymase can also convert the latent form of TGF-β to its active form (25). These direct and indirect effects of chymase could play a major role in producing vascular IH and fibrosis (26). The role of chymase in IH formation is supported by the ability of chymase inhibition to attenuate intimal proliferation in canine models of balloon injury (27), vein grafting (28), and AV fistula (AVF) creation and AV graft (AVG) insertion (29,30).
Given that uremia is a state of chronic inflammation and oxidative stress, and because chymase is believed to play a role in venous IH formation, we hypothesized that IH occurs before AVF creation, and that chymase is highly expressed in veins of chronic kidney disease (CKD) and ESRD patients with IH. We also sought to determine whether mast cell chymase expression would be reflected by elevated plasma chymase concentration.
Methods
Study Population
Control Subjects
Three men and two women between 20 and 49 years of age with normal renal function were enrolled to serve as control subjects.
CKD Subjects
Patients with Stage 4 and 5 CKD were identified between July 14, 2008, and July 31, 2010, who had no prior AV access creation (including central venous catheters), were six or more months from initiating hemodialysis, and were suitable candidates for AVF based on upper extremity venous mapping at the Emory Dialysis Access Center of Atlanta, Emory University Hospital and Emory Midtown Hospital. Subjects were enrolled as part of a study to evaluate the cardiovascular effects of AVF creation. A baseline visit occurred prior to AVF creation, at which time a blood sample, echocardiogram, and 6-minute walk test were obtained. Additionally, demographic data, clinical history, and medication use were obtained via direct patient interview. Study subjects underwent AVF creation within 2 days to 3 weeks following the baseline visit. The Institutional Review Board (IRB) of Emory University Medical Center approved the study protocol, and informed consent was obtained from each patient prior to study enrollment.
ESRD Subjects
We identified adult patients with ESRD receiving outpatient hemodialysis at Emory University dialysis units between March 24, 2009 and June 25, 2010, who were suitable candidates for AVF creation based on upper extremity venous mapping at the Emory Dialysis Access Center of Atlanta, Emory University Hospital, and Emory Midtown Hospital. Subjects were enrolled as part of a pilot study to evaluate the impact of vitamin Don AVF maturation, and all subjects included in this analysis were using a central venous catheter at the time of study enrollment. The average time on dialysis was 11.7 months (± 19.8 months). A baseline visit occurred prior to AVF creation, and demographic and clinical data were collected via direct patient interview, at which time a blood sample was obtained. Study subjects underwent AVF creation within 1–3 weeks following the baseline visit. IRB of Emory University Medical Center approved the study protocol, and informed consent was obtained from each patient prior to study enrollment.
Tissue Collection
Remnants of surgically excised cephalic and basilic veins were collected from 11 CKD and 12 ESRD subjects at the time of AVF creation at Emory University and Emory Midtown Hospitals between July 15, 2008, and July 30, 2010, by four vascular surgeons.
A peripheral vein from a non-CKD patient was used as control. Remnant vein segments, which are normally discarded, were harvested from the cephalic or basilic vein to be used as the AVF conduit by the surgeon. It was not possible to obtain vein segments from all study patients because of the limited length of the vessel in some cases. The vein remnants were carefully placed directly into 10% normal buffered formalin without use of forceps. The specimens were transferred to 70% ethanol 24 hours after initial placement in formalin. Within 1 week, venous segments were processed and embedded in paraffin blocks. Hematoxylin and eosin stain (H and E) was used to show cellularity and general morphological characteristics.
Tissue Immunohistochemistry
Paraffin-embedded vein segments were sectioned into 3.0-μm-thick serial sections. After heat-mediated antigen retrieval, slides were immunohistochemically processed in a DAKO Automated Immunostainer (DAKO Corp., Carpinteria, CA) using a labeled streptavidin–biotin method for interleukin-6 (IL-6; rabbit polyclonal antibody to IL-6 manufactured by Abcam, Cambridge, MA) and TGF-β1 (rabbit polyclonal IgG to TGF-β1 manufactured by Santa Cruz, Santa Cruz, CA). After processing, slides were coverslipped with a Leica CV5000 Coverslipper (Leica Microsystems, Inc., Buffalo Grove, IL). Each staining batch contained positive and negative slides; normal colon tissue was the positive control tissue for IL-6, and normal tonsil tissue was the positive control for TGF-β. The negative and positive control slides were treated identically to the patients’ slides except that antibody diluents were used rather than primary antibody on the negative slides.
Tissue Immunofluorescence and Confocal Laser Scanning Microscopy
Human veins were immersion-fixed in 4% paraformaldehyde and stored in 70% ethanol until paraffin embedding and sectioning. Sections (5 μm) were mounted on slides, deparaffinized in xylene, and rehydrated in ethanol. Sections were blocked with 5% goat serum in 1× PBS for 1 hour at room temperature. Primary antibodies (final concentration): ACE rabbit monoclonal (1:250; Sigma, St. Louis, MO) and Chymase (affinity bioreagents, 1:750) were combined in an appropriate volume of 5% goat serum in 1× PBS and applied to sections by overnight incubation at 4°C. The sections were incubated with Alexa Fluor 488 or Alexa Fluor 647 goat anti-rabbit to visualize the specific stains. All secondary antibodies were from Molecular Probes (Invitrogen Corp., Carlsbad, CA). 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) was used to visualize nuclear DNA. Image Acquisition was performed on a Zeiss LSM510 META confocal laser scanning microscope (Zeiss, Thornwood, NY) with a LSM510 system cooled CCD camera and software Zeiss ZEN 2008 (Zeiss). In showing chymase staining (micrographs in Fig. 1), chymase was pseudocolored in green and α-smooth muscle actin (α-SMA) in red. In micrographs 2 and 3, chymase was pseudocolored in pink and AC Erendered in green.
Fig. 1.
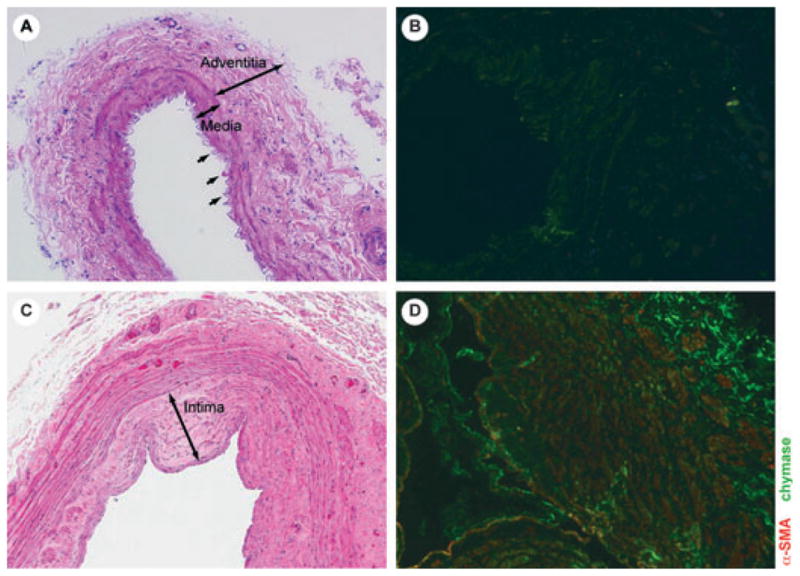
Non-CKD vein (A, B) with no intimal hyperplasia and minimal chymase activity (green); CKD vein with intimal hyperplasia (C) and abundant chymase (D), co-localized with α-smooth muscle actin (α-SMA).
Plasma Chymase Elisa
Plasma from normal, CKD, and ESRD subjects were diluted in 50 mm of sodium carbonate buffer, pH 9.6, (Sigma) and plated in 96-well flat bottom PolySorp plates (Nalge Nunc International, Rochester, NY) overnight at 4°C. The next day, the samples were washed in PBS containing 1% bovine serum albumin (BSA) and 0.05% Tween 20 (Sigma) and blocked for 2 hours with PBS containing 3% goat serum (Sigma) at 37°C. After three washes with PBS containing 1% BSA and 0.05% Tween 20, primary polyclonal antibody specific for chymase (Affinity Bioreagents, Golden, CO) was diluted to 1:1000 in PBS containing 3% goat serum and applied to the plate followed by a 37°C incubation for 1 hour. After three washes with PBS containing 1% BSA and 0.05% Tween 20, bound antibody was detected with a secondary horseradish peroxidase (HRP)-conjugated antibody (Sigma) and incubated for 1 hour at 37°C. The plate was washed three times with PBS containing 1% BSA and 0.05% Tween 20 and developed with Ultra TMB (Pierce Biotechnology, Rockford, IL). The reaction was stopped using 2 m sulfuric acid (Sigma), and its OD was read at 490 nm by the microplate reader (ELX 808; Bio-Tek instruments, Winooski, VT). Standard curve for chymase was prepared by measuring optical absorption from known concentrations of the respective.
Results
Clinical data for 29 CKD and 13 ESRD subjects were available for the analysis. Venous tissue was available for analysis on 11 CKD subjects and 12 ESRD subjects. Overall, the average patient age was 58 years, 67% of patients were Black, and 67%men. Mean SBP was 140, 93% had hypertension, 43% had diabetes, 59% had a history of tobacco use, and 17% had a history of myocardial infarction (MI; Table 1). Patients with CKD were older, less likely to be Black, with lower systolic and diastolic blood pressure compared to patients with ESRD. There was no difference between patient cohorts in gender, BMI, prevalence of hypertension, diabetes, smoking history, or history of MI.
TABLE 1.
Clinical characteristics of study subjects
| Characteristics | Control (n = 5) | CKD (n = 29) | ESRD (n = 13) | p-value |
|---|---|---|---|---|
| Age | 26 ± 12.9 | 61.2 ± 12.2 | 51.4 ± 7.9 | 0.004 |
| Black | 0 (0) | 15 (51.7) | 13 (100) | 0.002 |
| Women | 2 (40.0) | 11 (37.9) | 3 (23.1) | 0.485 |
| BMI | 22.6 ± 2.3 | 31.1 ± 8.2 | 29.6 ± 6.1 | 0.508 |
| SBP | 113 ± 6.0 | 135 ± 18.8 | 154 ± 27.8 | 0.042 |
| DBP | 71 ± 7.3 | 72 ± 11.9 | 86 ± 18 | 0.025 |
| Hypertension | 0 (0) | 28 (96.6) | 11 (84.6) | 0.222 |
| Diabetes | 0 (0) | 12 (41.4) | 6 (46.2) | 1 |
| Smoking | 0 (0) | 17 (58.6) | 8 (61.5) | 1 |
| History of myocardial infarction | 0 (0) | 4 (13.8) | 3 (23.1) | 0.657 |
Results are % or standard deviation. p-value compares patients with CKD versus ESRD.
Preexisting IH varied; 10 of 11 (91%) CKD vein segments exhibited IH and luminal stenosis prior to surgical AVF creation, demonstrating that IH exists in patients with CKD prior to AVF creation, and cannot be attributed solely to the effects of flow and pressure alterations resulting from AVF creation, and 9 of 12 (75%) ESRD vein segments had IH. Abundant chymase was present in vessels with IH and was diffuse, within the intima and adventitia, and colocalizing in the vessel media with α-SMA, reflecting myofibroblast formation (Fig. 1). The same finding was observed in vein samples from ESRD subjects. By contrast, the non-CKD control subject lacked IH, with little evidence of mast cell chymase and no evidence of α-SMA within the vein sample. Given the limited number of venous specimens, correlation of chymase and IH was not performed.
The distribution of vascular chymase was then compared with two factors that induce IH and are associated with AVF stenosis; transforming growth factor-β (TGF-β) (31,32) and IL-6 (33,34) (Fig. 2). TGF-β expression was greater in vessels with IH compared with those without IH and found to be greater in vessel intima and media layers, indicating that TGF-β production is greater in diseased vessels with IH. Veins with IH expressed more IL-6, which was more pronounced in the venous intimal and medial layers. The intensity of TGF-β and IL-6 expression was similar in CKD and ESRD veins with IH.
Fig. 2.
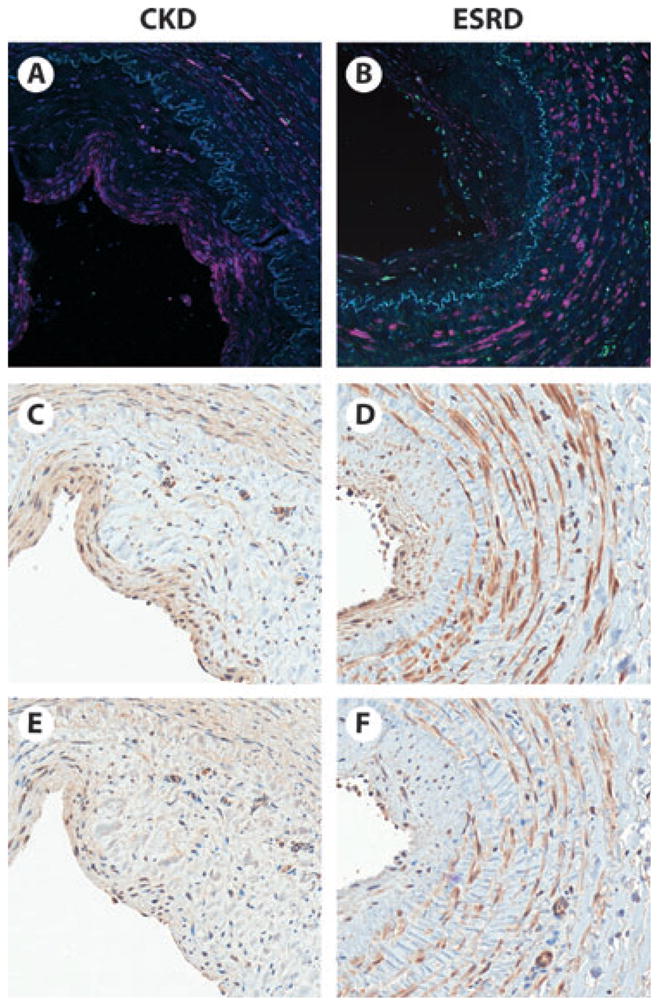
Serial vein segments from the cephalic vein of a pre-dialysis CKD (A, C, E) and an end-stage renal disease (ESRD) (B, D, F) patient. Immunoflourescence (A, B) shows abundant chymase (pink) expression in the thickened vessel intima and media (separated by elastic lamina, blue) of both the CKD and ESRD patient. Immunohistochemistry shows localization of TGF-b. (C, D) (brown) and IL-6 (E, F) (brown) primarily within the intima and media, in a similar distribution as chymase.
After finding abundant mast cell chymase in the veins of CKD and ESRD subjects preparing for AVF creation, we examined the relationship between mast cell chymase and plasma chymase concentrations (Fig. 3). The non-CKD control subjects had undetectable plasma chymase concentrations (<8 ng/ml), which corresponded with the virtual lack of mast cell chymase. CKD subjects demonstrated a bimodal distribution of plasma chymase, with plasma chymase concentrations that were as much as 6-fold greater than the lowest group, and abundant mast cell chymase expression. Finally, ESRD subjects had a different plasma chymase distribution than CKD subjects, suggesting that reduced creatinine clearance may be responsible for elevated plasma chymase levels in CKD subjects. Clearly, dialysis had an effect on plasma chymase levels; however, it is unlikely that this was because of direct removal of chymase from the bloodstream by dialysis, because chymase exists as a 200-kDA complex with alpha-2 macroglobulin in plasma (35). This led us to speculate that plasma markers of systemic inflammation may be elevated among patients with CKD with the greatest plasma chymase concentrations. However, we found no association between plasma c-reactive protein and plasma chymase concentrations (data not shown). Moreover, the plasma chymase bimodal distribution was not associated with statin use. We did, however, find that IL-6, a local marker of inflammation, was present in vascular IH lesions that express high levels of chymase (Fig. 2).
Fig. 3.
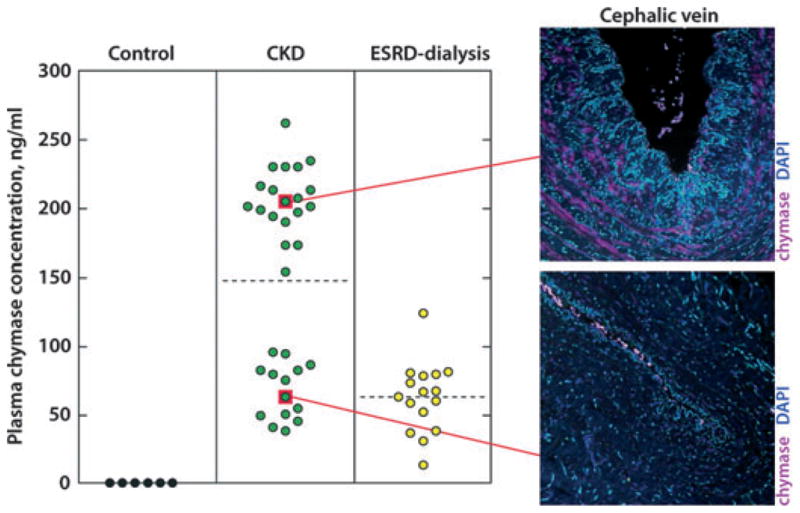
Plasma chymase concentration by patient cohort. Note bimodal distribution of plasma chymase in CKD patients. Elevated plasma chymase is shown with corresponding mast cell chymase expression in veins of same patients with intimal hyperplasia. ESRD patients have a different plasma chymase concentration, suggesting that creatinine clearance may influence plasma chymase.
Discussion
Progressive IH restricts AVF blood flow, preventing adequate dialysis, and increases the likelihood of AVF thrombosis. It is widely believed that IH develops after surgical AVF creation secondary to alterations in blood flow and pressure. Here, we show not only that venous IH is present prior to AVF creation, but also that important contributors to IH development such as chymase are present in veins of patients with CKD and ESRD. We show for the first time that the expression of mast cell chymase in vessels is reflected by as much as a 33-fold increase in plasma chymase concentration. The finding that chymase inhibition attenuates IH in animal models, and our finding that chymase levels are elevated in venous IH lesions of patients with CKD and ESRD, leads us to speculate that chymase inhibition could have therapeutic value in humans.
Early studies on mast cell chymase report that it is localized to the adventitial layer of blood vessels (36,37), and recently, it has been shown that chymase inhibition limits IH development in animal models of vascular injury (27,29,30). In patients with CKD, we find that chymase expression is diffuse, with greater chymase immunoreactivity in the vessel intima and media. TGF-β is a well-recognized promoter of VSMC proliferation and induces fibrosis and ECM production in the blood vessel (31,38,39). We find that TGF-β colocalizes with chymase in veins with IH and is most prominent in the intima and media layers of the blood vessel. One possibility for this colocalization is that chymase increases the expression of TGF-β indirectly via the promotion of Ang II formation and directly by activating pro-TGF-β1 (26,40,41). Another possibility for the chymase/TGF-β/IL-6 colocalization in these diseased vessels could be the finding that mast cells store and release all of these mediators into the vascular interstitium (42). Although our studies do not provide a causal link between chymase and TGF-β in vessels with IH, their combined presence, in addition to IL-6, not only indicates an inflammatory state but also suggests their involvement in IH progression.
Our findings indicate that IH is present before dialysis initiation; therefore, it is important to consider whether increased vascular chymase in patients with CKD is associated with elevated plasma chymase levels in these patients. We find that chymase levels were increased in all patients with CKD between 4- and 33-fold. Interestingly, chymase distribution was bimodal, yet there were no significant differences in comorbidities between the groups except BMI. The significance of the bimodal plasma chymase distribution in patients with CKD needs to be confirmed using a larger sample size. It is tempting to speculate that circulating chymase levels may be a predictor of extant IH in patients with CKD. We find significantly greater plasma chymase levels than previous studies on subjects with mastocytosis and aortic aneurysm (35,43). Although there are several endogenous inhibitors of chymase, such as alpha-1 antitrypsin, Raymond et al. (35) have shown that circulating chymase exists in a protected state in complex with alpha-2 macroglobulin such that it is capable of converting angiotensin I to angiotensin II in the circulation. Further, angiotensin receptor blockers (ARB’s) but not ACE inhibitors have been shown to attenuate IH after vascular injury (16–19). It is therefore tempting to suggest that elevated chymase levels could underlie the therapeutic advantage of ARB’s versus ACE inhibitors in this setting.
In conclusion, IH exists in veins of patients with CKD and ESRD prior to AVF creation. This suggests important consequences for early and late AVF failure. Further, chymase is significantly increased in veins with IH with known contributors to IH formation, IL-6 and TGF-β. Because chymase inhibitors effectively attenuate IH development in animal models, and our finding that chymase expression is elevated in venous IH lesions of patients with CKD and ESRD, we hypothesize that chymase inhibition could have therapeutic value in humans.
Acknowledgments
This study was supported by an NIH K23 Award (H.W) and a PHS Grant (UL1 RR02008, KL2 RR025009 or TL1 RR025010) from the Clinical and Translational Science Award program, National Institutes of Health, National Center for Research Resources.
References
- 1.Roy-Chaudhury P, Arend L, Zhang J, Krishnamoorthy M, Wang Y, Banerjee R, Samaha A, Munda R. Neointimal hyperplasia in early arteriovenous fistula failure. Am J Kidney Dis. 2007;50:782–790. doi: 10.1053/j.ajkd.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Roy-Chaudhury P, Kelly BS, Miller MA, Reaves A, Armstrong J, Nanayakkara N, Heffelfinger SC. Venous neointimal hyperplasia in polytetrafluoroethylene dialysis grafts. Kidney Int. 2001;59:2325–2334. doi: 10.1046/j.1523-1755.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- 3.Beaulieu MC, Gabana C, Rose C, MacDonald PS, Clement J, Kiaii M. Stenosis at the area of transposition—an under-recognized complication of transposed brachiobasilic fistulas. J Vasc Access. 2007;8:268–274. [PubMed] [Google Scholar]
- 4.Quinn SF, Schuman ES, Hall L, Gross GF, Uchida BT, Standage BA, Rosch J, Ivancev K. Venous stenoses in patients who undergo hemodialysis: treatment with self-expandable endovascular stents. Radiology. 1992;183:499–504. doi: 10.1148/radiology.183.2.1561357. [DOI] [PubMed] [Google Scholar]
- 5.Vorwerk D, Guenther RW, Mann H, Bohndorf K, Keulers P, Alzen G, Sohn M, Kistler D. Venous stenosisocclusion in hemodialysis shunts: follow-up results of stent placement in 65 patients. Radiology. 1995;195:140–146. doi: 10.1148/radiology.195.1.7892456. [DOI] [PubMed] [Google Scholar]
- 6.Gray RJ, Horton KM, Dolmatch BL, Rundback JH, Anaise D, Aquino AO, Currier CB, Light JA, Sasaki TM. Use of Wallstents for hemodialysis access-related venous stenoses and occlusions untreatable with balloon angioplasty. Radiology. 1995;195:479–484. doi: 10.1148/radiology.195.2.7724770. [DOI] [PubMed] [Google Scholar]
- 7.Stone GW, Rizvi A, Newman W, Mastali K, Wang JC, Caputo R, Doostzadeh J, Cao S, Simonton CA, Sudhir K, Lansky AJ, Cutlip DE, Kereiakes DJ. Everolimus-eluting versus paclitaxel-eluting stents in coronary artery disease. N Engl J Med. 2010;362:1663–1674. doi: 10.1056/NEJMoa0910496. [DOI] [PubMed] [Google Scholar]
- 8.Kedhi E, Joesoef KS, McFadden E, Wassing J, van Mieghem C, Goedhart D, Smits PC. Second-generation everolimus-elutingpaclitaxel-eluting stents in real-life practice (COMPARE): a randomised trial. Lancet. 2010;75:201–209. doi: 10.1016/S0140-6736(09)62127-9. [DOI] [PubMed] [Google Scholar]
- 9.Goldman S, Zadina K, Moritz T, Ovitt T, Sethi G, Copeland JG, Thottapurathu L, Krasnicka B, Ellis N, Anderson RJ, Henderson W. Long-term patency of saphenous veinleft internalmammary artery grafts after coronary artery bypass surgery: results from a Department of Veterans Affairs Cooperative Study. J Am Coll Cardiol. 2004;44:2149–2156. doi: 10.1016/j.jacc.2004.08.064. [DOI] [PubMed] [Google Scholar]
- 10.Garg S, Serruys PW. Coronary stents: current status. J Am Coll Cardiol. 2010;56(10 Suppl):S1–S42. doi: 10.1016/j.jacc.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 11.Brilakis ES, Saeed B, Banerjee S. Drug-eluting stents in saphenous vein graft interventions: a systematic review. Euro Intervention. 2010;5:722–730. doi: 10.4244/eijv5i6a119. [DOI] [PubMed] [Google Scholar]
- 12.Schwab SJ, Harrington JT, Singh A, Roher R, Shohaib SA, Perrone RD, Meyer K, Beasley D. Vascular access for hemodialysis. Kidney Int. 1999;55:2078–2090. doi: 10.1046/j.1523-1755.1999.00409.x. [DOI] [PubMed] [Google Scholar]
- 13.Daemen MJ, Lombardi DM, Bosman FT, Schwartz SM. Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ Res. 1991;68:450–456. doi: 10.1161/01.res.68.2.450. [DOI] [PubMed] [Google Scholar]
- 14.Griffin SA, Brown WC, MacPherson F, McGrath JC, Wilson VG, Korsgaard N, Mulvany MJ, Lever AF. Angiotensin II causes vascular hypertrophy in part by a non-pressor mechanism. Hypertension. 1991;17:626–635. doi: 10.1161/01.hyp.17.5.626. [DOI] [PubMed] [Google Scholar]
- 15.Su EJ, Lombardi DM, Siegal J, Schwartz SM. Angiotensin II induces vascular smooth muscle cell replication independent of blood pressure. Hypertension. 1998;31:1331–1337. doi: 10.1161/01.hyp.31.6.1331. [DOI] [PubMed] [Google Scholar]
- 16.Hermans WR, Rensing BJ, Foley DP, Deckers JW, Rutsch W, Emanuelsson H, Danchin N, Wijns W, Chappuis F, Serruys PW. Therapeutic dissection after successful coronary balloon angioplasty: no influence on restenosis or on clinical outcome in 693 patients. The MERCATOR Study Group (Multicenter European Research Trial with Cilazapril after Angioplasty to prevent Transluminal Coronary Obstruction and Restenosis) J Am Coll Cardiol. 1992;20:767–780. doi: 10.1016/0735-1097(92)90171-i. [DOI] [PubMed] [Google Scholar]
- 17.Faxon DP. Effect of high dose angiotensin-converting enzyme inhibition on restenosis: final results of the MARCATOR Study, a multicenter, double-blind, placebo-controlled trial of cilazapril. The Multicenter American Research Trial With Cilazapril After Angioplasty to Prevent Transluminal Coronary Obstruction and Restenosis (MARCATOR) Study Group. J Am Coll Cardiol. 1995;25:362–369. doi: 10.1016/0735-1097(94)00368-z. [DOI] [PubMed] [Google Scholar]
- 18.Peters S, Gotting B, Trummel M, Rust H, Brattstrom A. Valsartan for prevention of restenosis after stenting of type B2/C lesions: the VAL-PREST trial. J Invasive Cardiol. 2001;13:93–97. [PubMed] [Google Scholar]
- 19.Peters S, Trummel M, Meyners W, Koehler B, Westermann K. Valsartan versus ACE inhibition after bare metal stent implantation—results of the VALVACE trial. Int J Cardiol. 2005;98:331–335. doi: 10.1016/j.ijcard.2004.05.062. [DOI] [PubMed] [Google Scholar]
- 20.Chung IM, Gold HK, Schwartz SM, Ikari Y, Reidy MA, Wight TN. Enhanced extracellular matrix accumulation in restenosis of coronary arteries after stent deployment. J Am Coll Cardiol. 2002;40:2072–2081. doi: 10.1016/s0735-1097(02)02598-6. [DOI] [PubMed] [Google Scholar]
- 21.Nabel EG, Shum L, Pompili VJ, Yang ZY, San H, Shu HB, Liptay S, Gold L, Gordon D, Derynck R. Direct transfer of transforming growth factor beta 1 gene into arteries stimulates fibrocellular hyperplasia. Proc Natl Acad Sci US A. 1993;90:10759–10763. doi: 10.1073/pnas.90.22.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulick AH, Taylor AJ, Zuo W, Qiu CB, Dong G, Woodward RN, Agah R, Roberts AB, Virmani R, Dichek DA. Overexpression of transforming growth factor beta1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc Natl Acad Sci US A. 1998;95:6983–6988. doi: 10.1073/pnas.95.12.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibbons GH, Pratt RE, Dzau VJ. Vascular smoothmuscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest. 1992;90:456–461. doi: 10.1172/JCI115881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease- 9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–9296. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 25.Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiainen M, Kokkonen JO, Keski-Oja J, Kovanen PT. Activation of paracrine TGF-beta1 signaling upon stimulationdegranulation of rat serosal mast cells: a novel function for chymase. FASEB J. 2001;15:1377–1388. doi: 10.1096/fj.00-0273com. [DOI] [PubMed] [Google Scholar]
- 26.Zhao XY, Zhao LY, Zheng QS, Su JL, Guan H, Shang FJ, Niu XL, He YP, Lu XL. Chymase induces profibrotic response via transforming growth factor- beta 1/Smad activation in rat cardiac fibroblasts. Mol Cell Biochem. 2008;310:159–166. doi: 10.1007/s11010-007-9676-2. [DOI] [PubMed] [Google Scholar]
- 27.Takai S, Sakonjo H, Fukuda K, Jin D, Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y, Miyazaki M. A novel chymase inhibitor, 2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-N-[[,4-dioxo-1-phenyl-7-(2-pyridyloxy)]2-heptyl]acetamide (NK3201), suppressed intimal hyperplasia after balloon injury. J Pharmacol Exp Ther. 2003;304:841–844. doi: 10.1124/jpet.102.042580. [DOI] [PubMed] [Google Scholar]
- 28.Nishimoto M, Takai S, Kim S, Jin D, Yuda A, Sakaguchi M, Yamada M, Sawada Y, Kondo K, Asada K, Iwao H, Sasaki S, Miyazaki M. Significance of chymase-dependent angiotensin II-forming pathway in the development of vascular proliferation. Circulation. 2001;104:1274–1279. doi: 10.1161/hc3601.094304. [DOI] [PubMed] [Google Scholar]
- 29.Jin D, Ueda H, Takai S, Muramatsu M, Furubayashi K, Ibaraki T, Kishi K, Katsuoka Y, Miyazaki M. Roles of chymase in stenosis occurring after polytetrafluoroethylene graft implantations. Life Sci. 2007;81:1291–1300. doi: 10.1016/j.lfs.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Jin D, Ueda H, Takai S, Okamoto Y, Muramatsu M, Sakaguchi M, Shibahara N, Katsuoka Y, Miyazaki M. Effect of chymase inhibition on the arteriovenous fistula stenosis in dogs. J Am Soc Nephrol. 2005;16:1024–1034. doi: 10.1681/ASN.2003121009. [DOI] [PubMed] [Google Scholar]
- 31.Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC. TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol. 2009;297:H540–H549. doi: 10.1152/ajpheart.91478.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heine GH, Ulrich C, Sester U, Sester M, Kohler H, Girndt M. Transforming growth factor beta1 genotype polymorphisms determine AV fistula patency in hemodialysis patients. Kidney Int. 2003;64:1101–1107. doi: 10.1046/j.1523-1755.2003.00176.x. [DOI] [PubMed] [Google Scholar]
- 33.Marrone D, Pertosa G, Simone S, Loverre A, Capobianco C, Cifarelli M, Memoli B, Schena FP, Grandaliano G. Local activation of interleukin 6 signaling is associated with arteriovenous fistula stenosis in hemodialysis patients. Am J Kidney Dis. 2007;49:664–673. doi: 10.1053/j.ajkd.2007.02.266. [DOI] [PubMed] [Google Scholar]
- 34.DeMarchi S, Falleti E, Giacomello R, Stel G, Cecchin E, Sepiacci G, Bortolotti N, Zanello F, Gonano F, Bartoli E. Risk factors for vascular disease and arteriovenous fistula dysfunction in hemodialysis patients. J Am Soc Nephrol. 1996;7:1169–1177. doi: 10.1681/ASN.V781169. [DOI] [PubMed] [Google Scholar]
- 35.Raymond WW, Su S, Makarova A, Wilson TM, Carter MC, Metcalfe DD, Caughey GH. Alpha 2-macroglobulin capture allows detection of mast cell chymase in serum and creates a reservoir of angiotensin II-generating activity. J Immunol. 2009;182:5770–5777. doi: 10.4049/jimmunol.0900127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laine P, Kaartinen M, Penttila A, Panula P, Paavonen T, Kovanen PT. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation. 1999;99:361–369. doi: 10.1161/01.cir.99.3.361. [DOI] [PubMed] [Google Scholar]
- 37.Richard V, Hurel-Merle S, Scalbert E, Ferry G, Lallemand F, Bessou JP, Thuillez C. Functional evidence for a role of vascular chymase in the production of angiotensin II in isolated human arteries. Circulation. 2001;104:750–752. doi: 10.1161/hc3201.094971. [DOI] [PubMed] [Google Scholar]
- 38.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 39.Wolf YG, Rasmussen LM, Ruoslahti E. Antibodies against transforming growth factor-beta 1 suppress intimal hyperplasia in a rat model. J Clin Invest. 1994;93:1172–1178. doi: 10.1172/JCI117070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koibuchi Y, Lee WS, Gibbons GH, Pratt RE. Role of transforming growth factor-beta 1 in the cellular growth response to angiotensin II. Hypertension. 1993;21(6 Pt 2):1046–1050. doi: 10.1161/01.hyp.21.6.1046. [DOI] [PubMed] [Google Scholar]
- 41.Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–4696. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- 42.Hugle T, Hogan V, White K, van Laar JM. Mast cells are a source of transforming growth factor (TGF) beta in systemic sclerosis. Arthritis Rheum. 2011;63(3):795–799. doi: 10.1002/art.30190. [DOI] [PubMed] [Google Scholar]
- 43.Sun J, Zhang J, Lindholt JS, Sukhova GK, Liu J, He A, Abrink M, Pejler G, Stevens RL, Thompson RW, Ennis TL, Gurish MF, Libby P, Shi GP. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation. 2009;120:973–982. doi: 10.1161/CIRCULATIONAHA.109.849679. [DOI] [PMC free article] [PubMed] [Google Scholar]