

Summary
To successfully colonize and persist within a host niche, bacteria must properly regulate their gene expression profiles. The marine bacterium Vibrio fischeri establishes a mutualistic symbiosis within the light organ of the Hawaiian squid, Euprymna scolopes. Here, we show that the repressor NagC of V. fischeri directly regulates several chitin- and N-acetyl-D-glucosamine-utilization genes that are co-regulated during productive symbiosis. We also demonstrate that repression by NagC is relieved in the presence of N-acetyl-D-glucosamine-6-phosphate, the intracellular form of N-acetyl-D-glucosamine. We find that gene repression by NagC is critical for efficient colonization of E. scolopes. Further, our study shows that NagC regulates genes that affect the normal dynamics of host colonization.
Introduction
Vibrio fischeri is a Gram-negative bacterium utilized by several species of marine organisms as a source of bioluminescence. Like many of these animals, the Hawaiian squid Euprymna scolopes has evolved an organ that harnesses this bioluminescence while providing shelter and nutrients specifically to this beneficial microbe (McFall-Ngai, 2008). The mutualistic symbiosis is established shortly after squid hatch, and is maintained throughout the lifetime of the animal. A recent transcriptome analysis of the mature symbiosis of adult animals has revealed certain correlated patterns of bacterial gene expression, particularly with metabolic genes predicted to be involved in the utilization of chitin and its monomer, N-acetyl-D-glucosamine (GlcNAc) (Wier et al., 2010).
Chitin is produced in large quantities by many invertebrates to provide structural integrity to their exoskeletons (Keyhani et al., 1999). These chitin polymers are insoluble and as a result, are nutritionally accessible to microbes only by means of chitinases that break the polymer down into its subunits. GlcNAc and chitobiose (GlcNAc)2 are released by exochitinases, which hydrolyze the glycosidic bonds between GlcNAc or chitobiose subunits. As an aminosugar, GlcNAc serves as a source of both carbon and nitrogen, and appears to be a preferred carbon source by many vibrios (Hunt et al., 2008).
Our knowledge of the uptake and metabolism of GlcNAc in Gram-negative bacteria is primarily due to studies of four genes (nagE and nagBAC) within the nag locus of Escherichia coli (Alvarez-Anorve et al., 2009; Plumbridge, 1991; Rogers et al., 1988). Briefly, the nagE gene encodes enzyme IICGlcNAc, IIBGlcNAc, and IIAGlcNAc components of a PTS transporter specific for GlcNAc. During its transport into the cytoplasm, GlcNAc is phosphorylated on the 6-carbon within the glucose subunit, resulting in N-acetyl-D-glucosamine-6-phosphate (GlcNAc-6P). GlcNAc-6P is the substrate for the deacetylase NagA, which removes the acetyl group to yield glucosamine-6P. The amine group is then cleaved from glucosamine-6P by the deaminase NagB. NagC is a transcription factor that represses the nag locus by binding to two distinct operator sites upstream of the divergently transcribed nagE and nagBAC operons. Repression of the nag genes by NagC is relieved by allosteric binding of GlcNAc-6P.
Here we show that NagC directly regulates genes involved in the utilization of chitin and GlcNAc in V. fischeri. Mutants unable to repress genes controlled by NagC were outcompeted by wild-type cells during colonization of the squid light organ. The initial selection for strains of V. fischeri that repress genes via NagC could ensure that the squid host retains a symbiont population with the ability to regulate chitin- and GlcNAc-utilization genes as the symbiosis matures.
Results
NagC regulates chitin- and GlcNAc-utilization genes in V. fischeri
A previous study of the squid-vibrio system revealed that numerous bacterial genes predicted to be associated with the utilization of chitin and GlcNAc are co-regulated during symbiosis (Wier et al., 2010). We hypothesized that this pattern of gene expression arises from the activity of a single bacterial regulator. We selected VF_1598, which is predicted to encode a secreted exochitinase, as a representative gene from the group of co-regulated genes because this gene showed the most pronounced dynamics among the chitin- and GlcNAc-utilization genes in the study mentioned above. Furthermore, VF_1598 is conserved in the V. fischeri fish symbiont MJ11 (VFMJ11_1708), but has diverged in Allivibrio salmonicida LFI1238 to lose the chitin-binding domain (ChtBD3), suggesting that this biological activity of VF_1598 is specific to V. fischeri species. To monitor VF_1598 expression, we constructed a reporter plasmid (pTM314) by cloning the intergenic region upstream of VF_1598, which presumably contains the corresponding promoter, upstream of gfp in pTM267 (Miyashiro et al., 2010). The plasmid pTM267 constitutively expresses mCherry, and has an origin of replication that is maintained in V. fischeri. In cells harboring pTM314, the GFP/mCherry fluorescence ratio is a quantitative measure of VF_1598 gene expression.
To identify potential regulators of VF_1598, we conjugated pTM314 into a library of approximately 105 transposon mutants of the wild-type V. fischeri strain ES114. We screened 500,000 colonies by fluorescence, and isolated 160 mutants with elevated levels of GFP. Sequencing of the insertion in six of the candidate strains revealed four unique mutants in which the transposon had inserted into either VF_0807 (WPK54) or VF_0806 (WPK84) (Fig. 1A). VF_0807 and VF_0806 are co-transcribed and encode N-acetylglucosamine-6-phosphate deacetylase (NagA) and GlcNAc transcriptional repressor (NagC), respectively. Although transposon insertions in either gene resulted in elevated VF_1598 expression compared to wild-type (Fig. 1B), we assumed that the insertions in nagA are polar on nagC.
Figure 1. Regulation of exochitinase gene VF_1598 by NagC.
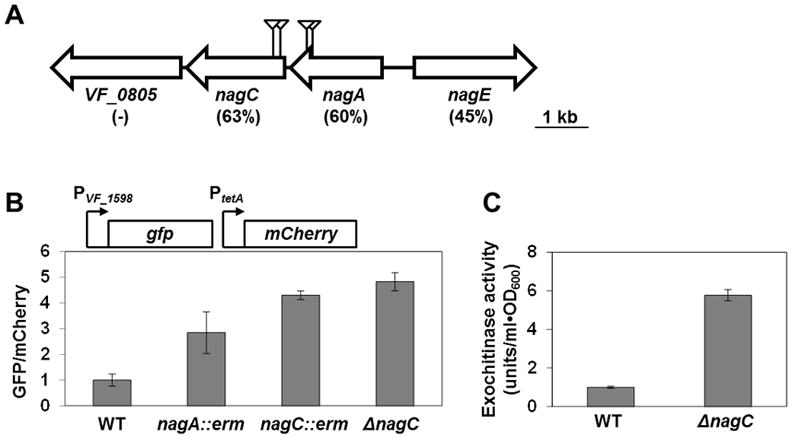
(A) The nag locus of V. fischeri strain ES114. Triangles indicate the positions of unique transposon insertions in mutants with elevated VF_1598 gene expression. Percentages below gene IDs are amino acid identities in comparison to E. coli homologues. Unlike in E. coli, in Vibrio sp., nagB is located at a distance from the nag locus.
(B) GFP/mCherry fluorescence ratios of various V. fischeri strains harboring the VF_1598 promoter reporter plasmid pTM314. WT = ES114; nagA::erm = WPK54; nagC::erm = WPK84; and ΔnagC = WPK100. Error bars indicate ± one SD.
(C) Exochitinase activities in supernatants of V. fischeri cultures. WT = ES114; and ΔnagC = WPK100. Error bars indicate ± one SD.
To determine whether NagC regulates other chitin- and GlcNAc-utilization genes in V. fischeri, we constructed an in-frame deletion of nagC in ES114 (ΔnagC). As with the transposon mutants described above, the level of VF_1598 expression in ΔnagC was higher than wild-type (Fig. 1B). Consistent with enhanced VF_1598 expression, we measured approximately 6-fold higher levels of exochitinase activity in the supernatants of ΔnagC cultures compared to wild-type (Fig. 1C).
Using quantitative reverse-transcriptase PCR (qPCR), we analyzed the effect of NagC on other chitin- and GlcNAc-utilization genes that exhibit an expression pattern correlated with the diel rhythm of the symbiosis (Wier et al., 2010). Consistent with our results using pTM314, VF_1598 transcript levels are elevated in a ΔnagC strain compared to wild-type (Fig. 2). Furthermore, overexpression of nagC in trans was sufficient to decrease VF_1598 transcription compared to ΔnagC. While VF_0655 and VF_A0013 transcript levels are independent of NagC, VF_2139, VF_A0143, and VF_A0715 are transcriptionally repressed by overexpression of nagC. Examination of the promoter region of these genes revealed potential binding sites upstream of VF_1598, VF_2139, and VF_A0715 but not upstream of VF_A0143 (Fig. S1). Together, these results suggest that NagC can regulate some, but not all, genes predicted to be associated with utilization of chitin and GlcNAc.
Figure 2. Regulation of chitin- and GlcNAc-utilization genes by NagC.

Expression levels of several V. fischeri genes measured by qPCR in wild-type (ES114) and ΔnagC (WPK100) harboring pTM214 (vector control) and ΔnagC (WPK100) harboring pTM360 (pTrc-nagC). VF_2254 = RpoD; VF_0655 = endochitinase; VF_0807 = NagA; VF_0808 = NagE; VF_1598 = exochitinase; VF_2139 = chitoologosaccharide-binding protein; VF_2357 = NagB; VF_A0013 = chitin-binding protein; VF_A0143 = N-acetylglucosamine-binding protein; and VF_A0715 = chitodextrinase precursor. Values are of triplicate biological replicates and normalized by wild-type levels. Error bars indicate ± one SD. One-way ANOVA and Tukey HSD comparisons with significance are shown with *, p-value < 0.05; and **, p-value < 0.005, where p-values are adjusted using FDR’s correction.
NagC controls responses to extracellular GlcNAc
Studies with E. coli have shown that NagC de-represses gene expression in response to the presence of extracellular GlcNAc. Among the genes repressed by NagC are nagE, nagA, and nagB, which encode, respectively, the GlcNAc PTS enzyme, deacetylase, and deaminase that are required to uptake and convert GlcNAc to fructose-6P (Plumbridge, 1991). Using qPCR, we have found that nagE, nagA and nagB are regulated by NagC in V. fischeri (Fig. 2). Furthermore, nagB is required for V. fischeri to preferentially utilize GlcNAc, suggesting that the metabolism of GlcNAc is similar to that of E. coli (Fig. S2). We attribute the growth of the nagA mutant WPK54 on GlcNAc to the presence of two additional copies of nagA (VF_0665 and VF_A0999) within the genome of ES114.
To monitor nagA expression in vivo, a two-color fluorescent reporter plasmid (pTM355) was constructed by cloning the nagA promoter upstream of gfp in pTM267. The exposure of wild-type cells grown under steady-state conditions to medium containing 20 mM GlcNAc was sufficient to induce the expression of nagA 15-fold compared to the medium blank alone (Fig. 3). Compared to wild-type cells, nagA expression in the ΔnagC mutant was 13-fold higher, and induced less than 1.5-fold in the presence of 20 mM GlcNAc. Together, these results show that NagC controls the response of the nag operon to extracellular GlcNAc in V. fischeri.
Figure 3. Effect of GlcNAc on expression of nagA in various V. fischeri strains.
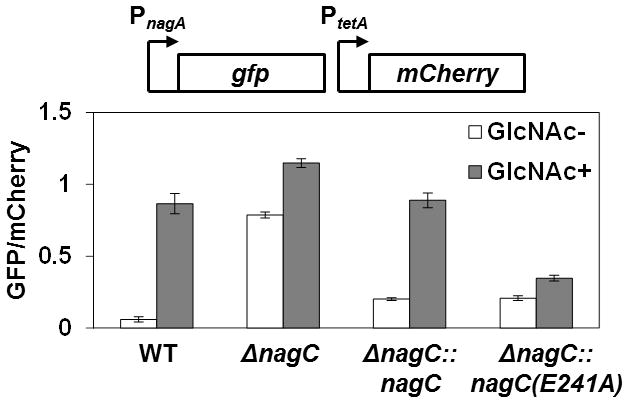
GFP/mCherry fluorescence ratios of various V. fischeri strains harboring the nagA promoter reporter plasmid pTM355 grown in the presence or absence of 20 mM GlcNAc. WT = ES114; ΔnagC = WPK100; ΔnagC::nagC = TIM377; and ΔnagC::nagC(E241A) = TIM381. The nagC alleles in TIM377 and TIM381 have myc and 6x-His affinity tags at the C-terminal ends. Error bars indicate ± one SD.
NagC specifically binds upstream of nagA and VF_1598
In E. coli, NagC represses expression of the nag operon by directly binding two operator sites between the nagB and nagE genes (El Qaidi et al., 2008). When bound by NagC, these operator sites, which are separated by 71 bps, lead to the formation of a DNA loop that strengthens the repression of the nagB and nagE promoters (Plumbridge et al., 1998). By examining the corresponding region within the V. fischeri nag locus, we identified two putative binding sites with sequences similar to the consensus sequence of the NagC operator site in E. coli (Fig. S1, VF_0807-VF_0808, boxes 3 and 4). In V. fischeri, as well as in all fully sequenced Vibrionacaeae members, the distance between the two putative operator sites is also 71 bp. This observation not only provides further bioinformatics support of the binding sites in V. fischeri, but also suggests that the cooperativity between the NagC dimers bound at the two sites is a conserved regulatory element of the nag locus. Using 5′-RACE, we have also determined that the transcriptional start site of nagA is located within the proximal NagC operator site (Fig. 4A), which suggests that NagC blocks transcription of the nagAC operon. Interestingly, there are also two potential NagC operator sites located further within the untranslated region of the nagA transcript (Fig. S1, VF_0807-VF_0808, boxes 1 and 2), but their effect on the nagA promoter is unknown at this time.
Figure 4. in vitro DNA binding by NagC.
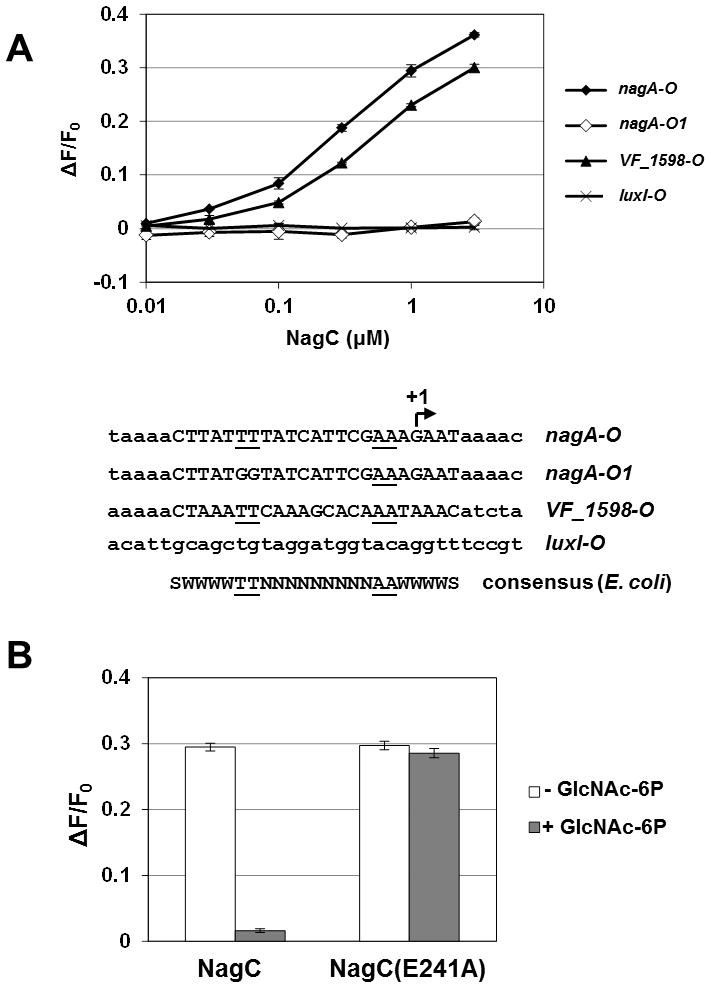
(A) in vitro NagC binding assay. The fractional change in anisotropy of fluorescently-labeled, double-stranded probes is plotted against the concentration (in μM) of NagC added. Points and bars represent mean and range of two independent binding reactions. Sequences of the probes are shown below the plot. The arrow indicates the transcriptional start site of nagA determined by 5′ RACE.
(B) in vitro binding of 1 μM wild-type (NagC) or mutant (NagC(E241A)) NagC protein, as determined by fluorescence anisotropy, in response to the absence (white bars) or presence (gray bars) of 10 mM GlcNAc-6P. Error bars indicate ± one SD.
To study the DNA-binding properties of the V. fischeri NagC, we cloned, expressed, and purified NagC with C-terminal myc and His tags. Previous studies with NagC in E. coli have shown that the C-terminus is not involved in DNA binding (Pennetier et al., 2008). Using anisotropy to detect protein binding of fluorescently-labeled DNA probes, we found that NagC bound the operator sequence proximal to nagA (Fig. 4A). In addition, replacement of two thymine bases in the consensus sequence with two guanine bases abrogated the specific binding of the operator by NagC. Consistent with this result, we found that mutating the same bases in the nagA promoter-reporter plasmid eliminated NagC regulation of nagA (Fig. 5).
Figure 5. Impact of NagC operator site on nagA expression.
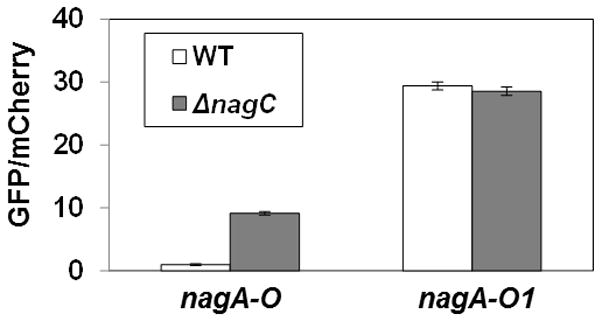
GFP/mCherry fluorescence ratios of V. fischeri strains harboring nagA-promoter reporter plasmids containing wild-type (nagA-O, pTM355) or mutated (nagA-O1, pTM361) NagC operator sites. WT = ES114; and ΔnagC = WPK100. Error bars indicate ± one SD.
Although we identified two putative NagC binding sites upstream of VF_1598 (Fig. S1, VF_1597-VF_1598, boxes 1 and 2), only box 2 is conserved upstream of the VF_1598 homologue in MJ11. Using a DNA probe containing this operator sequence, we found that, as predicted, the presence of NagC increased the anisotropy of the fluorescent probe (Fig. 4A). Together, these results demonstrate that NagC directly binds sites upstream of both nagA and VF_1598.
NagC repression is relieved in response to GlcNAc-6P
In E. coli, DNA binding by NagC is relieved in the presence of GlcNAc-6P (Pennetier et al., 2008). To determine whether the V. fischeri NagC also responds to GlcNAc-6P, we measured binding of NagC to the nagA operator site in the presence or absence of GlcNAc-6P. The presence of 10 mM GlcNAc-6P was sufficient to release NagC (Fig. 4B). In contrast, a variant of NagC containing the mutation E241A, which corresponds to the E244A mutation in the NagC of E. coli that disrupts the binding pocket for GlcNAc-6P (Pennetier et al., 2008), remained bound to the nagA operator site even at 10 mM GlcNAc-6P (Fig. 4B), thereby functioning as a ‘super-repressor’. The E241A mutation did not affect the affinity NagC has for the nagA operator site in the absence of GlcNAc-6P (data not shown).
To test whether the E241A mutation disrupts NagC regulation in vivo, we measured the response of the nagA promoter reporter plasmid pTM355 in cells exposed to GlcNAc. Cells containing nagC-myc-6xHis at the wild-type nagC locus (strain TIM377) displayed a 4.4-fold increase in nagA expression (Fig. 3). In contrast, the nagC(E241A)-myc-6xHis allele (TIM 381) resulted in only a 1.7-fold increase in nagA expression. These results demonstrate that NagC repression is relieved by GlcNAc-6P.
NagC is required for efficient colonization of E. scolopes
The proper regulation of chitin- and GlcNAc-utilization genes is predicted to be important for the long-term interactions between V. fischeri and its natural host E. scolopes (Wier et al., 2010). While the technical aspects of studying long-term interactions within the squid-vibrio symbiosis under laboratory conditions remain challenging, many details concerning the establishment of the symbiosis are known. Therefore, we investigated whether regulation by NagC is important during the initial steps of bacterial colonization of the squid light organ.
A 3-hour exposure to seawater containing 30,000 CFU/ml of wild-type V. fischeri cells results in the successful colonization of the squid light organ over 90% of the time (Fig. 6). In contrast, only 40% (6/15) of the squid were colonized by the ΔnagC mutant under the same conditions. In two of these six animals the level of colonization (<104 CFU/squid) was insufficient to result in detectable luminescence (Fig. 6). Increasing the time that the squid are exposed to the inoculum resulted in more successfully colonized animals (Fig. S3). The colonization defect of the ΔnagC strain was not observed in strains expressing the myc-6xHis-tagged variants of NagC (Fig. S3).
Figure 6. Single-strain, 3-h, light-organ colonization assay.
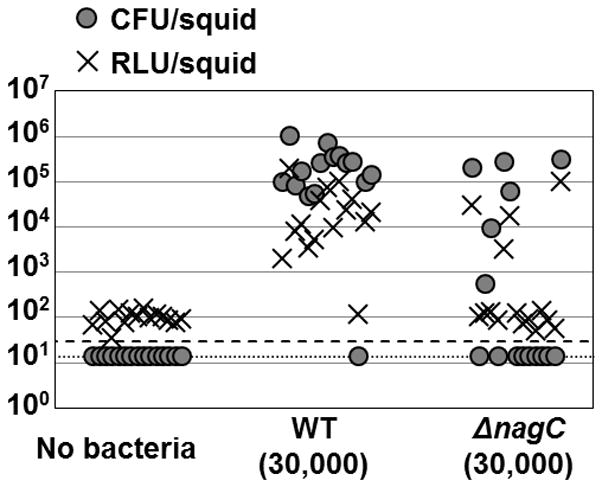
Symbiont population levels (CFU), and relative luminescence levels (RLU), of individual animals at 48 h post-inoculation with either wild type (ES114) or the ΔnagC mutant (WPK100) are shown as circles and crosses, respectively. The detection limit for CFUs is indicated by the dotted line, and for luminescence is indicated by a dashed line.
To determine whether the presence of wild-type V. fischeri could abrogate the colonization defect exhibited by the ΔnagC mutant, we performed co-colonization experiments using a GFP-labeled, wild-type strain of V. fischeri as the control strain. Squid were exposed to the inoculum for at least 20 h to allow each strain multiple attempts to colonize the light organ. When co-colonized with the control strain, the ΔnagC mutant was outcompeted by wild-type cells (Fig. 7 and Table 2). This result demonstrates that the presence of wild-type cells fails to complement the colonization defect of the ΔnagC mutant. Furthermore, the de-repressed genes in the ΔnagC mutant (e.g., the secreted exochitinase VF_1598) do not impede the ability of wild-type cells to colonize and dominate the light organ. In addition, our observation that a ΔnagB mutant, which cannot convert glucosamine-6P to fructose-6P, colonizes as well as wild-type (Table 2) suggests the colonization defect of the ΔnagC mutant is independent of GlcNAc metabolism. Increasing the abundance of the ΔnagC mutant 15-fold relative to wild-type cells within the inoculum was necessary to observe a 2-fold ΔnagC-dominant population within the light organs (Fig. 7 and Table 2). By comparison, the same inoculum bias with wild-type V. fischeri as the test strain led to a 35-fold bias in the V. fischeri population of symbionts.
Figure 7.
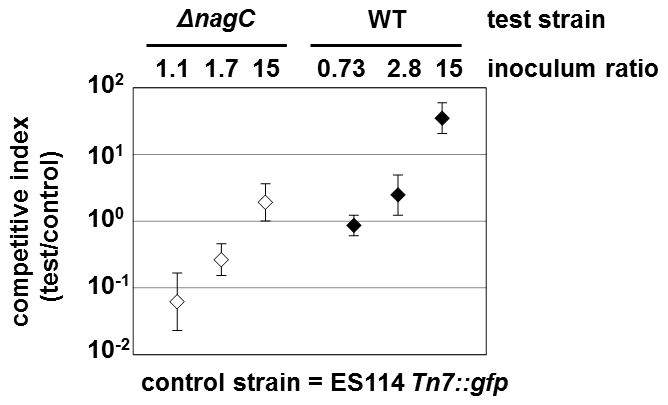
Competition experiment comparing pairs of V. fischeri strains during colonization of E. scolopes Competitive indices of ΔnagC (WPK100, open symbols) and wild-type (ES114, closed symbols) strains in animals co-colonized with the GFP-labeled, wild-type strain TIM302. The competitive index is defined as the ratio of test strain to control strain CFUs present in the light organ. Error bars indicate 95% confidence intervals with co-colonized animals.
Table 2.
Light-organ co-colonization assays
| Test straina | Inoculum ratio (test/control) | Total Inoculum (× 104 CFU) | Test onlyb (animals) | Control onlyb (animals) | Co- colonizedb (animals) | Competitive indexc (test/control) |
|---|---|---|---|---|---|---|
| WT | 0.73 | 1.1 | 0 | 1 | 29 | 0.86 |
| WT | 2.8 | 4.0 | 11 | 0 | 19 | 2.5 |
| WT | 15 | 7.5 | 17 | 0 | 13 | 35 |
| ΔnagC | 1.1 | 0.74 | 0 | 26 | 3 | 0.062 |
| ΔnagC | 1.7 | 2.5 | 0 | 6 | 24 | 0.26 |
| ΔnagC | 15 | 6.9 | 0 | 3 | 27 | 1.9 |
| ΔnagB | 1.5 | 1.1 | 0 | 0 | 29 | 0.98 |
| ΔnagC +GlcNAc | 1.0 | 0.32 | 4 | 0 | 26 | 1.5 |
| nagC(E241A) −GlcNAc | 1.2 | 0.63 | 0 | 3 | 27 | 0.56 |
| nagC(E241A) +GlcNAc | 1.2 | 0.46 | 6 | 0 | 24 | 2.6 |
For each competition, about 30 animals were exposed for 20 h to mixed inocula containing both the strain indicated in the column (test) and the GFP-labeled, wild-type strain TIM302 (control). At 48 h post-inoculation, light organs were homogenized and plated on LBS. Resulting CFUs were scored for GFP fluorescence. WT = ES114, ΔnagC = WPK100, ΔnagB = JAS101, and nagC(E241A) = TIM381.
Light organs were scored as singly colonized by the test (non-fluorescent) or by the control (fluorescent) strain, or as co-colonized by both.
The competitive index is defined as the average ratio of test strain to control strain CFUs present in co-colonized light organs.
We predicted that if wild-type cells were exposed to GlcNAc during colonization, then they would phenotypically mimic the ΔnagC mutant; i.e., de-repress any NagC-regulated genes. Consistent with this prediction, in squid exposed to seawater containing 20 mM GlcNAc and a mixed inoculum of the ΔnagC mutant and a GFP-labeled wild-type strain, the competitive advantage of wild type was lost (Table 2). We further hypothesized that the mutant harboring the super-repressor form of NagC (NagC E241A) would out-compete the wild-type strain in the presence of exogenously added GlcNAc because it would still maintain repression of NagC-regulated genes. When exposed to wild-type and super-repressor strains in the absence of GlcNAc, co-colonized squid contained a slight bias towards the wild-type strain (Table 2). However, as hypothesized, the presence of GlcNAc during colonization was sufficient to reverse the bias of the population within co-colonized squid towards the super-repressor strain (Table 2).
Discussion
In this study, we have identified NagC as an important bacterial regulatory element that functions during the initial colonization of E. scolopes by V. fischeri. NagC disassociates from DNA in response to intracellular GlcNAc-6P, leading to changes in gene expression. In addition to catabolic genes such as nagA and nagB, NagC also controls genes that are involved in the conversion of environmental chitin to GlcNAc, such as VF_1598 (exochitinase), VF_2139-VF_2148 (chitobiose-utilization genes), VF_A0143 (N-acetylglucosamine-binding protein), and VF_A0715 (chitodextrinase precursor). Because free monosaccharide sugars are of low abundance (<100 nM) in seawater (Mopper et al., 1992), we hypothesize that the environmental source of GlcNAc for V. fischeri is derived from invertebrate-associated chitin (Keyhani et al., 1999).
In E. coli, a mutation in nagA results in derepression of NagC due to the accumulation of GlcNAc-6P via peptidoglycan remodeling (Plumbridge, 2009; White, 1968). While it is possible that GlcNAc-6P levels are elevated in the nagA transposon mutant WPK54, the ability of this mutant to grow on GlcNAc (Fig. S2) suggests that the other two nagA genes are functional and can catabolize GlcNAc-6P. In V. cholerae, removal of the two copies of nagA is required to demonstrate sensitivity to GlcNAc (Ghosh et al., 2011).
This study contributes to our growing awareness of the nutritional and signaling roles GlcNAc plays in several host-microbe interactions. Recently, transcriptional evidence that the V. fischeri population in the mature squid light-organ symbiosis is exposed to chitin as a nutrient has been supported by the discovery of host blood cells carrying particulate chitin in the tissue surrounding the symbionts (Heath-Heckman et al., 2011). In addition, for both V. cholerae and V. parahaemolyticus, exposure to GlcNAc leads to the induction of genes associated with GlcNAc utilization (Meibom et al., 2004; Thompson et al., 2011). Recently, the nagA and nagB genes have been implicated in having an impact on host colonization by V. cholerae (Ghosh et al., 2011). In the pathogen Pseudomonas aeruginosa, the presence of GlcNAc leads to production of the antibiotic pyocyanin (Korgaonkar et al., 2011). Interestingly, in this organism, gene expression is activated by the LacI-family transcriptional regulator NagR in response to GlcNAc, highlighting the disparate strategies different microbes have employed to sense and respond to environmental sources of GlcNAc. Finally, the chitin-related Nod factors produced by rhizobia are key signaling molecules throughout the development of the Rhizobium-legume symbiosis (Hamel et al., 2010).
At this time, we are uncertain which NagC-controlled factor(s) is responsible for the significant colonization defect associated with the ΔnagC (i.e., unrepressed) mutant. However, our results from co-colonization experiments with wild-type and ΔnagC strains show: 1) the presence of wild-type cells does not abrogate the colonization defect of the ΔnagC mutant, and 2) the ΔnagC mutant does not impede the ability of wild-type cells to colonize the light organ. These findings suggest that the squid host is able to actively and specifically discourage bacterial cells expressing NagC-repressed genes while recruiting potential symbionts.
We initiated this study to determine whether specific bacterial regulatory proteins control the patterns of gene expression that were observed in a mature symbiosis. Interestingly, the transcriptomics study of the adult symbiosis did not reveal changes in the expression of nagA or nagB (Wier et al., 2010), which suggests that symbiont metabolism of GlcNAc is independent of the diel cycle. Nevertheless, we have discovered that the GlcNAc-6P-responsive repressor NagC is important for colonization of the squid light organ by its specific symbiont, V. fischeri. We hypothesize that the squid host selects for symbionts containing a functional NagC during the initial steps of colonization, thus ensuring that the bacterial population in the mature symbiosis retains the capacity to regulate the expression of the chitin- and GlcNAc-utilization genes. Future investigation of mature symbioses established with the NagC mutants generated in this study will reveal the role of NagC during the initial development and maintenance of the squid-vibrio symbiosis.
Experimental Procedures
Growth and media
V. fischeri strains were grown at 28°C with aeration in LBS broth (Graf et al., 1994) without supplemented glycerol. When necessary, chloramphenicol and erythromycin were used at 2.5 μg/ml and 5.0 μg/ml, respectively.
Strains and plasmids
The strains and plasmids used in this study are listed in Table 1, and additional details of their construction are located in the Supporting Information.
Table 1.
Strains and plasmids used in this study
| Strain | Genotype | Reference |
|---|---|---|
| EC100 | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ− rpsL (StrR) nupG | Epicentre Biotechnologies |
| ES114 | Wild-type V. fischeri | (Mandel et al., 2008; Ruby et al., 2005) |
| CA42 | ES114 ΔnagB | This study |
| JAS101 | ES114 ΔnagB Tn7::pEVS107 | This study |
| TIM302 | ES114 Tn7::gfp erm | (Miyashiro et al., 2010) |
| TIM377 | ES114 ΔnagC::nagC-myc-6xHis | This study |
| TIM381 | ES114 ΔnagC::nagC(E241A)-myc-6xHis | This study |
| WPK54 | ES114 nagA::erm | This study |
| WPK84 | ES114 nagC::erm | This study |
| WPK100 | ES114 ΔnagC | This study |
| Plasmid | Genotype | Reference |
| pBAD-myc-His | Protein expression vector | Invitrogen Corp. |
| pEVS79 | pBC SK (+) oriT cat | (Stabb et al., 2002) |
| pEVS104 | R6Kori RP4 oriT trb tra kan | (Stabb et al., 2002) |
| pEVS107 | R6Kori oriT mini-Tn7 mob erm kan | (McCann et al., 2003) |
| pRSETB-mCherry | mCherry expression vector | (Shaner et al., 2004) |
| pTM214 | pVSV105 lacIq Ptrc-mCherry | This work |
| pTM267 | Promoterless-gfp reporter plasmid | (Miyashiro et al., 2010) |
| pTM314 | pTM267 PVF_1598-gfp | This work |
| pTM322 | pEVS79 ΔnagC | This work |
| pTM331 | pBAD nagC-myc-6xHis | This work |
| pTM345 | pBAD nagC(E241A)-myc-6xHis | This work |
| pTM346 | pEVS79 ΔnagC::nagC-myc-6xHis | This work |
| pTM350 | pEVS79 ΔnagC::nagC(E241A)-myc-6xHis | This work |
| pTM355 | pTM267 PVF_0807-gfp | This work |
| pTM360 | pVSV105 lacIq Ptrc-nagC | This work |
| pTM361 | pTM267 PVF_0807-O1-gfp | This work |
| pTrc99A | lacIq Ptrc-MCS bla | (Amann et al., 1988) |
| pUX-BF13 | R6Kori tns bla | (Bao et al., 1991) |
| pVSV105 | R6Kori ori(pES213) RP4 oriT cat | (Dunn et al., 2006) |
| pXDC34 | pEVS79 ΔnagB | This work |
Transposon Mutagenesis and Screen
To generate a transposon-mutant library of V. fischeri, the vector pMJM10 (Studer et al., 2008), which contains a Tn5 transposon encoding oriVR6Kγ and Erm resistance, was conjugated into ES114 via pEVS104 (Stabb et al., 2002), and plated onto LBS with 5.0 μg/ml erythromycin. Approximately 100,000 ErmR V. fischeri colonies that resulted from multiple independent conjugations were pooled and stored in 17% glycerol at −80°C.
To isolate transposon mutants with enhanced VF_1598 expression, the reporter plasmid pTM314 was conjugated into the transposon-mutant library described above and plated onto LBS with 2.5 μg/ml chloramphenicol. The resulting colonies were screened for GFP levels using a Leica MZFLIII fluorescence dissecting microscope (Leica Microsystems, Wetzlar, Germany), equipped with a GFP2 filter set.
Fluorescence Assay
Overnight cultures were diluted 1:100 and grown aerobically in LBS broth at 28°C. At OD600 ~0.7, cultures were cooled quickly using an ice-slurry mix, and 1-ml samples were centrifuged at 4°C for 5 min at 10,000 g. Cell pellets were resuspended in 350 μl of minimal media without a supplemented carbon source. The OD600, GFP fluorescence, and mCherry fluorescence of 100-μl samples were measured in triplicate using a Tecan Genios Pro plate reader (Tecan Group, Mannedorf, Switzerland) as previously described (Miyashiro et al., 2010).
Exochitinase Assay
Exochitinase activities of 10-μl samples of overnight-culture supernatant were determined using 4-Nitrophenyl N,N′-diacetyl-β-D-chitobioside (Sigma-Aldrich Corp., St. Louis, MO, USA) according to the manufacturer’s instructions.
qPCR
Total RNA extraction and qPCR were performed with cultures grown to OD600 ~0.7 as previously described (Miyashiro et al., 2010). Significance between samples within each gene was determined by one-way ANOVA and Tukey HSD (SPSS software, version 19). The p-values were adjusted using false discovery rate’s correction (FDR, R software, version 2.12). Primer sequences are listed in Table S1.
Protein Expression and Purification
To purify NagC and NagC(E241A), overnight cultures of E. coli strain EC100 harboring pTM331 and pTM345, respectively, were diluted 1:100 into 500 ml Luria-Bertani (LB) broth containing 100 μg/ml carbinicillin, and grown at 37°C. After 4 h, cells were harvested, lysed by sonication and spun at 4°C for 15 min at 30,000 × g. The soluble fraction was incubated 4:1 with 50% Ni-NTA agarose (Qiagen, Valencia, CA, USA) at 4°C. After 1 h, the agarose bed was washed twice and eluted into four 0.5 ml fractions. Protein levels were determined by OD280. The fraction containing the highest level of NagC or NagC(E241A) was dialyzed into storage buffer (50 mM NaH2PO4 [pH 8.0], 600 mM NaCl, 10% glycerol) and stored at 4°C.
NagC-DNA Binding Measurements
5′ Texas Red-labeled, HPLC-purified DNA oligonucleotides and their unlabeled complements were obtained from Integrated DNA Techonologies, Inc. (IDT, Coralville, Iowa, USA). To generate the double-stranded DNA probes, equimolar levels of complementary single-stranded oligonucleotides were combined in annealing buffer (IDT) and heated to 94°C. After 2 min, the annealing reactions were slowly cooled to room temperature. Each 400-μl binding reaction contained 10 nM double-stranded DNA probe in binding buffer (20 mM Tris-HCl [pH 7.5], 300 mM KCl, 2.5 mM MgCl2, 0.5 mg/ml BSA, 6 μg/ml salmon sperm DNA). When present, GlcNAc-6P (Sigma-Aldrich Corp.) was used at 10 mM. NagC or NagC(E241A) was added to final concentrations between 10 nM and 3 μM. Reactions were incubated at 28°C for 10 min. Fluorescence polarization (P) of each reaction was measured using a Beacon 2000 fluorescence polarization system (Panvera Corp., Madison, WI, USA). Fluorescence anisotropy (F) was calculated as F = (2 × P)/(3 − P), where P is the polarization of each sample.
5′ RACE
Six micrograms of total RNA was extracted from a culture of ΔnagC (WPK100) grown to OD600 = 0.6, and subjected to dephosphorylation by Tobacco Acid Pyrophosphatase (TAP; Epicentre Biotechnologies, Madison, WI) for 30 min at 37°C. The RNA oligo RNA-linker was ligated to total RNA using T4 RNA ligase (New England Biolabs, Inc., Ipswich, MA) according to the manufacturer’s instructions. cDNA was synthesized using AMV RT (Promega Corp., Madison, WI) according to the manufacturer’s instructions with the nagA-specific primer nagA-L. PCR amplification was performed using the nested primers Adapter and nagA-nested. The single band that was present within the reaction containing TAP, but absent from the TAP-minus control reaction, was subcloned using the TOPO TA Cloning Kit (Invitrogen) and sequenced as recommended by the manufacturer.
Squid Colonization Experiments
Except for the details described within the text, squid colonization experiments were performed as previously described (Miyashiro et al., 2010).
Supplementary Material
Acknowledgments
We thank R. Kerby for assistance with the fluorescence aniosotropy measurements. We thank N. Kremer for assistance with statistical analysis of the qPCR data. We thank M. McFall-Ngai, and members of the Ruby and McFall-Ngai labs for valuable advice throughout the course of this study. We also thank two anonymous reviewers for their suggestions. This work was supported by the NIH grant RR12294 to E.G.R. and M. McFall-Ngai, by the NSF grant IOS-0817232 to M. McFall-Ngai and E.G.R., by 5F32GM084620 and 1K99GM097032 Awards from the NIGMS to T.M., by an NSF Graduate Research Fellowship to J.S., and by a China Scholarship Council’s State Scholarship Fund Award to X.C.
References
- Alvarez-Anorve LI, Bustos-Jaimes I, Calcagno ML, Plumbridge J. Allosteric regulation of glucosamine-6-phosphate deaminase (NagB) and growth of Escherichia coli on glucosamine. J Bacteriol. 2009;191:6401–6407. doi: 10.1128/JB.00633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann E, Ochs B, Abel KJ. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene. 1988;69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- Bao Y, Lies DP, Fu H, Roberts GP. An improved Tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene. 1991;109:167–168. doi: 10.1016/0378-1119(91)90604-a. [DOI] [PubMed] [Google Scholar]
- Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol. 2006;72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Qaidi S, Plumbridge J. Switching control of expression of ptsG from the Mlc regulon to the NagC regulon. J Bacteriol. 2008;190:4677–4686. doi: 10.1128/JB.00315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Rao KH, Sengupta M, Bhattacharya SK, Datta A. Two gene clusters co-ordinate for a functional N-acetylglucosamine catabolic pathway in Vibrio cholerae. Mol Microbiol. 2011;80:1549–1560. doi: 10.1111/j.1365-2958.2011.07664.x. [DOI] [PubMed] [Google Scholar]
- Graf J, Dunlap PV, Ruby EG. Effect of transposon-induced motility mutations on colonization of the host light organ by Vibrio fischeri. J Bacteriol. 1994;176:6986–6991. doi: 10.1128/jb.176.22.6986-6991.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel LP, Beaudoin N. Chitooligosaccharide sensing and downstream signaling: contrasted outcomes in pathogenic and beneficial plant-microbe interactions. Planta. 2010;232:787–806. doi: 10.1007/s00425-010-1215-9. [DOI] [PubMed] [Google Scholar]
- Heath-Heckman EA, McFall-Ngai MJ. The occurrence of chitin in the hemocytes of invertebrates. Zoology (Jena) 2011;114:191–198. doi: 10.1016/j.zool.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DE, Gevers D, Vahora NM, Polz MF. Conservation of the chitin utilization pathway in the Vibrionaceae. Appl Environ Microbiol. 2008;74:44–51. doi: 10.1128/AEM.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyhani NO, Roseman S. Physiological aspects of chitin catabolism in marine bacteria. Biochim Biophys Acta. 1999;1473:108–122. doi: 10.1016/s0304-4165(99)00172-5. [DOI] [PubMed] [Google Scholar]
- Korgaonkar AK, Whiteley M. Pseudomonas aeruginosa enhances production of an antimicrobial in response to N-acetylglucosamine and peptidoglycan. J Bacteriol. 2011;193:909–917. doi: 10.1128/JB.01175-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel MJ, Stabb EV, Ruby EG. Comparative genomics-based investigation of resequencing targets in Vibrio fischeri: focus on point miscalls and artefactual expansions. BMC Genomics. 2008;9:138. doi: 10.1186/1471-2164-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann J, Stabb EV, Millikan DS, Ruby EG. Population dynamics of Vibrio fischeri during infection of Euprymna scolopes. Appl Environ Microbiol. 2003;69:5928–5934. doi: 10.1128/AEM.69.10.5928-5934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall-Ngai M. Host-microbe symbiosis: the squid-Vibrio association--a naturally occurring, experimental model of animal/bacterial partnerships. Adv Exp Med Biol. 2008;635:102–112. doi: 10.1007/978-0-387-09550-9_9. [DOI] [PubMed] [Google Scholar]
- Meibom KL, Li XB, Nielsen AT, Wu CY, Roseman S, Schoolnik GK. The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci U S A. 2004;101:2524–2529. doi: 10.1073/pnas.0308707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashiro T, Wollenberg MS, Cao X, Oehlert D, Ruby EG. A single qrr gene is necessary and sufficient for LuxO-mediated regulation in Vibrio fischeri. Mol Microbiol. 2010;77:1556–1567. doi: 10.1111/j.1365-2958.2010.07309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mopper K, Schultz CA, Chevolot L, Germain C, Revuelta R, Dawson R. Determination of Sugars in Unconcentrated Seawater and Other Natural-Waters by Liquid-Chromatography and Pulsed Amperometric Detection. Environ Sci Technol. 1992;26:133–138. [Google Scholar]
- Pennetier C, Dominguez-Ramirez L, Plumbridge J. Different regions of Mlc and NagC, homologous transcriptional repressors controlling expression of the glucose and N-acetylglucosamine phosphotransferase systems in Escherichia coli, are required for inducer signal recognition. Mol Microbiol. 2008;67:364–377. doi: 10.1111/j.1365-2958.2007.06041.x. [DOI] [PubMed] [Google Scholar]
- Plumbridge J. An alternative route for recycling of N-acetylglucosamine from peptidoglycan involves the N-acetylglucosamine phosphotransferase system in Escherichia coli. J Bacteriol. 2009;191:5641–5647. doi: 10.1128/JB.00448-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge J, Kolb A. DNA bending and expression of the divergent nagE-B operons. Nucleic Acids Res. 1998;26:1254–1260. doi: 10.1093/nar/26.5.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge JA. Repression and induction of the nag regulon of Escherichia coli K-12: the roles of nagC and nagA in maintenance of the uninduced state. Mol Microbiol. 1991;5:2053–2062. doi: 10.1111/j.1365-2958.1991.tb00828.x. [DOI] [PubMed] [Google Scholar]
- Rogers MJ, Ohgi T, Plumbridge J, Soll D. Nucleotide sequences of the Escherichia coli nagE and nagB genes: the structural genes for the N-acetylglucosamine transport protein of the bacterial phosphoenolpyruvate: sugar phosphotransferase system and for glucosamine-6-phosphate deaminase. Gene. 1988;62:197–207. doi: 10.1016/0378-1119(88)90558-6. [DOI] [PubMed] [Google Scholar]
- Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, Gunsalus R, et al. Complete genome sequence of Vibrio fischeri: a symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A. 2005;102:3004–3009. doi: 10.1073/pnas.0409900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Stabb EV, Ruby EG. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 2002;358:413–426. doi: 10.1016/s0076-6879(02)58106-4. [DOI] [PubMed] [Google Scholar]
- Studer SV, Mandel MJ, Ruby EG. AinS quorum sensing regulates the Vibrio fischeri acetate switch. J Bacteriol. 2008;190:5915–5923. doi: 10.1128/JB.00148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson FL, Neto AA, Santos Ede O, Izutsu K, Iida T. Effect of N-acetyl-D-glucosamine on gene expression in Vibrio parahaemolyticus. Microbes Environ. 2011;26:61–66. doi: 10.1264/jsme2.me10152. [DOI] [PubMed] [Google Scholar]
- White RJ. Control of amino sugar metabolism in Escherichia coli and isolation of mutants unable to degrade amino sugars. Biochem J. 1968;106:847–858. doi: 10.1042/bj1060847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier AM, Nyholm SV, Mandel MJ, Massengo-Tiasse RP, Schaefer AL, Koroleva I, et al. Transcriptional patterns in both host and bacterium underlie a daily rhythm of anatomical and metabolic change in a beneficial symbiosis. Proc Natl Acad Sci U S A. 2010;107:2259–2264. doi: 10.1073/pnas.0909712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.