

1. Introduction
Phosphate anions are one of the most important constituents of living systems. Together with heterocyclic bases and sugars, phosphates make up DNA, the hereditary element of living systems.1,2 In addition, phosphate ions and their derivatives play pivotal roles in energy storage and transduction in biological systems. Protein phosphorylation is a key mechanism for signaling, and phosphorylated proteins are a significant source of ingested phosphate. Furthermore, phosphates are industrially important components of both medicinal drugs and fertilizers. Pollution from phosphate and phosphorylated compounds is in part responsible for the eutrophication of natural water sources, leading to a dangerous increase in toxic algal blooms.3,4
Phosphate also has important medicinal implications. Normally, excess phosphate is cleared through the kidneys; however, controlling the blood phosphate levels of patients with kidney failure is a difficult problem for which there is no adequate solution. Hyperphosphatemia, defined as an abnormally high concentration of serum phosphate levels, is a condition that affects nearly all hemodialysis patients.5,6 The adverse effects of hyperphosphatemia include the development of hyperparathyroidism, soft tissue calcification, cardiovascular complications resulting from the development of metastatic calcifications at the cardiac level, and increased morbidity and mortality.7-18
Given the central role phosphate plays in the environment and in biology, it is perhaps not surprising that efforts have been made to achieve the selective binding of phosphate and phosphorylated molecules for almost 100 years. In 1914, Taylor and Miller developed a colorimetric method for the estimation of phosphorous in biological material.19 This method, now much improved,20-34 relies on a colored molybdenum(IV) phosphate complex to assay inorganic phosphate. This complex is produced from sodium molybdate and a strong reducing agent and involves a procedure that is often time consuming. It also generates toxic metal waste and suffers from interference. Other traditional assays for phosphorylated molecules have involved derivatization-based protocols35-44 or biological recognition elements, such as enzymes, that often require special handling or that suffer from poor stability and complicated, costly production procedures.45-51
The investigation of synthetic (abiotic) phosphate receptors seeks to provide improved methodologies for the detection, extraction, and transport of biologically, chemically and environmentally important phosphates. While it has been extensively pursued, the sensitive and selective binding of phosphorylated molecules remains a challenging area of research in supramolecular chemistry. The large size of the phosphate anion and its high hydrophilicity place it near the bottom of the Hofmeister52 selectivity series, which is based on the Gibbs free energy of hydration. In the case of monobasic anions, for instance, the hydration energies (given in parentheses) increase as follows: ClO4- (-205 kJ/mol) > I- (-275 kJ/mol) > CN- (-295 kJ/mol) > NO3- (-300 kJ/mol) > Br- (-315 kJ/mol) > Cl- (-340 kJ/mol) > HSO4- (-330 kJ/mol) > HCO3- (-335 kJ/mol) > OAc- (-365 kJ/mol) > > H2PO4- ≈ F- (-465 kJ/mol).53 These values increase substantially as the charge of the anion increases. For instance, the hydration energy of PO43- is -2765 kJ/mol. As a result, phosphate binding is generally inefficient. The design of phosphate receptors is further complicated by the acid-base properties of phosphate anions. The pKa values of inorganic phosphate in water are 2.12, 7.20, and 10.9, meaning that two dominant anionic protonation states (monohydrogenphosphate and dihydrogenphosphate) are present at neutral pH.54 Receptor deprotonation by phosphate anions must also be considered, particularly at hydrogen bond donor sites and in organic media. In addition to the more common anion-hydrogen bond donor interactions, protonated phosphate anions can also interact with hydrogen bond acceptors. This property can be exploited to impart selectivity for inorganic phosphate over other tetrahedral anions such as sulfate that are not protonated near neutral pH; however, the dual donor-acceptor nature of phosphate anions can also lead to anion-anion interactions in less polar media. This behavior has been manifested in some cases through the binding of phosphate aggregates by organic hosts.55,56 More recently, Gale and co-workers reported anion-anion proton transfer between dihydrogenphosphate species in the presence of a high affinity phosphate receptor in mixed organic / aqueous media.57
Given these challenges, a variety of recognition interactions, such as hydrogen bonding, electrostatic interactions, van der Waals forces, π-surface interactions, shape complementarity, and metal coordination, have been employed alone or in concert to generate phosphate receptors.58-62 Such phosphate receptors have been developed for several different applications, including phosphate ester hydrolysis, ion selective electrodes and membrane transport. The goal of this review is to provide a comprehensive summary of these efforts with an emphasis on the fundamental molecular recognition processes involved rather than the potential or realized applications.
This review will specifically focus on small molecule phosphate receptor systems. Such systems offer several advantages over enzymatic and inorganic-based recognition and sensing protocols. In spite of these advantages, which will be highlighted in the sections that follow, a thorough review of the major progress in this area has not yet appeared. Although several recent works cover general aspects of anion binding and the associated recognition motifs,54,62-78 only a few reviews have specifically examined synthetic phosphate receptors,61,79-82 with these examples focusing on selected aspects of phosphate binding. Here, we have attempted to survey synthetic phosphate receptors reported through 2009 in order to codify the most relevant results within a single reference. Efforts have been made to include both advances in sensing methods and progress in the area of binding, providing an up-to-date survey of the basic subunits used to achieve phosphate recognition.
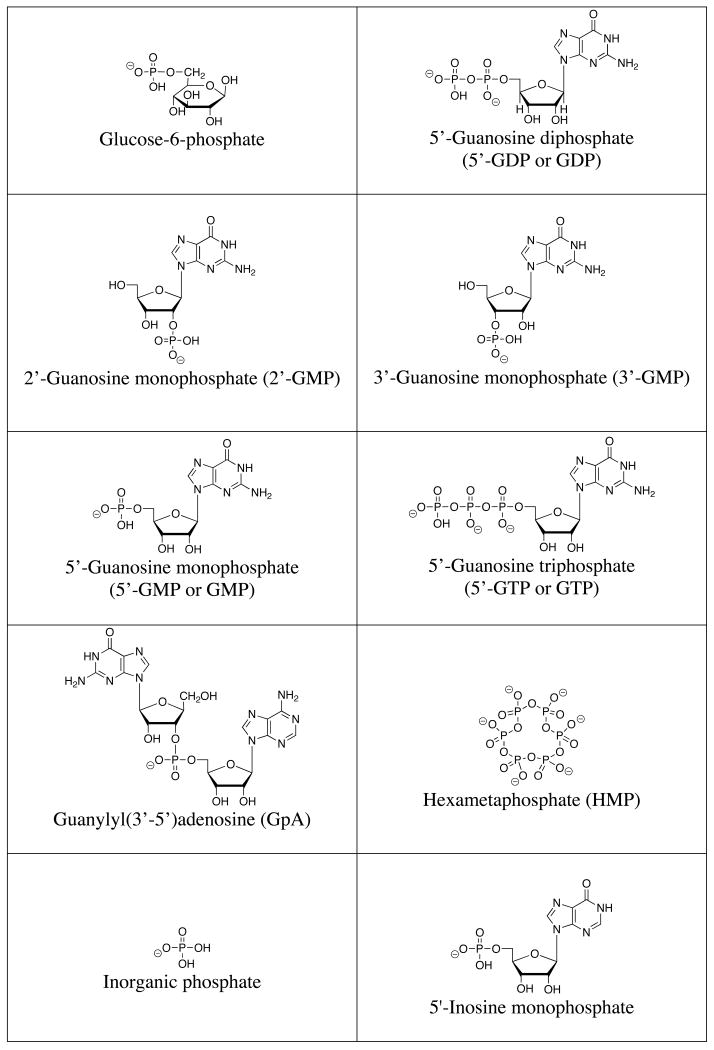
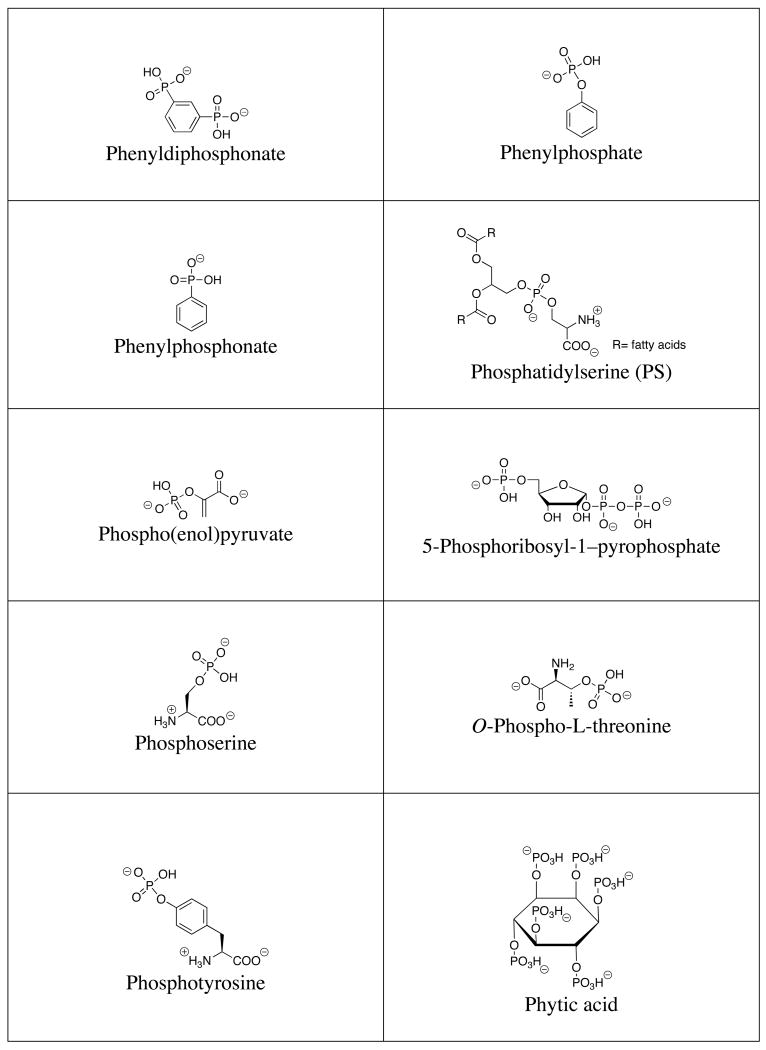
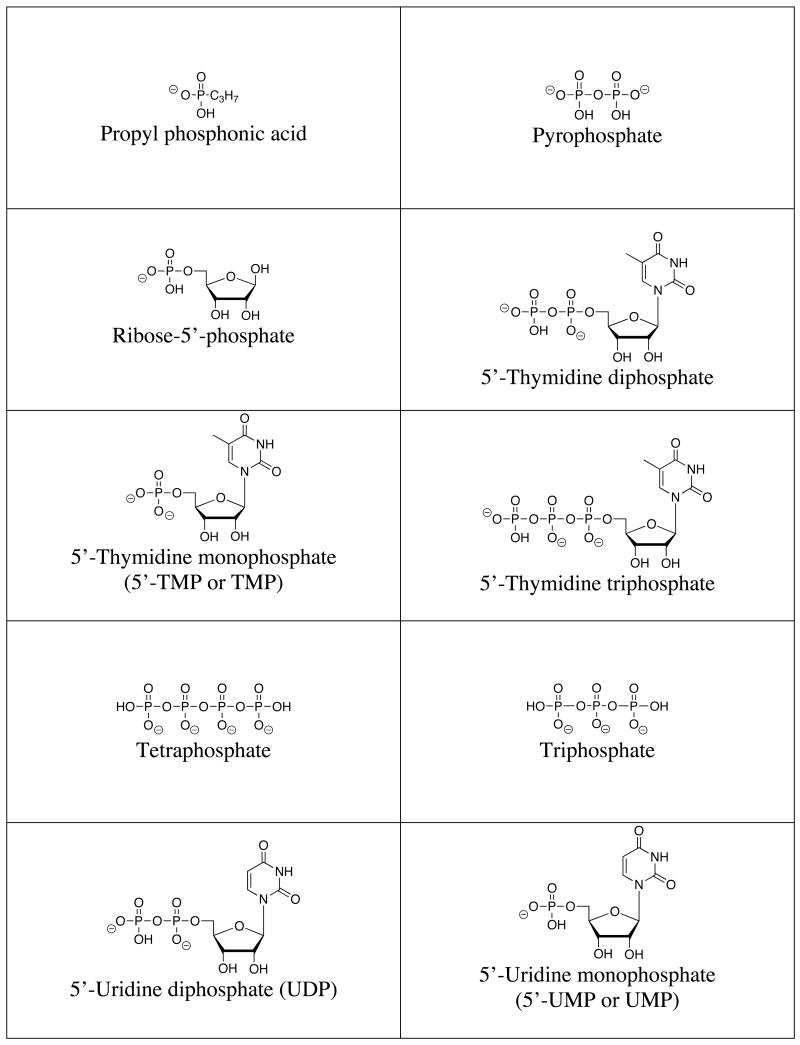
A list of the full names, common names, abbreviations and structures of the commonly targeted phosphates covered in this review is shown in Table 1. Discussions will focus on phosphate and small phosphorylated molecules rather than macromolecules (e.g., DNA or phosphorylated proteins).
Table 1.
Structures of commonly targeted phosphates.
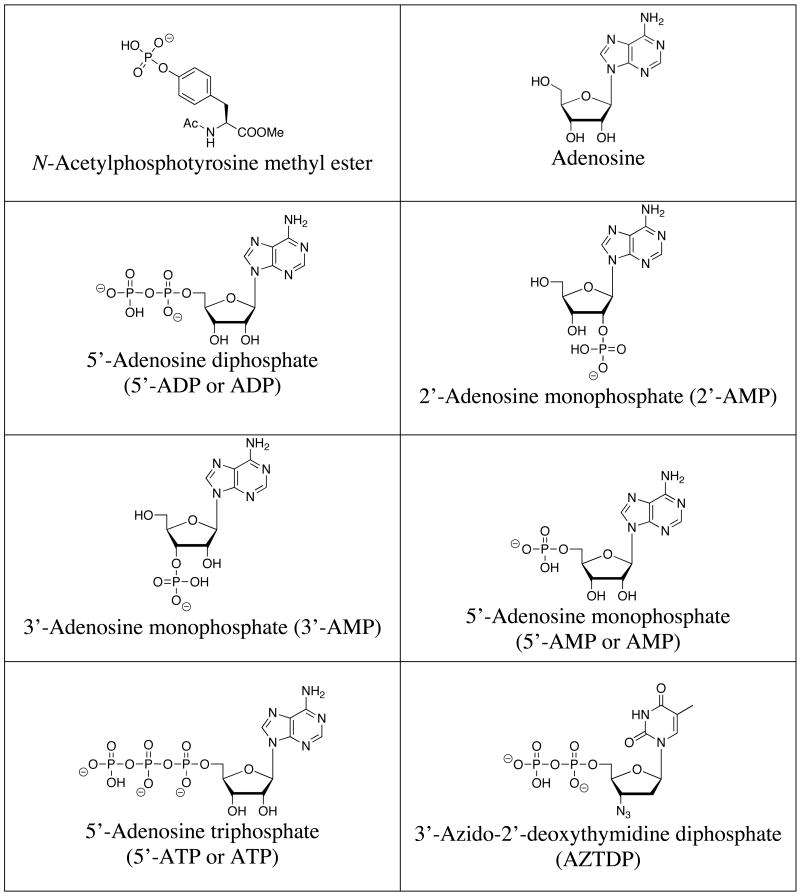 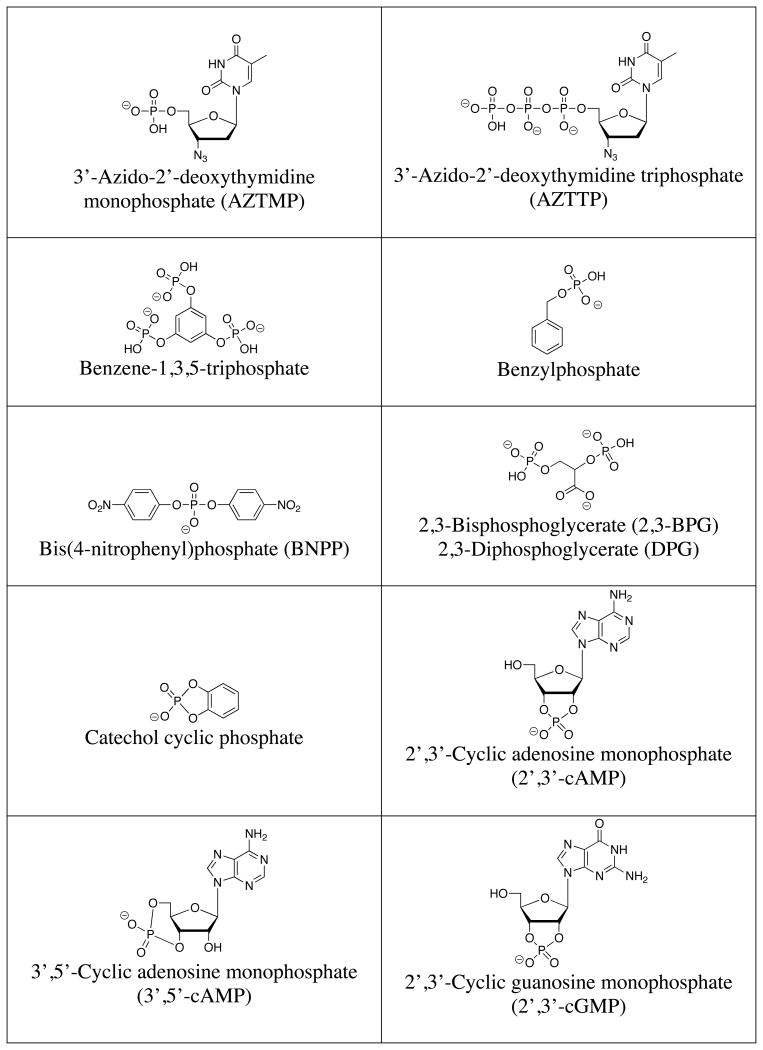 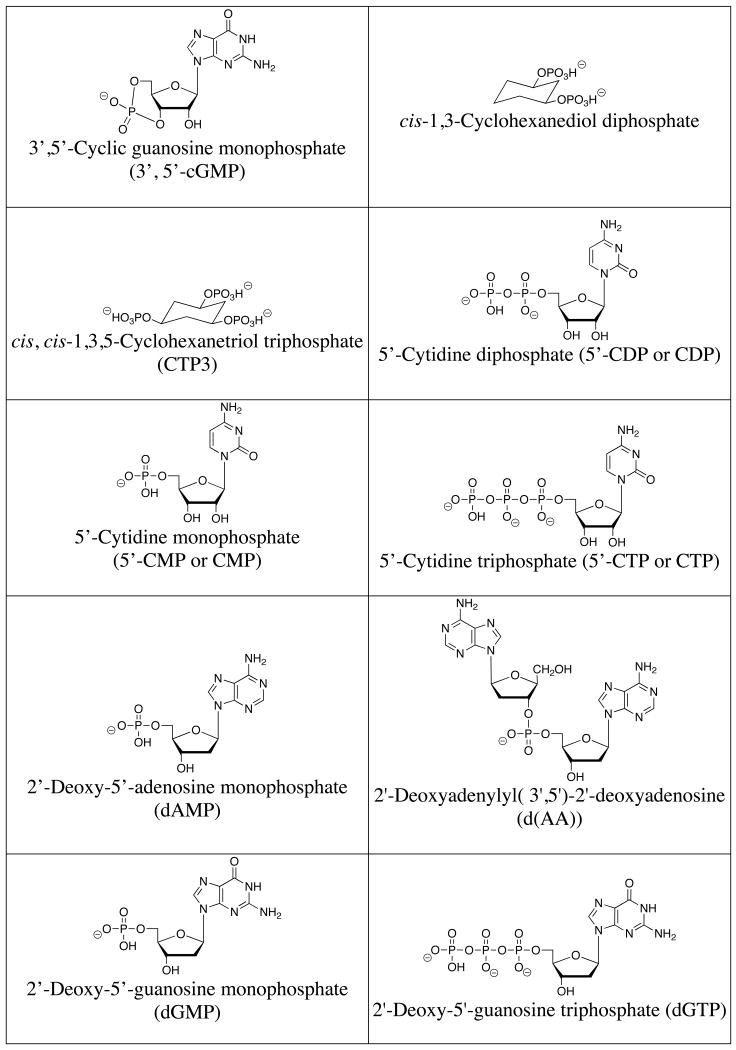  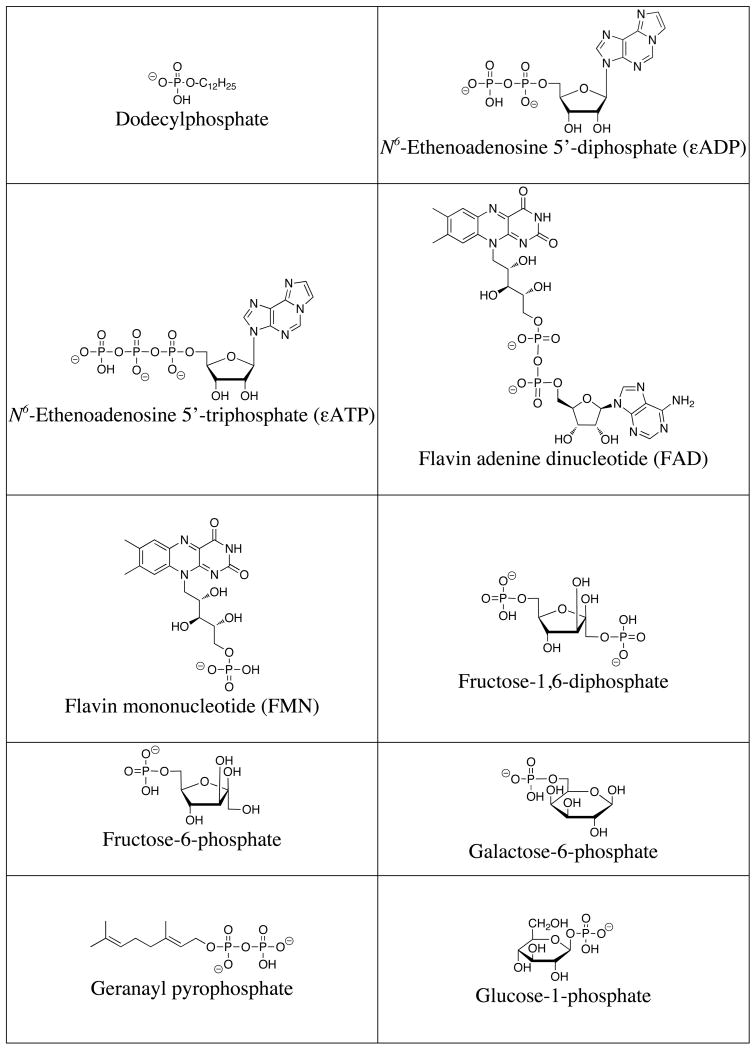     
|
2. Detection Methods
The interaction of phosphate and phosphorylated molecules with artificial receptor systems has been monitored using several methods, the choice of which has been largely dependent on the nature of the receptor in question. These methods will be outlined here in order to facilitate the ensuing discussion of receptor design.
2.1. Nuclear Magnetic Resonance
The detection of host-guest interactions by nuclear magnetic resonance (NMR) spectroscopy relies on guest-induced 1H NMR spectroscopic shifts of the synthetic receptor and/or 31P NMR spectroscopic shifts specific for phosphorylated molecules. This method is particularly useful for hydrogen-bonding moieties and can be used to gain both structural and thermodynamic information. For example, the interaction of phosphate anions with amide hydrogen atoms can often be conveniently followed by monitoring changes in the 1H NMR spectrum. The receptor can also be modified with groups such as aromatic moieties whose 1H NMR signals are highly sensitive to changes in the local environment, or which can interact with additional structural elements present in the targeted phosphorylated compounds. These modifications often increase both the sensitivity and specificity of the system. Phosphorous NMR spectroscopic studies can also lend valuable insight into host:phosphate interactions. Such analyses are often straightforward due to the high abundance of the 31P isotope and the high gyromagnetic ratio (17.235 MHz/T) of the phosphorous nucleus. Phosphorous chemical shifts, which are generally referenced to an external phosphoric acid standard, are often changed upon complexation to a host compound. In addition, the individual phosphorous atoms of nucleotide di- or triphosphates display characteristic signals that when monitored allow for the determination of which phosphate units participate most strongly in complexation. In well-behaved systems, the association constants corresponding to complex formation can often be elucidated using standard curve fitting programs, such as the EQNMR software.83 Stoichiometries can often be deduced from the method of continuous variation (so-called Job plots). Thus, considerable insights can be obtained from NMR spectroscopic analysis. However, it should be noted that the utility of 1H NMR spectroscopic methods is limited by the fact that relatively high concentrations (usually 10-2-10-3 M) are often required to obtain well-resolved signals. This leads to rapid saturation of the response during the titration in the case of strong binding. As a consequence, the dynamic range accessible by NMR spectroscopic methods is generally limited to K ≤ 104 M-1. Further limitations to these methods occur when the exchange rate is slow. In such instances, it is necessary to integrate the signals for both the bound and unbound forms.
2.2. Optical Methods
Optical methods have several advantages compared to those based on NMR spectroscopy. While much lower concentrations are often required, a change in the spectrum is also needed. To achieve this change, systems containing a combination of substrate-recognition functionality (receptor) and optical-signaling capacity (chromophore) are often prepared. These moieties may be either directly linked or appropriately associated in a noncovalent manner. Such designs permit the detection of substrates via binding-induced changes in the absorption or emission properties. Increasingly, these generalized approaches have been used to effect both qualitative and quantitative analysis. As such, optical methods have been instrumental in the creation of colorimetric sensors (“chemosensors”), as discussed in subsection 2.2.1 below. Other common methods of detection, including fluorescence spectroscopy and isothermal titration calorimetry (ITC), will be described in later sections.
2.2.1. Colorimetric Sensing
2.2.1.1 Covalently Attached Chromophores
As noted above, many non-colored receptors have been functionalized with chromophores to produce covalent frameworks that undergo a pronounced color change when treated with an appropriate guest (Figure 1).84 These color changes can have their origin in analyte binding-induced changes in the HOMO-LUMO gap or modification in key charge-transfer (CT) bands.85
Figure 1.
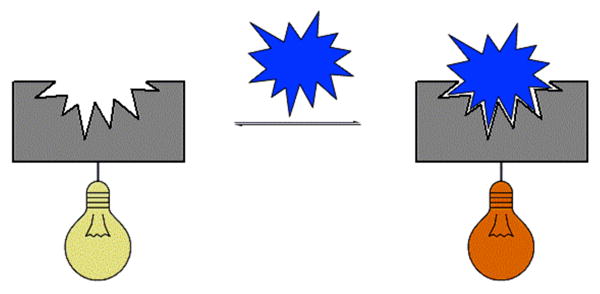
Covalently attached reporter-receptor system. The reporter unit is represented by the light bulb graphic with the receptor in gray and analyte in blue.
2.2.1.2 Indicator Displacement Assay (IDA)
Indicator displacement assay (IDA) is a competition method for the sensing of analytes (Figure 2).86 The molecular ensemble employed consists of a recognition unit designed for selective interaction with a desired analyte along with an external indicator that associates with the recognition unit in the absence of the analyte. When the analyte is added, the indicator is displaced from the binding cavity, producing a measurable change in the optical properties of the indicator. This method exhibits several useful features. For example, it can be applied to a variety of receptors without the need for covalent attachment of a reporter group. The use of noncovalent indicators makes each system amenable to both fluorescence and UV-Vis spectrophotometry following the selection of appropriate indicators. Furthermore, the use of indicators with varying association constants allows tuning of the system to analytes with a range of binding affinities. Finally, several indicators may be used with the same host-guest system as a means of corroborating results.
Figure 2.
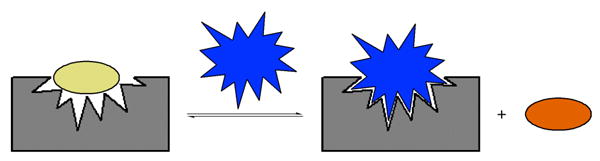
Indicator displacement assay (IDA). The indicator is represented by the oval, while the receptor unit is gray and the analyte blue.
2.2.2. Fluorescence Sensing
Fluorescence spectroscopy is an attractive analytical method due largely to its high sensitivity and submillisecond temporal resolution.87,88 Reported fluorescence anion sensors have utilized competitive binding,86 photo-induced electron transfer (PET),89-91 electronic energy transfer (EET),92 metal-to-ligand charge transfer (MLCT),93 excimer/exciplex formation,94,95 internal charge transfer (ICT),96 and, less frequently, excited-state proton transfer (ESPT).97,98 Many of the structural features that modulate fluorescence efficiency have been determined, including double-bond torsion, low energy n-π* levels, “heavy” atoms, weak bonds, and the availability of subunits that allow for PET or EET.87 As a result, many opportunities exist for modulating structural features at the molecular level in order to produce receptors that allow emission spectroscopic properties to be exploited in an analytically useful way.
2.3. Electrochemical Redox Activity
2.3.1. Cyclic Voltammetry (CV)
Another method for detecting the interaction between a receptor and a substrate involves monitoring changes in the redox properties of the system. Often the receptors themselves are not redox active, at least not within a useful electrochemical window. Therefore, redox-active groups (cobaltocenium, ruthenium(II) bipyridyl groups, ferrocenyl moieties, etc.) are often attached to receptors near the substrate binding sites.99 Analyte binding then shifts the redox potential of the reporter group. For example, in the presence of an anion, the voltammetric behavior of a metallocenium moiety is shifted toward that of the corresponding metallocene. This cathodic perturbation is rationalized in terms of anion-induced stabilization of the positively charged metallocenium moiety relative to the metallocene form. However, often a poor correlation exists between the electroactive changes and the actual binding affinity; i.e., the largest cathodic perturbation does not imply the strongest association. This is because the redox changes include contributions from at least two oxidation states, only one of which reflects binding to the parent form of the receptor.
2.3.2 Ion Selective Electrodes (ISEs)
A variety of anion-selective electrodes have been introduced based on recent advances in the host-guest chemistry of anions.100-110 While significant progress has been made, it remains a challenge to obtain systems that are selective for the strongly hydrophilic phosphate species. This reflects in large measure the unfavorable standard free enthalpies of transfer of phosphate anions from aqueous milieus to ISE membranes. This energetic cost, which again reflects the Hofmeister bias, needs to be overcome by selective complexation. While workable phosphate-selective membranes can be achieved by using homogenously distributed ionophores, such as organotin, organovanadyl, polyamine, or guanidinium-based cations, in the polymer membrane, the resulting systems often suffer from poor stability or low detection limits.111
2.4. Isothermal Titration Calorimetry
In recent years, isothermal titration calorimetry (ITC) has emerged as one of the more powerful methods for studying anion-receptor interactions. ITC is especially attractive since, in well-behaved cases, it provides ready access to the individual thermodynamic parameters corresponding to the proposed binding interactions (i.e. ΔG, ΔH, and ΔS). Although temperature-dependent studies (e.g., NMR, UV-Vis) can be used to derive these same thermodynamic parameters, such analyses are generally laborious, insensitive, and error-prone. ITC, on the other hand, allows the dissection of the free energy of association into its individual enthalpic and entropic components via a measurement carried out at a single temperature. However, ITC does not provide direct insights into the underlying chemistry (in contrast to, e.g., NMR spectroscopy) and requires fitting the data to a presupposed binding model. In other words, because calorimetric measurements reflect all processes occurring in solution, it is important to find and study simple systems whose host-guest interactions can then be extrapolated to more complex systems. Even then, great care needs to be exercised lest an incorrect assessment of the underlying chemistry be made.
3. Phosphate Recognition in Nature
A number of design principles for selective synthetic phosphate receptors can be derived from an examination of naturally occurring phosphate receptors.54,80 Over half of all proteins in living systems are thought to bind phosphorylated guests, particularly protein kinases and phosphatases, which regulate a large range of inter- and intracellular signaling processes. Analysis of phosphate binding sites has been made possible from a number of X-ray diffraction structural studies. For example, Quiocho and co-workers have examined a periplasmic phosphate binding protein that binds inorganic phosphate with high affinity and specificity over sulfate (Figure 3).112,113 Both dihydrogenphosphate and monohydrogenphosphate were found to be bound through an extensive hydrogen bonding network as well as through the formation of a salt-bridge with the guanidinium side chain of an arginine residue (Arg 135). The specificity of this protein against sulfate was attributed to a short hydrogen bond between the protonated oxygen of the phosphate guest and the carboxylate side chain of an aspartate residue (Asp 137) within the binding pocket.114,115 Specifically, it has been suggested that other anionic guests, such as sulfate, would be not be able to participate in this interaction and would thus be repulsed based on charge-charge interactions. The mutation of the Arg 135 and Asp 137 residues led to decreased selectivity but little change in the binding affinity of this protein.116 It was thus proposed that the strength of phosphate:protein binding was dominated by hydrogen bonding or local dipolar interactions.117
Figure 3.
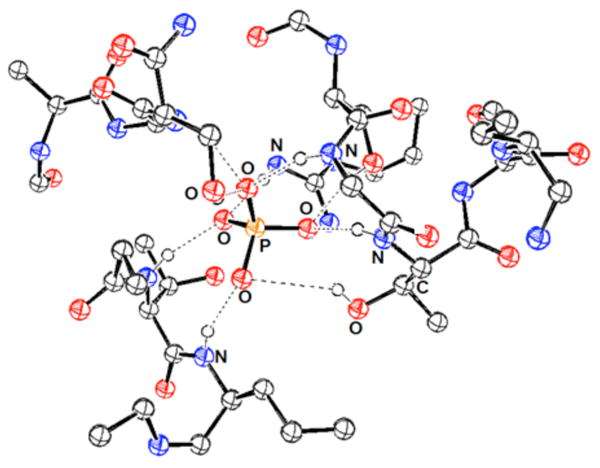
View of the phosphate binding site of the phosphate binding protein. Drawing generated from X-ray diffraction data obtained from the RCSB Protein Data Bank and originally published in reference 115.
Diederich and co-workers recently examined the Protein Data Bank for structural elements common to proteins that bind inorganic phosphate and other naturally occurring phosphorylated molecules.80 Over 3000 phosphate-binding protein structures were identified, and several trends were reported. For example, nearly all protein binding sites contained a large number of glycine residues located in loop motifs that allowed the phosphate guest to be encircled by a number of electrostatic or hydrogen bonding groups. This recognition element was compared to the encapsulation of a phosphate guest by flexible synthetic macrocycles. Hydrogen bonding interactions between the anion and the amide backbone or polar residues were also common. In fact, nearly one third of the studied protein structures relied solely on these interactions (i.e. without assistance from metal chelation or electrostatic interactions). Of the remaining structures, approximately 50% utilized electrostatic interactions with basic lysine or arginine residues, and approximately 20% of the studied proteins incorporated metal chelation interactions. Furthermore, cationic charges were found largely on the exterior of the binding pocket, while neutral polar residues predominated the interior of the cavity. It can thus be concluded that encapsulation, hydrogen bonding, and electrostatic interactions are critical for successful phosphate recognition in nature. As will be seen in the ensuing discussion, these themes are also common among synthetic phosphate receptors.
4. Major Phosphate-Binding Functionalities
In this section, the most common binding subunits employed to create phosphate selective receptors are introduced with the aim of providing a general overview of the motifs that are emanating from this area of research. While we have attempted to organize receptors according to their main binding moiety, it should be noted that many recent receptors combine multiple recognition subunits to increase functionality in complex systems, which makes their classification less than straightforward. It is also important to note that the association constants discussed in this review are taken directly from the referenced reports, and the accuracy of these values will depend heavily upon the detection method and mathematical model employed. While a thorough discussion of these limitations is beyond the scope of this manuscript, we encourage the reader to evaluate critically the methods used for binding constant determination in papers of particular interest.
4.1. Charge-Charge Interactions
4.1.1. Polyammonium Systems
The first synthetic receptors for phosphate to be described in the literature were polyammonium cations. Such systems, first reported over 30 years ago, owe their efficacy in large measure to strong electrostatic interactions generated between the negatively charged phosphates and the protonated polyammonium systems near neutral pH. Nevertheless, it is important to appreciate that systems containing closely spaced nitrogen atoms are not always fully protonated at pH 7. The protonation constants are often greatly reduced, presumably due to charge-charge repulsion. Such effects may explain why amine nitrogen atoms are often separated by 3 or 4 methylene units in naturally occurring systems, such as spermine and spermidine; presumably this spacing ensures maximum protonation near neutral pH. A careful determination of the degree of protonation for a given polyammonium receptor is critical to a discussion of its binding efficacy. Generally, this is accomplished through careful pH titrations. Such titrations are also used to determine binding constants, since the effective pKa of an ammonium site generally shifts in the presence of a bound anion.
In addition to the important role of electrostatic interactions, amine hydrogen bonding can also affect complex stability and selectivity.118 Other relevant factors include macrocycle formation, macrocycle size, nitrogen methylation and metal coordination (a factor discussed in greater detail in Section 4.3). The addition of other receptor moieties, such as aromatic rings and amide groups, can further increase selectivity between various phosphorylated species.
Interest in polyammoniums and their analogs as phosphate binding systems first arose from the discovery that biologically important acyclic polyamines, such as putrescine (1), spermidine (2), and spermine (3), bound 5′-adenosine monophosphate (AMP), 5′-adenosine diphosphate (ADP), and 5′-adenosine triphosphate (ATP).119 Early 1H and 13C NMR spectroscopic studies of the spermidine (2):5′-AMP complex supported a binding mode in which the amine ligand interacted with both the anionic phosphate group and the adenine base.120 In studies of the interaction of spermine (3) with AMP, ADP, and ATP, potentiometric titrations and 1H, 13C, and 31P NMR spectroscopic experiments led to the conclusion that hydrogen bonding interactions between the host and the adenine ring of the nucleotides were critical to complex formation.121,122 On the other hand, similar studies with putrescine (1) and 5′-uridine monophosphate (UMP) revealed interactions only with the phosphate group of the nucleotide.123 The apparent affinity constants for the interaction between these three naturally occurring polyammonium cations and 5-phosphoribosyl-1–pyrophosphate and 2,3-diphosphoglyceric acid (2,3-DPG) were determined using anion affinity chromatography.124 Values for K1 ranged from 300 to 2700 M-1 and increased as the number of amine units increased. Spermidine (2) and spermine (3) were also shown to bind pyrophosphate with association constants of 6.4 × 102 M-1 and 2.7 × 103 M-1, respectively, at pH 7.5, as inferred from resin competition experiments.125 Calorimetric studies yielded standard Gibbs energies of formation of -15.4 and -20.0 kJ/mol, respectively, for the complex formed between amines 2 and 3 and tetra-anionic pyrophosphate at this pH.126 Single crystal X-ray diffraction analysis of the 3:pyrophosphate complex revealed extensive hydrogen bonding interactions between the NH units of the host and the oxygen atoms of the guest.127
Felcman and co-workers later compared the ATP binding ability of spermidine (2) with other small linear polyammonium receptors (4 - 6).128 In potentiometric titrations, the diprotonated derivatives of these receptors were found to bind ATP in the order 6 > 2 > 5 ≈ 4. The stability constants were found to correlate with the number of nitrogen atoms available for electrostatic and hydrogen bonding interactions. These receptors were also found to form ternary complexes with ATP and copper(II), but these complexes were generally less stable than the binary receptor:ATP complexes.
In the early 1980's, the Kimura and Lehn research groups reported monocyclic polyamines, such as 7 - 11, that interact with biologically important polyanions, such as inorganic phosphate, AMP, ADP, and ATP, in aqueous media and at neutral pH.129,130 A wide range of macrocyclic amine receptors were used for these and later studies. Many contain regularly spaced nitrogen atoms. These will hereafter be referred to as [n]aneNm, where n represents the number of atoms in the ring and m the number of nitrogen atoms (e.g., 8 can be represented as [15]aneN5). In the Kimura systems, polarographic methods and 1H NMR spectroscopic shift measurements were used to provide support for a 1:l host:guest stoichiometry for receptors 7-11 and the polyanionic forms of several phosphates.129 In such cases, phosphate complex formation was found to be governed mainly by electrostatic forces. Thus, more negative nucleotides were bound more strongly by the protonated forms of these polyamine receptors. For instance, the stability sequence was found to be ATP4- > ADP3- > AMP2- for systems 7-11, with the corresponding association constants falling in the range of 102 - 106 M-1. The strongest association occurred between ATP and receptor 7, the only macrocycle that was found to bear four positive charges at neutral pH. Smaller polyamine macrocycles containing only two protonated nitrogen atoms at neutral pH did not bind appreciably to these test phosphates. Despite the fact that AMP and inorganic phosphate bear the same negative charge at pH 7, AMP was found to form complexes with 9 and 11 ([18]aneN6) that were approximately 10-100-fold more stable than those formed with inorganic phosphate. The added stability was thought to be due to an additional interaction involving the adenine base. Proton NMR spectroscopic shifts were observed that are consistent with the base bending back to complex the macrocycle, lending support to this hypothesis.
Receptors 9 and 11 were also found to solubilize Ca3(PO4)2, Ca(C2O4), and human calculi in acidic solution, presumably as a result of their anion chelating properties.131 At pH 4.4, polyamine 11 was found to dissolve calculi better than EDTA, a system commonly used to treat calculi. Alkyl functionalization of 11 led to receptor 12, which was found to dissolve Ca3(PO4)2 more effectively than EDTA or 11, displaying optimum activity at pH 7.0.132 In a separate study, macrocycle 9 was functionalized with a lengthy alkyl chain (13) to give a derivative that was then used as the active component of an ATP selective electrode.133 The electrode obtained in this case was found to function from pH 3.0 to 7.0, with a dynamic range of 10-7 to 10-3 M.
Compounds 14-16 were introduced and studied by Lehn using pH-metric methods. These measurements revealed that the fully protonated forms of these compounds formed stable complexes with the nucleotide anions AMP, ADP, and ATP. Log stability constants (log KS) were found to range from 3.4 to 9.1.130 As with Kimura's systems (i.e. compounds 7-11 discussed above), the stability of the complexes formed by a given receptor increased with the charge of the anion. Likewise, for anions of a given charge, the ion-receptor complex stability was found to increase as the degree of protonation (and hence the positive charge) of the receptor increased. Receptors 14 - 16 were studied in their fully protonated forms. In addition to 1:1 complexes, the hexaprotonated receptor 14 ([24]aneN6) was found to form 1:2 host:guest complexes with ADP, and ATP. Likewise, the octaprotonated system 15 ([32]aneN8) was found to form 1:2 complexes with AMP, ADP, and ATP. Although receptors 14 and 16 both contain six protonated nitrogen atoms in their fully protonated forms, the ether-derivative 16 was generally found to bind nucleotide substrates more effectively than compound 14. This finding was attributed to the higher local charge density permitted by the ethylene spacers as compared to the corresponding propylene systems. Later on, 31P NMR spectroscopic studies were used to probe further these stoichiometries.134-136 Interestingly, these studies revealed that the [18]aneN6 (11) and the acyclic polyamine 17 formed mixtures of 1:1 and 1:2 host:guest complexes when treated with ATP near neutral pH. In contrast, the [24]aneN6 macrocycle (14) was found to form 1:1 complexes with ATP and ADP near neutral pH, while the [32]aneN8 (15) formed 1:2 complexes. The oxygen-containing macrocycle 18 (commonly named OBISDIEN) was found to form stable 1:1 complexes with AMP, ADP, ATP, and pyrophosphate. The corresponding log Ks values ranged from 2.85 to 11.00 as inferred from potentiometric measurements. The anion binding properties of OBISIDEN have also been extensively studied in the case of its metal complexes, as discussed in Section 4.3. Based on this early work, both Lehn and Kimura suggested a “macrocyclic effect on anion binding,” a conclusion supported by the fact that macrocyclic analogues of naturally occurring polyamines 1 - 3 were observed to bind anions one to two orders of magnitude more strongly than the linear systems.
Hosseini expanded the chemistry of these systems by covalently attaching receptor 18 to polystyrene beads.137 The resulting solid-supported systems were found to interact with fluorescent N6-ethenoadenosine-5′-diphosphate (εADP) and N6-ethenoadenosine-5′-triphosphate (εATP) in solution at pH 4 where the hexamine is fully protonated. At pH 11 the receptor was no longer protonated and released the guests. The best guest interaction was achieved between 1-3 minutes after polymer immersion. In addition, the expected preference for ATP was observed, as inferred from competitive experiments.
In addition to the biologically important analytes discussed above, these simple macrocycles were also found to bind nicotinamide adenine dinucleotide in its oxidized form (NAD) and the oxidized form of nicotinamide adenine dinucleotide phosphate (NADP).138 In the case of tetraprotonated [21]aneN7 (19), potentiometric studies revealed log Ks values of 4.27 and 4.75 for these two substrates, respectively. While the differing basicities of NAD and NADP complicated the analyses, competition studies showed that the NADP:19 complex was indeed more stable over a wide pH range. Molecular dynamic simulations involving the two complexes in question gave rise to a minimum energy structure in which the NADP guest is bent, presumably to allow strong electrostatic interactions between all three phosphate groups and the four charged nitrogen atoms as well as to permit the formation of 11 hydrogen bonds.
Wilson and Williams examined the phosphate binding affinities of both acyclic triethylene tetraamine (trien, 20) and its cyclic variant [12]aneN4 (21) using 31P NMR spectroscopy.139 The linear trien was found to bind pyrophosphate, triphosphate, ADP, ATP, and hexametaphosphate (HMP) in D2O over a range of pH values. The highest association constants were found in the case of trien interacting with HMP, giving a log K value near 6 when triprotonated at pH 8.0. A similar value (within error) was found for the cyclic [12]aneN4 (21) and HMP, though at a pH (8.5) where the receptor was only monoprotonated. These results provide yet additional support for the conclusion that significantly higher association constants are seen for ring systems as compared to linear systems. It was also found that polyammonium receptors must contain four protonated nitrogen atoms in order to compete with Mg+2 and Ca+2 ions for phosphate complexation in biological systems.
A closer look at the protonation states of polyamine receptors revealed important guidelines for obtaining an optimal charge density at neutral pH.140-144 These guidelines reflect several key structure-function features of polyamines. In particular, the protonation constants for the macrocyclic systems were generally lower than those of linear systems, presumably due to higher charge density. Similarly, smaller, conformationally constrained macrocycles often had lower pKa's than larger rings. While ethylene spacing gave higher charge density than propylene spacing, pKa's for adjacent groups between ammonium residues were often far below 7 (ranging from, e.g., <1 in the triamine macrocycle, [9]aneN3, to 4.09 in [18]aneN6 (11)).141 This effect has the consequence that a full supplement of positive charges is only obtained at a lower pH. Fortunately, it can be mitigated somewhat by separating individual ethylenediamine units by non-basic spacers, as in for instance hexamine 16. In this latter system, where ether oxygen atoms “replace” an NH moiety, the final three protonation constants were 6.80, 5.65, and 5.55.140 The use of propylene spaced systems also served to alleviate much of this problem; in this case the amine sites displayed higher pKa's (i.e., between 6-7).
At a given protonation state, the effects of differences in charge density can be significant. A binding study of a series of larger ethylene spaced rings, such as [18]aneN6 (11), with ATP, inorganic phosphate and pyrophosphate revealed that, for a given protonation state, smaller macrocycles generally bound anions more strongly.144 For example, both [21]aneN7 (19) and [24]aneN8 (22) were tetraprotonated at neutral pH, but in this state the former bound ATP with a log Ks of 4.54 while the latter displayed a log Ks of 3.74. This effect was attributed to the reduced charge density present in the larger rings systems.
Electrostatic analyses and structural studies involving simple polyamine receptors opened the door for the synthesis of what may be considered as tunable polyamine systems. For example, it was found that the basicities of simple polyammonium systems could be modulated through nitrogen methylation, a modification that generally served to lower the protonation constants of polyammonium macrocycles.142,145,146 Methylation also led to charge localization, especially in partially methylated polyammonium receptors, such as 23-25. In macrocycle 23, for example, NMR spectroscopic studies revealed that the triprotonated species contained protons on alternating nitrogen atoms (one tertiary and two secondary).146 In the corresponding tetraprotonated species, four protons are present on secondary nitrogen atoms, which leads to significant charge localization. In the case of macrocycle 24, all protons of the triprotonated species were found to be located on secondary nitrogen atoms. Because of the symmetry of the compound, this selective protonation served to divide effectively the macrocycle. Crystal structure analysis of several complexes between pyrophosphate and the macrocycles 23 and [18]aneN6 (11) revealed these two receptors to be nearly planar in the solid state, with 23 adopting an almost elliptical shape.147
Methylation patterns can also affect the anion binding properties of polyammonium receptors. In preliminary competition studies, the tetramethylated macrocycle 25 was found to bind to ATP more strongly than the corresponding unsubstituted [18]aneN6 (11).148 Further studies revealed that the methylation pattern on 23 led to stronger ATP binding than is seen in the case of 24, 25, [21]aneN7 (19), and [18]aneN6 (11), at least above pH 5.3.146 Macrocycle 24 was found to be the weakest receptor. Similar trends were observed for the interactions of 23 and 24 with AMP, ADP, and pyrophosphate.149 The strong binding of tetraprotonated 23 was attributed to the localization of the cationic charge on the secondary nitrogen atoms, where as the charge was found to be delocalized around the ring in unsubstituted [3k]aneNk systems.
In an effort to explore the properties of more rigid systems, the protonation and phosphate binding behavior of methylated macrocycles 26 and 27 were analyzed. Receptors 26 and 27 contain only tertiary nitrogen atoms. Thus, it is of interest that in 26 protonation occurs first at the four benzylic nitrogen atoms, followed by the methylamine nitrogen atom between the piperazine rings.150,151 A similar pattern of protonation, made simpler by its symmetry, was observed in the case of 27. This latter system was found to interact with ATP and ADP as inferred from potentiometric and NMR spectroscopic methods.151 The NMR spectroscopic studies led to the suggestion that the main driving force for the formation of ATP and ADP complexes with 27 was electrostatic in nature, with the contribution from hydrogen bonding and π-surface interactions being minimal. An examination of structures from single X-ray diffraction analysis led to the suggestion that the conformational requirements of the piperazine rings in 26 act to orient the nitrogen atoms on opposite sides of the ring, thus decreasing the effective local charge. NMR spectroscopic studies of the substituted macrocycles 23, 24, 25, and 27 as well as [21]aneN7 (19) and [18]aneN6 (11) provided support for the notion that, while all macrocycles in the series bind inorganic phosphate and pyrophosphate in a 1:1 manner, no significant redistribution of charge takes place upon binding of the anion.147 Interestingly, these studies led to the conclusion that the charges in unsubstituted macrocycles remained delocalized on the NMR time scale when bound to these test anions. No obvious trends in stability were observed for the binding of inorganic phosphate and pyrophosphate in this series of macrocycles.
In a study that can be considered as a variation of investigations involving the alkylated polyammonium macrocycles discussed above, the anion binding properties of receptor 28, containing aminoethyl groups as substituents, were analyzed.152 This receptor was found to be tetraprotonated near neutral pH, with NMR spectroscopic studies leading to the inference that the first four protonation events involve the primary amines. Potentiometric studies provided support for the conclusion that ATP, P2O74-, [Fe(CN)6]4-, and [Co(CN)6]3- are bound to this receptor, with the strongest anion-receptor interactions being present at pH 6. Under these latter conditions, the 1:1 H6(28) 6+:ATP complex was considered to be the predominate species in solution. Comparisons of this macrocycle to [24]aneN8 (22), which would also be tetraprotonated near neutral pH, revealed that 28 is a less effective anion binding agent at an equivalent protonation state. This difference was attributed to the effective higher charge density of [24]aneN8 (22) compared to 28.
To avoid the complexities associated with protonation, Bianchi and co-worker prepared a series of quaternary ammonium receptors. These systems are a departure from earlier receptors in that the net charge was pH independent. However, the nitrogen atoms, being quaternized, were no longer able to participate in hydrogen bonds. Despite its high charge density, macrocycle 29 was found to have no appreciable interaction with ATP, as inferred from potentiometric measurements.153 These results were consistent with the hypothesis that the hydrogen bonding ability of the protonated 2° and 3° amine moieties played a major role in regulating the interactions of these receptors with ATP and like species. On the other hand, it proved difficult to make a direct comparison between 29 and various protonated ammonium receptors. For instance, it was noted that [12]aneN4 (21) did not to interact with ATP, even though it is of similar ring size to 29; however, receptor [12]aneN4 (21) was also found to be only doubly protonated near neutral pH.129 It thus makes a poor reference. In contrast, tetraamine 30 was found to be tetraprotonated near neutral pH and did interact with ATP when studied in aqueous media.153 However, the larger ring size of 30 compared to 29 could be responsible for the enhancements in anion binding. Thus, it too represents a poor reference for 29.
In the case of the quaternary ammonium cage compounds of general structure 31, effective binding with phosphate and nucleotides was achieved.154-156 Partial inclusion complexes with 1:1 stoichiometry were also inferred for inorganic phosphate, glucose-1-phosphate, glucose-6-phosphate, AMP, ATP, and NAD through CPK modeling.156,157 Dissociation constants with 31a were measured potentiometrically via bromide ion displacement, giving log KD values ranging from 2.0 to 2.5. Dissociation constants for 31b were measured spectroscopically via displacement of 2,4-dinitrophenolate, giving log KD values from 0 to 1.4. Both receptors exhibited little selectivity among the phosphates. The lower values observed for the larger receptor (31b) were attributed to the greater distance between the positively charged ammonium centers. This greater distance is expected to lead to a reduced positive electrostatic potential within the cavity. In general, the increased rigidity and lack of hydrogen bonding interactions in the methylated polyammonium systems dominate over the effect of charge density. Although the latter is increased through methylation, the net effect is a reduction in stability and selectivity.
Related to the problem of developing effective quaternary polyammonium receptors is the application of tetraquaternary 1,4-diaza[2.2.2]-bicyclooctane (DABCO) derivatives (32-40) as nucleotide phase transfer agents. Compounds 32-34 bear lipophilic alkyl chains that were meant to impart phase transfer properties, leading in some cases to systems capable of effective transport of nucleotides from an aqueous phase into an organic phase. Early efforts in this area were carried out by Tabushi, et al., who found that attaching stearyl chains to DABCO gave rise to a system (32) that allowed transport of AMP and ADP from aqueous solutions into chloroform with efficiencies that were much improved over traditional phase transfer and micellar reagents (trioctylmethyl ammonium chloride and stearyltrimethylammonium chloride, respectively).158 Transport across a chloroform membrane was up to 40-fold faster for ADP than AMP and could be driven by both pH and salt (NaBr) gradients.159 While ATP was extracted to the greatest extent at equilibrium, transport of ADP occurred at the fastest rate.160 The transport rate of ATP only matched the level of ADP in the presence of a coordinating cation (Na+ or K+), presumably due to the creation of a neutral species such as would be expected from 32:ADP. Similar trends in exchange efficiencies were observed for guanosine and uridine phosphates; however, transportation rates were much slower for uridine and cytidine phosphates as compared to the corresponding adenine systems.
Diederich, et al. investigated the structural optimization of DABCO carriers for the transport of dideoxynucleotide triphosphates (3′-azido-2′-deoxythymidine triphosphate (AZTTP) and dideoxythymidine 5′-triphosphate (ddTTP)). This was done by preparing and studying compounds 33-40.161,162 Transport studies were conducted in a standard U-tube cell with a chloroform liquid membrane. Receptor 33 proved to be insoluble in both organic and aqueous media, while receptor 34 was found to leak into the ATP-containing aqueous phase.161 In both cases, transport was precluded. On the other hand, the branched compound 35 proved to be a highly effective carrier for ATP, 5′-cytidine triphosphate (CTP), ddTTP, and AZTTP, even when compared to the original system, 32. Interestingly, compound 36 was found to be effective only for ddTTP under the conditions studied. Careful extraction studies suggested a 2:1 35:ATP binding stoichiometry, perhaps indicating a need for cooperation among four ammonium centers to effect nucleotide triphosphate transport. In line with this latter argument, receptors 34, 37, 38, and 39 each were found to form 1:1 complexes with ATP.162 However, only 39 gave rise to complexes that were sufficiently soluble in the organic layer to allow for transport. Even then, the transport rate proved to be an order of magnitude lower than that observed in the case of receptor 35. In liposomal studies, carriers 34, 35, and 39 largely acted as detergents. Specifically, these receptors were found to break up the liposomal structure, thus giving rise to nonspecific leakage from the liposomal interior. As part of a separate study, DABCO-based cyclophanes (40) were synthesized. This set of receptors was then analyzed in an attempt to correlate the intracavity encapsulation ability with receptor size.163,164 In this case, a 1:1 binding stoichiometry was established for ATP with log K's near 4.1 - 4.2 for all cavities. Such a finding is consistent with the fact that no evidence was found for encapsulation of the guest.
Smith, Duggan, and co-workers examined the effect of adding a boronic acid-based sugar-binding carrier (41) to the membrane transport system.165 The boronic acid moiety present in 41 is known to interact only with cis-diols. In accord with expectations, the transport of ribonucleoside-5′-monophosphates (AMP, 5′-guanosine monophosphate (GMP), and UMP) was facilitated when 41 was used in combination with ammonium-based carrier 34 as compared to when transport was carried out using 34 alone. Carrier 41 did not effect transport when used on its own. Nor did it effect the transport of 2′-deoxyribonucleoside-5′-monophosphates when used in conjunction with 34. The combination of 34 and 41 was found to transport 5′-GMP roughly 10-fold more effectively than either 3′-GMP or 2′-GMP. This study remains historically important because it highlights the benefit of combining multiple functional groups to tune selectivity and improve efficacy. Both are a recurring theme in supramolecular chemistry.
Direct attachment of ancillary binding motifs to polyammonium receptors has frequently been employed in attempts to both increase binding affinities and impart selectivity among other anions and within phosphate derivatives. Kimura and co-workers first incorporated amide groups, which are known to participate in hydrogen bonding, into polyamine macrocycles. Among the systems these researchers prepared are the diamides 42. While macrocycle 42a is only monoprotonated at pH 7.5, it was found to bind AMP, ADP, and ATP with affinities on the same order of magnitude as the naturally occurring polyamines 2 and 3. Compound 42a was also found to display a slight selectivity for AMP.129 Derivatives 42b and 42c, however, did not interact with the phosphates studied, likely due to steric interference. In later studies by Carrey and Riggan, the N3-cyclic amine 43 was prepared and found to be an effective ionophore for dibasic phosphate when employed in an ion-selective electrode. It proved more effective than other cyclic amine derivatives (i.e. the corresponding N4, N5, and N6 analogues).166 The electrode based on 43 exhibited submicromolar sensitivity and high selectivity. This finding was attributed to appropriate size and charge matching between the N3-cyclic amine ionophore and the HPO42- ions. Later, Ebdon and co-workers created an electrode with better stability and robustness by covalently linking ionophore 43 to the electrode.167 The resulting electrode displayed a higher stability while exhibiting similar selectivity toward HPO42-. This electrode operated from pH 6 to 8 over a working range from 3.9 × 10-3 to 1.0 × 10-6 M with a detection limit of 1.0 × 10-6 M.
The acyclic and cyclic receptors 44-46, which also combine amide and amine groups within their structure, were reported by Smith and co-workers.168 Based on potentiometric titrations carried out in aqueous media, receptors 44-46 were found to have a high affinity for phosphate anions (i.e. log K > 5). In addition, these receptors were found to interact with inorganic phosphate anions more strongly than with organic phosphate anions. This preference was attributed to the higher charge density and the smaller size of the inorganic phosphate analytes relative to the organic congeners.
The functionalized chiral polyamines 47 and 48 were prepared by the Burrows group, who studied their interactions with ATP.169,170 The triammonium macrocycle 47 was prepared to test whether a tripodal arrangement in a fully protonated macrocycle would provide a geometry suitably disposed to bind a trigonal oxyanion. However, analysis of the binding abilities of 47 and 48 revealed that tetraprotonated receptor 48 was more effective. Such observations further underscore the importance of charge-charge interactions in mediating the anion binding behavior of polyammonium receptors.
The Sessler group examined the combination of hydrogen bonding between complementary base pairs and the electrostatic interactions of polyammonium groups in an effort to enhance the affinity and selectivity of nucleotide recognition. This led to the synthesis of receptors 49-51.171,172 Their interactions with GMP were studied by 1H NMR spectroscopy in DMSO-d6 with the goal of analyzing both the strength of complexation and the relative contributions of each subunit.171 As expected, the alkyl receptor 49d formed 1:1 complexes of relatively low stability (i.e., with K values comparable to those displayed by simple guanine-cytosine base-pairs). On the other hand, the amine-containing receptors 49a-c, ditopic systems ostensibly capable of stabilizing both base pairing and electrostatic interactions, displayed higher affinities than 49d. In this latter case, 1H NMR chemical shifts were consistent with the conclusion that the additional nitrogen site also served to strengthen the hydrogen bonding interactions between the guanine and phosphate moieties of GMP. Receptors 49 also displayed 2:1 host:guest binding stoichiometries, presumably as the result of amines from two different receptors binding to a single dibasic GMP. The binding constants increased with increasing alkyl chain length, with 49c displaying association constants of 1300 M-1 (K1) and 1200 M-1 (K2). By comparing these values to that for triethylamine, which would interact with GMP only through electrostatic interactions, the contribution of hydrogen bonding effects to the overall affinity was estimated to be approximately 2.5-fold. A much stronger first association constant was observed for 50 and GMP as compared to 49c, with the K for 50 (2.60 × 103 M-1) being 20-fold higher than the first association constant and 17-fold greater than the second association constant for 49c. Analysis of the NMR chemical shifts led to the suggestion that the outer two nitrogen atoms of 50 chelate the phosphate group of GMP while the base pair participates in hydrogen bonding. Comparisons with N,N-tetramethylbutyldiamine revealed that the enhancement afforded by hydrogen bonding (2.7-fold) was similar to that observed in the case of 49c. Furthermore, substitution with long alkyl chains produced 51, which was then used to generate an ion-selective electrode.172,173 While little selectivity for phosphate or GMP was observed relative to other anions, the electrode was highly selective for guanosine nucleotides (5′-guanosine triphosphate (GTP) and 5′-GMP) over adenosine nucleotides (ATP, AMP) of the same charge. An electrode using a monotopic alkyl cytosine host (52) was found to have no response to nucleotides, while azamacrocycle 13 displayed no selectivity between nucleotide bases. This finding emphasizes the importance of using more than one binding mode. Consistent with this conclusion was the observation that selectivity for GMP over AMP could be achieved when receptor 53 was used in combination with either 54a or 54b.
The introduction of aromatic moieties into polyammonium macrocycles gives rise to structures with increased rigidity relative to aliphatic analogues. It also produces systems capable of supporting charge-dipole, π-surface, and hydrophobic interactions, features that have proved particularly important for nucleotide recognition. One early system reported was the pyridine macrocycle 55, which was investigated by Kimura.129 Receptor 55 is doubly protonated near neutral pH and was found to bind AMP with moderate selectivity over ADP and ATP. However, the overall stability constants proved to be significantly lower than analogous simpler polyammonium macrocycles (log KS ≈ 2.2-2.5).
The related 1,4-benzo macrocycle 56 was shown to be triprotonated near pH 7 and to bind ATP and pyrophosphate with log KS values of 2.58 and 3.32, respectively.174 A comparison between 56 and the larger systems 57 and 58 revealed similar binding constants, with 57 showing the highest affinity for inorganic phosphate and 58 showing the highest affinity for pyrophosphate (log KS from 2.60-5.79 over a range of protonation states).147 A comparison between an aryl-containing macrocycle and a less rigid control was made using the 1,3-phenylene receptor 59 and the acyclic polyamine 60. These systems exist in their tetra- and pentaprotonated forms near neutral pH.175,176 Both receptors bound ATP > ADP > AMP, with macrocycle 59 showing greater stability constants than 60 (log KS (ATP) = 5.2 and 4.6 for 59 and 60, respectively). This finding was rationalized on the basis of 1H NMR spectroscopic studies, which provided support for significant π-surface interactions between the phenyl unit of 59 and the adenine moiety of the guests. In the case of AMP and ADP, partial inclusion complexes were proposed wherein the adenine moieties participate in hydrogen bonding interactions with the benzylic nitrogen atoms. Further studies compared the binding of receptors 59 and 60 to the ortho- (61) and para-(62) substituted derivatives.177 Similar selectivities were observed among the nucleotides studied (ATP > ADP > AMP). Receptor 61 was found to form the most stable complexes over a wide pH range, followed by receptors 59 > 62 > 60. The stronger complexation of the ortho-derivative was presumably due to the formation of a more favorable conformation for nucleotide binding. Further studies compared the nucleotide binding of linear polyammonium receptor 60 with shorter derivatives 63 and 64.178 All receptors were found to form 1:1 adducts with ATP, ADP, and AMP, with the complex stabilities again being found to correlate with the charge of the guest. Interestingly, the ethylene-spaced receptors (63, 64) displayed more effective binding behavior than the propylene-spaced receptor 60 at the same pH (pH 2 - 10); presumably, this reflects the lower charge density of the latter receptor. Receptor 64 was also found to form ternary complexes with Cu(II) and AMP over a wide pH range.
The binding of phosphate to the bis-aromatic macrocycles 65 - 68 was investigated by Martell and co-workers. Comparisons to the ether-containing versions 18, 69, and 70 were also carried out. In the case of 65, a macrocycle prepared as a more rigid version of OBISDIEN (18), lower overall basicity was found for the furan-containing system, perhaps as a result of the electron withdrawing nature of the furan moieties.179 Measured association constants with pyrophosphate, however, proved similar for the two receptors. A structural analysis of diffraction grade crystals of the 65:pyrophosphate complex (grown at pH 3.5) revealed that the macrocycle adopts a bowl conformation. This provides a cavity that encloses one of the phosphate groups, which is ligated via hydrogen bonds to all three oxygen atoms. A hydrogen bond with the neighboring oxygen atom of the “outside” phosphate group is also reported. Macrocycle 66, in which the furan rings are replaced with phenyl rings, is characterized by a basicity that falls between that of 18 and 65; it was found to bind inorganic phosphate similarly to 18.180 The bis-phenyl macrocycle 66 was observed to bind pyrophosphate more strongly than did the bis-furan macrocycle 65, with triphosphate being complexed even more effectively. Single crystal X-ray diffraction analysis of the 66:pyrophosphate complex revealed a twisted chair-type shape with the anion completely enclosed within the cavity. Interestingly, the oxygen atoms on each end of the guest formed hydrogen bonds with the benzylic nitrogen atoms. As a consequence, the guest was found to bind perpendicular to the plane of the macrocycle in contrast to the co-planar arrangement typically found with nucleotides. In the course of efforts to explore the ability of receptor 65 to interact with nucleotides, it was demonstrated by 31P NMR spectroscopy and potentiometric studies that macrocycle 65 displays a preference for ATP over ADP and AMP; presumably, this reflects the large negative charge of ATP.181 Detailed 31P NMR spectroscopic studies provided evidence for ATP interacting with receptor 65 via insertion of the terminal phosphate group into the receptor cavity. As with the mono-phenyl macrocycle 59, receptor 66 was found to bind ATP > ADP > AMP through π-surface interactions, as inferred from 1H NMR spectroscopy.182 However, receptor 66 did not bind the nucleotides better than their inorganic analogues, despite the presence of moieties that could provide for additional π-surface interactions. This finding can be rationalized in terms of the reduced basicity of the nucleotides. Replacing the ethylenic spacers of 66 with propylenic spacers gave receptor 67. This latter system, as expected, proved a more basic macrocycle than 66. However, it displayed similar binding trends.183 Single crystal X-ray diffraction analysis of 67 with pyrophosphate revealed a facial interaction in which the macrocycle adopts a relatively planar arrangement with the phenyl rings perpendicular to the plane and facing opposite directions. While receptor 66 bound most phosphate anions more strongly than 67 at a given protonation state (presumably reflecting an increased charge density), the larger macrocycle (67) out competed 66 for phosphate anions near neutral pH. The binding of inorganic phosphate and pyrophosphate with macrocycle 67 was later compared to the para-substituted derivative 68.184 These receptors displayed similar binding affinities for inorganic phosphate; however, receptor 67 was found to bind pyrophosphate more strongly than receptor 68 at equivalent protonation states. This trend was attributed to a better geometric complementarity between the smaller ring of receptor 67 and pyrophosphate.
The phosphate binding properties of this class of macrocycle were further compared with those of the expanded OBISDIEN (18) derivatives. Among the latter, the ether-containing system 69 is analogous to 67, while 70 can be considered as an intermediate between 18 and 69.185 Both 69 and 70 proved to be more basic than their phenyl counterparts. While similar general trends were observed within the ether-containing series, competition experiments between 69 and 67 revealed that the ether-bridged macrocycle 69 bound inorganic phosphates more strongly than the phenyl-containing macrocycle 67. On the other hand, this latter macrocycle was found to bind nucleotides more strongly than 69. The metal complexes of many of these macrocycles were also studied as phosphate anion receptors. This chemistry will be presented in a later section (4.3).
In subsequent work, Schneider and co-workers investigated cyclophanes 71-73, which included, biphenyl, bipyridyl and para-benzyl bridging elements.186 Proton NMR titrations, carried out in D2O, provided evidence that receptor 71 interacts more strongly with nucleotide monophosphates than do either 72 or 73. The increased affinity observed in the case of 71 was attributed to its larger cavity size, which allowed for more efficient π-surface interactions between the phenyl rings of the receptor and the nucleobase of the guest, as inferred from studies of molecular models. Receptor 71 displayed a preference for AMP over other nucleotides, binding this particular nucleotide with an association constant of ca. 2200 M-1 in D2O. Macrocycle 71 also exhibits a selectivity for 5′-AMP over 3′-AMP and appears to form an inclusion complex with 5′- thymidine monophosphate (TMP). Macrocycle 73 was found to bind GMP in preference to other nucleotides, displaying an association constant of 540 M-1 in D2O. Proton NMR spectroscopic studies led to the inference that π-surface interactions were not the main driving force for binding in this latter instance.
Recently, Llobet and co-workers studied the anion-binding behavior of the anilinic compounds 74-75.187 In this case, 1H NMR spectroscopic and potentiometric studies carried out in aqueous solution provided support for the conclusion that the receptors with more rectangular cavities, 74a-b, bound triphosphate and ATP anions more strongly than 75. This finding was ascribed to size and shape complementarity.
Recently, Kumar and co-workers demonstrated by means of UV-Vis studies carried out in acetonitrile that the mixed amine ether macrocycle 76 interacted with sodium dihydrogenphosphate in preference to other anions.188 The incorporation of ionophore 76 into a polyvinyl chloride (PVC) membrane allowed for the creation of a phosphate-selective electrode with a dynamic concentration range of 2.1 × 10-7 to 1.0 × 10-2 M.
In 2004, Bowman-James and co-workers reported the anion binding properties of a simpler, acyclic phenyl-polyammonium receptor (77).189 This tripodal receptor was based on a tris(aminoethyl)amine (tren, 78) scaffold functionalized with benzyl units. Proton NMR spectroscopic studies in chloroform revealed a strong selectivity for dihydrogenphosphate and hydrogensulfate (log K > 3) over nitrate, chloride, and bromide (log K < 2). All anions were studied as the tetrabutylammonium (TBA) salts. Further studies of the 77:H2PO4- complex were conducted through single crystal X-ray diffraction analysis (Figure 4). These experiments led to the conclusion that extensive hydrogen bonded networks were present in the solid phase. In addition, a 1:3 host:guest ratio was inferred on the basis of these analyses.
Figure 4.
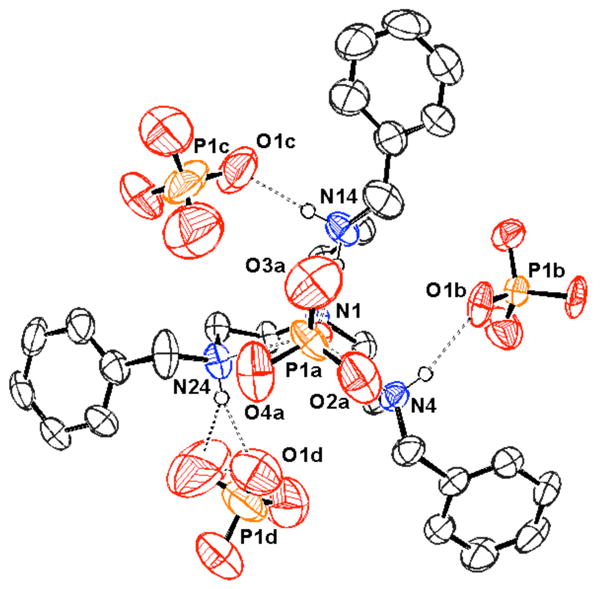
View of the H3(77):(H2PO4)3•H3PO4 complex. Drawing generated from X-ray diffraction data originally published in reference 189. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
García-España and co-workers reported the AMP binding ability of a series of similar systems (79) in 2006.190 In this case, potentiometric titrations revealed a number of host:guest stoichiometries in aqueous media. For example, receptors 79a and 79b were both found to form 1:2 and 1:3 host:guest complexes. However, only 1:1 stoichiometries were observed with receptor 79c. Significantly higher stability constants were measured for the receptors containing aromatic substituents (79b,c) relative to the alkyl polyammonium receptor 79a. This increase in binding was attributed to a combination of π-surface interactions and hydrophobic interactions between the aromatic units of the receptors and the adenine ring of AMP. The highest log stability constant among the hexaprotonated forms of these receptors was found to be 7.65 in the case of AMP and receptor 79b. Interestingly, the presence of AMP was observed to facilitate metalation of this receptor by copper(II).
A variety of other receptors with extended π-surfaces and positively charged bridges have been used to bind nucleotides. Among these is macrocycle 80. This system contains four quaternary ammonium centers along with long alkyl chains and displays a strong interaction with adenosine phosphates.191-193 Proton NMR spectroscopic studies and theoretical calculations of the complex formed between 80 and ATP provided support for the nearly complete inclusion of the adenine base within the lipophilic macrocycle, as well as an electrostatic interaction between the phosphate group and a quaternary nitrogen. Presumably as the result of this combination of factors, the equilibrium constant (derived from 1H NMR spectroscopic studies) proved to be on the order of 104 M-1 (ca. 26 kJ/mol) in D2O. Equilibrium constants were consistently higher for phosphate derivatives relative to the corresponding unsubstituted nucleobases, with each electrostatic interaction contributing ca. 5 kJ/mol to the overall binding energy. Receptor 80 was found to have a preference (approximately five-fold) for adenosine derivatives over other common nucleotides derivatives, possibly due to the higher polarizability of the adenine base. The free energy of binding for the open, cleft-like receptor 81 interacting with AMP proved to be ca. 9.5 kJ/mol lower than the corresponding value in the case of 80. This reduction in binding energy could reflect a lower level of preorganization, the absence of lipophilic inclusion, or the presence of fewer positively charged binding sites.
The inclusion of polyaromatic heterocycles, including known DNA intercalators, has been shown to increase the binding affinity for nucleotides, presumably due to π-surface interactions between these moieties and the nucleobases. For example, the Lehn group studied the synthetic incorporation of acridine into the OBISDIEN macrocycle (18) to give the ditopic nucleotide receptor 82a.194 In this case, the addition of ATP, ADP, and AMP produced measurable changes in the 31P NMR, 1H NMR and fluorescence spectra of 82a (studies carried out in D2O or H2O, respectively). The interaction of 82a with nucleotides produced a significant fluorescent enhancement (>30% with ATP) and a slight bathochromic shift. At the same time, the addition of pyrophosphate and triphosphate to 82a led to a modest quenching of the acridine fluorescence. Acridine units substituted with single amine groups (instead of full incorporation into a macrocycle) were found to undergo no change in emission upon the addition of ATP or ADP. Evidence for the presumed π-surface interactions between the π-surfaces of the nucleotide and the receptor was found through 1H NMR spectroscopy. Furthermore, competition experiments led to the suggestion that 82a bound ATP twice as strongly as 18, and that the binding of 82a with ATP as compared to simple triphosphate anion was also favored by a factor of two. Further studies revealed that the emission of 82a is constant over the pH range 4.0 to 7.6, ruling out protonation as a cause of the fluorescence change.195 Little to no change in fluorescence was observed with inorganic phosphates or simple nucleobases, confirming the necessity of the covalent linkage between the two units. An increase of the emission of 82a was also observed with CTP, and a slight decrease was observed with GTP. These findings were interpreted in terms of the receptor having a degree of inherent selectivity. The binding of the fluorescent derivative εATP was also studied, and it was found to bind five-fold more strongly than ATP. The combined data supported a dual binding mode in which the phosphate group of ATP is bound to the polyammonium macrocycle while the adenine moiety interacts with the acridine substituent.
The binding of nucleotides was further studied using the bisacridine receptor 82b.196 In this case, both NMR and fluorescence studies provided support for the suggestion that the two acridine units interact closely in the absence of an analyte, leading to a slight decrease in fluorescence. Consistent with this notion, the emission increase upon the addition of ATP to 82b was found to be more dramatic than the corresponding increase in the case of 82a. While 1:1 complexes with GTP, CTP, and 5′-uridine triphosphate (UTP) were also produced in the case of 82b, the emission increase was much smaller than with ATP. The complexation of dinucleotide phosphates NAD, NADH, NADP, and NADPH by 82a and 82b was also investigated. Proton NMR spectroscopic studies of 82b and NADH led to the suggestion that receptor 82b interacts with both the nicotinamide and adenine moieties of the guest. Both 1H NMR and fluorescence spectroscopic studies revealed that positively charged guests NAD and NADP were bound significantly less well than their neutral counterparts. This result is rationalized in terms of charge-charge repulsion with the protonated acridine near neutral pH. At the same time, the phosphorylated species (NADP and NADPH) bound more strongly than the non-phosphorylated species. Indeed, NAD and NADH produced no change in the fluorescence spectrum of either receptor. Fluorescence titrations revealed stable 1:1 complexes of both receptors with NADP and NADPH (log KS = 5.5 to > 8.4). The contributions to the overall stability of the complexes were investigated through competition experiments. In the context of these analyses it was found that the binding constants for 82a and 82b were similar for each analyte studied. However, a comparison with the unsubstituted receptor 18 revealed that the acridine units present in 82a and 82b served to increase the stability of the complexes by more than one order of magnitude. On the other hand, ATP, triphosphate, and NADPH were found to bind to both receptors with similar stabilities. This is consistent with electrostatic interactions again being the most significant factor in terms of defining complex stability. Interestingly, a 600-fold increase in selectivity was observed for NADPH over NADP.
Lehn and co-workers further examined a series of cyclic and acyclic aromatic receptors based on acridine derivatives and a variety of other substituents (83-85).197,198 A strong hypsochromic shift was observed in the UV-Vis spectrum upon addition of nucleotides to 83-85 in aqueous media, a finding consistent with the presence of π-surface interactions. Binding constants, determined by absorbance titration methods, revealed 1:1 binding stoichiometries and log KS values ranging from 2.04 to 5.05 for the binding to AMP (aqueous media, pH 6). Monomeric receptors 83 were found to bind AMP 2-3 orders of magnitude less well than the corresponding dimeric analogues 84. In addition, the acyclic dimeric receptors 84 were found to bind AMP one order of magnitude more strongly than the macrocyclic derivatives 85, possibly due to the rigidity of the cyclic systems. No large differences in stability were observed for the different substitution patterns of each receptor. Interestingly, in the case of 85b no significant differences in stability constants were observed between AMP, ADP, and ATP. This is consistent with electrostatic interactions playing only a minor role in terms of the overall binding energetics.
Similar studies focused on the use of phenanthridinium (ethidium), also a DNA intercalator, as a receptor subunit.199 This led to the synthesis and study of compounds 86 - 88. In all cases, fluorescence quenching was observed upon the addition of nucleotides, with the log KS values ranging from 4-6. All complexes were determined to be of 1:1 stoichiometry, with receptor 86 proving to be the weakest receptor. The dimeric, acyclic receptor 87 proved to be a slightly weaker receptor than 88. Apart from these general trends, little selectivity was observed. Specifically, for any given receptor no appreciable differences were observed among nucleotides. The lack of selectivity among differently charged phosphates led to the suggestion that electrostatic interactions may not have been significantly influential. Additional studies with this class of hosts as well as other intercalator-based receptors that were found to depend nearly exclusively on π-surface interactions were carried out but are not discussed further in this review.
Recently Piantanida and co-workers reported strong nucleotide binding by the phenanthridine-containing receptor 89 in aqueous media.200 Binding constants were determined through fluorescence titrations performed at pH 5 and 7 in a 0.05 M sodium cacodylate buffer. Among the series of AMP, GMP, and 5′-cytidine monophosphate (CMP), stronger binding constants were observed with the purine nucleotides at both pH values. Higher selectivity was observed at pH 7 than at pH 5. This trend was attributed to a significant contribution from π-surface interactions (which are known to favor purine binding) that were expected to be more pronounced for complexes with the unprotonated phenanthridine (pH 7) than the positively charged phenanthridinium (pH 5). Greater stabilization was also observed upon an increase in phosphorylation of the nucleotide (AMP < ADP < ATP). This latter trend supported the contribution of electrostatic interactions to the binding affinity. The highest stability constant for this series was measured for the complex of 89 and ATP at pH 7 (log KS = 3.67).
The previous studies by Lehn and co-workers led to the design of a series of inclusion receptors based on polyammonium macrocycles containing polycyclic aromatic moieties (90-92), as well as their acyclic analogues (93, 94).201-203 Based on the measured pKa values, the diethylenetriamine moieties of 90-92 were expected to be doubly charged near neutral pH, leading to tetraprotonated macrocycles. This high degree of protonation was expected to lead to strong electrostatic binding in addition to interactions involving the aromatic subunits of the receptors and those present on various test nucleotides. The naphthalene in receptor 90 was used to explore nucleotide binding. This was done using both 1H NMR and fluorescence spectroscopy in aqueous media, from which log KS values ranging from 3-5 at pH 6 were inferred. As expected based on charge considerations, 90 was found to bind ATP > ADP > AMP. Moderate selectivity for purines was observed among different nucleotide monophosphates, with GMP > AMP > UMP > CMP. The singlet emission features of the acridine derivative (91) proved to be a sensitive indicator of substrate binding. Specifically, purine nucleotides were found to quench the fluorescence, while pyrimidine nucleotides enhanced the fluorescence.202 Binding a pyrimidine substrate likely increases the distance between the two acridine units, giving rise to a strong fluorescence signal similar to that of the acyclic species 93. An electronic interaction between the purines bases and the acridine subunits likely overrides this latter effect, resulting in overall quenching. Stability constants (log KS) derived from these measurements were found to range from 3.8 to 8.4 at pH 6. Selectivities were similar to those obtained with 90, with the combination of 91 and ATP displaying the strongest interaction. Nucleotide binding by 91 was generally stronger than that of the acyclic derivative 93. This finding led to the suggestion that both hydrophobic and van der Waals inclusion interactions were critical for substrate binding interactions. Similar binding affinities and selectivities were inferred from emission studies involving the tris-acridine receptor 95.204
Still larger aromatic systems, specifically the quinacridine receptors 92 and 94, were designed with the expectation that the expanded aromatic system would allow for the binding of two nucleotides, possibly associated through traditional Watson-Crick base pairing.203 Addition of nucleotides to 92a produced a decrease in both absorption and fluorescence intensity. Complexes of 2:1 (host:guest) stoichiometry were observed with several nucleotide monophosphates. A strong preference for guanine bases was observed through potentiometric titrations in aqueous media (first stability constant, log KS1 = 4.1, second stability constant, log KS2 = 4.5), with a selectivity order of GMP > AMP > CMP > UMP ≈ 3′,5′-cGMP ≈ 2′,3′-cGMP. Such findings, evidence of cooperative binding, were rationalized in terms of the co-bound GMP entities being better able to interact with one another via hydrogen bonds. Complexes of 1:1 stoichiometry were observed for di- and triphosphates, with triphosphates exhibiting the highest binding constants, followed by diphosphates and monophosphates. The selectivity for guanine over adenine was retained for the higher phosphates. Interestingly, the KS1 values corresponding to the interaction of nucleotide monophosphates with 92a were lower than the corresponding binding constants in the case of 91. This was rationalized in terms of the cavity of 92a being too large to bind efficiently a single nucleotide monophosphate. Receptor 92b was found to display similar binding behavior. However, 92c was found to have reduced binding interactions with monophosphates. While the side chain of 92c provided an additional positive charge that was expected to enhance binding, on the basis of this finding it was concluded that the cavity size plays a dominant role in terms of regulating the anion binding affinities.
In the same report, the monomers 94 were described. These systems were found to be less effective nucleotide receptors than the corresponding dimeric macrocycles. Interestingly, the second association constant was found to be higher than the first association constant when GMP and 3′,5′-cGMP derivatives were tested as substrates. This result was rationalized in terms of strong G-G interactions within the complex. Indeed, no mixed ternary complexes were observed when mixtures of CMP and GMP were tested with 94. In addition, GMP apparently acts to displace CMP from the cavity to allow for G-G binding. Similar trends in stoichiometry and binding trends were noted when electrospray ionization (ESI) mass spectrometry was used to analyze mixtures of this receptor and various putative substrates. It was concluded by the authors that combining π-surface interactions, especially those associated with intercalation, and electrostatic interactions can greatly increase binding affinities and selectivities for nucleotides.
A similar insertion binding mode was inferred in the case of the polycationic pyrenophane receptors 96, prepared by Inouye and co-workers.205,206 In this case, preliminary studies with receptor 96a in 3:1 water:ethylene glycol revealed a change in the absorption spectrum upon the addition of a variety of guests, including nucleotide monophosphates AMP, UMP, GMP, and CMP. Among this latter class of substrates, a modest selectivity was observed for GMP (K = 3.5 × 104 M-1). Improved water solubility was achieved with receptor 96b. With this latter system, it was found that the addition of nucleotides led to a change in the receptor-based excimer emission spectrum in water. Receptor 96b exhibited a higher response toward nucleotide triphosphates over the mono- and diphosphates. For instance, association constants of 1.9 × 103 M-1, 5.3 × 103 M-1, and 1.0 × 106 M-1 were seen for AMP, ADP, and ATP, respectively. This differential response was attributed to a combination of the large charge of the triphosphate guests and the inclusion of the guest into the cavity formed between the pyrenophane and the ammonium groups present on the crown ether portion of the receptor.
Bencini and co-workers have included phenanthroline heterocycles within polyamine frameworks to form receptors 97 - 100. The interactions of 97 and 98 with ADP, ATP, pyrophosphate, and triphosphate were studied through potentiometric titrations.207 In general, 98 was found to bind phosphates more strongly than 97 for a given protonation state, as seen previously for smaller macrocycles. The stability constants with pyrophosphate and triphosphate, however, were much lower than those with ADP and ATP, respectively, with no binding observed between 98 and triphosphate. While guest basicity must be considered, these receptors were found to bind the nucleotides over analogous inorganic phosphates in competition experiments over a wide pH range. These results led to the proposal that interactions between the adenine base and the phenanthrolenic receptor are important. Support for this suggestion came from 1H NMR spectroscopic analyses. Specifically, chemical shift differences were seen that were consistent with the presence of strong π-surface interactions and partial inclusion of the guests. In addition, more dramatic shifts for ADP over ATP, as well as increased shifts for the larger cavity of 97 over 98, were observed.
More recently, these researchers reported the nucleotide binding properties of larger phenanthroline macrocycles 99 and 100.208,209 Hexaamine receptor 100 displayed higher binding affinity toward nucleotide triphosphates (ATP, CTP, GTP, and 5′-thymidine triphosphate (TTP)) than the pentaamine macrocycle 99 at the same pH, as inferred from potentiometric titration studies. The increased binding affinity for the former receptor was attributed to the increased charge density of the larger macrocycle at a particular pH. This increased charge density would be expected to augment electrostatic interactions with anionic guests. The complex of receptor 100 with ATP was found to be the most stable as judged by potentiometric titration analyses as well as 1H NMR and fluorescence spectroscopic methods. Both 1H NMR spectroscopic studies and molecular modeling studies supported an increased π-surface interaction between the adenine base and the phenanthroline moiety in the 100:ATP complex over the other studied complexes. Interestingly, 31P NMR spectroscopic studies and single crystal X-ray diffraction analysis (Figure 5) provided support for a binding model wherein the triphosphate moieties of nucleotide guests were encapsulated within the macrocyclic cavity. Receptor 100 thus proved to be a highly selective sensor for ATP in aqueous media in the pH range of 4.5-7.
Figure 5.
View of the 100:TTP complex. Drawing generated from X-ray diffraction data originally published in reference 209. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
Bencini, Bianchi, Paoletti, and co-workers later investigated the phosphate binding properties of an acyclic analogue (101) of the previously described phenanthroline polyammonium macrocycles.210 As inferred from potentiometric titrations, stability constants were found to depend heavily on electrostatic interactions, with the strongest complexes found near neutral pH. The anion recognition properties of the bipyridine derivative 102 were also investigated. This latter receptor displayed a similar trend but slightly higher binding affinity than receptor 101. Higher binding affinities for nucleotides relative to inorganic phosphates was seen for both receptors. Proton NMR spectroscopic studies provided support for the presence of π-surface interactions in the nucleotide:receptor complexes. The most stable complex in these studies was found to be H4(102)4+:ATP3- (log K = 5.45). These researchers further investigated anion binding by a bicyclic derivative of receptor 102 (103).211 Higher stability constants were observed with a greater degree of protonation of the receptor. At equivalent charge states, macrocyclic receptor 103 was found to bind pyrophosphate and triphosphate more strongly than ADP or ATP, respectively, in contrast to the binding behavior of receptor 102. This reversed selectivity was investigated through molecular modeling, which supported a folded conformation of the macrocycle. The predicted conformation featured a cleft appropriate for the binding of smaller, inorganic phosphates over nucleotides. This finding was further supported by a lower degree of π-surface interactions with receptor 103 compared to receptor 102, as inferred through 1H NMR spectroscopy. The thermodynamic analyses of these interactions revealed a strongly favorable entropic contribution in the binding of anions by receptor 103, which was also consistent with an encapsulation-type binding event.
The aminoanthracene unit has also allowed for sensitive fluorescent detection of anion binding. The Czarnik group reported anthrylpolyamine compounds (104, 105) that exhibited chelation-enhanced fluorescence (CHEF). Increased fluorescent intensity was observed for the binding of inorganic phosphate and ATP to receptor 104 at pH 6, giving log KS values of 0.82 and 4.2, respectively.212 It was proposed that, while the lone pair of the neighboring nitrogen atom quenched the anthracene fluorescence in the absence of an anion (presumably through PET), the change in protonation state of the nitrogen upon anion binding interfered with the quenching and led to the large enhancements in fluorescence. The expanded chemosensor 105 was found to bind pyrophosphate over 2000-fold more tightly than monophosphate.213 As binding analyses can be complicated by the chelation of pyrophosphate to adventitious transition metals, these studies were conducted in the presence of cyclen (106). This azamacrocycle strongly chelates transition metals but does not interfere with phosphate binding.
García-España and Piña later investigated a series of cyclic and acyclic polyaminoanthracene derivatives as possible ATP chemosensors.214 Receptor 107 was found to bind AMP, ADP, and ATP with log KS values ranging from 2.9 to 7.2 according to potentiometric titrations, with a 600-fold selectivity for ATP over ADP being observed. Receptors 108a-108c were found to have similar binding affinities. This series of receptors displayed guest-dependent changes in the fluorescence intensity below pH 4 (aqueous NaClO4), with a decrease in fluorescence being observed for ATP and ADP across the full series 107 – 108c. This quenching and inferred binding was attributed to interactions between the π-faces of the anthracene and the protonated adenine base of ATP. No change in fluorescence was observed when 107-108c were treated with AMP, adenosine, or triphosphate under the same conditions. Receptor 109 did not show a change in fluorescence with any of the tested analytes. Further studies served to demonstrate that receptors 110-113 also bound ATP and that, in general, receptors that contained one anthracene unit interacted more strongly with ATP than those containing a naphthalene group.215
Anthryl-functionalized open-chain polyammonium-alkanes 114-116 were studied by the Martínez-Máñez group.216 Due to solubility incompatibilities, in the case of 114 only the binding of inorganic phosphates could be studied by potentiometric methods (log KS = 3.99 - 15.52 in 7:3 MeCN:H2O). Both 114 and 115 could be studied using fluorescence methods. In both cases, the emission intensity was selectively quenched in the presence of ATP at acidic pH (MeCN/H2O 7:3), although no stability constants could be calculated. The addition of other anions (bromide, inorganic phosphates, sulfate, ADP, GMP) did not change the emission intensity. Interestingly, in water the emission intensity was found to increase at neutral pH in the presence of ADP and ATP but not with any other anions studied. This result was considered consistent with the conclusion that hydrogen bonding interactions with the nitrogen atoms nearest the anthracene lead to a reduction in PET quenching and therefore an emission increase. At lower pH values and in a solvent such as MeCN/H2O with a lower dielectric constant (where the degree of protonation relative to pure H2O is higher), the electrostatic interactions between the positively charged receptors 114 and 115 with guest anions were thought to be more significant. Such increased interactions were expected to favor PET resulting in a quenching of the fluorescence as seen by experiment. The most significant quenching in the presence of ATP in mixed aqueous-organic media was observed below pH 4. In this pH range, both 114 and 115 were fully protonated, maximizing the expected electrostatic interactions. On the other hand, no change in the fluorescence intensity was observed upon the addition of anions to 116, perhaps due to steric constraints imposed by the diazolidine moiety. All anions were present as the TBA salts in these studies. Further studies revealed that an anthrylmethylamine (117) anchored in mesoporous solids permitted the fluorescent sensing of ATP in aqueous media.217,218 The addition of ATP to solids with low loading levels of this anthracene derivative led to a quenching of the anthracene signal with a micromolar detection limit being noted at pH 2.8.
In 2007, García-España, Alcarón, and co-workers investigated the ATP binding properties of polyammonium naphthalene receptor 118.219 Log binding constants in the range of 4-5 with ATP were determined through pH-metric titrations. The binding affinity was found to correlate with the number of electrostatic interactions. Despite this high binding ability, quenching of the naphthalene fluorescence was only observed below pH 5. This pH dependence was attributed to protonation of the adenine moiety of the guest under more acidic conditions. The protonated adenine ring was expected to be a more capable acceptor for PET from the naphthalene excited state. ATP-dependent quenching was achieved over a broad pH range (2-12) by immobilizing this receptor on a boehmite solid support (119).
The rigidifying properties of aromatic rings can been used to effect specific preorganization. These effects were studied by the Anslyn group who designed the polyammonium receptors 120 - 122 in order to determine the optimum binding cavity size and hydrogen bonding arrangement for the complexation of phosphoric acid diesters. This series of receptors also allowed for the determination of the strength of interactions arising from individual hydrogen bond donors and acceptors in the host systems.55,56 Both 120 and 121 are cleft-like systems that contain three hydrogen bond donation sites and one hydrogen bond acceptor site. These receptors were designed to be complementary to the three oxygen atoms and one acidic hydrogen atom of a tetrahedral phosphoric acid diester. Compound 122 was designed to be a cleft-free control. Investigations by isothermal 31P or 1H NMR spectroscopy in chloroform led to the suggestion that mixtures of 1:1 and 1:2 complexes are formed in the case of receptors 120 and 122 with dibenzyl hydrogenphosphate, while receptor 121 exhibited strong 1:1 binding with this analyte. The complex equilibria of 120 and 122 were attributed to the dimerization propensity of phosphoric acid diesters in organic media. The dimerization constant of hydrogenphosphate derivatives was determined to be 6.5 × 104 M-1 through fluorescence titrations of dinaphthyl hydrogenphosphate with dibenzyl hydrogenphosphate. A lower substrate concentration was therefore needed to measure binding constants for receptor-phosphate interactions reliably in the absence of dimerization. In view of this need, optical methods were considered the most practical method of analysis. Using UV-Vis methods, 1:l binding constants of 7.8 × 103 M-1 and 8.9 × 104 M-1 could be determined in chloroform for the interaction of dibenzyl hydrogenphosphate with receptors 120 and 121, respectively. Using fluorescence methods, a binding constant of 1.3 × 103 M-1 was measured for the interaction of dinaphthyl hydrogenphosphate and receptor 122. On the basis of these analyses, it was concluded that the extent to which phosphoric acid diesters were complexed by this set of polyammonium clefts increased with both the number of hydrogen bonds formed (each hydrogen bond contributing roughly 1.2-1.7 kcal/mol) and the size of the cleft (-ΔG° being approximately 1.4 kcal/mol larger in the case of 121 than 120). These trends were in line with molecular modeling experiments.
Benzene and pyridine units were used to preorganize the cyclen (106) units of receptors (123 - 125) in systems developed by Handel and co-workers.220-222 The interactions of these receptors with inorganic phosphate, pyrophosphate, and triphosphate in aqueous media were studied through potentiometric and NMR spectroscopic methods. Only 1:1 anion:receptor complexes were found to be formed under these conditions. In studies with the bis-macrocycle systems 123, the highest stability constants were observed with ortho-substituted receptor 123a and the pyridine-substituted receptor 123c over a large pH range (pH 2 - 8).220 These trends were attributed to a greater degree of organization in the former case and to the additional binding site available in the latter case. In all cases, the three anions were bound with affinities that were seen to match their overall charge (inorganic phosphate > pyrophosphate > triphosphate). Interestingly, a reversal of this trend was observed for triphosphate at higher pH values (pH 9-12), and higher stability constants were reported for the meta- and para-substituted phenyl receptors 123b and 123d. It was proposed that these latter receptors were better organized for hydrogen bonding interactions, which would be expected to play a more important role under more basic conditions. Modeling studies of receptors 123a and 123c led to the suggestion that these receptors formed a cavity with cyclen-cyclen distances consistent with the size of the triphosphate anion. The more rigid tri-macrocycle systems 124 displayed generally higher stability constants compared to the bis-macrocycle systems 123, particularly at slightly acidic pH values.221 Significant changes in selectivity were also observed. For example, the ortho-substituted receptor 124a was found to bind inorganic phosphate and pyrophosphate more strongly than the larger receptors 124b and 124c. These latter receptors strongly bound triphosphate, with the para-substituted receptor 124c displaying the greatest selectivity for this anion. This selectivity was attributed to the large size of the receptor 124c cavity compared to the other receptors. Phosphorous NMR spectroscopy and modeling studies of the various 124:triphosphate complexes were consistent with a model in which the two terminal phosphate subunits were bound by the cyclen moieties while the central phosphate unit was forced outside the cavity.
The effects of pyridine incorporation were studied through a comparison of receptors 125 to receptor 124b.222 Pyridine receptors 125 displayed a stronger selectivity and affinity for triphosphate over inorganic phosphate and pyrophosphate, particularly at acidic pH values where the pyridine nitrogen is found to be protonated. The three studied receptors bound triphosphate in the order 125a > 125b > 124b over the entire pH range studied (pH 2 - 12). It was proposed that the mono-pyridine receptor 125a bound triphosphate most strongly due to a relatively higher charge density at the pyridine unit as compared to receptor 125b (containing two pyridine units) at an equivalent protonation state. Phosphorous NMR spectroscopy and modeling studies yielded similar results as those for receptors 124. These studies thus serve to underscore the importance of electrostatic interactions and geometric complementarity in receptor design.
Further studies compared the anion binding of receptor 123d and the linear analogue 126.223 As with the previous examples, only 1:1 anion:receptor complexes were observed. Interestingly, the more flexible linear analogue 126 displayed significantly stronger binding ability with triphosphate as compared to receptor 123d, particularly near neutral pH. This finding was attributed in part to the greater degree of protonation in the case of the linear derivative at equivalent pH values. In addition, 1H NMR spectroscopy and modeling studies provided support for a binding mode in which the polyammonium arms of receptor 126 were able to wrap around the anion for highly efficient electrostatic and hydrogen bonding interactions with the bound anions. In this particular case, increased receptor flexibility proved advantageous over preorganization.
These same researchers later reported the recognition of nucleotides (AMP, ADP, ATP) by receptors 123b and 123c under similar conditions.224 As inferred from potentiometric titrations, only 1:1 host:guest complexes are stabilized. Within this general trend, receptor 123b displayed a preference for AMP over ADP and ATP, while receptor 123c most strongly bound ATP over a wide pH range. Proton NMR spectroscopy of the anion:receptor complexes were consistent with strong π-surface interactions near neutral pH. Phosphorous NMR spectroscopy and modeling studies of the 123c:ATP complex provided support for a binding model in which the β- and γ- phosphate units were complexed to the cyclen moieties while the adenine ring participated in π-surface interactions with the pyridine ring. A similar binding mode was inferred in the case of the linear derivatives 127 and ATP.225 Interestingly, similar binding affinities for both ATP and triphosphate were found with both of these receptors, a finding consistent with the conclusion that pyridine nitrogen atom did not play a large role in anion binding. A direct comparison between the cyclic and linear systems, however, was not made.
Simple pyridine receptors 128 - 131 were prepared by Araki and co-workers and found to interact with diphenylhydrogenphosphate in organic solvents of both low and high polarity.226 The amino bipyridine receptor 128 was found to bind this substrate strongly in both cyclohexane and acetonitrile, with log K values over 7.1 as determined by fluorescence spectroscopy. Due to spectral similarities between the protonated form of 128 and the anion:receptor complex, binding was thought to occur largely through electrostatic interactions involving the protonated pyridine of 128 and the bound diphenylphosphate moiety. Proton NMR spectroscopic studies led to the suggestion that the amino groups are involved in simultaneous hydrogen bonding to the phosphate ester. On the other hand, triethylphosphate (lacking an acidic proton) and hexanoic acid produced minor changes in the optical properties of 128. On this basis, it was concluded that these substrates are bound with binding constants less than 100 M-1. Control receptors 129-131 also displayed optical changes when treated with diphenylhydrogenphosphate but to a much lower extent, despite the nearly identical basicities of all four systems in question. To the extent these optical changes reflect binding, the fully alkylated receptor 129 was found to bind this particular phosphate ester 105-fold less effectively than 128 in cyclohexane. In addition, the monopyridine receptor 130 was found to bind the phosphate ester 100-fold less effectively than 128. These results provided support for the conclusion that the amino hydrogen atoms play an important role in regulating the binding process. These atoms were also considered to underscore the benefits that accrue from having a second pyridine subunit within the receptor, at least for binding studies carried out in cyclohexane. The effects of the amino protons and pyridine subunits were less pronounced in acetonitrile. For instance, in this latter solvent receptor 128 displayed affinities that were only four-fold higher than those of 129 and six-fold higher than those of 130. This solvent effect was attributed to a reduction in the strength of the underlying hydrogen bonding interactions in the more polar solvent, a reduction that in turn was expected to mask the differences between the different receptors.
Receptor 132, designed by the Anslyn group, relies on a rigid, conformationally locked benzene ring to orient both boronic acid and secondary ammonium groups within a well-defined “pinwheel” cavity.227 On the basis of both 1H NMR spectroscopic analyses and UV-Vis indicator displacement assays, it was found that this system bound glucose-6-phosphate selectivly. Presumably, this selectivity was the result of boronic acid-diol interactions and electrostatic attractions to the protonated amines. Binding constants on the order of 103 M-1 for glucose-6-phosphate and receptor 132 were derived in 7:3 CH3OH:H2O at pH 7.4. Neither the addition of glucose nor the addition of sodium phosphate effected displacement of the indicator in competition-type indicator displacement assays, even when these latter species were present in a 100-fold excess relative to the receptor.
In a more recent example, the rigid scaffold of the natural macrocycle tetrandrine (133) was investigated for anion binding by Eliseev and Yatsimirsky.228 Proton NMR spectroscopic experiments carried out in water demonstrated that while 133 interacted most strongly with dicarboxylate anions, it also bound nucleotides ATP, ADP, and AMP. In the case of ATP and ADP, 1:1 complex formation was observed, although a 1:2 host:guest stoichiometry was observed in the case of AMP. Binding constants were reported to be 110 M-1 for ATP and 40 M-1 for ADP, with a first association constant of 48 M-1 and second association constant of 55 M-1 being noted in the case of AMP. Interestingly, the binding affinity did not correlate with the charge on the anion. This was thought to reflect a strong contribution from donor-acceptor interactions in the recognition process.
In addition to aromatic moieties, the use of aliphatic spacers has been explored by Bowman-James, Alcock, and co-workers. 229 These researchers reported the binding interactions between the simple macrocycle 134 with inorganic phosphate, as inferred from potentiometric titrations and single crystal X-ray diffraction analysis. Stability constants determined by the former method for 1:1 complexes were found to correlate with electrostatic interactions. Near neutral pH, the strongest 1:1 complex was formed between the pentaprotonated form of macrocycle 134 and monohydrogenphosphate (log K = 6.01). Interestingly, 1:2 host:guest stoichiometries were observed in the pH range of 2-4, presumably reflecting the binding of one phosphate anion to each triamine binding site. The existence of ditopic binding modes at lower pH was supported by solid-phase studies.
Yet another effective structural theme involves the use of polymacrocyclic receptors. As a general rule, these kinds of receptors can provide additional binding sites as well as permit the encapsulation of phosphate guests. In the case of bis(macrocyclic polyamine) ligands 135 and 136, Kimura and co-workers combined two smaller macrocycles through either a tether (135) or through direct fusing (136).230 Both receptors were found to form stable 1:l complexes with AMP, ATP, HPO42-, and other anions as observed through polarographic and potentiometric methods. The comparison of these association constants with those of the parent monomeric polyamines (9 and 42a, respectively) revealed that the attachment of the second polyamine moiety consistently enhanced anion encapsulation and did so by at least an order of magnitude despite an overall lower charge density (135 being only tetraprotonated and 136 diprotonated near neutral pH).129 The greater complex stabilities exhibited by the bismacrocycles were explained in terms of ditopic interactions, such as sandwich complex formation, where the donor anion is located between the two protonated macrocycles. Presumably, this allows for maximum electrostatic and hydrogen bonding interactions.
Bencini and Lippolis investigated the anion binding character of receptor 137, which was found to be zwitterionic at neutral pH.231 Potentiometric studies showed that receptor 137 bound phosphate anions with an affinity order diphosphate < triphosphate < ATP. While the diphosphate to triphosphate selectivity could be attributed to charge-charge interactions among the phosphate and the amine subunits, 1H NMR spectroscopy led the researchers to suggest that π-surface interactions were involved in the binding of ATP. Molecular dynamics calculations predicted that the two macrocycles would indeed act concertedly to chelate polyphosphates.
In 2009, a tris-macrocycle polyammonium system (138) was reported by Bencini, Giorgi, Handel, and co-workers.232 This receptor was found to display both proton sponge and anion binding properties. Stable 1:1 complexes were formed between receptor 138 and inorganic phosphate, pyrophosphate, triphosphate, ADP, and ATP in aqueous media (0.1 M NMe4Cl) as inferred from potentiometric studies. The strongest stability constants were observed in the pH range of 3.5 - 4.5. Complex stability was found to increase in the order of inorganic phosphate < pyrophosphate < triphosphate ≈ ADP < ATP. While the stability trend of the unsubstituted phosphate derivatives can be attributed to increased anionic charge, the stronger binding affinities displayed in the case of the adenosine derivatives at equivalent charge states led to the suggestion that the adenine base was also participating in binding interactions with receptor 138. A weak π-surface interaction between the adenine base and the pyridine unit of the receptor was inferred on the basis of 1H NMR spectroscopic analyses. Molecular modeling studies supported a binding model in which electrostatic interactions with the terminal one or two phosphate groups of each of the studied guests is the main contributor to complex stability.
As illustrated in part by the previous examples, cryptand-type polyammonium polycyclic receptors have been studied for their ability to form inclusion complexes with anions. An additional system that falls within this generalized receptor class is cryptand 139. This receptor was studied as a three-dimensional version of OBISDIEN (18).233 In its penta- and hexaprotonated (pKa 5 = 7.00, pKa 6 = 5.90) forms, 139 was found to form stable complexes with a variety of anions, including inorganic phosphate, pyrophosphate, AMP, ADP, and ATP, in aqueous media. While no conclusive data was obtained for the inclusion of inorganic phosphate and pyrophosphate, a comparison of the relative stabilities of the AMP, ADP, and ATP complexes revealed that the hexaprotonated form of monocyclic “control” receptor 18 binds these three nucleotides more strongly than does the hexaprotonated polycyclic receptor 139 as inferred from potentiometric titrations in aqueous medium. These results were rationalized in terms of both the partial exclusion of the anion from the cavity due to the steric bulk of the adenine moiety, as well as the relatively lower charge density present in 139 as compared to 18 given equal protonation states.
Another multidentate cryptand-type system is receptor 140, which incorporates hydrogen bonding pyrrole units. This system was found to form high-affinity 1:1 complexes with inorganic phosphate as well as other anions near neutral pH.234 The interaction with inorganic phosphate was studied by potentiometric and ITC methods, from which association constants of nearly 107 M-1 were calculated. Simple calculations of the electrostatic contribution predicted affinities lower than those observed. This was reconciled by considering a strong contribution from additional interactions, such as hydrogen bonding and possibly anion inclusion. Analysis of the thermodynamic parameters revealed that, while the binding was exothermic at 298.2 K, the large negative ΔH value was partially compensated by markedly negative ΔS values. These unfavorable entropic contributions, in turn, were attributed to the additional inclusion of water molecules in the anion/receptor complexes.
In 2009, Bianchi and co-workers reported the anion binding properties of simple tren (78), which was used as a scaffold in receptor 140 as well as a variety of other multidentate receptors (e.g., 77, 95, and 139).235 These studies were intended to elucidate the contribution of this subunit to the anion binding affinities of more complicated systems. Stability constants determined through potentiometric titration generally correlated with the strength of electrostatic interactions, and a significant selectivity for triphosphate was observed among the other anions studied (pyrophosphate, inorganic phosphate, nitrate, sulfate, tosylate) near neutral pH. Thermodynamic analyses of these binding interactions generally revealed favorable entropic changes and negligible enthalpic changes, as would be expected for electrostatic interactions; however, the binding of monohydrogenphosphate was found to display a strong favorable enthalpic component with an unfavorable entropic change. This unexpected result was attributed to a partial anion-to-amine proton transfer between monohydrogenphosphate and diprotonated tren (78). Studies of anion binding with tren (78) led to the conclusion that this unit can significantly contribute to the anion binding of larger structures.
Using very different design principles, Severin and co-workers reported the self-assembly of piperazine-based cryptand 141 in 2007.236 Anion binding studies were conducted using 1H NMR spectroscopy in D2O at pD 6.6. This receptor selectively bound inorganic phosphate and acetate over chloride, bromide, iodide, nitrate, and sulfate in neutral aqueous solution. Significant sharpening of the piperazine methylene signals was observed while no changes were found for the pyridyl protons. Based on these results, the authors proposed that the anions were bound in the center of the cryptand though electrostatic and hydrogen bonding interactions with the piperazine nitrogen atoms. Further analyses revealed a binding constant of 950 M-1 and 1:1 binding stoichiometry for inorganic phosphate. The affinity for inorganic phosphate was found to be significantly stronger than that for acetate (270 M-1), leading to the suggestion that this receptor has a geometry that is complementary to the phosphate anion.
The anion binding properties of a 2,3,6-triethylbenzene-based polyammonium cryptand (142) were reported by Delgado and co-workers in 2009.237 Anion:receptor interactions were studied through potentiometric titrations in a 1:1 methanol:water mixture containing 0.1 M potassium tosylate. Significant interactions near neutral pH were observed with H2PO4-, AcO-, SO42-, S2O32-, and SeO42-. Overall complex stabilities were found to correlate with predicted electrostatic interactions between the receptor and anions. A strong selectivity for sulfate was observed below pH 5, and this anion was thus the focus of further studies. Similar studies were conducted with pyridine-containing cryptand derivative 143.238 The presence of hydrogen bond donor (ammonium) and hydrogen acceptor (pyridine) units was expected to impart selectivity for inorganic phosphate over other anions. Under conditions identical to those used for studies with receptor 142, a strong preference for hydrogenphosphate over sulfate, acetate, nitrate, and chloride was observed near neutral pH in the case of receptor 143. The selectivity for hydrogenphosphate over sulfate near pH 7 was also supported by competition studies carried out using 31P NMR spectroscopy.
4.1.2. Guanidinium Systems
While by definition guanidinium-based anion receptors are polyamines, this functional group has several features that have led to it being categorized as its own unique anion recognition motif. It has been studied extensively in the context of phosphate anion recognition.239-243 Guanidine is readily protonated to form the guanidinium ion, which is stabilized by resonance and charge delocalization. Guanidine groups are characterized by a pKa value of 13.6, compared to a value of ca. 10.5 for a typical secondary amine. As a consequence, guanidinium residues remain protonated up to high pH values, and, when incorporated into suitable frameworks, the group can be used to broaden the pH range over which an anion receptor operates. The guanidinium moiety also presents two parallel hydrogen donor sites, a feature that leads to strong interactions with oxo-anions. However, the exploitation of guanidinium residues in host-guest chemistry is hampered by the fact that such species are highly solvated in water. These units are also characterized by a lower charge density compared to ammonium cations. Since binding affinity is often primarily electrostatic, polyammonium salts generally form more stable complexes (at a given net charge) than do polyguanidinium salts. Nevertheless, an appreciable number of artificial guanidinium-based anion receptors have been reported in the last few decades.
Historically, the recognition that the guanidinium ion had a role to play in anion recognition can be traced back to 1964 when Watters and Matsumoto reported the interaction between the guanidinium ion (144) and tetraphosphate.244 Cotton and co-workers published the crystal structure of a methylguanidinium:dihydrogenphosphate complex in 1973,245 as well as that of a bis(methyl)guanidinium:dihydrogenphosphate complex in 1974.246 Both structures confirmed the planarity of the guanidinium moiety as well as the two parallel hydrogen bonds between the guanidinium hydrogen atoms and phosphate oxygen atoms. Springs and Haake measured the binding constant of the guanidinium ion (144) with inorganic phosphate in 1977.247 Relatively low association constants were obtained using potentiometric titrations in aqueous media. Although not particularly dramatic, these studies were among the first to demonstrate the potential utility of guanidinium motifs in phosphate anion recognition.
Lehn and co-workers were among the early pioneers using guanidinium subunits to create more elaborated receptor systems, including a series of cyclic and acyclic guanidinium analogues 145 – 150.248 In the context of this work it was found that the tris-guanidinium receptor 145 bound inorganic phosphate with the highest affinity in water within the series 145 – 150, as determined via pH-metric titrations (log KS = 2.4). As a general rule, the macrocyclic systems (145, 146, 148) exhibited greater affinities for phosphate than did their acyclic analogues (147, 149, 150, respectively). All recorded stability constants proved significantly higher than those for N,N′-diethylguanidinium or for guanidinium itself. These results were considered consistent with the presence of a macrocyclic effect similar to that described in the previous section, as well as specific chelation interactions, that served to increase the affinity for phosphate-type anions relative to other negatively charged species. As expected given the less competitive nature of the environment, stronger binding was observed in a methanol/water mixture than in water. This was a general trend, true across the whole series of receptors.
The Lehn group also developed a series of protonated amine and guanidinium receptors to permit a more direct comparison between these two anion recognition motifs.249 The interaction of each receptor with inorganic phosphate and pyrophosphate was studied potentiometrically. As expected, electrostatic interactions proved to be the most important determinant of complex stability. Thus, receptors with amine substituents (151b – 157b) consistently gave rise to stability constants that were higher than their corresponding guanidinium analogues. This effect was significant, with the difference being roughly one order of magnitude for the same overall charge. It was rationalized in terms of the higher charge density provided by protonated amines relative to guanidiniums. This increase serves to overcome the putative benefits provided by the two linear hydrogen bonds possible with guanidinium receptors. Among the guanidinium containing receptors (151a – 157a), those with additional ammonium sites (and therefore a higher electrostatic charge) generally displayed the highest affinities. At the same time, increasing the spacing between the individual guanidinium subunits was found to decrease the propensity to bind phosphates (the log KS order was as follows: 151a > 152a > 153a and 156a > 157a) when studied at equivalent protonation states in aqueous medium. The most stable guanidinium complexes were formed with the closely packed triguanidinium receptor 156.
Göbel and co-workers examined the binding of a series of guanidinium receptors (158 - 163) to catechol cyclic phosphate (tetramethylguanidinium salt) using 31P NMR spectroscopy in DMSO-d6.250 Anion interactions were found to be much stronger with meta-substituted benzene receptors 158 and 159 (K ≈ 95 and 75 M-1, respectively) than monosubstituted receptors 160-162 (K ≈ 15-20 M-1). These differences in binding affinities could reflect the presence of a bidentate binding mode in the case of the former receptors. Although all association constants were calculated assuming a 1:1 binding stoichiometry, the authors report that some evidence of higher order complexes was found. Further details regarding this result were not discussed. A more complex scaffold, decalin, was explored to examine the effects of rigidifying the guanidinium receptors.251 Little difference was observed between the two bisguanidinium receptors 158 and 163 (K = 110±15 M-1 and 95±10 M-1, respectively).
Attachment of pyrene to a guanidinium residue led to the synthesis of receptor 164, which was designed to act as a fluorescent sensor for pyrophosphate.252 Proton NMR and fluorescence spectroscopic measurements by Teramae and co-workers provided evidence that 164 formed 2:1 host:guest complexes with pyrophosphate in methanol. These sandwich-like complexes were characterized by an enhanced excimer emission relative to the monomer emission. This change in emission behavior was not observed with other anions, including H2PO4-, CH3CO2-, SCN-, Cl-, and Br-. At low concentrations of guest, the binding constant corresponding to the formation of the 2:1 receptor:pyrophosphate complexes was determined by fluorescence measurements to be 1.2 × 108 M-2. Addition of more than 0.5 equivalents of pyrophosphate led to a decrease in excimer emission, which was attributed to the formation of a 1:1 complexes at higher concentrations of guest.
Rigid chiral bicyclic guanidinium moieties have been exploited successfully in phosphate anion recognition. For instance, Schmidtchen and co-workers reported the series of receptors 165 - 166. Preliminary 1H NMR spectroscopic titrations involving the addition of p-nitrophenylphosphate and CMP to the ditopic chiral hosts 165a and 165b provided support for the formation of 1:1 host:guest complexes in methanol.253 Additional NMR spectroscopic analyses of host R,R-166b revealed the formation of 1:1 complexes with inorganic phosphate, 2′-AMP, 2′-deoxy-5′-adenosine monophosphate (dAMP) and 5′-AMP.254 Interestingly, nearly identical binding constants were obtained for inorganic phosphate and 2′-AMP (log K= 4.26) in methanol, a finding that was consistent with little interaction between the receptor and the adenine base. The binding affinity of R,R-166b for 2′-AMP was reduced by approximately one third in DMSO as compared to that in methanol. It was also found that, as compared to 2′-AMP, dAMP was found to be bound more strongly. This latter finding could reflect the fact that the phosphate group is less sterically constrained in dAMP than 2′-AMP. The “addition” of a hydroxyl group (i.e. 5′-AMP) led to even higher affinities. While the phosphate interactions with receptor R,R-166a were not easily characterizable in methanol, when the analysis medium was unbuffered water, clean 1:1 complexes were formed with inorganic phosphates and a number of phosphate esters, including 5′-AMP, p-nitrophenylphosphate, p-nitrobenzylphosphate, guanylyl(3′-5′)adenosine (GpA), NAD, and ATP. However, the binding affinities in aqueous media were considerably reduced as compared to what was found in methanol (40-fold with 5′-AMP). In unbuffered water, the most stable complex was formed between receptor 166a and inorganic phosphate (log K = 2.99). This reflects the formation of a complex that is stronger than that produced from the more charged species, ATP. In these studies, the organic moieties of the phosphate esters appeared to interfere with the phosphate interaction, in contrast to the contribution observed with polyammonium systems.
In an effort to avoid the influences of counteranions and the inherent lack of selectivity generally associated with strong electrostatic attractions, a closo-borane cluster was added to the bisguanidinium framework (165c).255,256 The closo-borane cluster in receptor 165c carries a double negative charge in a delocalized, hydrophobic environment. The presence of this cluster was expected to allow for neutralization of the two guanidinium groups without formation of a strong contact ion pair. However, 1H NMR spectroscopic analyses in DMSO-d6 revealed evidence of dimerization, with an association constant of 250 M-1. On the other hand, the use of UV-Vis titrations (and a lower concentration) allowed binding analyses to be carried out without having to account for aggregation effects. This method permitted a log K value of nearly 4 to be determined for the interaction between p-nitrophenylphosphate (TBA salt) and 165c in DMSO. Using ITC, thermodynamic parameters were derived for the binding of p-nitrophenylphosphate in acetonitrile for 165c, as well as those for control compounds 165d and 165e. While the three receptors displayed similar association constants (log K = 5), the negative entropy values found with receptors 165d and 165c, compared to the slightly positive value with 165e, served to underscore the importance of the substituents on the central phenyl ring. Receptor 165c was later used to prepare what was hoped to be a phosphate selective electrode.257 While micromolar sensitivity for phosphate in chloride solutions was observed, and the stability was adequate, the electrode was not particularly selective against other anions.
The Schmidtchen group also investigated the phosphate anion extraction properties of receptors S,S-166b, 165f, and 165g.258 Each receptor was tested by following its ability to effect the extraction of inorganic phosphate, AMP, ADP, ATP, and other anions from aqueous solution (pH 7.4-7.8) into chloroform. Receptor S,S-166b displayed a selectivity for sulfate over inorganic phosphate. This receptor was also found to extract nucleotides AMP, ADP, and ATP more efficiently than inorganic phosphate. Under defined conditions, ATP was found to be 94% extracted, while ADP and AMP were extracted to the extent of 54% and 6%, respectively. Bromide and iodide anions were also efficiently extracted (82% and 96%, respectively). The more flexible isophthalic acid host 165g was found to effect significantly less nucleotide extraction than S,S 166b. On the other hand, in the case of 165f, the increased rigidity and the additional hydrogen bonding sites provided by the amide moieties was thought to lead to significant extraction of all oxoanions studied, with selectivity even over iodide being observed.
Comparisons between 167a and 167b were used to determine the thermodynamic contribution of the four amide groups present in 167a for the binding of inorganic phosphate.259 A strong enthalpic contribution was expected in the case of 167a from the addition of four hydrogen bonding sites. Upon the addition of TBA dihydrogenphosphate to 167a in acetonitrile, ITC studies revealed an initial exothermic 1:1 complexation event followed by an endothermic 1:2 host:guest complexation process. Evidence for higher ordered complexes at high phosphate concentrations was also obtained. For receptor 167b, 1:1 complexes were observed. While the binding constant was slightly higher for the amido receptor 167a, the enthalpic contribution was actually smaller for this receptor. The enhanced affinity was thus ascribed to a large increase in the entropic component of binding with 167a over that associated with 167b. Based on a comparison of the binding of these receptors to other anionic guests, the cause of this effect was proposed to involve the weakened structural definition of the 167a:phosphate complex compared to that formed with 167b, as opposed to, e.g., differences in solvation. This conclusion was supported by the observation of higher order complexes in the case of 167a but not in the case of 167b. Taken in concert, these results help illustrate the importance of individual thermodynamic parameters in regulating the affinities of host-guest systems.
The complexation of bicyclic chiral guanidinium receptors (168-169) with AMP derivatives was studied by the de Mendoza group. The ability of these receptors to extract anions from aqueous media into chloroform was tested with S,S- and R,R-168a and S,S-168b.260 No chiral recognition was observed based on interactions with 5′-AMP, 3′-AMP, 3′,5′-cAMP, or 2′,3′-cAMP. The cyclic AMP derivatives were extracted by 168a less efficiently than the linear derivatives, presumably due to steric effects that precluded π–π donor-acceptor interactions involving the naphthalene ring. On the other hand, in the case of 168b, the more lipophilic cAMP derivatives were found to be extracted more effectively than the more hydrophilic linear derivatives. Overall, 168a extracted this set of nucleotides more efficiently than 168b. Finally, receptor S,S-168c was designed to interact with nucleotides through both π–π donor-acceptor and base pairing effects. Such interactions were clearly observed through 1H NMR spectroscopy in DMSO-d6 in the case of 3′-AMP but not 3′,5′-cAMP. Anion interactions with 168c were not quantified.
Macrocyclic receptor 169 combines the bicyclic guanidinium unit with two urea moieties as additional hydrogen bonding sites and with amino acids to impart additional chirality.261 The xanthene unit was included to add rigidity and aid with preorganization. Molecular mechanics calculations led to the prediction that a diphenylphosphate ion would be located inside the cavity, stabilized by hydrogen bonds involving all of the phosphate oxygen atoms. The two phenyl rings would thus protrude to each side. Proton NMR spectroscopic studies of the isolated 169:diphenylphosphate salt in CDCl3 were consistent with a complex of C2 symmetry. However, the splitting of the signals seen at 213 K led to the suggestion that the ion was most likely residing outside the cavity and rapidly exchanging between both sides of the macrocycle at room temperature. Bulkier phosphates displayed similar 1H NMR spectral features, providing support for the conclusion that the phosphate esters were not bound within the cavity as predicted. No association constants were reported in this study.
The Rebek group also studied a series of guanidinium receptors that incorporate additional functionalities. For example, receptor 170 was designed to combine the phosphate recognition ability of guanidinium subunits, the π-electron donor capabilities of aromatic carbazoles, and the hydrogen bonding ability of amides in order to create a receptor selective for dinucleotide phosphates.262 In preliminary studies, receptor 170 was found capable of extracting a full equivalent of a dinucleotide (2′- deoxyadenylyl(3′,5′)-2′-deoxyadenosine, d(AA)) from aqueous solution into dichloromethane. Two-dimensional NMR spectroscopic experiments led to the suggestion that, while most of the binding was electrostatic in nature, significant hydrogen bonding existed between the base pairs of the dinucleotide and the receptor. The energetic contribution of the electrostatic interaction was examined by comparing the affinity of several adenine derivatives for receptor 171a and control system 171b in aqueous media at pH 6.263 In these studies, adenosine and 9-ethyladenine were found to bind equally well to both 171a and 171b. This finding was considered consistent with the conclusion that the guanidinium moiety of 171a did not interact with the nucleotide base and that the sugar moiety did not influence the binding process significantly. Furthermore, all four adenine derivatives (adenosine, 9-ethyladenine, 2′,3′-cAMP, and 3′,5′-cAMP) were found to bind to receptor 171b with similar affinity; again, this is as would be expected were the adenine moiety interacting only with the carbazole portion of this receptor and its more elaborate analogue 171a. In the case of 171a, relatively higher binding affinities were observed for the cAMP derivatives, although this effect was somewhat mitigated at higher ionic strengths. Little dependence on ionic strength was observed for complexes lacking an electrostatic interaction. The free energy of each system was then calculated from 1H NMR spectroscopic titration experiments with the goal of sorting out the energetic contribution of the electrostatic interactions. On this basis, the average phosphate-guanidinium interaction energy in this system was calculated to be 0.6 kcal/mol at an ionic strength of 51 mM (NaCl) and 0.3 kcal/mol at an ionic strength of 501 mM (NaCl).
Transport studies made using simple U-tube apparatus (two aqueous phases separated by a dichloroethane “membrane”) with receptors 172 revealed a preference for adenine over other nucleotide bases.264-266 Transport was observed for 2′,3′-cAMP, 3′5′-cAMP, and 3′-AMP but not for 2′,3′-cGMP and 3′5′-cGMP. Interestingly, transport was also not observed in the case of 5′-AMP. The more effective transport seen for adenosine nucleotides relative to guanosine nucleotides was thought to reflect the loss of one hydrogen bonding interaction in the corresponding complexes. Receptors 172a and 172b were found to transport dinucleotides containing adenine, although not those lacking an adenine moiety. Receptor 172d did not transport any of the nucleotides studied. For the adenine nucleotides, receptor 172c displayed the best transport properties among the set of receptors 172. This was ascribed to the favorable combination of interactions provided by the hydrophobic naphthalene moiety and the carbazole moiety acting as a hydrogen bond donor.
Mandolini, de Mendoza, and co-workers combined a bicyclic guanidinium moiety with a calixarene (173) with the goal of facilitating the binding of dioctanoyl-L-α-phosphatidylcholine (DOPC), a transition state analogue that acts as an inhibitor for the enzyme-mediated hydrolysis of acetylcholine.267-269 In the case of 173, the guanidinium moiety was expected to bind the anionic phosphate group while the calixarene moiety was expected to bind the ammonium group through cation-π interactions with the calixarene in its cone conformation. Proton NMR spectroscopic studies and molecular modeling supported these assumptions, although an analysis of the data led to the conclusion that the phosphate group was involved in hydrogen bonding with the amide and proximal guanidinium protons instead of both guanidinium protons as originally expected. In the absence of a guest, receptor 173b was found to have a stronger preference for the cone conformation, leading to a more preorganized receptor. Proton NMR binding studies carried out in CDCl3 allowed log K values of 4.9 for DOPC:173a and 5.0 for DOPC:173b to be derived. These quantitative analyses revealed no significant difference in binding affinity based on the presence or absence of the cyclohexane substituent, even though the presence of this latter subunit was expected to increase the extent of preorganization. The addition of one percent CD3OD to the CDCl3 solvent served to reduce the binding constants by an order of magnitude.
The monocyclic and acyclic guanidinium receptors 174 and 175 were studied by the Hamilton group.270 These systems contain neighboring acyl groups, a feature that was expected to enhance the rigidity of the receptors through intramolecular hydrogen bonding interactions. Acylation was also expected to decrease the basicity of the receptors. Despite this expected lowering in the pKa values, both receptors were found to remain protonated under all conditions explored in these studies. Proton NMR spectroscopic studies of the receptors in CD3CN provided support for the proposed rigid structure as evidenced by the lack of spectral splitting or line broadening. Spectral changes seen upon the addition of TBA diphenylphosphate were considered consistent with the formation of four hydrogen bonds to the anionic guest, which, in turn, was proposed to reside inside the cleft in the case of both receptors. Compound 175 displayed a first association constant of 5 × 104 M-1, a binding event that was considered to be followed by the binding of two additional phosphate ions as inferred from 1H NMR spectroscopic analyses. Only 1:1 binding was observed with compound 174, a difference that was rationalized in terms of the absence of an additional hydrogen bond donor as compared to 175.
Additional basic moieties were incorporated to produce receptors such as 176. These latter systems were prepared in pursuit of catalysts that would facilitate transesterification reaction of 2-hydroxypropyl-para-nitrophenylphosphate.271 The interactions between receptors 176b and 177 with diethylphosphate in CD3CN were studied using 31P NMR spectroscopy. Receptor 176b was found to form strong 1:2 host:guest complexes, while 177 formed weaker 2:1 host:guest complexes.
Several guanidinium receptors have been investigated by the Anslyn group. The first of these were the cleft-like monocyclic guanidinium receptors 178 and 179.272,273 Within this pair, it was considered likely that the binding affinity and selectivity could be tuned through variations in the cavity size and the overall conformational flexibility, brought about by linking guanidinium moieties to cyclopenteno and cyclohexeno rings in the case of 178 and 179, respectively. Both the meso- and d,l-diastereomers were isolated and tested in the form of various counter anion salts. Binding constants for the interaction with dibenzylphosphate were determined through 31P NMR spectroscopic titrations carried out in various mixtures of water and DMSO. Low concentrations of water led to a mixture of 1:1 and 1:2 complexes, while concentrations near or above 20% led to the formation of only 1:1 complexes. This was true for both receptors. In pure DMSO and with the receptors studied in the form of their tetraphenylborate salts, stronger binding was found in the meso-forms of both receptors. This finding was attributed to enhanced cooperativity between the two guanidinium groups constrained on the same side of the cleft-like receptor. The putative increased flexibility present in receptor 179 was thought to underlie the slightly higher binding affinities observed for the meso-form of 179 as compared to meso-178. In addition, increased binding constants were observed in the presence of LiCl, NaCl, and KCl. However, decreased binding affinities were observed in the presence of NaClO4 and NaSCN. These results were tentatively attributed to a “salting out” effect rather than an ionic strength effect. With dibenzylphosphate, the strongest measured binding constant was recorded when the chloride salt of meso-178 was studied in pure DMSO. In this case, values of K1 = 4.0 × 103 M-1 and K2 = 1.0 × 102 M-1 were recorded.
The guanidinium-mediated complexation of several phosphate anions with preorganized C3v symmetric receptors was also studied by the Anslyn group. These receptor systems, embodied by 180 and 181b, were created in order to achieve geometric complementary in addition to allowing for hydrogen bonding, electrostatic interactions, and metal coordination. While highly selective for citrate, receptor 180 was also found to interact with ATP, displaying a binding constant of 1.2 × 103 M-1 as inferred from 1H NMR spectroscopic analyses (D2O, pH 7.4).274 Receptor 181b, in which six guanidiniums are sterically geared to converge and create a cavity, was used to effect the selective detection of inositol triphosphate (IP3).275 Using an indicator displacement assay, 181b was shown to bind IP3 more strongly than a variety of other phosphorylated molecules including ATP and fructose-1,6-diphosphate in an aqueous medium (HEPES, pH 7.4). Of the compounds tested, only benzene-1,3,5-triphosphate and phytic acid were found to bind with similar or greater affinity. The protonated amine receptor 181a was tested for comparison. Although slightly higher binding constants were observed with 181a compared to 181b, evidence for non-specific interactions was also obtained. Binding constants between 181b and IP3 were found to be 4.7 × 105 M-1 in water and 1.0 × 108 M-1 in methanol. In neither case was a significant ionic strength dependence observed. Using fluorescence spectroscopy, IP3 could be detected at 1 μM concentrations in water and 2 nM in methanol. These results led to the suggestion that significant binding can be achieved through geometric complementarity. In further studies, improved binding to IP3 was observed when this assay was performed in the presence of 2% Triton X detergent at pH 4.0 (formate buffer).276 This increased stability was attributed to the inclusion of the neutral host:guest complex within a micelle core.
A small library of guanidinium chemosensors (182a,b) was also developed by the Anslyn group and based on the preorganized “pinwheel” scaffold. This library was screened for selective binding to ATP.277,278 Control receptor 183 was found to bind ATP with an association constant of 3.5 × 102 M-1 in water. However, it was not found to be selective among nucleotides.277 The ability of the more elaborate receptors 182 to effect differentiation was expected to derive from the peptide arms (prepared in a combinatorial manner). Library 182a was first screened for its ability to bind to a fluorescently labeled ATP analogue. Several receptors were observed to interact strongly with the fluorescent guest, and the peptide arms of a selection of these high affinity receptors were sequenced. A small subset of these receptors in the form of 182b was then synthesized based on the sequences obtained. Fluorophores were then used in conjunction with 182b so as to allow for direct fluorescent sensing of ATP. Among the sequences used to create the 182b library, Ser-Tyr-Ser exhibited strong binding (3.4 × 103 M-1) and high selectivity for ATP over AMP and GTP. The lack of response to AMP and GTP provided support for the notion that specificity was dependent on both the length of the phosphate chain and the nucleobase. The specific subunits involved, namely tyrosine and serine, led to the consideration that binding modes involving interactions of the π-surfaces and hydrogen bonding were important. The library of peptides represented by structure 182a was later used to create an array in which ATP, AMP, and GTP could be differentiated via pattern recognition analysis. Here, the relevant studies were carried out by noting the response of each library member to the analyte in an indicator displacement assay.278 In this way, one array could be used to identify selectively these three different phosphorylated analytes.
Schmuck and Schwegmann used a variation on the guanidinium pinwheel to bind phosphorylated sugars through both ion pairing interactions and hydrogen bonds.279 Receptor 184 was found through 1H NMR spectroscopic titration to bind mono-substituted anionic sugars. With this receptor, a two-fold preference for phosphorylated sugars over carboxylated sugars and a three-fold preference for phosphorylated sugars over methylphosphate was reported. The highest binding affinities near neutral pH were found between 184 and mannose-1-phosphate, with an association constant of 1.25 × 103 M-1 being recorded in aqueous DMSO (30% water). Two-dimensional NMR spectroscopic experiments and molecular modeling analyses led to the suggestion that the anionic groups interacted with the guanidiniums, while the sugars participated in hydrogen bonds with the pyrrole and amide units. Receptor 185 was also found to bind sugar phosphates more strongly than sugar carboxylates.280 In addition, this receptor was found to interact with AMP and 3′,5′-cAMP. While no obvious trends were observed among phosphorylated compounds, UV-Vis titrations carried out in aqueous DMSO (20% water) at pH 6 revealed that receptor 185 bound methylphosphate with an association constant of K = 3.8 × 103 M-1.
The Kunitake group developed guanidinium-functionalized monolayers for the binding of nucleotides at the air-water interface.281,282 The guanidinium amphiphiles 186 were found to form a monolayer on pure water that was then observed to bind AMP and ATP.281 Langmuir isotherm analysis of X-ray photoelectron spectroscopy (XPS) data yielded binding constants of 1.7 × 107 M-1 and 3.2 × 106 M-1 for ATP and AMP, respectively. Interestingly, ATP was found to effect saturation at a concentration of 10-5 M at a 1:3 ratio of ATP:186a, whereas with AMP saturation was seen at a 1:1 ratio. Such findings are consistent with each phosphate interacting with one guanidinium moiety. In addition, changes in the IR spectrum were observed that supported the notion that hydrogen bonding plays an important role in the recognition process. Amphiphiles 186b and 186c were mixed with equimolar ratios of 186a in order to aid in discrimination between nucleotides.282 Mixtures with adenine-substituted 186b were found to have a slight preference for UMP over AMP, while the opposite trend was observed with the thymine-containing receptor 186c.
A three-component monolayer consisting of the diaminotriazine derivative 187, the guanidinium derivative 188, and the orotate derivative 189 was used in an effort to bind the isoalloxazine, the phosphate, and the adenine subunits of flavin adenine dinucleotide (FAD).283 Binding of FAD by the monolayer (1:2:1 187:188:189) was nearly stoichiometric at concentrations as low as 10-6 M. Significantly lower binding was observed with combinations of only two components or with other guest molecules such as ADP, AMP, and flavin mononucleotide (FMN). However, each of these multi-component systems suffered from competing hydrogen bonding interactions between the amphiphilic components. Binary mixtures were thus explored later on in an effort to bind FMN.284 In the context of this work, it was found that a 1:1 mixture of 188 and melamine derivative 190 bound FMN in stoichiometric fashion with saturation being observed at ca. 5 × 10-6 M. FTIR analysis provided support for interactions between the guanidinium subunits and the phosphate moiety and between the melamine units and the isoalloxazine moiety. A monolayer mixture made up from 190 and 191 was found to behave similarly. These studies demonstrated that appropriate combinations of individual guanidinium species and additional recognition motifs can be used to effect the selective binding of appropriately chosen phosphorylated substrates.
Recently, Mellet and Fernández reported a small library of monosaccharide pseudo-amide oligomers containing guanidinium, urea, or thiourea recognition units (192-193). These elaborated cores were found to bind both dimethyl and phenylphosphate in pure D2O.285 The relative binding affinities were found to depend on the nature of the pseudo-amide receptor. Specifically, as the acidic character of the receptors increased, the binding affinity was found to improve. Guanidinium receptors 192c and 193c were found to interact most strongly with these two test anions, with binding constants in the range of 48-60 M-1 being calculated in the case of sodium dimethylphosphate, as inferred from 1H and 13C NMR spectroscopic analyses.
4.1.3. Other Charged Systems
In addition to the ammonium and guanidinium receptors previously discussed, a number of other charged binding moieties have been investigated in the context of phosphate recognition. For example, receptor 194 was created by Göbel and co-workers in 1992 using an amidinium group, a chemical entity bearing considerable resemblance to the guanidinium cation.286,287 While the measured association constant was small with catechol cyclic phosphate (200 M-1 in DMF as inferred from 1H NMR spectroscopic titrations), this system was found to catalyze the hydrolysis of the phosphate unit of the guest. Presumably, this reactivity was the result of the neighboring alcohol group.
Thiouronium units have also been utilized for phosphate recognition. In 1998, the Hong group explored the use of thiouronium groups in creating receptors 195 and 196.288 The interactions of receptors 195 and 196 with a variety of TBA:oxoanion salts were studied via 1H NMR spectroscopy in DMSO. Binding constants were found to correlate well with basicity except for dihydrogenphosphate (K = 1.1 × 103 M-1), which bound receptor 195 nearly twice as strongly as benzoate despite having a significantly lower basicity. This difference was attributed to an increased number of oxygen atoms available to hydrogen bond to the host in the case of dihydrogenphosphate. When constrained in similar receptor geometries, the thiouronium groups were estimated to be more effective anion recognition motifs than thiourea groups. However, the thiouronium-based systems proved much less effective than guanidinium-derived receptors. The latter observation was attributed to the larger size (and hence lower charge density) of sulfur compared to nitrogen.
As with many of the previous examples, additional functionalities can be combined with thiouronium motifs to give receptors with improved selectivity. This has been nicely demonstrated by the Hong group. For example, these researchers attached a thymine subunit to thiouronium-based skeletons to obtain receptors 197-198. This allowed for the selective transport of 5′-AMP, presumably in U-tube type experiments using a chloroform membrane.289 In terms of design, the use of charged receptors was expected to lead to enhanced transport by creating charge neutrality in the host:guest complex, while the thymine was expected to increase selectivity for substrates containing the complementary nucleobase (i.e., adenine). As predicted, receptors 197a and 198a displayed significantly higher transport rates for 5′-AMP as compared to receptors 197b and 198b, at both pH 5.0 and pH 7.0. Bis(thiouronium) receptors 197 were also significantly more effective than receptors 198, particularly at pH 7.0 where 5′AMP exists primarily as a dianionic species. Lower transport rates were observed for the non-complementary nucleotide 5′-GMP. These results provide yet another demonstration of how substrate specificity may be achieved through an appropriate combination of multiple binding moieties.
A fluorescent thiouronium receptor 199 was developed by the Kubo group.290,291 A dramatic increase in the fluorescence of compound 199 was observed upon the addition of both TBA acetate (K > 106 M-1) and TBA dibutylphosphate (K = 5.6 × 104 M-1) in acetonitrile, with binding affinities corresponding well with the basicity of the anions in question. Based on control experiments, the authors suggested that the thiouronium moiety quenched the naphthalene emission through a PET process involving electron transfer from the fluorophore to the attached cationic center. This process is then precluded upon anion binding to the thiouronium moiety, which results in a higher fluorescence intensity. A more dramatic response was observed for receptor 200. However, in this case precipitation at higher concentrations prevented determination of a binding constant for the dibutylphosphate anion. Similar results to those obtained with receptor 199 were reported with the 1,3-bis(isothiouronium)-derived naphthalene receptor 201.292 Here, addition of HPO42- and acetate (as the corresponding [K+-[18-crown-6] salts) led to the restoration of the fluorescence emission of receptor 201 in acetonitrile (6% water). Job plot analysis provided support for the proposed formation of a 2:1 (host:guest) complex in the case of HPO42-.
Teramae and co-workers developed a system ostensibly similar to 199 but where the naphthalene fluorophore was attached to the thiouronium anion recognition moiety through the sulfur atom instead of the nitrogen atom (202).293 An emission increase was again observed in the presence of select anions, which were studied as their K+-[18-crown-6] salts in methanol. Strong binding was observed with HPO42- (K = 1.1 × 104 M-1). On the other hand, acetate was found to bind approximately one order of magnitude less well. The addition of dihydrogenphosphate or chloride anions led to little or no change in the emission spectra of receptor 202. A direct comparison between the Teramae and the Kubo systems (199) was not possible as different solvents (methanol vs. acetonitrile) were used by the two research groups.
A more elaborate thiouronium receptor system 203 was prepared by Ahn and co-workers using a tripodal core. 294 The interactions of these receptors with dianionic sulfate and trianionic phosphate were measured in methanol using ITC. Strong 1:1 complexes were observed with sulfate. However, titrations with phosphate revealed complex equilibria that could not be resolved, although evidence of binding was inferred.
Kubo, Nakhara, and co-workers examined thiouronium groups organized in monolayers at the air-water interface in efforts to sense dihydrogenphosphate.295 In preliminary studies, dihydrogenphosphate was observed to expand monolayers of receptor 204 to a much greater degree than acetate or chloride anions. Interestingly, picrate also expanded the monolayers. In Langmuir-Blodgett (LB) films, picrate was used as an indicator and allowed the sensing of dihydrogenphosphate via absorption spectroscopy. Indeed, a large decrease in absorbed picrate was observed in the presence of the dihydrogenphosphate anion. On the other hand, little change was observed in the presence of acetate or chloride. This method allowed for the simple detection of dihydrogenphosphate in a film sensor system.
The Kubo group also examined isothiouronium groups assembled on gold nanoparticles (205). These studies were carried out in an effort to develop an optical sensing system for oxoanions.296 While little color change was observed upon the addition of chloride, red-shifts in the absorption spectrum were observed in the presence of acetate, monohydrogenphosphate, and malonate in 10% water/methanol. The addition of monohydrogenphosphate led to a 13 nm shift in the plasmon absorption band of the nanoparticle, presumably due to increased aggregation. Malonate, however, produced an even greater shift (as well as an observable color change), indicating a lack of specificity for monohydrogenphosphate in these receptors.
Imidazolium groups, which contain cationic hydrogen bond donor sites, have been studied extensively as anion binding motifs in recent years. Compound 206, for example, was designed by K. S. Kim to investigate (C-H)+ - X- hydrogen bonds.297 However, this system was also found to bind dihydrogenphosphate. The nitro groups were intended to decrease the electron density of the ring. By means of 1H NMR spectroscopic titrations carried out in DMSO, a binding constant of 2.5 × 103 M-1 was measured for dihydrogenphosphate (TBA salt). A higher association was recorded for chloride. Receptor 207 was designed to have a larger cleft and provide a fluorescent response upon exposure to targeted anions.298 In this case, a tweezer-like binding mode was inferred, while an association constant of 1.3 × 106 M-1 for the binding of TBA dihydrogenphosphate in acetonitrile was calculated from fluorescence titrations. Interestingly, receptor 207 displayed selectivity for dihydrogenphosphate over chloride. A more rigid receptor, the macrocyclic system 208, was found to bind dihydrogenphosphate with an affinity that was similar to that of 207. However, a much great selectivity for dihydrogenphosphate over chloride and fluoride was reported (all anions added as TBA salts).299 A water-soluble version, receptor 209, was then prepared. This derivative could selectively sense GTP in aqueous solutions at pH 7.4 through a chelation-enhanced quenching mechanism.300 The greatest change in the fluorescence (a notable increase) was seen with GTP. From the spectral changes involved, a 1:1 binding stoichiometry and a binding constant of 8.7 × 104 M-1 was inferred. Addition of ATP and ADP also engendered an increase in the fluorescence intensity, while anions such as pyrophosphate, dihydrogenphosphate, fluoride and chloride produced no appreciable change in the fluorescence. Based on molecular modeling, the authors suggested that the selectivity for GTP over ATP was due to enhanced π-H interactions between anthracene and the respective nucleobases. Further fluorescence studies, carried out in acetonitrile, revealed that receptors 207, 208, 210, and 211 displayed a strong preference for pyrophosphate (TBA salt).301 The highest pyrophosphate interaction was found with receptor 208, for which a binding constant of ca. 1.01 × 108 M-1 was inferred. An extensive 1H NMR spectroscopic study provided support for the inference that the dominant interaction mode between receptor 208 and pyrophosphate involved ion pairing.
Recently, the imidazolium receptor 212, prepared by J. Yoon and co-workers, was studied as a fluorescent chemosensor.302 This receptor was found to have a high affinity for dihydrogenphosphate (TBA salt) in acetonitrile with a binding constant of 2.2 × 105 M-1 being reported. The quinoxaline-imidazolium receptors 213-216, prepared by K. S. Kim, were found to act as fluorescence sensors, allowing for the detection of pyrophosphate as well as acetate in acetonitrile (TBA salts).303 However, binding constants for pyrophosphate could not be determined due the insolubility of the resulting adducts. These researchers also investigated the anion binding properties of the acridine derivative 217.304 The effects of a number of TBA anion salts (pyrophosphate, dihydrogenphosphate, hydrogensulfate, acetate, iodide, bromide, chloride, and fluoride) on the emission spectrum of this receptor were studied in acetonitrile. Interestingly, addition of pyrophosphate led to significant fluorescence quenching, while the addition of dihydrogenphosphate led to strong emission enhancement. The other anions tested produced only small spectral changes. An association constant of 4.9 × 107 M-1 was determined for the binding of pyrophosphate to receptor 217 through fluorescence titrations. Job plot analysis confirmed a 1:1 binding stoichiometry, and 1H NMR spectroscopic analyses in DMSO-d6 proved consistent with the presence of strong imidazolium C-H hydrogen bonding interactions in this complex. The emission increase observed upon the addition of dihydrogenphosphate was attributed to the formation of a hydrogen bond between the acridine nitrogen atom and one of the OH groups of the phosphate anion. Job plot analysis was consistent with a 1:2 host:guest binding stoichiometry, and the binding constant was estimated to be greater then 108 M-2. In competition experiments, it was found that the emission response of receptor 217 to pyrophosphate was not effected by a 10-fold excess of dihydrogenphosphate, acetate, or fluoride. However, no response to anions was observed under aqueous conditions (9:1 CH3CN:H2O).
K. S. Kim, J. Yoon, and co-workers reported the anion sensing abilities of receptor 218, containing two imidazolium recognition units and two pyrene reporter units, in 2007.305 Quenching of both the monomer and excimer pyrene emission peaks was observed upon the addition of a variety of anions (H2PO4-, HSO4-, AcO-, I-, Br-, Cl-, and F- as TBA salts) in acetonitrile. A modest preference for dihydrogenphosphate was observed with receptor 218a, while a high selectivity for dihydrogenphosphate was found with receptor 218b. Similar dihydrogenphosphate binding affinities for both receptors were found, as determined through fluorescence titrations (K ≈ 2 × 105 M-1).
These same researchers recently reported the tetraimidazolium receptor 219, a system put forward as a selective fluorescent sensor for ATP.306 In neutral aqueous solution (20 mM HEPES, pH 7.4), unique ratiometric changes in the monomer and excimer peaks of the pyrene units of receptor 219 were observed upon the addition of ATP. From these spectral changes, a binding constant of 1.03 × 104 M-1 was calculated for the ATP:219 interaction. Little to no change in the emission peak ratio was reported upon the addition of dihydrogenphosphate, pyrophosphate, CTP, UTP, TTP, GTP, AMP, and ADP. Calibration curves developed for ATP worked well even in the presence of 40 equivalents of other nucleotide triphosphates. One- and two-dimensional 1H NMR spectroscopic studies carried out in DMSO-d6 provided support for the suggestion that the adenine moiety of ATP was inserted between the pyrene units upon complex formation. On the other hand, it was inferred that the guanine nucleobase of GTP was bound to one pyrene unit on the outside of the cleft. The significant changes in the emission spectra of receptor 219 with ATP were attributed to this difference in nucleobase-pyrene interactions. In the case of both ATP and GTP, the triphosphate unit was found to be bound to the positively charged imidazolium moieties, while the ribose subunit was considered not to interact with this receptor (219), as inferred from NMR spectroscopic analyses. These binding modes were further supported by density functional theory calculations of the host:guest complexes. Finally, preliminary studies in cell cultures supported the use of receptor 219 as a real-time monitoring system for ATP levels.
In 2003, Sato and co-workers reported the anion binding ability of the imidazoliophane receptor 220.307 Proton NMR spectroscopic titration studies carried out in DMSO-d6 revealed a strong size and shape selectivity when anions were added as their TBA salts. Binding affinities with receptor 220 decreased in the order hydrogensulfate > bromide > dihydrogenphosphate > chloride > iodide > perchlorate. A binding constant of 1350 M-1 was reported for dihydrogenphosphate.
A series of imidazolium receptors with varying symmetry was reported by Schatz and co-workers (221 - 223).308 All receptors were found to have a similar binding affinity for dihydrogenphosphate (K ≈ 2000 M-1) in DMSO-d6 as inferred from 1H NMR spectroscopic studies. This observation led these researchers to propose that only two imidazolium units of each receptor are involved in phosphate anion recognition. The highest selectivity for dihydrogenphosphate against other anions (chloride, bromide, and hydrogensulfate) was observed for receptor 223b. All anions were studied as the TBA salts.
Multifunctional imidazolium-binol receptors 224 - 225 were reported by Yu and co-workers in 2009.309 Receptor 224a displayed red-shifted emission intensity peaks upon addition of acetate and fluoride and a quenching of emission intensity upon addition of dihydrogenphosphate, chloride, bromide, and hydrogensulfate (TBA salts) in acetonitrile. The spectral shifts observed for fluoride and acetate anions were attributed to deprotonation and charge-transfer effects, respectively. Binding constants were found to be on the order of acetate ≈ dihydrogenphosphate ≫ hydrogensulfate ≈ chloride > bromide. The association constant for receptor 224a and dihydrogenphosphate was determined to be 2.46 × 105 M-1 under these conditions. Anion-induced quenching was also observed upon the addition of these anions to receptors 224b and 225; however, no binding constants were reported.
In work also involving an imidazolium subunit, the Chang group screened a small library of chloro-substituted benzimidazolium receptors of general structure 226 for their ability to bind GTP.310 Of this set, two compounds, 226a and 226b, were found to display an enhanced fluorescence in the presence of micromolar GTP at pH 7.4 (HEPES buffer, 1% DMSO). These turn-on sensors demonstrated little to no response to all other nucleotides and nucleosides included in the test set. However, compound 226b displayed significant photobleaching and was not subject to follow-up study. On the other hand, more detailed studies involving 226a revealed that it binds GTP more strongly than 2′-deoxy-5′-guanosine triphosphate (dGTP), a finding that provided support for the sugar moiety being involved in GTP binding. Due to its visible green emission, compound 226a was dubbed “GTP green” by this group of researchers.
Ghosh and co-workers recently developed a series of benzimidazolium-based cleft receptors (227 - 228).311 Preliminary studies with mono-anthracene receptors 227 and 228 revealed quenching of the emission spectra by several anions (acetate, dihydrogenphosphate, propanoate, benzoate, mandelate, pyruvate, and fluoride) when added as the TBA salts in DMSO. The strongest signal changes were observed for acetate and dihydrogenphosphate with the urea-based receptor 228. Spectral changes were also observed through UV-Vis spectroscopy, which was used to determine 1:1 binding constants for the host:guest complexes. Receptors 227 and 228 were found to bind dihydrogenphosphate with binding constants of 1.73 × 103 M-1 and 1.12 × 104 M-1, respectively. While dihydrogenphosphate displayed one of the highest signal changes, carboxylate anions were generally found to have the strongest binding affinities. Proton NMR spectroscopic experiments carried out in DMSO-d6 led to the conclusion that all N-H groups and the benzimidazolium C-H groups in these receptors were involved in anion-binding interactions.
The bis-anthracene receptor 229 was reported later.312 In this case, fluorescence titrations revealed a strong preference for TBA dihydrogenphosphate over acetate, propanoate, benzoate, chloride, bromide, iodide, and hydrogensulfate in acetonitrile (K (H2PO4-) = 5.41 × 103 M-1). Job plot analysis revealed a 1:2 host:guest binding stoichiometry for dihydrogenphosphate. Fluoride also produced a strong decrease in emission intensity, presumably due to deprotonation. However, the sensing ability of receptor 229 was significantly diminished in aqueous methanol solution (4:1 CH3OH:H2O). Support for the proposed N-H and C-H hydrogen bonding interactions with dihydrogenphosphate came from standard 1H NMR spectroscopic studies carried out in DMSO-d6.
Triazolium recognition units were used by Pandey to produce deoxycholic acid-containing scaffolds 230-231.313 Here, click chemistry was used, and this allowed for a facile synthesis of these receptors. The binding properties were studied via 1H NMR spectroscopic titrations carried out in chloroform using various TBA anion salts. While meta-substituted receptor 230a displayed no appreciable binding for dihydrogenphosphate, receptors 230b and 231 displayed binding constants of 1.10 × 103 M-1 and 1.92 × 103 M-1, respectively. These receptors also displayed a significant selectivity for dihydrogenphosphate over the halide anions (F-, Cl-, Br-, and I-) as well as over acetate. This binding specificity was found not to correlate with basicity, which is noteworthy. Rather, the observed selectivity trend was attributed to the slightly larger cavity of receptor 230b as compared to compound 230a, as well as to the greater flexibility of receptor 231. The ease of preparation and the observed selectivity for dihydrogenphosphate makes it safe to predict that triazolium receptors have a future in the area of phosphate recognition.
A relatively novel anion recognition functionality, aminothiazolinium, was investigated in the context of the previously described “pinwheel” scaffold by T. H. Kim in 2009.314 On the basis of ITC studies carried out in methanol, receptors 232a and 232b were observed to interact with AcO-, SO42-, and PO43-. Despite the higher charge of the trianionic phosphate guest, the lowest binding affinity for these two receptors was reported for this analyte, with apparent binding constants of 1.34 × 104 M-1 and 1.81 × 104 M-1, respectively. A complex binding stoichiometry was inferred for both receptors based on ITC titration data and ESI mass spectrometric analyses.
Another example of charged receptor units comes from the group of Fabbrizzi. These researchers reported the pyridinium receptors 233 and 234, which were expected to bind with anions via a combination of electrostatic and hydrogen bonding interactions.315 In fact, spectrophotometric studies in DMSO led to the conclusion that receptor 233 undergoes deprotonation in the presence of basic anions such as acetate, fluoride, and dihydrogenphosphate (TBA salts). The less acidic receptor 234 was only deprotonated by the fluoride and acetate anions. In contrast, 1:1 adducts were produced with dihydrogenphosphate (log K = 3.11).
Beginning in 1996, Powell and co-workers began a series of studies involving the fluorescent pyridinium receptor 235, a system characterized by an excited state with charge-transfer character.316,317 An increase in fluorescence upon anion binding was observed in phosphate buffered solution at pH 7.2. This increase was interpreted in terms of the formation of twisted intramolecular charge-transfer (TICT) complexes when receptor 235 was treated with purine nucleotides; however, no interaction was observed for pyrimidine nucleotides. Receptor 235 permitted 3′,5′-cAMP and 3′,5′-cyclic guanosine monophosphate (3′,5′-cGMP) detection in aqueous solution but did not discriminate between the two purine guests (i.e., K = 212 M-1 for 3′,5′-cAMP and 285 M-1 for 3′,5′-cGMP).
In 2006, Steed and co-workers reported the binding of pyridinium receptor 236 to ATP.318 Significant shifts in the 1H NMR spectral peaks corresponding to the pyridyl aromatic protons were observed upon the addition of Na2ATP to receptor 236 in 1:1 acetonitrile:water (deuterated). A binding constants of 70 M-1 was determined using this method. No 1H NMR spectral changes were observed upon the addition of chloride, bromide, iodide, hydrogensulfate, sulfate, dihydrogenphosphate, acetate, or nitrate in deuterated acetonitrile. These results led these researchers to suggest that receptor 236 binds ATP through a combination of electrostatic interactions and π-surface interactions.
In the same year, the Steed group examined the ability of a preorganized urea-pyridinium hybrid (237) to bind simple inorganic anions.319 Binding constants were determined through 1H NMR spectroscopic titrations carried out in DMSO-d6 with all anions added as the corresponding TBA salts. A preference for H2PO4- over Cl-, Br-, I-, NO3-, AcO-, HSO4-, CF3SO3-, and ReO4- was observed. Log binding constants for the 1:1 and 1:2 host:guest complexes with H2PO4- were found to be 3.70 and 3.68, respectively.
A pyridinium-amide receptor (238) bearing anthracene substituents has recently been reported by Ghosh.320 UV-Vis and fluorescent studies revealed that 238 bound strongly basic anions such as acetate, dihydrogenphosphate, and fluoride, with an affinity constant on the order of 104 M-1 in the case of dihydrogenphosphate in acetonitrile. These researchers further reported selective dihydrogenphosphate sensing with the biphenyl-based pyridinium receptor 239.321 A strong increase in the emission intensity was seen upon the addition of dihydrogenphosphate (TBA salt) to a solution of receptor 239 in CHCl3 containing 2% CH3CN. Significantly smaller changes were observed upon the addition of acetate, hydrogensulfate, and a variety of dicarboxylates as the TBA salts. The observed increase in emission was attributed to a conformational change of the receptor upon anion binding. It was proposed that dihydrogenphosphate interacts with both pyridinium moieties, thus bringing the two anthracene arms closer together and increasing excimer emission. A binding constant of 1.22 × 104 M-1 was measured under these conditions. At higher concentrations of dihydrogenphosphate (greater than 4 equivalents relative to host 239), the emission intensity was seen to decrease. This decrease was attributed to the formation of 1:2 host:guest complexes at higher concentrations. Significant changes were also observed in the UV-Vis and 1H NMR spectra upon the addition of dihydrogenphosphate to receptor 239. However, no excimer formation was observed upon the addition of anions in mixtures of water and acetonitrile, a more competitive environment.
Another charged recognition functionality that is commonly employed for phosphate anion recognition is viologen. In 2005, Ramaiah and co-workers reported the selective binding of ATP by viologen - anthracene receptor 240.322 This receptor displayed reduced absorption intensity upon the addition of ATP in 10 mM phosphate buffer. UV-Vis titrations were used to measure a 1:1 binding constant of 4.04 × 103 M-1 for the 240:ATP complex. No significant changes in the absorption features were seen upon the addition of adenosine, AMP, or ADP. These results provided support for the suggestion that strong electrostatic interactions contribute to the binding process. Proton NMR spectroscopic experiments were consistent with strong π-surface interactions between the adenine base of ATP and the viologen moieties of receptor 240. In addition, complex formation was inferred from the observed shift in the reduction potential of this receptor in the presence of ATP as measured using differential pulse voltammetry.
Receptor 240 exhibited negligible fluorescence, presumably due to a PET process from the anthracene units to the viologen units. In efforts to develop a fluorescent nucleotide sensor, an indicator displacement assay was developed using receptor 240 and 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS) as the indicator.323 In this system, the fluorescence of HPTS was quenched by complexation with receptor 240. Addition of GTP, ATP, and inosine triphosphate (ITP) led to a recovery of the HPTS fluorescence, which was attributed to displacement of the indicator. A significantly stronger response to GTP (150-fold emission increase) was observed as compared to ATP (45-fold) and ITP (50-fold). The addition of other guests (adenosine, AMP, ADP, CTP, UTP) did not result in changes to the HPTS emission. The selectivity exhibited by this system allowed for the discrimination of GTP in the presence of other nucleotides and in biological fluids.
Schiller and Singaram reported that viologen receptors 241 and 242 were able to sense glucose-6-phosphate via fluorescent IDA in 2009.324 Binding constants were measured using HPTS as the indicator in a phosphate buffered aqueous solution at pH 7. Receptor 241 bound glucose-6-phosphate (K = 7.5 × 102 M-1) more strongly than fructose (K = 5.6 × 102 M-1), presumably due to a charge-charge interaction between the pyridinium and phosphate groups. This additional interaction thus overcomes the inherent fructose > glucose selectivity of boronic acids. However, this selectivity was reversed in the case of receptor 242, leading to the suggestion that the charge-charge interaction was not as strong in this case. Interestingly, no binding interactions were observed for either receptor with glucose-1-phosphate or sucrose. Receptor 241 and HPTS were then employed in a bioanalytical assay for the activity of phosphoglucomutase (PGM), which converts glucose-1-phosphate to glucose-6-phosphate.
The use of larger aromatic moieties was explored through the use of N,N-dialkyl-diazapyrenium units, which represent known DNA intercalators. For example, nucleotide binding was studied with receptors 243 and 244.325 Addition of nucleotides to the more complex system 243 in neutral aqueous solution produced significant fluorescence quenching, allowing for the derivation of binding constants. Among the set of potential guests, adenine, AMP, ADP, and ATP, the binding constants were found to increase as the number of phosphate groups increased (e.g., log K = 2.5 and 3.1 for adenine and ATP, respectively). Among nucleotide monophosphates AMP, GMP, CMP, and TMP, the observed binding affinity was found to increase with decreasing oxidation potential. These trends led the authors to suggest that both electrostatic and charge-transfer interactions influence the overall binding affinity.
As discussed elsewhere in this review (e.g., Section 4.1.1), hydrogen bonding between complementary nucleobases has been used in several instances to create selective receptors. Receptors 245 and 246 combine this strategy with one that allows for charge neutralization by appending a phosphonium center to a clip-like framework. These systems were found to extract and transport GMP and AMP effectively through a chloroform membrane.326 Concentration-dependent extraction studies provided support for the conclusion that 1:1 host:guest complexes were found at pH 5.0 and 2:1 host:guest complexes at pH 7.0. In accord with design expectations, better transport rates were observed with these combined receptors than was observed with either the phosphonium or nucleobase component alone or when the individual components were both contained separately within the organic phase.
Sapphyrins, a class of expanded porphyrin derivatives, generally bind phosphates in their protonated forms and charged receptors based on these motifs are known. They are treated in Section 4.4, which includes a discussion of several other macrocyclic receptor systems.
4.2. Hydrogen Bonding
Hydrogen bonding is a stabilizing interaction that exists between an electronegative atom and a hydrogen atom bound to another electronegative atom. This type of bonding always involves hydrogen atom(s) and is generally directional. The strength of a hydrogen bond is largely determined by electrostatic attraction (e.g., dipole-dipole interactions and charge-dipole interactions), although significant contributions are also observed from charge-transfer, dispersive, and covalent forces. Therefore, the typical hydrogen bond is stronger than van der Waals forces but weaker than covalent or ionic bonds.
The use of multipoint hydrogen bonding is commonly observed in the molecular recognition of biological compounds. In supramolecular chemistry, a variety of scaffolds have been used to preorganize the hydrogen bonding components with the goal of enhancing interactions between the receptor and the functional groups of the target compound.327,328 Highly selective systems have been developed based on these interactions, although for the most part the systems in question have been proven effective in less-competitive organic solvents. In the specific case of phosphate binding, amide and urea-based binding moieties have been the most commonly employed, reflecting their known ability to act as strong hydrogen-bond donors.329,330 However, other subunits have been employed, as have systems containing several different hydrogen bonding functionalities. In this section, we present a discussion of neutral receptors that have been used for phosphate anion recognition. They are organized to the best of our ability according to the specific functional group involved.
Prior to commencing a discussion of individual receptor systems, it is important to point out that many neutral hydrogen bond-based phosphate receptors are plagued with low solubility. As a general rule, these receptors also display low binding affinities in competitive media (as the result of what are effectively weaker receptor-anion hydrogen bonds as compared to those involving solvent). As a result, a variety of solvents and solvent mixtures have been used in the analysis of neutral hydrogen bond-based phosphate anion receptors. Such solvent differences, however, generally prevent direct comparison of the anion binding properties of different receptor systems. It is to be further noted that, as a general rule, studies with neutral receptors have relied on the use of tetraalkylammonium salts. In most cases, these salts allow for solubility in organic media. As such, it has long been assumed that tetraalkylammonium cations are innocent and do not interfere in the recognition process. However, the validity of this assumption is starting to be called into question.331,332 Nevertheless, unless otherwise stated, all studies in this section were carried out using tetraalkylammonium salts.
4.2.1. Amide Systems
The amide group has a time-honored place in anion recognition chemistry. This presumably reflects its ease of preparation, as well as its effective hydrogen bond donating ability. On the other hand, amide groups can also serve as hydrogen bond acceptors, and this can lead to complications in terms of receptor design.333 Nevertheless, to date amide subunits have been incorporated into a wide variety of both cleft-like and macrocyclic receptors. Amides have also been studied on their own. For instance, the study of a simple system, N-methylacetamide, revealed an association constant of 26 M-1 for TBA dihydrogenphosphate in CDCl3 as inferred from 1H NMR spectroscopic analyses.334 This modest affinity for a single amide unit provides a baseline against which the success of other more elaborate receptors may be judged.
In 1995, phosphate receptors 247 and 248 incorporating amide moieties were reported by Raposo and co-workers.335 These triangular receptors were designed to be complementary to tetrahedral phosphate anions. The binding properties were studied using 1H NMR spectroscopic methods, with the actual analyses being carried out in CDCl3, CD3OD and DMSO-d6 as dictated by solubility considerations. In chloroform, the solubility of phenylphosphonic acid was found to be enhanced in the presence of receptor 247, leading to the suggestion of a host:guest interaction. However, the specifics of the putative binding process were not fully characterized. No interaction was observed between 247 and a variety of phosphates in mixtures of chloroform and methanol. On the other hand, evidence for a weak interaction was found upon addition of tris(tetramethylammonium) phosphate in DMSO. In the case of receptor 248, measurable complex stabilities were recorded in chloroform/methanol mixtures, as well as in DMSO and pure methanol. Presumably, this reflects the presence of the additional hydrogen-bond donors present in 248 as compared to 247. Phenylphosphonic acid, propylphosphonic acid, bis(tetrabutylammonium) phenylphosphonate and tris(tetramethylammonium) phosphate were all tested as possible substrates for receptor 248. In DMSO, the strongest interactions were observed with the most negatively charged targets, dianionic phenylphosphonate and trianionic phosphate (K = 1.5 × 104 M-1, K > 105 M -1, respectively). In methanol, trianionic phosphate was also found to be complexed well by 248, with an association constant of 1.2 × 104 M-1 being calculated for this interaction.
In 1992, the Hamilton group examined the phosphate interactions of a set of related cleft and macrocyclic receptors containing pyridine bis-amide cores.336 Specifically, the pyridine bis-amide clefts 249 and 250 and the corresponding macrocycle (251) were studied through UV-Vis and NMR spectroscopy in chloroform. While the addition of phosphotriesters resulted in no significant change in the absorption spectrum, the addition of a phosphodiester, bis-(4-nitrophenyl)phosphoric acid, led to spectroscopic changes similar to those caused by simple pyridine protonation. In contrast to what is true for pyridines, however, the spectroscopic changes could be reversed via the addition of methanol. This then led to the conclusion that a hydrogen-bonded complex was formed following protonation. Proton NMR spectroscopic and a preliminary single crystal X-ray diffraction analysis provided support for a structure wherein the amide NH and protonated pyridine protons act to stabilize a bidentate phosphate complex similar to those observed with guanidinium receptors (discussed in detail in section 4.1.2). Shifts in the NMR spectrum were observed for the amides protons of 250b and 251 that are closest to the phenyl spacer. Presumably, this is due to inclusion of the guest. This latter conclusion was supported by NOE studies. In addition, a Job plot constructed for the interaction of 250a and 251 with the phosphodiester provided support for the proposed 2:1 guest:host binding stoichiometries. In analogy to what was true for the diesters, the phosphomonoester dodecylphosphoric acid was found to bind to 249 with a binding constant of 2.8 × 104 M-1 in chloroform. However, addition of the monoester to 250a led to minor changes in the absorption spectrum. Further, NMR spectroscopic studies provided support for a more symmetrical complex wherein both amide protons and both pyridine nitrogen atoms participated in hydrogen bonding interactions (as hydrogen-bond donors and acceptors, respectively). However, little evidence of interactions with the proximal phenyl spacer was found. A binding constant of 1.0 × 105 M-1 in chloroform and a stoichiometry of 1:1 were then inferred. These results provided support for the conclusion that the four hydrogen bonds present in the complex stabilized by the cleft system of 250a led to a stronger interaction than would be obtained using the simpler system 249.
A series of convergent macrocyclic receptors containing amide moieties, specifically systems 252, 253, and 254, were also analyzed by these researchers.97,337,338 These rigid structures provided convergent C3 symmetric hydrogen bonding groups in the interior of the macrocyclic scaffold, a design feature that was thought to enable the macrocycles to bind tetrahedral anions with size and shape selectivity. Tetrahedral anions, such as p-TsO-, HSO4-, and H2PO4-, were observed to bind strongly, as inferred from 1H NMR spectroscopic analyses, even in more competitive hydrogen bonding solvents, such as DMSO. At the same time, the binding of halides and nitrate anions proved dramatically less effective in this latter solvent. All anions were studied as the TBA salts.
Receptor 252b displayed a 1.5 × 104 M-1 binding affinity toward dihydrogenphosphate in DMSO, while the linear analogue 253 displayed an affinity of only 500 M-1.337,338 The equilibrium between dihydrogenphosphate and receptor 252a, however, proved too slow on the NMR time scale to yield an accurate binding constant. Nevertheless, these results provided strong support for the intuitively appealing notion that increasing the preorganization of a macrocyclic receptor (relative to the corresponding open-chain system) serves to increase the binding affinity substantially.
A coumarin fluorophore was attached to the basic scaffold in order to generate a selective fluorescent sensor (252c).97 While system 252c displayed only a small change in the emission spectrum in the presence of tetrahedral anions, binding affinities of 5.9 × 105 M-1 and 2.7 × 106 M-1 could nevertheless be determined for the binding of phenylphosphonate and dihydrogenphosphate (TBA salts) in a solvent mixture consisting of a 1:1 DMSO:1,4-dioxane. The fluorophore was later covalently incorporated into the macrocycle structure, producing 254, in an effort to achieve greater spectral changes. As expected, receptor 254 displayed more dramatic spectral changes, including ones that were anion dependent. On the other hand, the actual binding affinities proved similar to those recorded in the case of 252c. Control studies led to the suggestion that the spectral changes observed for receptor 254 were due to proton transfer from the fluorophore excited state to the anion (ESPT). A sensitive fluorescent sensor for phosphate anions was thus achieved.
Simpler but related systems were later analyzed by Gale and Paver.339 While the simple bis-amide 255, a compound earlier developed by Crabtree as a halide anion receptor,340,341 was not found to bind dihydrogenphosphate, the coordination of two metal carbonyl fragments to the phenyl groups (256) led to a weak interaction with dihydrogenphosphate. A binding constant of 119 M-1 was determined from 1H NMR spectroscopic titrations carried out in CD3CN. The increased binding was attributed to the electron withdrawing nature of the coordinated metals, which rendered the amides better hydrogen bond donors. However, the highest interactions were observed with halide anions.
In further studies, Gale and co-workers examined the effect of incorporating an anthraquinone moiety at the central phenyl ring giving receptor 257.342 The interactions of this latter receptor and the control compound 258 with TBA anion salts were then studied via 1H NMR spectroscopic titrations carried out in DMSO-d6-0.5% water. Under these conditions, receptors 257 and 258 displayed significantly higher binding constants for dihydrogenphosphate than the chloride, benzoate, hydrogensulfate, or bromide anions. The anthraquinone derivatives generally bound dihydrogenphosphate more strongly than the phenyl derivative. It was proposed that receptors 257 adopted a twisted conformation as a result of steric clashes with the anthraquinone oxygen atoms and that this conformation was appropriate for hydrogen bonding interactions with dihydrogenphosphate. Particularly strong binding was observed upon the incorporation of electron withdrawing substituents in receptor 257c, which displayed a 1:2 host:guest stoichiometry (K1 = 1520 M-1, K2 = 65 M-1).
Lees and co-workers studied the behavior of the amide receptors 259 - 261, which incorporated bis(4,4′-di-tert-butylbipyridine) rhenium complexes.343,344 Receptor 259a, a luminescent cleft-like receptor, was found to act as fluorescent anion sensor, displaying a high preference for halides, cyanide, and acetate anions as inferred from fluorescence quenching studies carried out in dichloromethane. On the other hand, the interaction with dihydrogenphosphate was found to be weak, characterized by a binding constant of 147 M-1 in this same solvent. Interestingly, dihydrogenphosphate complex formation was slightly weaker with the 2-substituted bipyridyl receptor system 259b (K = 110 M-1) as compared to 259a. On the other hand, much stronger binding was observed with the phenyl-linked receptor 259c (K = 1.8 × 105 M-1), linear receptor 260 (K = 5.5 × 104 M-1), and monotopic receptor 261 (K = 1.6 × 104 M-1). This trend was not observed with any of the other anions tested.
The multifunctional amidopyridine receptor 262 and several other two-arm receptors (e.g., 263 and 264) were studied by the Fang group.345,346 It was proposed that the central pyridine moiety serves to restrain the rotation of the appended amide bonds through hydrogen bonding, leading to the formation of the cleft-like structure shown explicitly in the case of 262. This conformation was confirmed through a single crystal X-ray diffraction analysis of 262a, a study that revealed a pseudo-tetrahedral geometry characterized by multiple hydrogen bonding sites. Proton NMR spectroscopic studies with TBA anion salts revealed that both 262a and 262b bound H2PO4- in DMSO-d6, with association constants of 549 M-1 and 1374 M-1 being recorded in the case of these two receptors, respectively. The corresponding association constants for acetate were found to be 3 to 6-fold lower than those of H2PO4-. Other studied anions (NO3-, Br-, I-, SCN-, ClO4-, H2SO4-) displayed little response.
The pyrene-bearing receptor 262b was found to display both monomer and excimer emission in THF in the absence of anion. Upon addition of H2PO4- or PO43- in THF, a reduction in the excimer emission and an increase in the monomer emission was observed. Based on control studies, the authors suggested that this change was due to an increase in the distance between the two arms that occurred upon phosphate anion complexation. Association constants for complexes of 262b with H2PO4- and PO43- were calculated to be 1.8 × 105 M-1 and 3.3 × 105 M-1, respectively. While weaker binding was generally observed in DMSO, it was found that hydrogenpyrophosphate displayed significant binding (K = 1.1 × 104 M-1) in this highly competitive solvent. The fluorescence response proved selective for phosphate anions over all other anions tested.
More recent studies examined the ability of receptor 262d to bind the metabolite geranyl pyrophosphate (GPP) in DMSO.347 Among the series of receptors 262b-e, receptor 262d displayed the strongest binding affinity for GPP (3300 M-1) and di(hexadecyl)phosphate (DHP, 1830 M-1) (TBA salts), as inferred from 1H NMR spectroscopic experiments. The increased affinity of receptor 262d for these analytes relative to 262c (a system containing four aliphatic substituents) was attributed to preorganization of the receptor cavity by the pyrene moieties of 262d. Receptor 262b displayed the strongest binding affinity for dihydrogenphosphate among this series. In emission studies, no change in the fluorescence of receptor 262d was observed upon the addition of GPP. A FRET-based displacement assay was then developed in which coumarin phosphate was first complexed with receptor 262d to form a strong FRET pair. Upon displacement of coumarin phosphate from the receptor by GPP, a decrease in FRET efficiency was observed. This change was used to determine a binding constant of 2.3 × 104 M-1 in DMSO, a significantly higher value than that observed through 1H NMR spectroscopic methods. This sensing ensemble was selective for the binding of GPP over simple anions (fluoride, chloride, bromide, thiocyanate, acetate, nitrate, and perchlorate) as well as carboxylate ions of fatty acids. The increased signal for GPP over other anions was attributed to hydrophobic interactions between the host and guest aliphatic tails and multi-point hydrogen bonding interactions between the phosphate unit and the amide units of receptor 262d.
In separate work, Fang and co-workers synthesized receptor 263, a system that contains both electrochemical and fluorescent sensing units.348 A control receptor bearing a single arm (264), was also prepared and analyzed. Proton NMR titrations of receptor 264 with a variety of TBA anions in DMSO provided evidence for 1:1 binding interactions. The associated affinity constants were found to range from 1.5-2.5 × 102 M-1, with a preference for dihydrogenphosphate being seen over acetate, iodide, nitrate, perchlorate, and hydrogensulfate. In contrast to what was seen for the control system 264, the two-armed receptors (263) displayed a 1:2 host:anion stoichiometry. Based on ITC measurements carried out in THF, the authors suggested that the binding behavior of both types of receptors was favorable in both enthalpic and entropic terms, with the association constants of 263b (K1 = 1.97 × 105 M-1, K2 = 8.30 × 103 M-1) being much higher than those of 264b (K = 9.10 × 103 M-1). In the end, both receptors proved to be effective sensors with a fluorescence response to dihydrogenphosphate being observed for receptor 263c and an electrochemical response to dihydrogenphosphate being observed in the case of 264a.
In 2004 Kovalchuk and co-workers reported a different pyridine bis-amide cleft-type receptor. The system in question, 265, was designed to serve as a charge-transfer type fluorescent sensor.349 As designed, the central amidopyridine unit was expected to act as the anion receptor while the two styryl chromophores would participate in donor-acceptor interactions. Of the anions tested, only H2PO4- and acetate (TBA salts) led to an enhancement in the receptor fluorescence, at least when tested in DMSO:water (95:5) mixtures. This enhancement allowed binding constants of 55 M-1 and 74 M-1 to be determined for acetate and dihydrogenphosphate, respectively. Since other potentially complexing anionic species, including halide, sulfate, nitrate, cyanide and carboxylate anions, did not produce such an enhancement, it was proposed that, in spite of the low binding constants, the relatively specific, on-off fluorescence response displayed by 265 would make it a potentially useful anion sensor.
A slightly different combination of amides and pyridine was used to create receptors 266 and 267, reported by Kondo and co-workers in 2005.350 Within this set, the strongest interaction with dihydrogenphosphate (added as the TBA salt) was observed in the case of 267c. This finding provided support for the notion that the pyridyl groups participated in the binding event. It was further postulated that the four amide protons bound the carbonyl-type and anionic oxygen of the bound phosphate while the pyridyl hydrogen atoms interacted with the protonated oxygen groups of the H2PO4- guest. The corresponding binding affinity was determined via standard UV-Vis titrations, yielding a binding constant of 1.20 × 105 M-1 in 10% DMSO-CH3CN. Receptor 267c also displayed high selectivity, binding dihydrogenphosphate 85-fold more strongly than either acetate or sulfate.
The inclusion of a carbazole moiety within a cleft-like arrangement was used by Jurczak and co-workers to generate receptors 268a and 268b. Here, the goal was to create a receptor system containing an additional hydrogen bond donor compared to the earlier systems, while at the same time retaining a high level of rigidity.351 The chlorine substituents present in 268 were meant to increase the hydrogen bond-donating ability by withdrawing electron density. The binding behavior of 268a and 268b was studied using standard 1H NMR spectroscopic methods and various TBA anion salts. In DMSO-d6 containing 0.5% H2O, both receptors 268a and 268b were found to be selective for dihydrogenphosphate, displaying relative affinities of H2PO4- > PhCOO- ≫ Cl-. The selectivity over chloride was attributed to the cleft being too large for the smaller anion, as judged from a single crystal X-ray diffraction analysis. Interestingly, the aliphatic amide system (268b) was found to bind anions more strongly than its aromatic analogue (268a). The reason for this difference was not, however, discussed in detail.
Sessler and co-workers have reported the diamidopyrazole receptor 269.352 However, quantitative 1H NMR spectroscopic analyses could not be carried out due to the formation of aggregates at the concentrations required for study. On the other hand, ITC titrations carried out in DMSO provided evidence that receptor 269 bound dihydrogenphosphate (K = 5.76 × 105 M-1) in preference over benzoate, perchlorate, cyanide and bromide; however, receptor 269 was also found to interact strongly with hydrogensulfate. All anions were studied as the TBA salts.
Early on, more flexible receptors, based on the use of diethylenetriamine and tren (78) scaffolds, were reported by Reinhoudt and co-workers.353 The systems in question, receptors 270-272, were described in 1993 and were found to display a selectivity for dihydrogenphosphate over chloride and hydrogensulfate anions, as inferred from 1H NMR spectroscopic analyses carried out in chloroform (all anions studied as the corresponding TBA salts). Host:anion 1:2 adducts were established with dihydrogenphosphate, and this stoichiometry was attributed to the formation of an intermolecular hydrogen bond between both dihydrogenphosphate guests. Receptor 272b displayed the highest affinity of the series (K(H2PO4-) = 1.4 × 104 M-1). This finding was rationalized in terms of the relatively increased preorganization of its binding pocket as the result of: 1) interactions involving the π-surfaces of the naphthyl units and 2) the more acidic character of the sulfonamide NH protons compared to the corresponding amide analogues.
Hiratani and co-workers incorporated a hydroxynaphthamide moiety in receptors 273 in order to provide additional hydrogen bonding sites as well as a signaling moeity.354 Upon addition of tetraethylammonium fluoride, TBA dihydrogenphosphate, and TBA acetate to 273a in acetonitrile, a strong increase in emission at 486 nm was observed. A red-shift in the emission maximum as well as an increase in intensity was also observed when these anions were added to a solution of 273b. However, in the case of dihydrogenphosphate the spectral changes could not be fit to a simple binding profile, precluding calculations of the association constants.
Phenylborate and anthracene moieties were included in macrocyclic 274, a system also prepared by Hiratani.355 Fluorescence titrations carried out in acetonitrile:chloroform (9:1) revealed that receptor 274 interacted with dihydrogenphosphate more strongly than with acetate, sulfate, chloride, bromide and iodide (all anions studied as their TBA salts). In this solvent mixture, the association constant was found to be on the order of 104 M-1 for dihydrogenphosphate. Proton NMR spectroscopic studies led to the conclusion that the hydroxy, amide, and ether groups all participated in hydrogen bonding interactions with the bound dihydrogenphosphate guest. On this basis, the increase in the fluorescence intensity was attributed to a Lewis acid interaction with the phenylborate that, in turn, served to overcome the otherwise inherent PET quenching of the anthracene fluorescence.
Cleft receptors based on 2,2-biimidazoles (275) with various amide groups at the 4- and 4′-positions were studied by the Allen group.356 The intrinsic fluorescence of the biimidazole unit was quenched, without an emission shift, upon addition of dihydrogenphosphate or chloride (TBA salts) in CH2Cl2. In this study, 1:1 complexes were formed with association constants on the order of 104 M-1 for both H2PO4- and Cl-. Receptor 275b proved to be the strongest binding receptor, while receptor 275f was found to be the weakest. The receptor-based differences in affinities were found to be small for dihydrogenphosphate (ca. 3-fold) but higher for chloride (ca. 100-fold). Bromide and nitrate anions produced a much smaller change. An effort was made to probe whether the syn or anti conformation of the amide groups gave rise to higher affinities. However, solution state 1H NMR spectroscopic studies proved uninformative. On the other hand, a single crystal X-ray diffraction analysis revealed that the anti conformation was the preferred conformer, at least in the solid phase.
The use of diphenylglycouril as a scaffold for the formation of concave phosphate binding clefts, such as receptors 276 and 277, was reported by Kang.357,358 While the strongest binding interactions were observed with acetate and benzoate (K values on the order of 104 M-1 in CH3CN), a binding constant of 1.6 × 103 M-1, corresponding to the interaction between receptor 276 and dihydrogenphosphate was measured by means of UV-Vis titrations carried out in acetonitrile. All anions were studied as the TBA salts. In the case of receptor 276, the binding affinities were found to correlate well with anion basicity. Receptor 277 contains a built-in fluorophore. It thus allowed anion binding to be monitored by following the substrate-induced emission quenching in acetonitrile. Using this receptor and this solvent, a variety of anions were found to be bound, although only bromide was found to bind more strongly than dihydrogenphosphate (K (H2PO4-) = 7.5 × 104 M-1). The relatively good selectivity for dihydrogenphosphate displayed by 277 was attributed largely to the size of this anion relative to others studied.
The fipronil-based receptor 278 was recently prepared by Qian and Li.359 This system was found to interact with highly basic TBA anion salts, such as acetate, fluoride and dihydrogenphosphate, through deprotonation. UV-Vis titrations allowed for the determination of what can be considered pseudo-association constants in acetonitrile, with a value of 2.27 × 105 M-1 being recorded for dihydrogenphosphate. However, the highest interaction was found with acetate.
Several macrocyclic amide systems have also been analyzed for their anion binding properties, in particular by Bowman-James and co-workers. Strong binding interactions with receptor 279a were observed for both dihydrogenphosphate and hydrogensulfate as determined by 1H NMR spectroscopic titrations carried out in CDCl3 (log K = 4.50 and 4.66, respectively), while other anions were found to bind two or three orders of magnitude less well (all anions added as the TBA salts).360 While 1:1 complexes were observed in solution for these two anions, single crystal X-ray diffraction analysis revealed 2:1 macrocycle:anion sandwich complexes. The binding affinities were attributed to efficient geometric complementarity between the amide hydrogen atoms and the anion oxygen atoms, as inferred from the solid-state structural studies.
A follow-up report detailed the anion binding ability of the pyridine and thioamide derivatives 279b-d as well as bicyclic derivatives 280.361 Proton NMR spectroscopic studies in DMSO-d6 revealed the formation of 1:1 complexes with a number of anions studied as the TBA salts. In general, the strongest binding interactions were observed for dihydrogenphosphate, hydrogensulfate, and fluoride with chloride, bromide, iodide, nitrate, and perchlorate being bound significantly less well if at all. Thioamide monocycles 279c-d were found to bind anions more strongly than the respective amide derivatives 279a-b. However, the opposite trend was observed for bicyclic receptors 280. Here, weaker binding affinities were observed for the thioamide derivative 280b relative to the amide derivative 280a. Such differences were attributed to increased intramolecular hydrogen bonding interactions between the thioamide groups on different arms of the receptor, thus leading to a more compact cavity. In support of this latter conclusion was the finding that fluoride was bound by receptors 280 with high selectivity over other anions.362 The highest measured phosphate affinity in these studies was seen for 279c and dihydrogenphosphate (log K = 4.97). Macrocycle 279c was also found to display significant selectivity for dihydrogenphosphate over hydrogensulfate (log K (HSO4-) = 3.15).
Separate analysis revealed that the corresponding quaternary ammonium receptors 281a and 281b displayed, in general, higher anion affinities than receptors 279. This finding was ascribed to the combination of hydrogen bond and electrostatic interactions available with the former macrocycles.363 Indeed, 1H NMR spectroscopic titrations performed in DMSO-d6 revealed that receptors 281a and 281b interacted well with dihydrogenphosphate (as the TBA salt), with log K = 4.06 and 5.32 for these two systems, respectively.
Optimization of cryptand-based binding was pursued through expansion of the core of receptor 280a producing receptor 282 and quaternization of the ammonium centers giving receptor 283.364 Single crystal X-ray diffraction analyses revealed a capsule shape for receptor 282 but a bowl conformation for receptor 283. Despite its expanded core, receptor 282 was found to be selective for fluoride, in analogy to what was seen for receptor 280a, as inferred through 1H NMR spectroscopic titrations carried out in DMSO-d6 using the TBA anion salts. Receptor 283, on the other hand, was found to be highly selective for dihydrogenphosphate under these conditions (K = 1.2 × 104 M-1). This latter receptor was found to bind dihydrogenphosphate with a four-fold selectivity relative to chloride and a 35-fold selectivity relative to hydrogensulfate. Expanded monocyclic structures (284) were also explored under these conditions.365 These analyses revealed that receptor 284b bound dihydrogenphosphate with an affinity constant of 4.4 × 103 M-1; however, this value was lower than the association constant obtained with the analogous receptor 279b. Macrocycle 284b was found to be selective for hydrogensulfate and displayed weak binding for fluoride, chloride, bromide, and acetate. Single crystal X-ray diffraction analysis of the complex of 284b and dihydrogenphosphate revealed the binding of two guest molecules in a dimeric fashion within the receptor cavity (Figure 6). In this case, each dihydrogenphosphate molecule participated in two hydrogen bonds with neighboring amide substituents as well as two hydrogen bonds with the other guest molecule. On the other hand, relatively small anion affinities were observed for N-substituted receptors 284a and 284c.
Figure 6.
View of the H2(284b):(H2PO4)2 complex. Drawing generated from X-ray diffraction data originally published in reference 365. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
Nearly contemporaneously with the studies of Bowman-James, the anion binding behavior of the amido-macrocyclic receptors represented by structure 285 was being studied by Jurczak and co-workers.366-368 This latter set of macrocycles allowed the effect of size on selectivity to be probed. It was found to be quite pronounced despite the relatively high conformational flexibility of the receptors in question. The 20-membered macrocycle (285b) was found to bind the tested anions (TBA salts of Cl-, Br-, CN-, PhCOO-, AcO-, H2PO4-, p-NO2-Ph-O-, and HSO4-), more strongly than 285a or 285d in DMSO-d6 as determined by 1H NMR spectroscopic analysis (K (285b:H2PO4-) = 7.41 × 103 M-1). It was proposed that receptor 285b embodied the best balance of flexibility and preorganization among the series. Receptor 285c was not sufficiently soluble in DMSO to allow for the determination of binding constants. Furthermore, dihydrogenphosphate selectivity was reported for receptors 285b and 285d while receptor 285a exhibited selectivity for acetate. These trends were attributed to differences in the geometric complementarity of the host:guest systems. The binding behavior of receptor 285b and the linear analogue 286 with chloride, acetate, and dihydrogenphosphate (TBA salts) was also compared. Similar selectivity but significantly lower binding affinities (ca. 20 - 40 fold) were observed. Proton NMR spectroscopy and single crystal X-ray diffraction analysis led to the suggestion that linear receptor 286 exists largely as a folded structure with significant intramolecular hydrogen bonding interactions in the absence of an anionic guest. The lower binding affinities displayed by 286 were attributed in part to the structural reorganization required for guest binding. The influence of the pyridine ring present in receptor 285b on anion binding was studied through the analysis of phenyl derivatives 287 and 288.369,370 On this basis it was determined that the bis-phenyl receptor 287 was a less efficient host. The reduced binding ability was attributed to intramolecular hydrogen bonds between the distal amide units in the free receptor. These interactions were thought to lead to a high energetic cost of reorganization upon anion binding. Hybrid receptor 288 was observed to binding anionic guests more strongly than 287 and 285b, presumably due to a more favorable conformation of the free host. Similar selectivity was reported for all three receptors (H2PO4- > PhCOO- > Cl- > Br- > HSO4-). On the basis of 1H NMR spectroscopic titrations, the binding constant corresponding to the interaction between receptor 288 and dihydrogenphosphate was estimated to be greater than 8.0 × 103 M-1 in DMSO-d6 and 2.6 × 103 M-1 in DMSO-d6 / 5% water.
The amide-based macrocyclic sensor 289 was also studied by the Jurczak group. This system, which contains a built-in chromophore, allowed for the colorimetric detection of H2PO4−, as well as F− and AcO−, in both DMSO and CH3CN solutions.371 The response was found to be both anion and solvent dependent for the aforementioned anions. In DMSO, the binding affinities followed the order of the anion basicities, such that F- ≫ AcO- > H2PO4-. However, dihydrogenphosphate was found to bind more strongly than acetate (K = 4.2 × 103 M-1 vs. 5.03 × 102 M-1, respectively) in acetonitrile. This effect was attributed to the additional hydrogen bond interaction available between the OH group of the phosphate and an ether oxygen of receptor 289. This hydrogen bond would be disrupted by the solvent to a much greater extent in DMSO than in acetonitrile. Interestingly, the three test anions, namely F-, H2PO4-, and A2O-, produced three distinct colors when added to colorless solutions of receptor 289 in both DMSO and acetonitrile. On the other hand, solutions of 289 remained clear when treated with Cl-, Br- and HSO4-. All anions were studied as the TBA salts.
In 2004, Costero and co-workers studied the anion binding of amide-containing macrocycles 290 and 291.372 While all of these receptors were found to bind fluoride anion strongly, only macrocycle 290 was found to interact appreciably with larger anions such as chloride, acetate, and dihydrogenphosphate as inferred from 1H NMR spectroscopic experiments carried out in CD3CN (all anions studied as the TBA salts). The results obtained were found to be consistent with the presence of hydrogen bonding interactions between the amide moieties near the pyridine unit and these larger anions.
He and co-workers studied the anion binding ability of hybrid amine-amide macrocycle 292 in the same year.373 Interactions of fluoride, chloride, dihydrogenphosphate, acetate, bromide, and iodide (as the TBA salts) were studied by fluorescence and 1H NMR spectroscopy in chloroform. Emission quenching was observed on the order fluoride > chloride > dihydrogenphosphate ≈ acetate while bromide and iodide did not affect the emission intensity. A similar trend was found in the binding constants determined through fluorescence titration. The dihydrogenphosphate binding affinity was measured to be 1.39 × 104 M-1. Proton NMR spectroscopic experiments were consistent with the presence of hydrogen bonding interactions with both the amine units and the amide units of these host:guest complexes.
Several recent studies have examined more complicated amido macrocycles. For example, ditopic receptors 293 and 294 were prepared by Morey and co-workers and analyzed using an indicator displacement assay.374 Naked eye anion detection was achieved via the addition of potassium picrate to a 1:1 DMSO:H2O solution of these receptors, followed by the addition of the anion salts of interest. In addition to sodium phytate, TBA salts of H2PO4-, GMP, and ADP could be visually detected in aqueous DMSO solution through a yellow-to-green color change. On the other hand, the TBA salts of Cl-, Br-, I-, NO3-, and HSO4- engendered no change in the yellow color of the solution.
Another amide macrocycle, the dithioxamide receptor 295, developed by Gupta, was used to produce a phosphate-selective electrode.375 This system, which involves incorporation of the receptor into a PVC membrane, was found to have a working detection range of 1.7 × 10-6 to 1.0 × 10-2 M at pH 8. Semi-empirical calculations supported the assumption that all four amide hydrogen atoms participate in phosphate binding.
A larger polyamide macrocycle, receptor 296, was synthesized by Cheng and co-workers. This system is noteworthy in that the closed ring contains a disulfide bridge derived from oxidize cystine groups.376 UV-Vis studies in acetonitrile provided support for the inference that this receptor binds dihydrogenphosphate, although a preference for acetate and fluoride anions was also observed (anions added as TBA salts). Due to interference from aggregation, the interactions with dihydrogenphosphate could not be assessed in a quantitative manner.
A different approach to anion detection was explored by the Watanabe group. In this work, a gold nanoparticle surface modified with amide ligands (297) was tested as a possible optical sensor for a number of different anions, added as the TBA salts.377 An increase in the plasmon absorbance band at 520 nm was observed with the addition of up to 0.5 equivalents of dihydrogenphosphate in dichloromethane. This effect was attributed to anion-induced aggregation of particle 297 as the result of hydrogen-bond interactions between the anion and interparticle amide ligands. Further addition of anions to the solution caused a decrease in absorbance, reflecting the deaggregation of the suprananoparticles composed of compound 297 and anions. Compound 297 was capable of acting as an optical sensor and detecting changes in the anion concentration at the 10-6 M level. This constitutes a three order of magnitude increase in the detection sensitivity relative to what can be obtained using the constituent, neutral amide ligands. A preference for HSO42- and H2PO42- over other anions was observed, a finding that was interpreted in terms of hydrogen-bonding interactions mediating the receptor-anion binding events and the nanoparticle aggregation phenomena.
4.2.2. Urea and Thiourea Systems
Urea and thiourea groups are widely used in the design of artificial anion receptors. These moieties provide two hydrogen bond donors within a single functionality, with the consequence that these motifs often provide for effective anion recognition. As a general rule, thiourea-based systems give rise to stronger H2PO4- binding than do those containing ureas. This can be rationalized by examining the pKa of the thiourea group. It is roughly 6-9 pKa units lower than the pKa of the urea group. This difference makes thioureas much stronger hydrogen bond donors than ureas but also makes the corresponding receptors more susceptible to deprotonation. The use of thiourea groups offers additional advantages, including generally enhanced solubility in lower polarity solvents and a lower propensity to undergo self-association. Presumably, these features reflect in part the lower hydrogen bond acceptor ability of sulfur versus oxygen.378
In 1992, the Wilcox group reported phosphate binding studies of the urea-based cleft receptor 298.379 Proton NMR spectroscopic studies supported the predicted anion-receptor hydrogen bonding interactions, while UV-Vis titrations carried out in chloroform were used to measure binding affinities. Binding constants were found to be on the order of 103 – 104 M-1, with diphenylphosphate binding less strongly than benzoate but more strongly than tosylate. Each anion was tested in the form of its TBA salt.
In 1994, Kelly and M. H. Kim reported the mono and bisurea receptors 299 and 300. These rather rigid acyclic systems were screened for their ability to bind a variety of TBA phosphate salts under different solvent conditions by means of 1H NMR spectroscopy.380 As expected, the binding constants recorded in DMSO-d6 proved significantly lower than those obtained in CDCl3. It was found that the selectivity correlated well with basicity, such that, in the case of 299, phenylphosphonate and phenylphosphate bound more strongly than phenylsulfonate but less well than benzoate. Similar results were observed with ditopic anions, such as phenyldiphosphonate and receptor 300. Here, however, much higher association constants were observed than would be expected given the increased charge of the anionic guests. For instance, receptor 300 was found to bind phenyldiphosphonate with an association constant of 3.4 × 105 M-1 in DMSO-d6 vs. 140 M-1 for phenylphosphonate.
The Alcázar group examined both tris(2-aminoethyl)amine and 1,3,5-tris(aminomethyl)cyclohexane as spacers for the construction of tripodal urea receptors. Using these building blocks, they produced systems represented by structures 301 and 302.381 An association constant of 1.1 × 104 M-1 was measured for tris(tetramethylammonium) phosphate with the more flexible receptor 301a as determined by 1H NMR spectroscopic titrations carried out in DMSO-d6. Studies with system 302, a receptor containing a more rigid spacer, resulted in a decreased association constant. This result was attributed to the cavity size being too large for this particular anion. Interestingly, the thiourea analogue of 301a (receptor 301b) also displayed a lower association constant for trianionic phosphate than did 301a, with the difference being roughly one order of magnitude. Finally, an association constant of 1.0 × 102 M-1 was measured for the water-soluble analogue 301c under conditions where trianionic phosphate was added to water. However, no pH or buffer conditions were reported for these latter experiments. Additional binding functionality was incorporated through the use of chromane groups. The resulting receptor, 303, was found to bind tris(tetramethylammonium) phosphate roughly five-fold more effectively than receptor 301a. It is important to note the use of trianionic phosphate as a guest in these studies, which is expected to give rise to binding affinities that are substantially enhanced relative to what would be expected for dihydrogenphosphate based on simple electrostatic considerations.
The pentafluorophenyl substituted receptor 301d, synthesized by Ghosh and co-workers,382,383 provided support for the intuitively reasonable supposition that the presence of the electron withdrawing groups on the phenyl rings would lead to higher anion binding affinities. In fact, receptor 301d was found to display a preference for dihydrogenphosphate (studied as the TBA salt), binding this substrate with a log K = 5.52 in DMSO-d6 as inferred from 1H NMR spectroscopic studies. A single crystal X-ray diffraction analysis revealed that dihydrogenphosphate was encapsulated within the C3v symmetrical cavity of receptor 301d, being held in place by a combination of seven hydrogen bonds and what were taken to be two additional intermolecular anion-π interactions.
Ganguly and Das recently reported receptor 301e that contains nitrobenzene as a built-in chromophoric subunit.384 In an acetonitrile/water mixture (95/5, v/v) this receptor was found to bind the dianionic sulfate anion in preference over other anions; however, strong interactions were observed with other tetrahedral anions including dihydrogenphosphate (log K = 4.26), as inferred from UV-Vis spectroscopic titrations. All anions were studied as the TBA salts, added as commercially available aqueous solutions.
Another tris (2-aminoethyl)amine-derived compound, the fluorescent tripodal urea receptor 304, was designed and tested by the Wu group.385 In this case, a significant enhancement in the fluorescence intensity was observed upon the addition of TBA anion salts in DMF, with the derived association constant being 1.1 × 104 M-1 for dihydrogenphosphate. Hydrogensulfate was found to bind significantly more weakly, and bromide and iodide anions displayed no spectral change. Based on control studies, the authors suggested that the spectral response and the strong binding affinity resulted from protonation of the central tertiary amine, an event that would lead to a reduction in the PET quenching and a strong charge-charge association within the resultant complex (304-H2PO42-).
A more rigid, tribenzyltrindane C3V -symmetric scaffold (305) was later reported by the Choi group.386 This scaffold allowed for the preorganization of all three urea arms on the same side of the receptor. The binding properties were analyzed via 1H NMR spectroscopy in DMSO-d6 using various TBA anion salts. On this basis, it was determined that receptor 305 binds dihydrogenphosphate with an association constant of 5.21 × 102 M-1. Other anions (nitrate, hydrogensulfate, chloride, bromide and carbonate) were found to bind with much lower affinities.
In 1997, the Umezawa group studied the anion binding ability of fluorescent urea receptor 306 through its incorporation into a PVC membrane that was immobilized on a glass slide.387 Presumably as the result of the appended cytosine moiety, this receptor was found to display a preference for guanosine derivatives over other nucleotides. Further, this system was found to function as an efficient fluorescent sensor for 5′-GTP, displaying specificity over 5′-ATP in an aqueous buffer (10 mM tris/ HCl at pH= 7.4). Receptor 306 was also incorporated into a monolayer for channel mimetic sensing.388 In this case, selective binding of 5′-GMP over 5′-AMP was manifested in terms of a reduced permeability of the monolayer to [Fe(CN)6]4- when it was analyzed by CV in the presence and absence of the anions. Interestingly, systems based on receptors containing a urea moiety were found to display an increased selectivity when compared to monolayers based on cytosine derivatives. Glassy electrodes based on receptor 306 were found to be unstable.
Reinhoudt and co-workers developed a series of multidentate urea receptors (307 – 308), which allowed them to probe the effect of substituents as well as the effect of ring closure on the anion affinities.389 All anion interactions were studied in DMSO-d6 by means of 1H NMR spectroscopic titrations using the corresponding TBA anion salts. Each receptor displayed significant spectral shifts with H2PO4- but little to no change in the presence of Cl-, Br-, NO3-, or HSO4-. While association constants could not be determined with receptors 307a or 307c due to what were considered to be indeterminate binding stoichiometries, 2:1 guest:host binding was observed for 307b and 307d with association constants near 107 M-2. In this case, the thiourea moiety of 307d did not improve binding for dihydrogenphosphate relative to 307b. On the other hand, its presence did lead to a modest affinity for the chloride anion. In contrast, systems 308, macroyclic analogues of 307, were found to bind H2PO4- with a 1:1 binding stoichiometry and with affinities near 103 M-1. This latter system displayed a low binding affinity for chloride. In fact, receptor 308b displayed a greater than 100-fold selectivity for dihydrogenphosphate over chloride. On the basis of NMR spectroscopic experiments, the relatively low affinity for chloride displayed by these receptors was rationalized as being due to the fact that only the outside urea moieties can participate in chloride binding, whereas all four urea moieties can participate in dihydrogenphosphate binding. Certainly, it appears safe to conclude that the number and spatial organization of the hydrogen bonding units within receptors 307 and 308 play a critical role in determining their selectivity.
Gale and co-workers later explored the anion binding ability of the internal bisphenylurea binding unit of receptors 307 and 308 (309a) as well as the related cleft receptors 310 and 311.390 The anion binding ability of these receptors was explored using 1H NMR spectroscopy in DMSO-d6 / 0.5% water with all anions added as the TBA salts (Cl-, Br-, AcO-, PhCOO-, H2PO4-, and HSO4-). Receptor 309a displayed a significantly lower binding affinity for dihydrogenphosphate (K = 7.32 × 102 M-1) than the larger systems 307 and 308, although the use of slightly different solvent systems must also be considered. This receptor further displayed significant selectivity for acetate and benzoate over dihydrogenphosphate. Receptor 310, containing only two hydrogen bond donor sites, produced the lowest binding affinities, while experiments with pyrrole-based receptor 311 led to intermediate values. The decreased binding of receptor 311 relative to receptor 309a was attributed to the formation of a smaller more closed binding cleft in the former receptor. Receptor 311 was also found to bind dihydrogenphosphate in DMSO-d6 as inferred from 1H NMR spectroscopic studies carried out by Cheng and co-workers.391 Further studies by Gale and co-workers focused on probing the effect of electron withdrawing substituents in receptors 309b-e.392 Within this series, receptor 309b displayed the highest binding affinities, with the largest increase being observed for the binding of dihydrogenphosphate (K = 4.72 × 103 M-1). The strong binding of receptor 309b relative to the other receptors in the series was attributed to increased intramolecular hydrogen bonding interactions between the carbonyl oxygen atoms and the nearby hydrogen atoms of the central phenyl ring that served to preorganize the binding cleft. It was also proposed that the ortho-nitro substituents of receptors 309d and 309e led to reduced binding through steric interactions within the binding site. The low binding of thiourea derivative 309f was also attributed to unfavorable steric interactions involving the relatively large sulfur atom and the central phenyl ring. This interaction was expected to distort the binding cleft. The incorporation of an additional hydrogen bond donor unit (as in receptors 312 and 313) also served to increase the anion binding ability relative to receptor 309a, particularly the ortho-substituted receptor 313. Receptor 313 displayed a 1:2 host:guest binding stoichiometry with dihydrogenphosphate and a high overall binding constant (1.85 × 105 M-2), presumably due to the interactions of this anion with the additional hydrogen bonding sites. These researchers also incorporated an ortho-phenylenediamine unit into a macrocyclic system (314).393 However, this receptor displayed a low binding affinity for dihydrogenphosphate (K = 1.42 × 102 M-1). Chemical shift analysis led to the conclusion that this anion only interacted with the amide hydrogen atoms closest to the pyridine ring. On the other hand, this macrocycle was found to bind the acetate anion well.
Schneider and co-workers examined the mono- and tri-dentate urea receptors 315-317. These systems incorporate aromatic units and were designed to permit the binding of nucleotide monophosphates.334 The relevant interactions were studied using 1H NMR spectroscopy (DMSO-d6) and fluorescence emission spectroscopy (H2O, unbuffered). System 315a, with a direct link between the urea and aromatic moiety, proved to be the strongest binder. In this case, 1:1 association constants on the order of 104 M-1 were recorded in DMSO-d6 for AMP, CMP, GMP and TMP (add as sodium salts). The corresponding association constants for receptor 315b could not be determined. Receptors 316 and 317 were designed to act as improved receptors. These compounds contain additional aromatic units, which were expected to improve the π-surface interactions. While receptor 317 displayed weak interactions, the monodentate receptors 316a and 316b were indeed found to bind various nucleotides well. The resulting association constants were approximately 103 M-1 in H2O, with lower affinities being noted in DMSO-d6. Interestingly, the free energies of association recorded in the case of unsubstituted pyrene were found to be only 2 – 5 kJ lower than the free energies of association with receptors 316a and 316b. This led to the suggestion that the contribution from the urea unit was small, possibly due to steric restraints that precluded the full benefit of concurrent full chelation and π-surface interactions.
A number of chromophores have been appended to urea receptors in order to facilitate optical signaling. For example, Jeon, Nam, and co-workers appended urea units to an anthraquinone unit in receptor 318.394 All three receptors displayed a chromogenic shift in the absorption spectrum upon the addition of TBA dihydrogenphosphate in DMSO. The strong color shift (ca. 100 nm) observed upon the addition of dihydrogenphosphate and acetate to receptor 318a was determined to reflect abstraction of the phenolic hydrogen atoms. However, the UV-Vis spectrum of receptors 318b and 318c was found to undergo a 30 nm shift upon the addition of dihydrogenphosphate but not acetate, chloride, or hydrogensulfate. The selectivity for dihydrogenphosphate was attributed to the formation of four hydrogen bonding interactions with the urea NH units. Binding constants were measured via 1H NMR spectroscopic titrations carried out in DMSO-d6. All three receptors were found to display similar affinities for dihydrogenphosphate (K ≈ 1 × 104 M-1), and lower affinities for the other tested anions. Significant changes in the chemical shifts of these receptors upon the addition of dihydrogenphosphate were only observed for the NH units closest to the anthraquinone unit, leading to the conclusion that these hydrogen bond donors were the most critical for binding. Less dramatic shifts were reported for the addition of acetate.
Receptor 319, recently developed by Fabbrizzi and co-workers,395 contains 4-nitrophenyl substituents. This design feature allowed its use as a colorimetric anion sensor. The electron withdrawing nitro groups also serve to enhance the acidity of the NH groups. UV-Vis titrations, carried out using a variety of TBA anion salts in acetonitrile, demonstrated that receptor 319 exhibits a preference for oxoanions, with a log K for dihydrogenphosphate of 5.37. The binding affinity was found to reflect the basicity of the anions in question (i.e., log K falling in the order AcO- > C6H5COO- > H2PO4- > NO2- > HSO4- > NO3-). The urea linker was also functionalized via the attachment of two naphthalimide groups. In this case, the resulting receptor, 320, proved effective for the detection of carboxylic acids, as determined through colorimetric and fluorescent experiments. Receptor 320 was also found to form a supramolecular adduct with H2PO4- at low concentrations. However, the addition of excess anion resulted in deprotonation. Deprotonation was also observed for fluoride and acetate anions. Fabbrizzi and co-workers later developed the chiral diurea receptor 321 based on a bridging (R,R)-cyclohexane-1,2-diamine subunit.396 Receptor 321 was studied via UV-Vis titrations carried out in DMSO. These analyses revealed a 1:2 (host:guest) binding stoichiometry. The formation of this 1:2 adduct was attributed to the presence of an extra hydrogen bond between both anions and an unexpected cooperative effect. The first and second log K values for dihydrogenphosphate were determined to be 2.96 and 3.46, respectively. The binding interaction with pyrophosphate was characterized by a log K of 4.63. Incorporation of a nitrobenzofurazan moiety (322) led to the production of more dramatic changes in the absorption spectrum upon anion addition in acetonitrile.397 In analogy to what was seen for other systems, a hydrogen bonding complex was observed at less than one equivalent of anion, and further additions led to deprotonation of the receptor. A log K value of 5.40 was calculated for the complex with dihydrogenphosphate on the basis of UV-Vis titration studies. Binding selectivities correlated with anion basicity in this case.
Gunnlaugsson and co-workers have also combined urea and naphthalimide moieties for anion recognition (producing, e.g., receptors 323).398 The interactions of acetate, dihydrogenphosphate, and fluoride with receptors 323 were followed by 1H NMR, UV-Vis, and fluorescence spectroscopy in DMSO. These spectral changes were attributed to a combination of hydrogen bonding interactions and deprotonation. No changes to the spectra of 323 were observed upon the addition of chloride or bromide. The UV-Vis titration data with acetate and dihydrogenphosphate could generally be fit to a 1:1 binding equation. A log K of ca. 3 was reported for the complexation of dihydrogenphosphate by receptors 323a and 323c; however, receptor 323b displayed more complex binding behavior with dihydrogenphosphate and could not be fit to a 1:1 model. All studies with fluoride were consistent with full deprotonation of the receptor. Importantly, a strong green-to-purple color change was observed upon the addition of acetate and dihydrogenphosphate to these receptors. All anions were studied as their TBA salts.
These researchers were also able to achieve anion sensing by combining urea binding subunits with anthracene fluorophores (giving receptors 324).399 Significant fluorescence quenching was observed upon the addition of acetate, dihydrogenphosphate, and fluoride anions as their TBA salts in DMSO. The reduced emission intensity was attributed to an increase in PET quenching of the anthracene unit by the urea nitrogen atom upon anion complexation. Similar binding constants were measured for interactions of all receptors with dihydrogenphosphate and acetate, with log K values being ca. 2; however, acetate exhibited more significant quenching. Fluoride was found to effect the largest quenching and to display the highest binding constants. The addition of chloride and bromide anions produced little to no effect on the fluorescence spectra of these receptors.
J. Yoon, J. S. Kim, and co-workers studied the anion binding of phenylurea-anthracene receptors 325 - 326.400 These receptors were found to be highly selective for fluoride and hydrogenpyrophosphate over dihydrogenphosphate, chloride, bromide, iodide, hydrogensulfate, and acetate in DMSO. The interactions of TBA anion salts with receptors 325a and 326a were followed through UV-Vis spectroscopy while the binding behavior of receptors 325b and 326b were followed through fluorescence spectroscopy. Using the latter method, binding constants of 6.0 × 103 M-1 and 2.6 × 103 M-1 were determined for the interaction of hydrogenpyrophosphate with receptors 325b and 326b, respectively. Interestingly, 1H NMR spectroscopic studies of receptors 325a and 325b in DMSO-d6 led to the conclusion that, in addition to the urea units, the 9-H atom of anthracene played a role in mediating the anion binding process.
A hybrid acridone - urea fluorescent sensing system (327) was reported by Gale and co-workers in 2007.401 Dramatic quenching of the receptor emission was observed upon the addition dihydrogenphosphate (TBA salt) in an acetonitrile:DMSO solution (94:6). Addition of chloride led to a much smaller decrease in emission intensity, and the addition of acetate and benzoate produced a slight increase in intensity. The emission enhancement observed with the latter anions was attributed to deprotonation of the receptor. The binding of dihydrogenphosphate was probed by 1H NMR spectroscopy in DMSO-d6 / 0.5% water. Interestingly, a combination of supramolecular complexation, deprotonation, and receptor tautomerization was inferred from these studies.
The use of a small series of urea-functionalized carbazole receptors (328) allowed for the differential recognition of several anions.402 These systems, reported by K. S. Kim and co-workers, combined the hydrogen bonding moieties of the carbazole and the urea units within one receptor. These receptors allowed the optical properties of a carbazole motif to be complemented by those of various substituents. In the case of receptor 328a, this combination produced an effective colorimetric receptor that displayed visible color changes in the presence of several TBA anion salts in 9:1 CH3CN:DMSO. The actual color change was found to depend on the identity of the anion. For example, dihydrogenphosphate caused a color shift from yellow to orange, while hydrogenpyrophosphate caused a color change from yellow to red. More dramatic changes were observed with the hydroxide and fluoride anions. The anion-based differences could also be monitored using UV-Vis and fluorescence spectroscopy. While binding constants were not reported, two-dimensional plots of the absorbance wavelength change vs. fluorescence intensity change allowed for the differentiation of most anions tested, including dihydrogenphosphate and pyrophosphate.
Gale and co-workers recently reported the anion binding properties of the monourea carbazole receptors 329 - 331.403 These receptors display a strong selectivity for acetate and bicarbonate. However, they also display a moderate affinity for dihydrogenphosphate as inferred through 1H NMR spectroscopy (DMSO-d6 / 0.5% water). All anions were studied as the TBA salts. Titrations of dihydrogenphosphate with receptors 329 and 330 did not allow for the determination of binding constants; however, a 1:1 binding constant was determined for receptor 331 (K = 6.14 × 103 M-1). Chloride was found to be weakly bound by these receptors.
Urea moieties have also been combined with a benzoxazole reporter unit, as embodied in receptor 332.404 It was proposed that in the case of this latter receptor, anion binding would disrupt the internal hydrogen bonding between the oxazole nitrogen atom and the proximal urea N-H. Fluorescence experiments carried out in DMSO revealed that while acetate followed the proposed model, fluoride led to deprotonation and the less basic dihydrogenphosphate participated in hydrogen bonding interactions without disrupting the internal hydrogen bonding of the receptor. All anions were studied as their TBA salts. Due to the presence of three different mechanisms, the additions of fluoride, acetate, and dihydrogenphosphate produced distinct signals and could thus be differentiated by receptor 332. No spectral changes were observed upon the addition of chloride, bromide, iodide, and hydrogensulfate. A more acidic derivative, sulfonamide receptor 333, was found to be deprotonated by fluoride, acetate, and dihydrogenphosphate in DMSO.
The liquid-liquid extraction ability of urea functionalized dendrimers (334) was reported by Stephan, Johannsen, Vögtle, and co-workers in 1999.405 Both second generation (G2) and third generation (G3) dendrimers functionalized with hexyl, octyl, or dodecyl alkyl chains were studied. Extraction of AMP, ADP, and ATP from aqueous solutions at pH 5.4 (2-(N-morpholino)ethanesulfonic acid (MES) buffer) and 7.4 (HEPES buffer) into chloroform was observed with second generation dendrimers of type 334a and 334c as well as third generation dendrimers of type 334b and 334c. Extraction efficiency was found to correlate with the charge of the guest (ATP > ADP > AMP) and the generation of the dendrimer (G3 > G2). In addition, dendrimers with shorter alkyl chains (hexyl and octyl) at the periphery extracted anions more effectively than dodecyl-functionalized dendrimers. As a result, the highest degree of extraction was observed for dendrimer 334b-G3. Mass spectrometry studies supported the formation of a 1:5 334b-G3:ATP complex.
Thioureas have also played an important role in anion recognition. In 1995, the interaction of TBA dihydrogenphosphate with a series of bis-urea and thiourea receptors (335 and 336) was studied by Umezawa. This study was motivated by a desire to compare the two functionalities, as well as the effect of the size of the appended hydrocarbon group. With such goals in mind, the binding behavior of 335a and 335b were first compared.378 Both 1:1 and 2:2 host:guest complexes were observed at high concentrations of dihydrogenphosphate in DMSO-d6 by 1H NMR spectroscopy. Significantly stronger binding was observed for the thiourea congener 335b (K = 820 M-1 versus 110 M-1 for 335a). These dihydrogenphosphate binding constants were nearly two-fold higher than those observed for acetate. NMR spectroscopic studies supported the conclusion that all four N-H groups in the receptor participate in hydrogen bonding interactions involving the bound phosphate. Anions Cl-, HSO4-, NO3- and ClO4- did not show significant binding interactions. Several other trends were analyzed through comparisons of 335b-d and the 336 series.406 All association constants were measured in DMSO-d6 via 1H NMR spectroscopy.
A first comparison can be made between 335b and 335c, wherein replacement of the butyl substituent with a phenyl substituent serves to increase the association constant by more than five-fold (K = 8.20 × 102 M-1 and 4.60 × 103 M-1, respectively). This effect was explained in terms of the electron-withdrawing nature of the phenyl groups, which increases the acidity of the thiourea. The naphthyl group in host 335d was not nearly as beneficial, likely due to steric interactions with the phosphate guest. Interestingly, the use of a xanthene spacer in 336a and 336b greatly increased the binding affinity toward dihydrogenphosphate (5.5 × 104 M-1 and 1.95 × 105 M-1, respectively). This increase in binding affinity was attributed to the enhanced rigidity of the spacer. High selectivities for dihydrogenphosphate were also observed in this series, with binding constants for dihydrogenphosphate being two-fold higher than those for acetate in most cases. Ion channel-mimetic sensors based on receptor 336a were then constructed. These displayed the largest relative changes for dibasic phosphate as compared to other common anions, while ion-selective electrodes based on the same receptor gave responses in line with the Hofmeister series.407
Teramae and co-workers later illustrated the use of receptor 336a in an amperometric ion selective electrode in which several anions, including dihydrogenphosphate, could be distinguished.408 Receptor 335e, containing nitrophenyl substituents to enhance acidity, was found to transport dihydrogenphosphate in a 1:2 host:guest stoichiometry across a nitrobenzene/water interface. The increased binding stoichiometry provided greater overall transport efficiency than that observed with receptor 336a, although the rates of transport were similar. Receptor 335e also displayed a high selectivity for dihydrogenphosphate over chloride. Ion transfer polarography studies of receptor 335e confirmed the transportation of dihydrogenphosphate across the nitrobenzene-water interface in the form of a 2:1 adduct.409
Macrocyclic derivatives of receptors 335 were investigated by Hong and co-workers in 2000.410 Receptor 337a was reported to be selective for dihydrogenphosphate over acetate and chloride (TBA salts) as inferred from 1H NMR spectroscopic experiments carried out in DMSO-d6. This selectivity was in agreement with that observed for acyclic derivatives 335. In contrast, receptor 337b was found to bind anions in order of basicity (i.e. acetate > dihydrogenphosphate > chloride). Binding constants determined in the case of receptor 337b were two-fold higher for dihydrogenphosphate and 15-fold higher for acetate then those reported with receptor 337a. This increase in binding affinity was attributed to increased preorganization of the macrocyclic core in the case of the ethyl substituents.
Thiourea based receptors with alkyl, pyrene, and crown ether moieties (338 - 341) were developed by the Teramae group. In preliminary studies, 338 (n = 1) was shown to bind dihydrogenphosphate and acetate with log K values of 4.3 and 5.7, respectively, in acetonitrile as determined via UV-Vis titrations with the corresponding TBA salts.411 The electron-withdrawing p-nitrophenyl group attached to the thiourea moiety was thought to provide an effective intramolecular charge transfer conjugate upon anion binding, a feature that was expected to result in an anion-induced color change. In a subsequent study, receptor 338 (n = 1-8) was mixed with a detergent to create vesicles.412 While association constants were not determined, the anion selectivities of this series of receptors were found to be highly dependent upon the length of the alkyl chains when studied under these conditions. However, none of these vesicle systems displayed a preference for dihydrogenphosphate.
Fluorescent pyrene receptors were also found to be effective. For example, the directly conjugated receptor 339 displayed a significant emission increase upon the addition of TBA acetate and TBA dihydrogenphosphate in acetonitrile. A binding constant of 8.70 × 102 M-1 was determined for the dihydrogenphosphate complex, but a strong preference for acetate was also reported.413 Receptor 340 was designed to contain a spacer between the recognition and the reporter subunits. This sensor displayed a quenching of the pyrene monomer emission followed by intramolecular exciplex formation between the thiourea and the pyrene in the presence of anions.95 The quenching was attributed to the increased electron donating ability of the thiourea when complexed to an anion. Based on the magnitude of this effect, it was concluded that receptor 340 binds acetate more effectively than either H2PO4- or Cl-. A binding constant of 5.2 × 103 M-1 was reported for the dihydrogenphosphate complex in acetone. However, the use of different solvents in these studies precludes a direct comparison between the receptors.
Studies of the putative ditopic receptor 341 revealed a decreased binding affinity for TBA dihydrogenphosphate in acetonitrile relative to phenyl thiourea. Nevertheless, the affinity for dihydrogenphosphate proved higher than that of the other anions tested.414 However, attempts to add sodium cation in the form of (NaBPh4) in an effort to increase the anion affinity through concurrent cation and anion complexation were stymied due to precipitation. Further studies compared the anion binding ability of receptors 336a, 338, and 341 at a liquid-liquid dichloroethane-water interface through the use of interfacial tension measurements.415 While receptors 336a and 338 did not show any activity, crown ether conjugate 341 displayed selectivity for sodium dihydrogenphosphate under these conditions. No changes in the tension were observed in the presence of CH3COO-, Cl-, Br-, or ClO4-. It is to be noted that the interaction was limited to the liquid-liquid interface as receptor 341 did not extract the Na+:H2PO4- ion pair from aqueous solution into the organic phase.
The synthetic addition of a thiouronium group and the removal of the crown ether moiety (leading to receptor 342) served to improve the binding of dihydrogenphosphate at the DCE-water interface, without leading to a reduction in the phosphate selectivity observed in the case of receptor 341.416 Based on control experiments, the authors suggested that both the thiourea and thiouronium arms contributed to the strong binding observed in the case of 342.
The C3-symmetric optically-active receptor 343 was prepared by the Wu group.417 A UV spectral response was observed for both TBA dihydrogenphosphate and TBA sulfate, whereas no change was observed with bromide, iodide, acetate or trifluoroacetic acid (all as the TBA salts). Among the two tetrahedral anions studied, receptor 343 displayed a 1:1 binding affinity for dihydrogenphosphate that was two orders of magnitude higher than that for sulfate in DMF (K = 1.1 × 105 M-1). Proton NMR spectroscopic analyses led the authors to conclude that the dihydrogenphosphate guest was tightly bound within the receptor cavity via hydrogen bonds. The observed binding affinity was attributed to the availability of six hydrogen bonding sites as well as the three-dimensional and flexible nature of the cavity.
Tobe and co-workers studied the influence of macrocyclic preorganization in a series of metacyclophane-based thiourea receptors, namely 344-347.418,419 Proton NMR spectroscopic titrations carried out in DMSO-d6 using various TBA anion salts revealed that these receptors bound H2PO4- in preference over other anionic species with the selectivity order being H2PO4- > CH3COO- > Cl- > HSO4- > Br-. In all cases, the binding stoichiometry was 1:1. These cyclic receptors displayed higher stability constants than the analogous acyclic compound 335b in this solvent system. For example, cycle 346 was found to bind dihydrogenphosphate with an association constant of 1.2 × 104 M-1, while receptor 335b displayed an association constant of 5.2 × 102 M-1. The strongest interactions were observed for the lariat-type receptor 347, for which the association constant proved too high to determine using standard 1H NMR spectroscopic methods.
Herges, König, and co-workers later reported the anion binding properties of a thiourea macrocycle containing ether linkages (348).420 Proton NMR spectroscopic experiments carried out in DMSO-d6 led to the suggestion that this macrocycle bound a number of anions (added as the TBA salts) with varying stoichiometries. Complexes of 1:1 host:guest stoichiometry were indicated for nitrate, acetate, cyanide, bromide, iodide, and hydrogensulfate, while 2:3, 2:1, and 1:2 complexes were found for sulfate, dihydrogenphosphate, and chloride, respectively. The overall binding constant for binding to dihydrogenphosphate was determined to be 5.3 × 104 M-2.
Tripodal thiourea fluorescence sensors 349 were reported by the Suzuki group in 2001.421 The pyrene substituted receptor 349a was found to display a fluorescence emission band that was shifted to longer wavelength upon the addition of anions in acetonitrile. The anthrylmethyl substituted receptor 349b, however, displayed a fluorescence signal whose intensity was quenched upon the addition of anions. The binding affinities for receptor 349a were generally greater than those of 349b. Presumably, this reflects the increased acidity of the thiourea groups that results from direct conjugation to the fluorophore. Both receptors displayed a slight selectivity for dihydrogenphosphate over acetate in acetonitrile, with binding constants of 3.7 × 105 M-1 and 1.9 × 104 M-1 being obtained for receptors 349a and 349b, respectively. In both cases, the relative change in emission intensity was found to be H2PO4-> CH3COO- > Cl- ≈ ClO4-. In control studies involving the constituent monomeric units, a selectivity for acetate over dihydrogenphosphate was observed. This latter result provided confirmation that geometry matching between the host and guest confers anion binding selectivity and enhances sensitivity in the case of receptors 349. All anions were studied as the TBA salts.
A number of thiourea receptors were studied by the Gunnlaugsson group beginning in 2001. In preliminary studies, the monothiourea receptors 350 were studied by monitoring the change in fluorescence seen upon the addition of TBA anion salts in DMSO.422,423 The addition of dihydrogenphosphate, acetate, and fluoride resulted in quenching of the anthracene-derived fluorescence intensity. The corresponding log K values were 3.35, 2.55, and 2.05 for the three anions, respectively, in the case of receptor 350a. No change was observed upon the addition of chloride or bromide to either 350 or 351 under these same conditions. The association constants were higher for 350a than those observed for receptor 350b, presumably due to an increase in acidity of the thiourea groups in the former due to the electron-withdrawing trifluoromethyl group. Consistent with this same trend, receptor 350c displayed the lowest overall binding affinities. Interestingly, however, this latter system displayed a reversed selectivity for dihydrogenphosphate over acetate. Photochemical studies of the urea version of 350b were carried out by the Arai group.424 The addition of phosphate to this latter receptor resulted in fluorescence quenching and a significant decrease in the fluorescent lifetime of the receptor. No binding constants were reported.
Changes in the fluorescence spectra of the bis-thiourea receptors 351a and 351b upon addition of monoanion TBA salts in DMSO were also analyzed by the Gunnlaugsson group.425,426 Dihydrogenphosphate, acetate, and fluoride all led to quenching of the fluorescence intensity. However, no signal modulation was observed upon the addition of chloride, bromide, or perchlorate. Both fluorescence and NMR spectroscopic data supported the formation of 1:2 receptor:anion complexes in the case of these monodentate anions. Even greater changes were observed upon the addition of bis-anions such as pyrophosphate, which bound to receptors 351a, 351b, and 351c with log K values of 3.40, 3.07, and 2.72, respectively (producing 1:1 complexes in all cases). Similar association constants were observed for bis-carboxylates. The differences in pyrophosphate binding affinity reflected the expected acidities of the hydrogen bonding donor units, as seen in other receptor systems and noted earlier in this review. Proton NMR spectroscopic studies and the absence of excimer/exciplex emission provided support for the formation of 1:1 bridging complexes in the case of pyrophosphate. The reduction in emission intensity seen in the case of all of these systems was attributed to an enforcement of PET quenching of the anthracene fluorescence by the thiourea/urea groups induced by anion binding.
Later, Gunnlaugsson, Pfeffer, and co-workers developed the series of naphthalimide-containing receptors 352 as fluorescent thiourea sensors for anion recognition.427 The naphthalimide moiety in these systems emits in the green spectral region and does so with a high quantum yield. However, the intensity of this signal was quenched in the presence of anions. This effect was attributed to an increase in electron transfer from the thiourea unit to the fluorophore as the result of anion binding. While chloride and bromide did not produce a change in the emission features, fluoride, acetate and dihydrogenphosphate were all found to quench the fluorophore. Dihydrogenphosphate displayed the lowest association constants among these three anions, with log K values of 2.9 and 3.7 being reported for 352a and 352b, respectively, in DMSO when the anions were added as the TBA salts. The higher association constant for receptor 352b was attributed to an increase in the acidity of the thiourea brought about as the result of the electron-withdrawing nature of the trifluoromethyl substituent. Further analysis of receptors 352b and 352c revealed strong colorimetric changes upon the addition of TBA anions in both DMSO and protic media.428 Naked-eye detection of dihydrogenphosphate in protic media was observed as a yellow-to-purple color change.
These same researchers later investigated receptor 353 with the expectation that the N-H unit of the naphthalimide moiety would be able to participate in hydrogen bonding as a result of the increased flexibility of the aliphatic unit compared to the phenyl spacer of 352a.429 Proton NMR spectroscopic analyses proved consistent with the participation of this third N-H proton in the binding of dihydrogenphosphate (TBA salt) in DMSO-d6. The presumed additional interaction was also supported by molecular modeling studies and the measurement of an increased binding constant (log K (353:H2PO4-) = 3.4) relative to that of the analogous receptor 352a. In studies of other anions, chloride and bromide anions did not significantly change the 1H NMR spectrum. Acetate displayed a similar association constant but interacted with only the thiourea protons (i.e., not the naphthalimide N-H), whereas the addition of fluoride resulted in deprotonation. However, no changes in the emission spectrum were observed upon the addition of anions to receptor 353 in DMSO, presumably due to the fact that the increased flexibility disrupted the fluorescence signaling mechanism observed in receptors 352.
In a subsequent design, an ortho-substituted phenyl linker was incorporated into the framework in order to organize better the anion binding site. This led to receptor 354.430 Proton NMR spectroscopic analysis in DMSO-d6 led to the conclusion that with this new system dihydrogenphosphate (TBA salt) was again bound to all three N-H atoms. Significantly stronger binding was observed for dihydrogenphosphate (log K ≈ 4) as compared to receptor 353, and a selectivity for dihydrogenphosphate over acetate was achieved. Acetate was observed to bind to only the thiourea protons, in analogy to what was found for receptor 353. The addition of both anions led to a decrease in the emission intensity of receptor 354, although a larger quenching effect was observed for acetate.
In 2008, Gunnlaugsson and co-workers reported the colorimetric anion sensing ability of amidothiourea receptor 355.431 This receptor was found to respond to acetate, fluoride, dihydrogenphosphate, hydrogenpyrophosphate, AMP, and ADP as inferred from UV-Vis experiments conducted in both DMSO and DMSO:H2O (4:1) solutions, whether the anions were present as their TBA or sodium salts (in the latter solvent system). These binding events were also reflected in a transition from colorless to yellow colored solutions. No alteration in the absorption spectrum was observed upon the addition of chloride, bromide, or ATP. UV-Vis titration data was fit to multiple binding models and led to the conclusion that dihydrogenphosphate (TBA salt) was bound in a 1:2 host:guest ratio with a log K value of 4.06. Pyrophosphate was found to form a 1:1 complex with a log K value of 6.53. Proton NMR spectroscopic titrations were consistent with a binding mode that involved both hydrogen bonding and deprotonation of the receptor.
The anion recognition properties of thiourea receptors 356 were investigated by Kruger and Gunnlaugsson in 2009.432 All three stereoisomers of 356 were investigated for their ability to bind acetate, dihydrogenphosphate, and fluoride (as the TBA salts) through a number of spectroscopic techniques. Binding constants were measured through 1H NMR spectroscopy in wet (ca. 2% H2O) DMSO-d6, with enantiomers 356a and 356b displaying similar association constants for acetate and dihydrogenphosphate (log K value ca. 2 for both receptors with both anions). Receptor 356c displayed a slight selectivity for acetate over dihydrogenphosphate (log K values of 2.86 and 1.83, respectively). This preference in anion binding was attributed to a tighter binding pocket in 356c relative to 356a and 356b that precluded binding of the bulkier dihydrogenphosphate anion. The presence of such a binding pocket was supported by both 1H NMR spectroscopic studies and single crystal X-ray diffraction analysis. Binding constants for fluoride could not be determined due to competing deprotonation of the thiourea protons. Interestingly, neither the absorbance nor emission spectra of these compounds was observed to change significantly upon the addition of the anions in question in acetonitrile. For the chiral receptors 356a and 356b, changes in the CD spectra were observed in the presence of acetate and dihydrogenphosphate in DMSO. No receptor-anion interactions were observed for chloride using either 1H NMR or CD spectroscopic techniques.
Hennrich, Resh-Genger, and co-workers reported the iminoylthiourea fluorescent sensors 357 and 358, along with their oxidized 1,2,4-thiadiazole derivatives 359 and 360 in 2001.433 Increases in the naphthalene emission were observed upon the addition of sodium salts of HCO3-, CO32- and HPO42- in methanol. This finding was attributed to a rigidifying effect brought about by anion binding. Little or no increase in emission intensity was seen in the presence of other anions, including H2PO4-. Significantly larger changes were generally seen with the reduced forms 357 and 358 as compared to the oxidized forms. This observation is consistent with the presence of a greater number of hydrogen bond donors in the reduced forms. No binding constants, however, were reported. It should be noted that a much greater fluorescent enhancement was observed for the bicarbonate and carbonate anions than for monohydrogenphosphate.
Also in 2001, the Hong group reported a class of chromogenic azophenol-thiourea based anion sensors (361 and 362) that permitted the naked-eye detection of anions. Receptor 361a is representative of this class. It was found to undergo a large red-shift in its absorbance maximum in the presence of H2PO4-, F-, and AcO- anions (TBA salts) in chloroform solution.434 A visible color change from light yellow to deep red was observed at micromolar concentrations of receptor in the presence of three molar equivalents of these anions. While other anions did not induce such a color change, no discrimination could be made among the H2PO4-, F-, and AcO- anions. While the color change was attributed to the participation of the phenolic hydrogen in hydrogen-bonding, no color change was observed in control systems in which the thiourea groups were absent. Receptors 361b and 361c were analyzed in an effort to tune the selectivity of the system.435 It was found that the presence of the phenyl substituents in 361b did not lead to a change in the observed selectivity. On the other hand, replacing these phenyl substituents with nitrophenyl groups, as in 361c, led to a red-shift in the absorbance band upon nitrophenyl anion complexation. In the case of 361c, addition of dihydrogenphosphate (as the TBA salt) produced the largest wavelength shift, allowing for the selective detection of this species among anions of similar basicity. This effect was attributed to the additional hydrogen bonding interactions that the phosphate anion can support in comparison to the fluoride and acetate anions. In any event, this serves to underscore how thiourea moieties can be used to tune the selectivity of an anion receptor.
The indoaniline receptor 362 was produced by the Hong group in 2002 with the goal of obtaining a receptor whose selectivity would differ from that predicted solely on considerations of anion basicity.436 In contrast to the azophenol receptors, a blue-shift of absorption was observed with receptor 362 in the presence of anions. This shift was attributed to a breakdown in the intramolecular hydrogen bonding interactions between the thiourea NH protons and the carbonyl of the receptor upon the addition of anions. This loss of internal hydrogen bonding serves to free the carbonyl group, leading to the observed blue absorbance shift. A preference was observed for tetrahedral anions, such as H2PO4- and HSO4-, over spherical or planar anions, likely due to a geometric complementarity between the receptor and these tetrahedral guests. Quantitative UV-Vis spectroscopic analyses revealed an order of magnitude difference in binding affinity for H2PO4- and HSO4- relative to other anions (104 M-1 vs. 103 M-1) in chloroform when the anions were studied as the TBA salts. A similar trend was observed in acetonitrile, although the absolute value of the respective binding constants proved to be considerably lower than in chloroform.
From 2002-2009, Jiang and co-workers reported a number of hybrid amidothiourea anion receptors. Preliminary studies examined the anion binding ability of receptor 363a.437 This host contained a combination of a thiourea recognition moiety and a p-dimethylaminobenzamide dual fluorescent reporter that were linked by a hydrazine spacer. Modeling studies (AM1) and 1H NMR spectroscopic studies in DMSO-d6 led to the conclusion that the receptor was held in a rigid conformation through the hydrogen bond network shown below. Addition of anions (as their TBA salts) to a solution of this receptor in acetonitrile served to perturb the emission spectrum significantly. Specifically, the charge-transfer state band was quenched while the locally excited state band was enhanced. In general, anions were observed to interact with this receptor in the order of increasing basicity, and a large preference for acetate was observed. Dihydrogenphosphate was found to bind the receptor with an association constant of 4.7 × 104 M-1.
The ability of this system to function as a chemosensor was further probed by varying the substituents on the benzene ring of the recognition motif (363b-h).438 Interestingly, the 1H NMR, absorption, and fluorescence spectra of these complexes were consistent with a lack of coupling between the thiourea recognition moiety and the reporter unit. This decoupling effect was presumably due to a twisted conformation of the hydrazine single bond in these receptors prior to anion binding. At the same time, strong changes in the absorption and fluorescence spectra were observed upon the addition of anions in acetonitrile. These changes were attributed to an anion-induced conformational change that facilitated electronic communication between the recognition and reporter units. The measured binding constants were found to correlate with the electronic-withdrawing nature of the benzene substituent as well as the basicity of the anionic guest. The highest binding constant for dihydrogenphosphate was reported for receptor 363h (K = 1.3 × 105 M-1), as determined via absorbance spectroscopy in acetonitrile. A 1:1 host:guest stoichiometry was inferred on the basis of Job plot analyses for all systems.
Pyrene was also studied as a fluorescent reporter in these systems (364).439 An enhancement in the PET quenching of the pyrene unit by the thiourea moiety was observed upon the addition of a number of anions as their TBA salts in acetonitrile. The degree of quenching was found to follow the order F- > AcO- > H2PO4- ≫ HSO4- > Cl- > Br- > I- > NO3- > ClO4-. Binding constants for dihydrogenphosphate were determined as 4.29 × 105 M-1, 1.63 × 106 M-1, and 8.86 × 106 M-1 for receptors 364a, b, and c, respectively. A 1:1 binding stoichiometry was confirmed through Job plot analysis. The binding affinity of acetate and fluoride was too high to be determined. These neutral sensors also responded to anions in mixtures containing up to 8% water in acetonitrile.
Further iterations varied the substitution of the benzamide reporter unit (365a - h).440 These receptors displayed a change in the absorption spectrum upon anion addition in acetonitrile. These experiments also supported an anion-induced coupling mechanism (vide supra). The binding constants generally correlated with the electron-withdrawing nature of the substituent and the basicity of the anion, although a few exceptions were observed. For example, the p-Br substituted receptor 365f displayed stronger binding to dihydrogenphosphate (K = 5.09 × 105 M-1) than the p-NO2 substituted receptor 365h (K = 1.38 × 105 M-1) despite the increased electron-withdrawing ability of the latter substituent. Receptor 365f was also observed to bind dihydrogenphosphate more strongly than fluoride. The studied benzamide receptors (365a-h) displayed higher binding affinities toward anions than simpler thio-derived counterparts (366a-h). Based on these findings, it was concluded that the contribution of the hydrogen bonding ability of the amide unit in receptors 365a-h was more significant than the increased acidity of the NH protons in receptors 366a-h. The effects of further expansion were investigated by preparing and studying the symmetrical receptors 367i-l.441 In general, these N,N′-bis(benzamido)thiourea receptors displayed increased anion binding affinities relative to the related N-benzamidothiourea receptors 365i-l. Within this class, the chloro-substituted receptors 367k and 367l were found to bind dihydrogenphosphate the most strongly (K ≈ 105 M-1).
Phosphate binding constants on the order of 106 M-1 have been achieved with N-benzamido-N′-benzoylthiourea receptors of general structure 368, reported by Liu and Jiang in 2008.442 Significant UV-Vis spectral changes were observed upon the addition of dihydrogenphosphate, acetate, and fluoride (as TBA salts) to receptors 368 in acetonitrile. Little to no changes were observed upon the addition of other monoanions (HSO4-, Cl-, Br-, I-, NO3-, ClO4-). A 1:1 host:guest stoichiometry was confirmed through Job plot analysis. As with previous systems, the signal mechanism was proposed to arise from the breaking of strong intramolecular hydrogen bonds by the binding of the anionic guest to the thiourea moiety. Interestingly, the binding affinity was found to show little dependence on the aryl substituents in the case of these receptors.
These researchers also prepared a series of N-(isonicotinamido)-N′-(substituted-phenyl)thiourea receptors 369.443 These acyclic receptors were not only found to bind carboxylate anions well but also to interact with dihydrogenphosphate with a binding constant on the order of 105-106 M-1 as inferred from UV-Vis titrations carried out in acetonitrile (anions studied as their TBA salts). It was suggested that the hydrophobic environment created around the thiourea group facilitated hydrogen bonding interactions with the bound anions. Spectral changes were also observed in water - acetonitrile mixtures, although no binding constant was reported for dihydrogenphosphate.
Jiang and co-workers further examined N-(acetamido)thiourea based anion receptors 370 and 371.444 Variations at the aceto-position were examined through studies with receptors 370a-d, and variations at the benzene unit were examined through studies with receptors 371a-f. Binding constants on the order of 104 - 107 M-1 in acetonitrile were reported for acetate, fluoride, and dihydrogenphosphate anions (studied as the TBA salts) in the case of receptors 370a-d and 371a-e. These values were measured by monitoring changes in the absorption spectra of the receptors upon anion addition. The large wavelength shift seen in the case of receptor 371f led to the conclusion that deprotonation of this receptor was occurring in the presence of anions. This result was ascribed to the increased acidity of this system relative to the other derivatives. Little to no changes in the absorption spectra were observed upon addition of hydrogensulfate, chloride, bromide, iodide, nitrate, and perchlorate. In these studies, a preference for acetate and fluoride over dihydrogenphosphate was observed.
In 2004, He and co-workers reported the colorimetric sensors 372, which also incorporated amidourea hydrogen bonding sites.445 The addition of anions (TBA salts) to receptors 372 in DMSO was followed through UV-Vis spectroscopy. Significant color changes were observed with fluoride, acetate, and dihydrogenphosphate. On the other hand, chloride, bromide, and iodide did not affect the absorbance spectrum. Interestingly, the presence of the pyridine ring in receptor 372b did not influence the binding affinity toward dihydrogenphosphate, and binding constants on the order of 104 M-1 were reported for receptors 372a and b. However, these two systems displayed a preference for acetate and fluoride over dihydrogenphosphate.
These researchers later studied receptor 373 containing p-nitrophenylthiourea groups and a spacer between the amide and thiourea units.446 Spectrophotometric titrations carried out in DMSO established that this mixed amide-thiourea system interacted similarly with acetate and dihydrogenphosphate anions (K(H2PO4-) = 1.41 × 104 M-1). However, the highest affinity was found for fluoride, in which case naked-eye detectable color changes were observed. This same group also reported systems 374.447,448 Preliminary studies with receptors 374a and 374b were performed using UV-Vis and fluorescence titrations, respectively, using various TBA anion salts. Binding affinities for the interactions with dihydrogenphosphate were determined in DMSO. Receptors 374a and 374b were found to bind dihydrogenphosphate with association constants of 1.85 × 104 M-1 and 1.61 × 103 M-1; however, binding was not selective against acetate, p-nitrophenolate, or p-nitrophenylphosphate. The incorporation of a pyridine unit led to the development of receptors 374c and 374d. Both acetate and dihydrogenphosphate were found to participate in hydrogen bonding interactions with all the NH units present in these receptors as inferred from 1H NMR spectroscopic measurements carried out in DMSO-d6. Anion-dependent changes in the UV-Vis spectrum of receptor 374c (DMSO) were also seen. The same proved true for the emission spectrum of receptor 374d (CHCl3). The use of different solvents for the two receptors, however, prevented comparison of the binding ability. Both receptor 374c and 374d displayed a high affinity for dihydrogenphosphate (K = 1.6 × 104 M-1 (DMSO) and 1.0 × 106 M-1 (CHCl3), respectively), but a preference for acetate. Other anions (bromide, chloride, iodide, p-nitrophenolate, p-nitrophenylphosphate) were found to bind more weakly.
Martínez-Máñez, Rurack, and co-workers reported the colorimetric detection of dihydrogenphosphate and other anions using monourea and thiourea receptors bearing an appended chromophore unit (375a,b).449 These receptors were investigated as part of a larger study of the 375 - 376 receptor series. Analysis of the binding behavior of these receptors was conducted through UV-Vis spectroscopy in acetonitrile using the TBA salts of all studied anions. While all receptors in this set were found to bind dihydrogenphosphate, only receptors 375a and 375b underwent a significant bathochromic shift (ca. 30 nm) upon the addition of this anion. These receptors also displayed the strongest binding constants with dihydrogenphosphate (K = 3.9 × 103 M-1 and 7.4 × 103 M-1, respectively). While lower binding affinities were reported for chloride, significantly higher binding constants were observed for the interaction of these receptors with acetate and benzoate as compared to dihydrogenphosphate. Little to no spectroscopic change was observed upon the addition of hydrogensulfate, thiocyanate, nitrate, bromide, and iodide anions. Deprotonation was often reported upon the addition of fluoride and cyanide.
In 2005, the Fabbrizzi group reported the results of an extensive study of the anion binding behavior of the urea and thiourea receptors 377.450 In the case of urea receptor 377b, UV-Vis titrations in DMSO demonstrated that 1:1 adducts were formed with all anions when added as the TBA salt, excluding fluoride for which deprotonation was observed. The binding constant corresponding to the interactions between receptor 377b and dihydrogenphosphate was found to be log K = 4.47. In analyzing the thiourea receptor 377a deprotonation was observed for fluoride, acetate, benzoate, and dihydrogenphosphate, a finding consistent with the more acidic character of the thiourea receptor 377a. This study provides a classic demonstration of the tenant that a balance must be maintained in receptor design if strong hydrogen bond donating systems are to be obtained that will not deprotonate under conditions of use. Since these latter can vary (anion, solvent, concentration, temperature, counter cation, etc) a caveat coming from this and other studies451-453 is that great care must be exercised in the analysis of new hydrogen bond-based anion receptors.
The effect of urea and thiourea moieties on anion binding was also directly compared using the anthraquinone receptors 378 reported by Ganguly and Das in 2005.454 Colorimetric responses were observed with both receptors in DMSO:CH3CN (1:9), although the urea derivative 378a required heating to 60°C before a color change was observed. This heating was thought to reflect a need to break strong intramolecular hydrogen bonds within the urea compound before binding the anion becomes competitive. UV-Vis titrations were consistent with 1:1 binding behavior when the anions were added as the TBA salts. Moderate selectivity for dihydrogenphosphate over acetate, benzoate and hydroxide was observed. Further, the binding affinities were higher in the case of the thiourea derivative than the urea derivative (K for H2PO4-= 1.0 × 106 M-1 and 1.3 × 104 M-1, respectively). Computational studies led the authors to suggest that the receptor binds anions in a tweezer-like fashion, wherein the dihydrogenphosphate anion is held via all four hydrogen bond donors. In contrast, acetate was thought to be held by only three hydrogen bond donors.
At the same time, the diurea and dithiourea receptors 379-380 were reported by Yoon and Kim.455 Proton NMR spectroscopic studies provided support for the conclusion that all members of this series stabilized 1:2 (host:guest) complexes when anions were added as the TBA salts. Across the board, these receptors displayed a strong affinity for adipate. An interaction with pyrophosphate was also observed. Only in the case of receptor 379a, however, was the affinity toward pyrophosphate substantial. In this case a binding constant of ca. 1 × 105 M-1 was recorded in CD3CN:DMSO-d6 (9:1).
In 2005, Pfeffer, Gunnlaugsson and co-workers first reported a family of [3]polynorbornane frameworks. These receptors were designed in order to obtain a set of conformationally preorganized aromatic thiourea receptors (381 and 382).456,457 While 381b displayed a colorimetric response to TBA anion salts, the binding behavior for all receptors was studied most extensively via 1H NMR spectroscopic titrations carried out in DMSO-d6. These receptors displayed an excellent affinity for the dihydrogenphosphate (H2PO4-) and dihydrogenpyrophosphate (H2P2O72-) anions, among others. Consistent with their respective charges, these anions were bound in 1:1 and 2:1 (host:guest) ratios, respectively. Based on this finding and other considerations, it was proposed that the cavity of the receptors was appropriately organized for the formation of four hydrogen bonds to one phosphate group, thus allowing for the formation of a sandwich-type complex in the case of pyrophosphate.
Receptors 381a and 381b exhibited similar binding affinities, with specific log K values of 3.9 and 7.9 being derived for the binding of TBA H2PO4- and TBA H2P2O72, respectively, to receptor 381a. The estimated binding constants for receptor 382 were slightly lower, a finding that was attributed to its additional flexibility. Receptors 381 and 382 were also found to bind acetate, although with a lower binding affinity and in a 1:2 host:guest ratio. Fluoride caused deprotonation of the receptors.
Functionalized norbornene scaffolds with smaller cavities (383-385) have also been prepared. As a general rule, these were found to interact with acetate and dihydrogenphosphate (TBA salts) to form 2:1 complexes as deduced from 1H NMR spectroscopic titrations carried out in DMSO-d6.458,459 Receptor 383a, for example, displayed a log K1 = 3.66 and a log K2 = 3.00 for the dihydrogenphosphate anion. Negligible binding was observed with bromide, chloride, and hydrogensulfate. Subtle differences in binding affinities were attributed to the varying steric constraints associated with each receptor.
Ramamurthy, Thirumalai, and co-workers developed a dual-fluorescent reporter system based on the acridinedione fluorophore (386).460 The effects of a number of anions on the UV-Vis and fluorescence spectra of receptor 386 were studied in acetonitrile (F-, Cl-, Br-, I-, HSO4-, ClO4-, AcO-, H2PO4-, and BF4-, added as their TBA salts). Only the addition of dihydrogenphosphate led to changes in the absorbance spectrum of receptor 386a; however, the addition of dihydrogenphosphate, acetate, and fluoride affected the fluorescence spectrum of receptor 386a. Interestingly, the addition of acetate and fluoride was found to quench the fluorescence of receptor 386a, while the addition of dihydrogenphosphate led to emission enhancement. It was proposed that acetate and fluoride participated in hydrogen bonding interactions with the thiourea moiety and thus increased the PET quenching of the fluorophore by the thiourea unit. On the other hand, dihydrogenphosphate was thought to participate additionally in a hydrogen bonding interaction with the nitrogen atom of the fluorophore in receptor 386a. This additional interaction was expected to facilitate an ICT process and a corresponding enhancement in emission intensity. Support for this mechanism came from studies with receptor 386b, which lacked the additional hydrogen bond donor. As expected, fluorescence quenching was observed upon the addition of dihydrogenphosphate. A 1:1 binding constant of 380 M-1 was determined for the complex 386a:H2PO4-.
In 2006, a receptor system containing a (benzylideneamino)thiourea unit (387) was reported by Fabbrizzi and co-workers.461 This receptor displayed a strong response to acetate, fluoride, and dihydrogenphosphate anions (TBA salts) as inferred from UV-Vis spectroscopic studies carried out in acetonitrile. Proton NMR spectroscopy led to the conclusion that addition of fluoride led to deprotonation, while acetate and dihydrogenphosphate participated in strong hydrogen bonding interactions with receptor 387. Similar log binding constants were observed for the 1:1 complexes of these anions (log K(H2PO4-) = 3.34, log K(AcO-) = 3.62).
A practical application for thiourea sensors was recently introduced by the Anzenbacher group. Specifically, these researchers demonstrated that an array of sensors (388-389) could be used to detect phosphate concentrations in blood serum.462 All of the receptors in this series formed 1:1 complexes with anions (TBA salts) according to 1H NMR spectroscopic studies, with 388b-d and 389b-d displaying an increase in the observed fluorescence intensity upon anion binding. This emission increase was attributed to a reduction in the conformational flexibility of the fluorophores as a result of interactions between the bound anions and each arm with the guest. Affinities were similar for each anion (K ≈ 106 M-1 in DMSO), but a general trend of H2PO4- > HP2O73- > AcO- ≫ Cl- > Br- was observed for each receptor. The low relative selectivities, coupled with the differential binding behavior observed for each receptor, allowed Anzenbacher and co-workers to exploit this series of receptors in a pattern recognition protocol. By immobilizing the receptors in a polyurethane matrix, and employing principle component analysis (PCA), AMP, ATP, phosphate, and pyrophosphate could be detected independently of one another in the complex medium of blood serum.
In 2008, the Jang group reported the hybrid receptor 390a. This receptor combined thiourea moieties with benzimidazole groups.463 Fluorescence studies of 390a revealed a quenching of the emission intensity only after the addition of PO43- in DMSO:water (8:2) with a binding constant of 1.0 × 104 M-1. No quenching was observed upon addition of other common anions (F-, Cl-, Br-, I-, CN-, ClO4-, H2PO4-, HPO42-, AcO-, NO2-, CO32-, HCO3-, SO42-, and HSO4-). All anions were studied as the sodium salts. The selectivity for PO43- was attributed to two factors, namely the geometrical complementary between receptor 390a and PO43- and the high negative charge of the anion. Receptor 390b, in which the NH groups of the benzimidazole entities were substituted by sulfur units, interacted more weakly with PO43-, resulting in a binding constant of 1.8 × 103 M-1 and a lack of selectivity. These results help illustrate the important role that additional hydrogen bond donor moieties can play in stabilizing anion-receptor complexes.
4.2.3. Pyrrole- and Indole-derived Receptors
Another hydrogen bonding motif that has been studied for the purpose of phosphate anion recognition is pyrrole. It and other nitrogen-containing heterocycles provide a source of NH donors that can be neutral or positively charged depending on the system in question. It should be noted that these heterocycles are only discussed here in the context of acyclic receptors. Polypyrrolic macrocycles, including calixpyrroles and expanded porphyrins, will be discussed in Section 4.4.
Some of the most interesting pyrrolic receptors developed for the purpose of anion recognition have been based on quinoxaline scaffolds. A variety of these receptors, which have been exploited for the colorimetric detection of anion binding, have now been designed and tested. The earliest contributions came from Sessler and co-workers, beginning in 1999. Nearly all of the quinoxaline receptors reported to date produce a strong colorimetric response to fluoride. However, some receptors were also found to bind dihydrogenphosphate. The first receptors synthesized, 391a-b, displayed a low affinity for dihydrogenphosphate (K < 100 M-1) as determined by fluorescence quenching measurements carried out in dichloromethane (using the TBA salt of this anion).464 The incorporation of electron withdrawing fluorine substituents at the pyrrole beta positions (391c) increased the affinity to 1.7 × 104 M-1 and also enhanced the selectivity of the receptor for dihydrogenphosphate over chloride.465 Receptor 391c was also found to undergo a visible color change (yellow to orange) upon the addition of dihydrogenphosphate. Additional hydrogen binding sites were incorporated in receptors 392 and 393a, which bound dihydrogenphosphate with binding constants of 4.3 × 103 M-1 and 5.0 × 105 M-1, respectively, as inferred UV-Vis spectroscopic titrations carried out in dichloromethane.466 Based on the observed increase in affinity relative to 391, it was concluded that the additional pyrrole units contribute strongly to phosphate binding. Interestingly, incorporation of electron withdrawing nitrile groups on the quinoxaline unit (393b) decreased the affinity for dihydrogenphosphate and reversed the selectivity.467
The Anzenbacher group sought to tune further this class of receptors through incorporation of aromatic substituents on the quinoxaline rings (394-395). While low binding was observed with dihydrogenphosphate, a strong selectivity for pyrophosphate was exhibited in preliminary studies with receptors 394a-c and 395a.468 Receptor 394a proved to have the strongest binding constant (9.4 × 104 M-1) as determined through fluorescence quenching studies carried out in dichloromethane using the TBA salts of pyrophosphate. Remarkably, the quenching could be seen using a simple UVA lamp, such as is commonly used in the laboratory for the analysis of TLC plates. The general anion binding properties of receptors 394 - 395 were further examined.469 These receptors were found to undergo both a visual colorimetric change as well as fluorescence quenching upon the addition of fluoride and pyrophosphate (TBA salts) in dichloromethane. An analysis of the binding constants revealed a slight preference for pyrophosphate over fluoride for these receptors. This stands in contrast to what is observed for the parent receptor 391a. The strongest binding constant was reported for the complexation of pyrophosphate by receptor 394a (K = 9.37 × 104 M-1). Incorporation of these receptors into a polyurethane matrix allowed for the colorimetric sensing of anions in aqueous solution near neutral pH.
Further studies by these researchers sought to improve the fluorescence response through the incorporation of pyrene fluorophores (396-397).470 These receptors indeed displayed a much stronger fluorescence increase compared to the parent receptor 391a. In the case of receptor 396, this enhancement was attributed to a resonance energy transfer mechanism from the pyrene (donor) units to the dipyrrolylquinoxaline (acceptor) unit. With receptor 397, it was proposed that the extended conjugation allowed for delocalization of the excited state, which was expected to increase the overall fluorescence intensity. In addition, in dichloromethane, enhanced anion binding affinities were observed for these receptors relative to receptor 391a. For example, pyrophosphate (TBA salt) was found to bind receptor 397 with a binding constant of 2.95 × 105 M-1.
Sessler and co-workers further investigated the anion-binding properties of a terpyrrolic analogue of dipyrrolylquinoxalines.471 Receptor 398 was observed to undergo a visual color change upon addition of dihydrogenphosphate, fluoride, and chloride (TBA salts) in dichloromethane. Quenching of the emission intensity was also reported in the presence of these anions. A strong increase in binding affinity was found for these anions as compared to the unfunctionalized dipyrrolylquinoxaline (391a). For instance, the K for the formation of 398:H2PO4- was determined to be 1.75 × 104 M-1. The increased binding affinity was attributed to the electron-withdrawing nature of the ester substituents. High selectivity for dihydrogenphosphate over chloride was also observed (K (Cl-) = 1.60 × 102 M-1).
Simpler pyrrole receptors, such as 399, in which the hydrogen bonding ability of the pyrrole moiety was combined with amide groups were designed and tested by Gale and co-workers. While both receptors 399a and 399b were produced as the result of this effort, these two systems displayed vastly different selectivities as determined from 1H NMR spectroscopic titrations.472,473 Specifically, the alkyl substituted receptor 399a displayed selectivity for benzoate in acetonitrile, while 399b displayed selectivity for dihydrogenphosphate in DMSO/H2O 0.5% (K = 1.45 × 103 M-1) among the various anions studied. Because receptor solubilities demanded the use of different solvents, direct comparisons could not be made. The importance of the pyrrole N-H was further supported by the vastly decreased binding of furan-derivatives 400 with acetate and dihydrogenphosphate (TBA salts studied in DMSO/H2O 0.5%).474
The putative ditopic receptor 399c was also prepared and analyzed. However, no improvement in binding affinity was observed in the presence of a metal cation.475 Thus, no direct benefit as the result of appending a crown ether could be inferred. Gale and co-workers also studied dipyrrolylmethane analogues of his basic receptor system (cf. structures 401-402). Of these, receptor 401b displayed a high selectivity for dihydrogenphosphate (studied as the TBA salt).476 The associated binding constant proved too large to be measured reliably via 1H NMR spectroscopic titrations in 5% water. However, analysis in 25% water in DMSO allowed a binding constant of 2.34 × 102 M-1 to be determined. The chemically more stable dimethyl substituted dipyrrolylmethane receptors 402 displayed similar selectivities but was characterized by reduced binding constants as compared to receptor 401.477 Proton NMR spectroscopy and computational studies of these receptors supported a linear binding cleft with hydrogen bonds between one phosphate oxygen atom and both the pyrrole and amide hydrogen atoms on each side of the receptor.
A small library of receptors derived from pyrrole-2,5-diacetic acid (403) were also reported by Gale.478 Proton NMR titrations in CD3CN revealed that receptor 403a interacted most strongly with dihydrogenphosphate and hydrogensulfate. These and other anions were studied as the TBA salts. This result was attributed to a proton transfer process from the oxoanions to the amine groups of the receptor. In addition, receptors 403b-403d were found to display the following selectivity order: Benzoate > dihydrogenphosphate > chloride, a sequence that correlates well with that obtained in the case of the parent systems 399. However, the anion affinities of the receptors 403a-403d were higher than those found with receptors 399. The presence of a methylene group between the pyrrole and amide units was thus credited with creating an improved cavity for anion binding. Amidourea substituted pyrroles, such as 404 – 405, were also analyzed in the hope that the additional hydrogen bonding functionality present in these systems would increase the binding affinity and selectivity.479 While these receptors were able to bind dihydrogenphosphate in pure DMSO (K = 103 – 105 M-1), little selectivity was observed among the anions tested. Additional pyrrolic units were also attached through synthetic means (giving systems 406). These receptors were then compared to receptor 399b.480 Based on 1H NMR spectroscopic studies carried out in DMSO, it was concluded that receptor 406 binds dihydrogenphosphate (as the TBA salt) more strongly than receptor 399b (K = 5.50 × 103 vs. 1.45 × 103 M-1). However, it displays a reversed selectivity toward benzoate, binding the latter anion twice as strongly as dihydrogenphosphate. It is evident that by varying the nature of the substituents, selective pyrrolic cleft receptors can be developed for dihydrogenphosphate, although the specific design criteria needed to achieve this objective have yet to be fully elucidated.
Even simpler pyrrole receptors, of general structure 407, were studied by the Jurczak group. These systems were prepared in the course of efforts designed to compare directly the binding ability of amide and thioamide recognition moieties.481 It was concluded that the amide-containing receptors bound to dihydrogenphosphate more strongly than their thioamide counterparts. The opposite trend was observed for binding interactions involving the benzoate and chloride anions. Interestingly, the lack of phenyl substituents on the pyrrole led to decreased binding affinities, with the highest affinity found to be 2.03 × 102 M-1 for dihydrogenphosphate and receptor 407b in DMSO/H2O 0.5% (all anions studied as the TBA salts). Single crystal X-ray diffraction analysis revealed that, at least in the solid state, the pyrrole amide receptors preferred an anti-anti conformation in the absence of anions. However, rotation to give a syn-syn conformation (thus forming a binding cleft) occurs in the presence of anions. Based on computational experiments, the authors suggested that thioamides have a stronger preference for the anti-anti conformation, which may account for their decreased binding affinities relative to their amide analogues.
A series of dipyrrolyldiketone difluoroboronate receptors (408) were produced by Maeda and co-workers.482 These receptors were found to bind various anionic guests via hydrogen bonding interactions that involve both the pyrrolic N-H and bridging C-H protons. While receptor 408a displayed a preference for the fluoride anion, absorption-based spectroscopic titrations revealed that this receptor also interacted strongly with dihydrogenphosphate (K(H2PO4-) = 1.3 × 104 M-1) in dichloromethane. It appeared that the incorporation of difluoroboronate groups into these receptors served to enhance the interaction with the anions, especially in the case of dihydrogenphosphate. The substitution of the β pyrrole positions with fluoride units (giving receptor 408b) led to an increase in the dihydrogenphosphate affinity (K = 1.9 × 105 M-1 in dichloromethane).483 However, the strongest binding interaction was observed with acetate (K = 9.6 × 105 M-1). Furthermore, the N-“blocked” receptors 409a and b and the C-modified receptor 410 displayed similar fluoride affinities as those displayed by receptor 408a.484 However, for the other anions subject to study, the binding constant was found to decrease as the result of such substitution (K(H2PO4-) = 1.4 × 103 M-1 for receptor 409a, 3.2 × 103 M-1 for receptor 409b and 250 M-1 for receptor 410). On the basis of these findings, it was concluded that in order to bind the small fluoride anion, only 1 or 2 binding sites are actually needed. However, with this type of receptor, more recognition interactions are needed to bind larger anions. Further studies involving the β-tetraethyl substituted dipyrrolylketones 408c and 408d were also conducted.485 It was found that receptor 408c displayed higher anion affinities (e.g., K(H2PO4-) = 9.1 × 104 M-1) than receptor 408a, with the exception of fluoride. These results were attributed to the presence of a relatively stable inverted conformation in the case of 408c. However, receptor 408d, which bears ethoxycarbonyl groups in the α-pyrrole positions, was found to display a lower anion binding affinity, presumably as the result of increased electrostatic and steric repulsions. All anions were studied as the TBA salts.
The aryl-substituted receptors 411 were also prepared and studied.486 They were found to display the following anion selectivities: F- > AcO- > H2PO4- > Cl- (all anions as the TBA salts). Further, the anion affinities were found to increase in the order 411c < 411b < 411a. This binding affinity trend was rationalized in terms of both the number of C-H units available as well as to differences in planarity. Receptors 412 and 408e, characterized by substituted α and β pyrrole positions, displayed the highest affinities for acetate. However, these systems were also found to interact with dihydrogenphosphate, with binding constants of 2.2 × 103 and 3.6 × 104 M-1 being derived for receptors 408e and 412, respectively, in dichloromethane.487
Changes in the boron substituents were also explored.488 Replacing the fluoride substituents of receptor 408c with pinacol (413a) served to decrease the anion affinity (K(H2PO4-) = 21,000 M-1) in dichloromethane. The binding affinity was partially restored upon substitution with a catechol group (413b, K(H2PO4-) = 67,000 M-1) and found to be comparable to receptor 408c in the case of nitrocatechol substitution (413c, K(H2PO4-) = 115,000 M-1).
Significantly stronger binding was achieved by incorporation of addition pyrrole substituents (414a).489 On the basis of UV-Vis titration experiments carried out in CHCl3 containing 5% ethanol, binding constants in the range of 1.2 - 3.0 × 106 M-1 could be determined for chloride, acetate, and dihydrogenphosphate. These values were two to three orders of magnitude higher than the binding constants obtained for receptors 408c and 408e under the same conditions. The anion binding affinities of the pyrrolic receptor 414a also proved larger than those seen for the furan (414b) and thiophene (414c) derivatives. This increased binding affinity was attributed to the additional hydrogen bonding interactions available in the case of receptor 414a, as well as to the extended π-conjugation present in this system.
The effect of alkyl substituents at the α-position of the pyrrole units (415b-k) was also investigated.490 In these studies, anion binding affinity was observed to decrease upon increasing the length of the alkyl substitution in dichloromethane. For example, unsubstituted receptor 415a was found to bind dihydrogenphosphate rather strongly (K = 2.7 × 105 M-1), but the binding affinity of the methyl-substituted receptor 415b fell to 1.5 × 105 M-1 and that of ethyl-substituted receptor 415c to 7.6 × 104 M-1. These reductions in affinity were attributed to the electron donating ability of the alkyl chains. Support for the critical role of these electronic contributions came from studies involving fluoroalkyl receptor 415k. Here, a strong increase in the binding affinity relative to that of the unsubstituted receptor 415a was observed (K(H2PO4-) = 5.6 × 105 M-1), with this enhancement attributed to the electron withdrawing nature of the fluoro-substituent. Interestingly, the binding affinity of receptor 415k for anions was found to about two-fold higher than the binding affinity of the β-fluoro-substituted receptor 408b.
In 2009, Maeda and Eifuku reported the anion binding properties of various alkoxy-substituted receptors of general structure 416.491 No significant differences in binding affinity were observed among the series of receptors 416c-f, and the affinities were found to be comparable to that of unsubstituted aryl receptor 411a. No appreciable binding was observed with ortho-substituted receptors 416a-b. Taking into account the wide range of dipyrrolyldiketone difluoroboronate receptors studied, it becomes apparent that seemingly small differences in the shape, size, and hydrogen bonding donor number can have significant effects on the anion binding affinities and selectivities within this series of receptors.
More recently, Cheng and co-workers examined the anion binding ability of the tripodal pyrrole receptor 417.492,493 This receptor was studied in DMSO-d6 using 1H NMR spectroscopy and various TBA anion salts. Of the anions tested, the highest binding affinity was observed for dihydrogenphosphate (K = 2.40 × 102 M-1), followed by fluoride, chloride, and hydrogensulfate. A single crystal X-ray diffraction analysis of the complex formed between receptor 417 and dihydrogenphosphate revealed the expected C3-symmetry and confirmed that the anion is held within the receptor cavity through hydrogen bonds.
A different set of amide-pyrrolic receptors 418-419 was studied recently by Yin and Cheng.494 In this case, proton NMR titrations performed in DMSO-d6 using various TBA anion salts revealed that receptor 419 is a less effective anion receptor than 418. Presumably, this reflects the presence of greater steric encumbrance in host 419 as the result of the bulky Boc substitution. Receptor 418a, containing a pyridine group, displayed the strongest interactions with anions within the set of receptors (K = 448 M-1 for H2PO42-). This relatively higher binding affinity was rationalized in terms of intramolecular hydrogen bonding interactions between the two amide protons and the pyridinic nitrogen, which led to a rigid, preorganized receptor even in the absence of an anion. Quantitative measurements revealed that receptors 418b-419 display a preference for the dihydrogenphosphate anion with the associated binding constants being on the order of 102 M-1. On the other hand, receptor 418a was found to interact most strongly with the fluoride anion.
Indole moieties are yet another N-heterocyclic aromatic functionality that have been successfully incorporated into phosphate anion receptors. Some of the first indole receptors tested were the acyclic systems 420 – 423.495 In this case, the interactions with TBA anion salts were monitored using UV-Vis spectroscopy in acetone. It was found that the strongest binding affinities were observed for benzoate followed by dihydrogenphosphate. In general, receptor 423 displayed the highest binding affinity in this solvent (log K = 5.3 for dihydrogenphosphate), followed by receptor 421. Addition of dihydrogenphosphate also led to significant fluorescence enhancement in the case of this latter receptor in acetone.
Receptor 424, reported by Jeong in 2006, was designed to contain both indole hydrogen bond donor and pyridine acceptor functionalities within a rigid scaffold.496 Changes in the absorption spectrum were observed in acetonitrile in the presence of TBA dihydrogenphosphate, from which an association constant of 1.1 × 105 M-1 could be derived. Based on experiments with control compounds, the authors suggested that each pyridine moiety increased the binding affinity by ca. 15-fold. Selectivity for dihydrogenphosphate over a variety of other anions was observed, with the closest competitor being acetate, a species that bound 5-fold more weakly. This study thus served to support the conclusion that good affinity and selectivity for phosphate species can be achieved via incorporation of an appropriate number and arrangement of hydrogen bond donors and acceptors. Later studies explored the ability of appended hydroxyl groups (425) to participate in hydrogen bonding interactions with anions.497 UV-Vis titrations in acetonitrile / 1% water led to the determination of a strong binding affinity between receptor 425 and dihydrogenphosphate (TBA salt, K = 2.9 × 104 M-1). Proton NMR spectroscopic experiments were consistent with this anion being bound through hydrogen bonding interactions involving both the indole NH units and the hydroxyl groups. Significantly lower association constants were seen with control receptor 426, a finding that was taken as evidence that the OH units play an important role in anion binding. Single X-ray diffraction studies revealed the formation of a 2:2 host:guest complex in the solid state. In this complex, the OH units participated in dihydrogenphosphate binding, acting as both a hydrogen bond donor and acceptor. Despite these strong interactions, receptor 425 was found to bind acetate and chloride more strongly than dihydrogenphosphate.
Biindole moieties were also incorporated into macrocycles by Jeong and co-workers.498 While the binding interactions with anions (added as the corresponding TBA salts) could also be observed using 1H NMR spectroscopy, association constants for receptors 427 and 428 were determined using UV-Vis spectroscopic titrations carried out in acetonitrile. Association constants for the binding of dihydrogenphosphate proved to be in the range of 106 M-1; however, similar values were observed for chloride and several other anions.
Several other biindole receptors (429-431) were prepared by the same group and tested in an effort to gauge the effect of incorporating additional hydrogen bond motifs into a given acyclic framework. 499 The binding constants of these receptors with TBA anion salts were measured by carrying out UV-Vis titrations in DMSO containing 0.1 - 0.2% water. While receptor 429 displayed a low affinity (K ≈ 102 M-1) for most simple anions, significantly higher affinities were observed for the elaborated receptors 430 and 431. Receptor 431 was found to bind most anions slightly more strongly than 430, although the effect was not dramatic (K (H2PO4-) = 7.5 × 105 M-1 and 1.4 × 105 M-1 for 431 and 430, respectively). Proton NMR spectroscopic studies carried out in DMSO-d6 led to the suggestion that the terminal NH groups of the urea functionalities present in receptor 431 did not participate fully in the anion binding process, thus accounting for the modest increase in observed binding affinity. However, receptor 431 was found to bind hydrogenpyrophosphate an order of magnitude more strongly than 430, providing support for the suggestion that all six hydrogen bond donors are involved in substrate recognition in this case. These receptors were found to bind acetate approximately two to three-fold less well than they bound dihydrogenphosphate. On the other hand, dicarboxylate anions were found to bind with affinities similar to those seen for dihydrogenphosphate.
To date, a number of symmetric indole-substituted clefts have been reported, and a number of groups have contributed to the development of this chemistry. For example, Sessler and co-workers combined indole moieties with quinoxaline scaffolds (vide supra) to obtain receptors 432a and 432b.500 The association of receptors 432a and 432b with anions was analyzed by carrying out UV-Vis spectroscopic titrations in dichloromethane. Reasonable selectivity was observed for dihydrogenphosphate over chloride, fluoride, benzoate, and hydrogensulfate (all studied as the TBA salts). The association constants for the binding of H2PO4- to receptors 432a and 432b were determined to be 6.8 × 103 M-1 and 2.0 × 104 M-1, respectively. In the case of dihydrogenphosphate binding to receptor 432b the corresponding constant was 5.0 × 102 M-1 in DMSO. The higher selectivities relative to those observed with dipyrrolylquinoxaline systems were attributed to the more open conformation created by attachment at the “beta” position of the pyrrolic portion of the indole subunit. As true for the nitro-substituted dipyrrolylquinoxalines (vide supra), receptor 432b displayed a visible color change in the presence of fluoride and dihydrogenphosphate.
Indole clefts containing a variety of ancillary recognition motifs were reported by Gale and coworkers. Receptor systems 433 and 434, for example, incorporate pyridine, amide, urea, and thiourea moeties.501,502 The interaction of receptors 433 with anions (TBA salts) was studied by 1H NMR spectroscopic methods in DMSO/water mixtures. The highest affinities were observed for fluoride. However, receptor 433b displayed an association constant of 1.14 × 103 M-1 with dihydrogenphosphate in 0.5% water. This value dropped to 2.6 × 102 M-1 in 5% water. Interestingly, a much lower association constant was observed for the interaction between receptor 433a and dihydrogenphosphate. The small library of compounds 434 was screened for binding to acetate, dihydrogenphosphate, benzoate, and chloride. The selectivity of receptors 434a-c was found to be a function of anion basicity, with each receptor displaying similar binding constants for dihydrogenphosphate (3 × 102 – 4 × 102 M-1) in DMSO/0.5% water as determined from 1H NMR spectroscopic titrations. Receptors 434d-f were all found to bind anions with essentially the same selectivity. However, as compared to the previous receptors, these compounds were found to be more effective binding agents. The actual binding affinities followed the trend 434d < 434f < 434e. The most effective receptor, the urea compound 434e, was found to bind phosphate with an association constant of 4.95 × 103 M-1.
Diindolylureas 435 were later synthesized by this same group and analyzed as anion receptors.503,504 In general, these urea-bridged receptors displayed a preference for dihydrogenphosphate as inferred from 1H NMR titrations carried out in DMSO-d6/0.5% water. The diindolylthioureas 435c and 435d were found to have only a moderate affinity for the H2PO4- anion (K = 3830 M-1 for receptor 435c and 1630 M-1 for receptor 435d (studied as the TBA salt)). On the other hand, the diindolylurea receptors 435a and 435b exhibited much stronger affinities for H2PO4-, with K > 104 M-1. The lower affinity of the thiourea compounds was attributed to the larger sulfur atom that served to destabilize the planar conformation in the case of receptors 435c and 435d.
Recently, amide receptors based on 1H-indole-7-amine (436-439) were prepared by Jurczak and co-workers; these systems were found to interact with dihydrogenphosphate as judged from 1H NMR spectroscopic studies carried out in DMSO-d6 / 0.5% water with TBA anion salts.505 Receptor 437 displayed the highest affinity for dihydrogenphosphate, with an association constant 2 × 103 M-1 being calculated for this solvent system.
In 2009, these researchers investigated diamido-diindolylmethane receptors 440.506 Stability constants were determined in DMSO-d6 / 5% H2O for dihydrogenphosphate, benzoate, chloride, and bromide (TBA salts). A very strong preference was observed for dihydrogenphosphate, particularly with receptor 440a. The dihydrogenphosphate:440a binding affinity proved too high to be accurately measured by this method in 5% water solutions. In 10% water DMSO-d6 solutions, however, a binding constant of 6.0 × 103 M-1 was measured for this complex; this value represented a 10-fold increase in affinity as compared to what was seen for benzoate using this same receptor (440a). Bromide and chloride displayed little interaction with any of the receptors, even in 0.5% water solutions. Receptors 440b and 440c displayed much weaker interactions with dihydrogenphosphate, a result that can be rationalized in terms of unfavorable steric interactions. The use of similar solvent conditions allowed direct comparison to the dipyrrolylmethane-based receptors 401, which were expected to share a similar hydrogen bond donor arrangement. Interestingly, receptor 440a bound dihydrogenphosphate with a significantly higher binding constant than any of the these latter systems.
Pfeffer and co-workers examined the effects of combining monoindoles with other H-bond donors. Towards this end, the Pfeffer group prepared receptors 441- 443.507 The interactions with H2PO4- (TBA salt) were then studied using 1H NMR spectroscopic titrations. This provided support for the suggestion that all available hydrogen bond donors interact with dihydrogenphosphate under these conditions. Binding constants for dihydrogenphosphate were similar (within error) for all three receptors (log K ≈ 3.5) and reasonably similar to those found for acetate. While the authors of this review are not certain, these titrations appear to have been performed in DMSO.
Later studies by Lin and co-workers demonstrated that an indole receptor bearing a phenylhydrazone moiety (444), binds dihydrogenphosphate well in DMSO (K = 2.25 × 104 M-1), as inferred from UV-Vis spectroscopic titrations carried out using the TBA salt.508 However, the highest affinity was seen for acetate, perhaps reflecting the geometrical constraints of this particular receptor.
Caltagirone and co-workers recently reported the anion binding ability of several bis-indole systems containing a bipyridine scaffold (e.g., receptors 445 - 446).509 It was expected that the coordination of platinum by the scaffold 445 would pre-organize receptor 446 so as to enhance anion binding. All anions were tested as their TBA salts. In the absence of platinum(II), receptor 445 displayed a slight selectivity for dihydrogenphosphate over fluoride, acetate, benzoate, and chloride as inferred from 1H NMR spectroscopic measurements carried out in DMSO-d6 / 0.5% water. A binding affinity of 90 M-1 for dihydrogenphosphate was reported under these conditions, and a 1:1 anion binding stoichiometry was proposed on the basis of a Job plot analysis. In the presence of platinum(II), a significant increase in binding affinity was observed (K(H2PO4-) = 3600 M-1 for 446). Phosphorous and 195Pt NMR spectroscopic studies led to the conclusion that the phosphate anion did not interact with the platinum center. As a result, it was proposed that coordination of platinum by the bipyridine moiety prevented free rotation around the pyridine-pyridine bond, which led to pre-organization of the hydrogen bonding indole cleft.
Bis-amidoindole systems 447 - 448 were recently reported by Gale and co-workers.510 Proton NMR spectroscopy titrations in DMSO-d6-0.5% water allowed for the calculation of binding constants between these receptors and a number of monoanions (TBA salts). On this basis, it was concluded that receptors 447a, 448a, and 448b bind dihydrogenphosphate over fluoride, acetate, benzoate, and chloride. Receptor 447b on the other hand displayed a preference for acetate over dihydrogenphosphate. This change in binding behavior was attributed to steric interactions between the dihydrogenphosphate anion and the nitro groups within the binding cleft of receptor 447b. This conclusion was further supported by the formation of 1:2 host:guest complexes with nitro-substituted receptor 448b while all other complexes displayed 1:1 stoichiometries. The strongest single binding constant was reported between receptor 447a and dihydrogenphosphate (K = 260 M-1). Interestingly, receptor 447a was found to bind a dihydrogenphosphate dimer in the solid phase as deduced from a single crystal X-ray diffraction analysis.
In 2009, Jurczak and co-workers reported the anion binding properties of a series of novel isoindole-based receptors (449).511 Intense color changes were produced upon the addition of basic anions (acetate, benzoate, dihydrogenphosphate, and fluoride) as the TBA salts in DMSO solution. No significant changes were observed for the less basic chloride anion. Titrations with the series of anions was performed by both UV-Vis spectroscopic and 1H NMR spectroscopy in DMSO / 0.5% water. Analysis by both methods led to the conclusion that the more basic anions serve to deprotonate the receptors under these conditions. This phenomenon prevented the determination of accurate binding constants.
4.2.4. Other Hydrogen Bonding Systems
Several other types of hydrogen bonding motifs have been used for phosphate recognition. Included in these are imidazolidone-based receptors 450-451 prepared by Kang and co-workers.512 Proton NMR and UV-Vis spectroscopic titrations carried out in DMSO:CH3CN (1:9) provided evidence that the urea-based receptor 450 binds anionic guests more strongly than does the corresponding amide receptor 451 (anions studied as the TBA salts). Such a finding is consistent with what was seen for other receptor systems (see previous discussions). Interestingly, receptor 450 was found to interact more strongly with acetate, whereas receptor 451 was found to display a preference for dihydrogenphosphate (K (H2PO4-) = 6.8 × 102 M-1).
Peng, Han, and co-workers incorporated a benzimidazole unit into a fluorophore (452) and used it to effect colorimetric and fluorescent anion sensing.513 UV-Vis and fluorescence spectral changes were observed upon the addition of fluoride, acetate, and dihydrogenphosphate (TBA salts) in acetonitrile. The degree of spectral change was found to correlate with basicity, such that fluoride > acetate > dihydrogenphosphate. No changes were observed upon the addition hydrogensulfate, chloride, and bromide. Proton NMR spectral studies carried out in CD3CN led to the conclusion that these anions formed hydrogen bonds with receptor 452 when up to one equivalent of anion was added. However, further anion addition was found to lead to deprotonation of the benzimidazole hydrogen atom.
A series of benzimidazole receptors were analyzed by the Jang group. Their first system, receptor 453, was found to undergo a quenching of fluorescence when exposed to dihydrogenphosphate and a variety of other anions added as the TBA salts.514 An association constant of 2.01 × 105 M-1 was determined for receptor 453 and dihydrogenphosphate in acetonitrile. This relatively high affinity was attributed to the formation of four hydrogen bonds between the receptor and the anion. Receptors 454, also produced by Jang and co-workers, were found to bind dihydrogenphosphate in addition to acetate and fluoride in a 9:1 CH3CN:DMSO solution.515 The interaction between H2PO4- and receptor 454a was quantified by UV-Vis spectroscopy, giving rise to a K of 3.3 × 103 M-1. On the other hand, receptor 454b displayed a significantly larger association constant of 3.1 × 105 M-1 for this same substrate. The stronger binding interaction with receptor 454b was unexpected. This is because the nitro groups on compound 454a were expected to increase the acidity of the hydrogen bonding functionalities. Proton NMR spectroscopic titrations led to the suggestion that DMSO may occupy half of the binding sites in receptor 454a, leaving fewer hydrogen bonds available for anion interactions.
In 2009, these researchers developed the benzothiazole-based cleft system 455.516 This receptor displayed strong fluorescence quenching upon the addition of dihydrogenphosphate in a solvent mixture of 98:1:1 CH3CN:DMSO:water containing a HEPES buffer. An association constant of 7.9 × 103 M-1 was determined using this method. Little change in emission intensity was observed upon the addition of fluoride, acetate, hydrogensulfate, hydrogenphosphate, trianionic phosphate, iodide, bromide, chloride, or nitrate. This high selectivity was attributed to the specific combination of both hydrogen bond donating and accepting moieties within the cleft. Strong binding within the cleft of receptor 455 was further supported by the absence of significant anion binding interactions with receptor 456 under these conditions. Molecular modeling studies and 1H NMR titration experiments also supported the proposed binding mode.
Concomitantly, T.H. Kim and co-workers reported colorimetric anion recognition using the benzothiazole-based receptor 457.517 A solution of this receptor in acetonitrile underwent a light to dark yellow color change upon the addition of acetate, fluoride, and dihydrogenphosphate (studied as their TBA salts). No color change was observed upon the addition of chloride, bromide, iodide, or hydrogensulfate. The intensity of the color change was observed to correlate with the basicity of the anion. Proton NMR spectroscopic studies in DMSO-d6 led to the suggestion that the resultant color change was due to deprotonation of the NH proton of receptor 457. ITC studies further supported this conclusion.
Another subunit that has been explored in dihydrogenphosphate anion recognition is oxadiazole. This moiety was used by Wang and co-workers to prepare the hydroxyphenyl-oxadiazole receptors 458.518 The free receptors displayed both short and long wavelength emission bands, with the long wavelength bands being attributed to an excited state intramolecular proton transfer (ESIPT) process wherein a phenol proton is transferred to an oxadiazole nitrogen atom. This long wavelength band becomes quenched upon the addition of TBA dihydrogenphosphate, while the short wavelength band increases in intensity. The intensity changes were attributed to a decrease in proton transfer due to hydrogen bonding between the anion and the phenolic proton. Higher binding constants and increased selectivity were seen for receptor 458b. In this case an association constant of 1.8 × 106 M-1 was recorded for the binding of dihydrogenphosphate (TBA salt) in DMF. Lower association constants of 4.1 × 104 M-1 and 5.0 × 102 M-1 were reported for the fluoride and chloride anions, respectively. Visual changes from colorless to yellow were observed for both receptors in the presence of dihydrogenphosphate and fluoride.
A series of azole containing peptide macrocycles (459-460) were studied by Haberhauer and co-workers in 2009.519 Proton NMR spectroscopy supported the presence of hydrogen bonding between the amide NH group and azole nitrogen atom, which was expected to preorganize the binding cavity in the conformation shown. The anion binding ability of these macrocycles was evaluated by means of 1H NMR spectroscopic titrations carried out in DMSO-d6 / 5% CDCl3 using the TBA anion salts. On the basis of Job plot analyses, all anions were found to bind as 1:1 host:guest complexes. Anion binding affinities of receptors 459 were shown to correlate with the acidity of the azole nitrogen atom such that the thiazole receptor 459c bound anions more strongly than oxazole receptor 459b, followed by the imidazole receptor 459a. The lowest binding affinities were generally observed with receptor 460, a finding that was attributed to the increased size of the macrocycle interior. In general, affinities for different anions were found to correlate with the size and basicity of the guest. As a result, the strongest binding constants were reported for dihydrogenphosphate, acetate, and fluoride over hydrogensulfate, toluene sulfonate, methyl sulfonate, chloride, nitrate, bromide, iodide, and perchlorate. The preference for dihydrogenphosphate over all other anions, however, did not fit this trend. The observed selectivity was attributed to the formation of addition hydrogen bonds between the phosphate hydrogen atoms and the azole nitrogen atoms. As a result, the highest dihydrogenphosphate:acetate selectivity (10-fold) was observed for the more basic imidazole receptor 459a. The strongest binding constant was recorded for the 459c:dihydrogenphosphate complex (K = 3.0 × 104 M-1).
As seen in previous sections, pyridine subunits have been extensively exploited in the design of phosphate anion sensors. Their utility reflects both acid-base and hydrogen bonding properties. The ability of pyridine (and related quinolines) to form salts was supplemented in early work by creating receptors, such as 461, 462, and 463, that provide for the preorganized placement of hydrogen bond donor groups. Preliminary studies provided a confirmation that receptor 461a facilitated the precipitation of FMN from a biphasic aqueous:chloroform mixture.520 Salt formation between 461a and FMN was inferred from the fluorescence quenching of FMN seen in the presence of receptor 461a in apolar media. Such quenching was not observed with compound 461b. Later, 1H NMR spectroscopic studies of receptor 462a and dodecylphosphoric acid in methanol led to the suggestion that salt formation involving the pyridine subunit, as well as hydrogen bonding interactions with the hydroxyl groups, contributed to the binding behavior. An association constant of 1.2 × 103 M-1 was calculated under these conditions.521 Titrations with receptor 462b produced similar results. In a separate study, analogous interactions were observed between receptor 463 and methyl phenylphosphonic acid.522 In this case, an association constant of 7.1 × 102 M-1 was calculated from 1H NMR spectroscopic titrations carried out in chloroform. However, direct comparisons between these receptors were made difficult due to the different targets and solvents employed. Single crystal X-ray diffraction analysis of receptor 463 and methyl phenylphosphonic acid confirmed complex formation and revealed the presence of three hydrogen bonds.
The phosphate binding properties of a bispyridine quinoxaline derivative were reported by Kruger and co-workers in 2001.523 The protonated form of receptor 464 (464-H+) displayed luminescence that was quenched by a variety of anions added as the TBA salts in acetonitrile. The proton was thought to be held between the pyridine units through a strong intramolecular hydrogen bond. This, it was suggested, would serve to increase the planarity of the receptor and enhance the preorganization of the anion binding cleft. Dihydrogenphosphate was the most strongly bound of the anions tested (including hexafluorophosphate and the halides), being bound with a K of 2.15 × 104 M-1 in acetonitrile. However, the emission intensity was greatly decreased in aqueous solutions and receptor 464-H+ was not a useful anion sensor in this medium.
Lu and co-workers developed the tetrathiofulvalene (TTF) amide system 465 with the goal of producing an electrochemical sensor.524 Proton NMR spectroscopic studies and computational modeling supported a 1:2 dihydrogenphosphate:465 binding stoichiometry. It also provided evidence for a complex in which each receptor contributes to three kinds of hydrogen bonds involving the pyridine nitrogen, amide NH, and the TTF C=C-H units. Cyclic voltammetry (CV) experiments carried out in dichloromethane revealed significant perturbation upon the addition of dihydrogenphosphate, while no significant change was observed in the presence of acetate, sulfate, or the halides (all species studied as the corresponding TBA salts). This lack of response in the case of competing species provides support for the high selectivity of this receptor for dihydrogenphosphate.
In early work, various heterocycles, tested as off-the-shelf, naked-eye detectable anion sensors by Sessler and Miyaji, were applied to phosphate detection.525 Of the indicator systems tested, compounds 466-475 all displayed a visible colorimetric change in dichloromethane upon the addition of dihydrogenphosphate as the TBA salt. For example, receptor 466 displayed a dramatic yellow to purple color change in the presence of 100 molar equivalents of dihydrogenphosphate. A color change was also seen with fluoride. This signal modulation was thought to be due to the formation of charge-transfer complexes as a result of hydrogen bonding. Although absolute specificity for phosphate over fluoride could not be achieved by a single readily available compound, a multi-sensor array was suggested as a possible approach to anion sensing at neutral pH.
The benzylidene malonitrile derivatives 477-478 were reported by Zhou and co-workers in 2005; these species were observed to interact via hydrogen bonding with dihydrogenphosphate, as determined from UV-Vis spectroscopic studies carried out in dichloromethane with TBA anion salts.526 As expected, control compound 476, which lacks a phenol or catechol hydrogen bond donor group, did not show any spectral change upon the addition of anions. In contrast, receptor 477 displayed a visible color change in the presence of fluoride and dihydrogenphosphate with association constants of 1.24 × 104 M-1 and 3.8 × 103 M-1 being derived for these two species, respectively. These two anions could be visually differentiated, however, via the addition of a small amount of protic solvent; this served to change the spectrum of the fluoride complex but had little effect on the spectrum of the dihydrogenphosphate complex. Little or no change was observed with other anions, including chloride, acetate, and hydrogensulfate. Compound 478 also displayed a color change in the presence of dihydrogenphosphate. However, a stronger color change was observed for the acetate and fluoride anions.
The dinitrophenylhydrazone sensors 479-480, produced by Ou and co-workers, also displayed significant color changes in the presence of dihydrogenphosphate and fluoride anions (TBA salts).527 The interactions were followed by UV-Vis titrations in DMSO. On the basis of these studies it was determined that receptor 479 bound fluoride more strongly than dihydrogenphosphate while the reverse was true for receptors 480. While the binding constants for dihydrogenphosphate were similar for all three 480 derivatives, the best selectivity was found with receptor 480c. This receptor bound dihydrogenphosphate 20-fold more strongly than fluoride, displaying a dihydrogenphosphate binding constant of 2.37 × 105 M-1. It is also noteworthy that receptors 480 displayed different wavelength shifts depending on the anion added, and this difference could be observed with the naked eye. Proton NMR spectroscopic studies led to the suggestion that fluoride deprotonated the receptors while dihydrogenphosphate only interacted through hydrogen bonding. Interestingly, chloride, bromide, iodide, and nitrate produced no observable change, as determined using UV-Vis or 1H NMR spectroscopies.
The anion recognition properties of hydrazone were investigated by Shang and Xu in 2009.528 UV-Vis and fluorescence spectroscopic studies in DMSO revealed interactions of dinitro-receptor 481c with F-, AcO-, and H2PO4- anions (added as TBA salts). Upon anion binding, a bathochromic shift in absorbance maxima was observed, along with a quenching of the inherent fluorescence of receptors 481. In addition, visual yellow-to-violet color changes were observed when F-, AcO-, and H2PO4- anions were added to receptor 481c. At the same time, receptor 481b displayed spectral changes only in the presence of F- and AcO-, and no anion interactions were observed for receptor 481a. The increased binding ability of 481c relative to 481a and 481b was attributed to an increased acidity of the hydrazone moiety, presumably due to the increased number of electron withdrawing nitro substituents. Addition of Cl-, Br-, and I- anions did not lead to spectral changes for any of the studied receptors. In the case of receptor 481c, the largest affinity constants were reported for AcO- (K = 2.1 × 105 M-1) and F- (K = 4.0 × 104 M-1), while H2PO4- was found to bind one to two orders of magnitude less well (K = 1.3 × 103 M-1). This selectivity was attributed to the high basicity of F- and geometric complementarity of the Y-shaped acetate anion. In addition, visual color changes were observed when F-, AcO-, and H2PO4- anions were added to receptors 481b and 481c.
Also in 2009, Lin and co-workers reported a system in which p-nitrophenylhydrazone moieties were appended to a ferrocene unit in hopes of forming a cleft for anion recognition (482).529 The absorption spectrum of receptor 482 was monitored upon the addition of a number of anions as the TBA salts in DMSO. A decrease in the intensity of the maximum absorption was observed upon addition of acetate, dihydrogenphosphate, hydroxide, and fluoride. No significant change was observed upon the addition of chloride, bromide, or iodide. The change in the absorption spectrum was then used to determine association constants, which were found to bind in the order of acetate > hydroxide > fluoride > dihydrogenphosphate. A 24-fold increase in the affinity was observed for acetate (K = 3.91 × 104 M-1) over dihydrogenphosphate (K = 1.6 × 103 M-1) in this system. The strong preference for acetate was attributed to the increased basicity of this anion relative to dihydrogenphosphate.
Another motif that has been used for phosphate recognition is squaramide. This subunit was incorporated into receptors 483-486 by Costa and co-workers. These systems were then tested for their anion recognition ability in a colorimetric indicator displacement assay (IDA) using Cresol red as the indicator.530 While the addition of the dianions HPO42- and SO42- both led to a positive IDA response, competitive UV-Vis titrations carried out in an ethanol:water mixture (9:1) confirmed that receptors 483-486 displayed selectivity toward sulfate as opposed to phosphate. In the specific case of 485, a relative SO42-/HPO42- selectivity ratio of 2.28 was found under these conditions. In this case, the anions were studied as their sodium salts.
Receptors that rely on oxygen-based hydrogen bond donor units have also been reported, specifically the carbonyl and hydroxyl motifs. For example, a reaction-based approach to phosphate detection was developed by Sancenón and co-workers.531 It relies on the use of the 1,5-pentanedione derivatives 487 and 488. These compounds normally undergo cyclization to form a pyrylium cation at pH 2-5, a transformation that results in a color change from yellow to magenta. However, in the case of receptor 487, the addition of ATP allowed this color change to be visible at pH 6 in 7:3 dioxane:water. Sulfate also produced a small color change, but no other anions tested, including chloride, bromide, phosphate, GTP, and ADP, produced such a visual change at this relatively high pH. In the case of 487, a linear response range was observed from 40 ppm to 1100 ppm for ATP. With receptor 488, dramatic visual responses were observed for sulfate, ADP, and ATP. These shifts were attributed to hydrogen bonding between the carbonyl units of the receptors and the anions, which in turn was thought to facilitate the ring closing reaction. The addition of several inorganic cations had no effect on these reactions. Nucleotides were studied as the sodium salts and inorganic anions were studied as the TBA salts.
A unique system was reported by Zhang and Wu in 2004. It relies on the use of the magnesium complex 489, which undergoes a naked-eye detectable color change upon treatment with TBA hydrogensulfate and TBA dihydrogenphosphate in acetonitrile.532 These findings were rationalized in terms of a combination of electrostatic interactions involving the pre-bound Mg(II) center and hydrogen bonding with the carbonyl oxygen. The binding behavior was examined using UV-Vis and fluorescence spectroscopy in acetonitrile. However, no association constant with dihydrogenphosphate was reported.
The hydroxyl group has also been investigated as a hydrogen bond donating group. For example, Davis and co-workers reported the solubilization of hydrogenphenylphosphonate (TBA+PhP(OH)O2-) in organic solvents by a number of cholic acid derivatives (490 - 491) in 1998.533 Preliminary proton NMR spectroscopic studies in benzene-d6 revealed solubilization of the guest in the presence of cholate 490a. In a deuterated 1:1 benzene:hexane mixture, increased solubilization was found to correlate with increasing hydroxyl substitution such that cholates 490b > 490c ≈ 490d and no solubilization was observed with cholate 490e. In addition, alkene derivative 491 was found to be as efficient as 490a at solubilizing the phosphonate guest in benzene. These results, as well as 1H NMR spectral shift analysis, were considered consistent with strong hydrogen bonding interactions between the anionic guest and the cholate OH groups with little participation from the carbonyl oxygen atom of cholates 490.
In 2002, Kondo and co-workers further investigated the hydroxyl group as a hydrogen bond donor in anion recognition.534 The anion binding ability of receptors 492 and 493 were compared through 1H NMR spectroscopic experiments carried out in deuterated acetonitrile. All anions tested (TBA salts of AcO-, H2PO4-, HSO4-, Cl-, Br-, I-, and ClO4-) displayed a 1:1 binding stoichiometry as inferred from Job plot analyses, with the exception of perchlorate for which no binding was observed. Significant shifts in the signals corresponding to the OH group were consistent with the participation of hydroxyl-derived hydrogen bonds in the recognition of anions by receptors 492. This conclusion was further supported by the stronger binding constants determined with receptor 492 as compared to receptor 493. The actual values were found to correlate well with the basicity of the anion in question, with a preference for acetate being reported. Binding constants for receptors 492 and 493 for dihydrogenphosphate were determined to be 1.89 × 103 M-1 and 2.09 × 102 M-1, respectively.
4.3. Metals in Phosphate Recognition
4.3.1. Metal Cation Coordination
Metals cations are commonly found in the binding sites of phosphate-binding proteins.535-545 Their presence and the interactions they sustain inspired many of the receptors in this section. As will be detailed in greater length below, many of these receptors effect phosphate anion recognition as the result of dative coordinative bonds between the metal cation of the receptor and a negatively charged oxygen atom present on the phosphate group of the guest of interest.546 Although additional functionalities can be applied to increase specificity, metal coordination of the anion is generally the greatest contributor to the stability of the complexes formed from this class of receptors. As a result, the discussion will center around the metal atom employed. Further, to provide an organizational structure, the metals will be discussed in order of atomic number, at least to the greatest extent possible.
We begin our discussion with vanadium complexes, which, like many other transition metal complexes, have been employed in the membranes of ion-selective electrodes. In 2003, Ganjali and co-workers found that inclusion of vanadyl salen complex 494 in a PVC membrane allowed for the creation of a monohydrogenphosphate selective electrode.547 A control electrode without ionophore 494 did not display a response to the anions tested (SCN-, SO4-, SO32-, ClO4-, I-, Br-, CO3-, tartrate2-, NO3-, HPO42-). UV-Vis binding studies in acetonitrile with compound 494 provided support for axial coordination of monohydrogenphosphate. No changes in the absorption spectrum were observed with the other anions tested. The electrode gave a linear response from 1.0 × 10-1 to 5.0 × 10-6 M at pH 8.2 with a response time of 25 seconds. The vanadyl salophen complex 495 was also analyzed as a component in a PVC membrane.548 The resulting electrode proved selective for monohydrogenphosphate, displaying a similar response range to that found for the electrode based on ionophore 494. However, it displayed an improved response time (less than 20 seconds). In addition, the lifetime of the electrode based on complex 495 was determined to be 14 weeks. Membranes containing complexes 494 and complex 495 were further used to measure successfully the percentage of monohydrogenphosphate in commercial fertilizer.
The manganese(II) dipicolylamine (Mn(Dpa)) derived receptor 496 was found to interact selectively with pyrophosphate and ATP in aqueous solution as reported by Yoon and co-workers.549 Upon addition of manganese to the free base Dpa receptor, chelation-enhanced fluorescence quenching (CHEQ) and a yellow-to-pink colorimetric response were observed in aqueous solution at pH 7.4. A reversal of these effects (i.e., enhanced fluorescence and a corresponding opposing color change) was observed when pyrophosphate or ATP were added to the Mn2+ complex. Little or no change was observed upon the addition of ADP, AMP, H2PO4-, HSO4-, or CH3CO2-. In addition, these researchers found that the measured association constant corresponding to the binding of pyrophosphate to receptor 496 (4.2 × 104 M-1) was unaffected by the presence of up to 50 equivalents of H2PO4-. While additional structural studies were recognized as necessary to determine the precise binding modes, control experiments led to the suggestion that the manganese remains coordinated to receptor 496 and that the metal center then facilitates binding of the phosphate anions. No spectral changes were observed when the free base form of receptor 496 was treated with pyrophosphate in the absence of a metal cation. It is noteworthy that while several other metal anions led to CHEQ, no spectral changes were observed when anions were added to complexes of metals other than manganese.
D. H. Kim, J. Chin, and co-workers studied the dimethylphosphate binding ability of cobalt(III) receptors 497.550 As determined from single crystal X-ray diffraction studies, the coordinated water of receptor 497a was found to be displaced by the negatively charged oxygen atom of dimethylphosphate (Figure 7). Evidence for a strong hydrogen bond between the anilinic NH group of receptor 497a and the coordinated phosphate oxygen was also found. This binding mode was further supported by 1H NMR spectroscopic analysis in D2O. Little binding was observed with receptor 497b, presumably due to the absence of the additional hydrogen bond donor group present in receptor 497a. In line with these observations, equilibrium constants for receptors 497a and 497b were found to be 210 M-1 and 6.2 M-1, respectively, via 1H NMR spectroscopy at 80 °C. Fluoride was found to bind receptor 497a with an affinity similar to that displayed by dimethylphosphate, while chloride and bromide interacted more weakly with this complex. All anions were studied as their sodium salts.
Figure 7.
View of the 497a:dimethylphosphate complex. Drawing generated from X-ray diffraction data originally published in reference 550. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
Through a series of detailed studies, the Martell group analyzed the phosphate binding behavior of the protonated states and metal complexes of the polyamine ligands 498 and 501.551 The associated interactions were studied over a pH range of 2 – 11 using potentiometric methods unless otherwise mentioned. In 1992, equilibrium constants in the range of log K = 1.5 – 7.0 were measured for the interaction of phosphate and several protonated forms of macrocycle 498.552 The most noteworthy results of these studies involved the complex of the hexaprotonated form of compound 498 and dihydrogenphosphate and the complexes of the di- through hexaprotonated forms of compound 498 with monohydrogenphosphate. When the copper(II) complexes of receptor 498 were studied, it was found that the mononuclear complex (499:Cu) displayed equilibrium constants an order of magnitude higher than the free ligand 498, at least when comparing systems bearing equivalent charges. The related dinuclear complex (500:Cu2) displayed similar equilibrium constants (log K ≈ 4.0) as did the diprotonated form of the mononuclear (499:Cu) complex which bears a like charge. Further studies were performed to investigate the binding behavior of complex 499:Cu and the dimetalated derivative 500:Cu2 with pyrophosphate.553 In this study, complex 500:Cu2 was found to interact with pyrophosphate more strongly (log K = 2.25) than the diprotonated (equally charged) form of complex 499:Cu. Molecular mechanics calculations involving complex 500:Cu2 led to the suggestion that the receptor was rather flexible and that the pyrophosphate guest resided above the plane of the ligand rather than between the two coordination sites. Similar studies analyzed the interactions between the cobalt(II) complexes of 498 (499:Co and 500:Co2) and dianionic phosphite and monohydrogenphosphate.554 The equilibrium constants corresponding to the formation of phosphite complexes with receptors 498 were found to be approximately an order of magnitude higher than those for the interaction with monohydrogenphosphate. In studies of the related cobalt(II) complexes, the equilibrium constants for the interaction between the monocobalt complex 499:Co and both dianionic phosphite and monohydrogenphosphate were found to be approximately 0.5 log units higher than the respective equilibrium constants for the diprotonated and hence equally charged forms of macrocycle 498.
The binding interactions between copper(II) complexes of furan-containing macrocycle 501 and its metalated species 502:Cu, 503:Cu2 with pyrophosphate were also investigated.555,556 Based on the results of a prior study,179 it was concluded that coordination of copper(II), giving rise to 502:Cu or 503:Cu2, led to a significant increase in the anion binding affinities. Specifically, the equilibrium constants for the copper complexes were found to be two to three orders of magnitude higher than the respective association constants for the protonated forms of receptor 501 (at equal charge). The equilibrium constant for the interaction of pyrophosphate with the dinuclear complex 503:Cu2 (log K = 7.73) was found to be approximately 0.4 log units higher than the equilibrium constant for the equivalent interaction with diprotonated 502:Cu.
Interestingly, the incorporation of the aromatic units appeared to significantly affect the binding affinities. For example, the equilibrium constants corresponding to the interaction of phosphate with complex 502:Cu were found to be two-fold higher than the corresponding equilibrium constants reported for complex 499:Cu with respect to the same anion. However, the dinuclear 500:Cu2 complex displayed a higher affinity for pyrophosphate then did the corresponding furan-containing 503:Cu2 complex. Single crystal X-ray diffraction analysis of the two complexes led to the suggestion that, in the absence of an anion, the distances between the two copper centers of receptors 500:Cu2 and 503:Cu2 were approximately equal, at least in the solid state. These results provided support for the conclusion that the steric bulk of the furan rings interfered with the binding behavior of complex 503:Cu2.
Later, single crystal X-ray diffraction analysis of complexes of free base furan-containing receptor 501 and dihydrogenphosphate, pyrophosphate, or triphosphate led to the suggestion that the observed anion binding reflected a favorable combination of electrostatic interactions and hydrogen bonds.557 Solution phase studies provided support for the conclusion that monophosphate interacts less well with receptor 501 than pyrophosphate. The interaction of pyrophosphate with receptor 501 was in turn weaker than that of triphosphate at a similar level of receptor protonation. The results were in line with the notion that electrostatic interactions played a critical role in regulating the binding affinity. Interestingly, this direct correspondence with electrostatic effects did not hold true across the board for the copper(II) complexes (502:Cu, 503:Cu2). In analogy to what proved true for receptor 501, the triphosphate complexes generally proved more stable at equal protonation states than the corresponding pyrophosphate complexes. On the other hand, the greater basicity of pyrophosphate led to higher equilibrium constants with the unprotonated and monoprotonated states of 499:Cu and with the monoprotonated state of 503:Cu2 as compared to the respective complexes with triphosphate. Only the dinuclear complexes 503:Cu2 were found to interact with monophosphate, and even here the association constants were found to be relatively low.
Copper(II) complexes of expanded macrocycle 504 were also found to interact with pyrophosphate.558 In the absence of copper, analysis of the free base macrocycle 504 revealed equilibrium constants approximately two orders of magnitude lower than those observed for the pyrophosphate complexes of the furan macrocycle 501. However, the copper(II) complexes of macrocycle 504 (505:Cu and 506:Cu2) were found to interact with pyrophosphate significantly more strongly than the respective complexes of 501.
Similar trends were found for phenyl-containing macrocycle 507 and its metal complexes.559 In addition, direct competition experiments led to the conclusion that the dinuclear complex 509:Cu2 interacts more favorably with pyrophosphate than it does with monophosphate near neutral pH. A slight selectivity for triphosphate over pyrophosphate by this complex was also found under these conditions. The mono- and dinuclear macrocycles (508:Cu and 509:Cu2) were also found to interact with ATP and ADP.182
In a review of the phosphate binding behavior of the protonated and copper(II) complexes of macrocycles 498, 501, and 507, Martell and co-workers proposed similar binding modes for all three macrocycles.82 In the mononuclear complexes, a covalent metal bond was available, as well as up to three hydrogen bonds, modes that were thought to give rise to strong receptor:anion interactions. The dinuclear complexes occupied all of the available nitrogen sites, leaving one or two available coordination sites at each metal center. The highest binding for mononuclear complexes was achieved when the three remaining nitrogen atoms were protonated and the bridging phosphate group remained unprotonated. Presumably, this finding reflects the fact that such an arrangement serves to maximize both coulombic and hydrogen bonding interactions. The dinuclear metal complexes were found to interact with phosphate anions most strongly when the phosphate group(s) were unprotonated and when a greater number of negative charges were available (i.e., triphosphate was found to bind more strongly than pyrophosphate). Similar trends were observed for the nucleotides ATP and ADP with respect to triphosphate and pyrophosphate. For example, it was predicted that only two of the available phosphate groups of both ATP and triphosphate interacted with the macrocyclic receptors based on distances measured during single crystal X-ray diffraction analysis. This conclusion was further supported by the observation that monoprotonation of these two anions did not produce a significant change in the equilibrium constant of the complexes.
Several research groups have expanded on these studies. For example, Herman and co-workers also examined the interactions between 508:Cu and 509:Cu2 and pyrophosphate and monophosphate.560 The use of different conditions from the Martell studies, however, precludes a direct comparison. Nevertheless, quantitatively similar trends were observed. In addition, a preference for pyrophosphate over malonate, maleate, and fumarate was observed.
Fabbrizzi and co-workers utilized receptor 509:Cu2 as the host in a pyrophosphate-selective indicator displacement assay in neutral aqueous solution.561 Three fluorometric indicators were used for these studies. The emission of all three was completely quenched when mixed with receptor 509:Cu2 in buffered water (HEPES, pH 7). For example, addition of inorganic phosphate or pyrophosphate to a complex of coumarin 343 and receptor 509:Cu2 led to a strong increase in emission. This displacement allowed for quantification of the total phosphate concentration. Fluorescein, on the other hand, was found to be displaced by only pyrophosphate. This produced a ca. 10-fold increase in the fluorescence intensity when pyrophosphate was tested in the 2-20 μM range. No interference was observed upon the addition of sulfate, chloride, cyanate, acetate, benzoate, azide, or inorganic phosphate. Eosine Y displayed an even stronger signal upon displacement. The equilibrium constant measured for the interaction of receptor 509:Cu2 and pyrophosphate (log K = 7.2) was at least three orders of magnitude higher than the log K values measured for inorganic phosphate or other tested anions.
Marcotte and Taglietti designed the larger macrocycle 510 with the goal of selectively sensing ATP using an indicator displacement ensemble.562 Through force field calculations, the copper-copper distance was estimated to be 10.5 Å. The authors proposed that this was both i) an appropriate distance for the two-point coordination of triphosphate and ii) a distance that was too large to allow for the two-coordinate binding of pyrophosphate without imposing severe strain. The most effective indicator for the sensing ensemble was found to be 6-carboxytetramethylrhodamine (6-TAMRA). The binding interactions between complex 510 and inorganic phosphate, pyrophosphate, triphosphate, and their corresponding adenosine nucleotides (AMP, ADP, ATP) were analyzed at pH 7 in aqueous media. The resulting binding affinities for complex 510 and inorganic phosphate or pyrophosphate were found to be similar to the binding affinities measured for these analytes in the case of complex 509:Cu2. The binding affinity of complex 510 and triphosphate, however, was found to be slightly higher (log K = 8.0) than the binding affinity measured for complex 509:Cu2 and triphosphate. The binding constants of the nucleotides and their corresponding phosphates were generally similar. An exception was observed for AMP, whose equilibrium constant with complex 510 was found to be nearly 0.5 log units higher than that for inorganic phosphate. This result led to the suggestion that the adenine plays a modest role in mediating the binding interactions of ATP and ADP but that, in the case of AMP, the base interacts with one of the copper centers. By using the indicator displacement assay described above, ATP could be selectively identified among other common neurotransmitters through visual inspection of the sample upon illumination from a simple laboratory UV lamp.
In 2003, the smaller azamacrocycle 511 (16aneN6:Cu2) was analyzed by Ren and co-workers for its phosphate binding properties.563 In HEPES buffer at pH 7.4, binding constants on the order of 103 M-1 were measured for inorganic phosphate and phosphate monoesters by monitoring the copper absorption bands via UV-Vis spectroscopy. No binding interaction was detected with fluoride, nitrate, bicarbonate, or benzoate under these conditions. Single crystal X-ray diffraction analysis with phosphate monoesters revealed two phosphate bridging structures, one on each side of the macrocyclic plane (Figure 8). However, the use of different buffer systems and different measurement methods precluded a direct comparison with the previous systems.
Figure 8.
View of the 511:glycerol 2-phosphate complex. Drawing generated from X-ray diffraction data originally published in reference 562. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
In the same year, Anslyn and co-workers found that the copper complexes 512 and 513 allowed the binding behavior of inorganic phosphate to be followed using UV-Vis spectroscopy and without the need for an indicator.564 Strong binding interactions were measured for complex 513 in neutral aqueous (98:2 water:methanol) solutions, with a K of 1.5 × 104 M-1 for inorganic phosphate. While similar binding was observed for arsenate, no appreciable binding was observed for a variety of other anions, including sulfate. Stronger binding interactions but lower selectivities were seen in the case of receptor 512. ITC was also used to analyze the thermodynamics of binding in the case of complexes 512 and 513. These studies provided support for the conclusion that metal ligation was the dominant contributor to the binding interaction. Interestingly, similar Gibbs free energies were measured for the interactions of both receptors with inorganic phosphate.565 The receptor-phosphate interactions for complex 512 were found to be entirely entropy driven while phosphate interactions with complex 513 were found to be primarily enthalpy driven. These differences in the thermodynamic parameters were attributed to solvation. Since the ammonium groups of complex 512 would be more effectively solvated, the binding of phosphate would result in a higher enthalpic cost as well as a higher entropic gain as a result of the release of these solvent molecules. Conversely, the lower charge density of the guanidinium groups and the reduced flexibility of complex 513 were assumed to lower both the enthalpic loss and entropic gain upon solvent release.
Using an indicator displacement assay with carboxyfluorescein, receptor 513 was later used in the colorimetric determination of phosphate in protein-free horse serum and human saliva.566 The concentrations of phosphate measured using receptor 513 were comparable to the concentrations determined using a commercially available kit.
The same group analyzed the highly-preorganized “clam-shell” receptor 514 to bind 2,3-bisphosphoglycerate (2,3-BPG).567 The cavity of this copper complex also contains four guanidinium units that were designed to provide geometric complementarity for the phosphate groups of 2,3-BPG. This receptor exhibited a strong binding affinity for 2,3-BPG (8 × 108 M-1 in 1:1 water:methanol at pH 7.4). Good selectivity was also observed. Guests containing a carboxylic acid and only one phosphate group displayed binding affinities that were ca. 80 - 180-fold lower. Simple carboxylate and phosphate analytes were found to be bound with affinities four to five orders of magnitude lower than those observed for 2,3-BPG. Furthermore, this high affinity receptor could be used to reduce the level of available 2,3-BPG in a solution of horse red-cell hemolysate. The reduction was measured by monitoring the oxygenation level of hemoglobin, which is known to display increased oxygen binding upon a reduction in the 2,3-BPG concentration. Specifically, addition of receptor 514 allowed the oxygenation levels of hemoglobin to be modified.
Yoon and co-workers also analyzed the pyrophosphate binding ability of the copper and zinc receptors 515 and 516.568 The addition of pyrophosphate to complex 515a in aqueous solution led to both an increase in the emission intensity of the receptor and a bathochromic shift of the emission spectrum. This is especially interesting as the structurally similar Mn(II) receptor 496, described earlier and studied by the same group, displayed an emission increase without a shift in the emission maximum. The addition of other test anions, including dihydrogenphosphate, led only to a slight quenching of the emission of 515a in aqueous solution. Similar spectral changes were observed with the copper-containing receptor 515b. Association constants for the interaction with pyrophosphate were determined to be 1.68 × 105 M-1 and 9.84 × 104 M-1 for complexes 515a and 515b, respectively, in buffered aqueous solution (HEPES, 7.4). The closely related copper-based receptor 516 exhibited fluorescence enhancement upon the addition of pyrophosphate but no change in the emission wavelength. The binding constant for the interaction between receptor 516 and pyrophosphate was determined to be 6.4 × 103 M-1 under the same conditions. It is noteworthy that all three receptors were found to interact selectively with pyrophosphate in 100% aqueous solution at pH 7.4. In addition, compounds 515a and 515b could be used as ratiometric sensors. In particular, the differential response at different wavelengths allowed for the creation of a calibration curve, which was considered to facilitate the use of these sensors in biological applications.
A simpler Cu(Dpa) system (517) was reported by Hong and co-workers in 2009.569 Both UV-Vis and fluorescence indicator displacement assays were developed using pyrocatechol violet and fluorescein indicators, respectively, in 10 mM HEPES buffer at pH 7.4. These ensembles were able to detect selectively pyrophosphate over inorganic phosphate, AMP, ADP, ATP, fluoride, chloride, bromide, iodide, nitrate, and acetate.
UV-Vis titrations were used to measure a binding constant of 3.93 × 107 M-1 for the complexation of pyrophosphate by receptor 517. The binding abilities of bis-Dpa metal complexes are described in more detail in the discussion of zinc-based receptors.
Phenolic receptor 518 and the corresponding 2:1 copper(II) complex 519 were reported by Lin and co-workers.570 On the basis of UV-Vis titrations carried out in DMSO, receptors 518 and 519 were found to interact with dihydrogenphosphate. Binding constants were measured to be 2.73 × 104 and 1.15 × 104 M-1 for these two receptors, respectively. Both receptors, however, displayed stronger interactions with acetate than phosphate. The phosphate binding behavior of these receptors was presumably due to a combination of metal coordination and hydrogen bonding interactions involving the amide functionalities.
Fluorescent IDAs employing receptors 520 and the indicator eosin Y were developed by the Ahn group. These receptors were found to display varying selectivities for phosphorylated compounds depending on the identity of the complexed metal.571,572 For example, the use of Cu(II) complex 520a allowed the selective determination of phytate concentration in aqueous media. Pyrophosphate anions were found to interact with receptor 520a to a lesser extent. In contrast, the use of the Zn complex 520b allowed for the detection of IP3. In this study, metal ions not only imparted additional binding affinity but also affected the selectivity of the host complexes.
In addition to the copper and cobalt complexes previously described, polyammonium macrocycles have also been used as ligands to produce zinc-based phosphate receptors. Many of these complexes were prepared in the context of efforts to obtain phosphoester hydrolysis catalysts. Kimura and co-workers were some of the first to study the phosphate binding behavior of such zinc complexes. For example, receptor 521 was found to interact with dianionic phenylphosphate and nitrophenylphosphate with log K values of 3.5 and 3.1, respectively, as inferred from potentiometric titrations.573 A significantly lower binding affinity was observed for acetate, while no appreciable affinity was seen for bis(phenyl)phosphate esters. Zinc complexes of the larger macrocycles 522 – 524 were found to bind nitrophenylphosphate with similar association constants.574 Incorporation of a pyridine substituent yielded receptor 525.575 This host was able to fluorescently sense select anions in aqueous media (HEPES buffer, pH 7.0). Significant binding was observed for dicarboxylate anions, a number of thiol derivatives, inorganic phosphate, phospho(enol)pyruvate, and O-phospho-L-threonine. Phosphorylated derivatives displayed log K values from 2.8 - 3.7. The observed effects on the emission spectrum were attributed to a conformational change around the amine-pyridine bond upon anion complexation. More recently, these researchers incorporated styrene-functionalized complex 526 into polymers for the separation of nucleotides by high performance liquid chromatography (HPLC).576 Interestingly, this receptor monomer displayed increased binding to 4-nitrophenylphosphate and dAMP compared to receptor 522, presumably due to hydrophobic or π-surface interactions with aromatic residues present in the guests.
The bridged receptor 527 was found to bind nitrophenylphosphate with a log K value of 4.0, a modest increase compared to what was observed in the case of the monomer 522(log K = 3.3).577 Despite the small apparent advantage in binding nitrophenylphosphate, this ditopic receptor, as well as the related receptor 528, could be employed in the selective binding of deoxythymidine (dT) derivatives, including anti-HIV drugs 3′ -azido-3′ -deoxythymidine 5′ -monophosphate (AZTMP) and 3′ -azido-3′ -deoxythymidine 5′ -diphosphate (AZTDP), as inferred from ITC, potentiometric, UV-Vis, and NMR spectroscopic experiments.578 Receptors 527 and 528 were found to have higher selectivities for thymidine derivatives over other nucleotides when compared to their monomer counterpart; presumably this is due to the additional binding interactions between the dibasic phosphate and the second Zn2+ center. The log apparent binding constants at pH 7.6 for receptors 527 and 528 for thymidine mono- and diphosphate derivatives ranged from 5.2-6.4. Stronger binding interactions were observed with para-substituted receptor 528, a finding that is ascribable to an enhanced level of geometrical complementarity.
Kimura and co-workers also reported that the tris(Zn(II)-cyclen) complex 529 is an effective receptor for C3-symmetric organic phosphates and phosphonates in slightly acidic aqueous solutions.579 Potentiometric titrations of receptor 529 and phenylphosphate were consistent with the formation of 1:1 host:guest complexes with the strongest interaction (log K = 6.6) being observed at slightly acidic pH. The reduced association at higher pH was attributed to competitive binding of hydroxide anions to the Zn(II) centers. Receptor 529 also bound nitrophenylphosphate, glucose-1-phosphate and phenylphosphonate with the relative affinities mirroring the substrate order of basicities. No evidence of binding interactions was found with phosphodiester monoanions. Phosphorous NMR spectroscopic titrations revealed that the interaction between phenylphosphate and the free-base polyammonium rings was negligible. This finding provided support for the conclusion that the phosphate associations resulted from interactions with the three zinc(II) ions. Further studies focused on the design of an IP3 sensor.580 In the context of this effort, receptor 529 was found to display strong binding to cis, cis-1,3,5-cyclohexanetriol triphosphate (CTP3), an achiral model system for IP3. A log apparent association constant of 8.0 for the 529:CTP3 complex at pH 7.4 was determined through pH-metric titrations. In efforts to develop a luminescent sensor, the phenyl core of receptor 529 was replaced with a tris(2,2′ -bipyridyl)ruthenium unit (530). Single crystal X-ray diffraction analysis of receptor 530 revealed a bifacial arrangement of the zinc(II) cyclen moieties, with three zinc centers making up each face. A Job plot analysis was performed for the binding of CTP3 to receptor 530 through 1H NMR spectroscopy and revealed a 1:2 host:guest stoichiometry (D2O, pD = 7.4). Based on these results, it was proposed that each face of the receptor bound one CTP3 molecule through coordination to the three zinc centers of that face. A strong blue shift and increase in intensity of the emission band were observed upon the addition of CTP3 to a solution of receptor 530 in 10 mM HEPES at pH 7.4. The enhancement of the emission intensity was attributed to the increased rigidity of the complex relative to the free receptors. Receptor 530 was further able to distinguish CTP3 from mono- and diphosphates (inorganic phosphate, phenylphosphate, glucose-6-phosphate, cis-1,3-cyclohexanediol diphosphate, and fructose-1,6,-diphosphate). The presence of a 10-fold excess of inorganic phosphate did not affect the luminescence sensing of CTP3 by this receptor. Analysis by pH-metric titration allowed for the determination of a log apparent association constant of 19.0 for the 1:2 530:CTP3 complex. The addition of IP3 also led to an increase in emission intensity, albeit a less dramatic increase than that observed for CTP3.
The ditopic zinc receptor systems 531 were developed by Benniston, Peacock, and co-workers. These systems were designed to facilitate interactions between the zinc-azamacrocycles and inorganic phosphate as well as interactions between the crown ether moiety and sodium or potassium cations. In preliminary studies with receptor 531a, sodium and potassium phosphate were found to bind more strongly to receptor 531a than to the zinc-azamacrocycle alone in aqueous media near neutral pH.581 As might be predicted by the size of the crown ether, potassium phosphate was bound more effectively (K = 9.3 × 104 M-1, pH 7.4, HEPES buffer) than sodium phosphate (K = 4.93 × 104 M-1). The proposed ditopic binding mode was further supported by the absence of any appreciable interaction with NaClO4 or KClO4. Interestingly, ITC studies revealed that binding to the simple zinc-azamacrocycle 522 was exothermic while binding to receptor 531a was endothermic. This led to the suggestion that binding to this ditopic receptor is driven largely by entropy. More detailed 1H NMR spectroscopic analyses revealed that the addition of Zn2+ ions strongly perturbed the signals of the azamacrocycle but produced only minor changes to the signals associated with the crown ether. Such observations support the conclusion that the zinc ion was bound primarily to the azamacrocycle.582 Phosphate interactions were also analyzed for receptor 531c, a zinc(II) bimetallic complex and the copper(II) receptor 531b. UV-Vis titrations of receptor 531c with monohydrogenphosphate supported the formation of 1:2 host:guest complexes; however, association constants could not be determined from ITC studies, presumably due to competing aggregation phenomena. No phosphate interactions were observed for receptor 531b.
Zn2+-cyclens were employed in material-based molecular recognition by Kalinina, König, and co-workers. In 2007, monotopic (532) and ditopic (533) Zn2+-cyclen amphiphiles were used in the formation of films with characteristics of both self-assembled monolayers and Langmuir-Blodgett films, which were immobilized on a gold surface.583 On the basis of surface plasmon resonance (SPR) measurements in aqueous solution (pH 7.5) it was concluded that films based on the monotopic 532 displayed much greater sensitivity for inorganic phosphate (10-8 M) than uracil (10-4 M). SPR measurements in solutions of uridine and adenosine nucleotides led to the suggestion of monovalent nucleotide-receptor interactions with 532. Films based on bis(Zn2+-cyclen) complex 533, however, were significantly more sensitive to nucleotides than 532-based films. This sensitivity was attributed to divalent intramolecular interactions in which the phosphate moiety and the nucleobase each interact with one of the two Zn2+-cyclen rings.
In 2009, this recognition chemistry was applied to self-assembled vesicular receptors based on 534 and 535.584 These materials were formed from diacetylene surfactants, which self-assemble into vesicles that can be photopolymerized to form polydiacetylene (PDA) in situ. Vesicles based on monotopic 534 displayed a colorimetric response (blue-to-red) upon addition of ATP and pyrophosphate anions in buffered aqueous solution (HEPES pH 7.2). No color change was observed upon addition of halide anions, dihydrogenphosphate, acetate, AMP, or ADP. These anion-binding interactions were also monitored through UV-Vis spectroscopy, which allowed for the determination of apparent binding constants for ATP and pyrophosphate to 534-functionalized vesicles (log K(ATP) = 2.2; log K(pyrophosphate) = 2.5). Stronger binding interactions were observed with vesicles based on ditopic 535 functionalization (log K(ATP) = 2.7; log K(pyrophosphate) = 3.2), presumably due to divalent interactions between these anions and the bis(Zn2+-cyclen) complex. The copper(II) complex 536 was also evaluated for a response to anions under these conditions. Interestingly, 536-functionalized vesicles displayed a change in absorption spectra only with pyrophosphate among the anions studied (log K(pyrophosphate) = 2.6). These trends were also found in paper test strip-based experiments. Interestingly, light microscopy images led to the conclusion that ATP and pyrophosphate caused aggregation of 535-functionalized vesicles under these conditions, but inorganic phosphate did not.
The Lin group investigated the binding behavior of ligand 537 and its metal complexes (538) with adenosine nucleotides.585 As seen with many of the previously described polyammonium macrocycles, the binding affinities with receptor 537 increased with increasing degree of protonation as inferred from potentiometric titration (pH 2-10.5). This receptor bound ATP > ADP > AMP in accordance with the relative anionic charge of the analytes. The ternary complexes formed by combination of ligand 537 with Zn2+ or Cd2+ (giving complexes 538) were also found to interact with ATP. Phosphorous NMR spectroscopic studies led to the conclusion that only the terminal phosphate of the ATP substrate interacts with the metal centers.
Another ditopic Zn(II) complex (539) was prepared by Kwong and co-workers.586 This receptor was proposed to interact with anions via a combination of metal-ligand and hydrophobic interactions. Fluorometric studies in water:methanol (1:4) at pH 7.4 revealed strong fluorescent quenching of receptor 539 by carboxylates, with less quenching observed in the presence of the dihydrogenphosphate, nitrate, bicarbonate, bromide, and iodide anions.
Bencini and Bianchi analyzed the multifunctional receptor 540.587 Proton and 31P NMR spectroscopic studies revealed strong binding to ATP. Both terminal phosphate groups of ATP were found to interact with the metal center. In addition, π-surface interactions were postulated to exist between the tripyridine unit and the adenine base.
The heteroditopic zinc-containing receptor 541 was prepared and analyzed by Gunning. This receptor displayed positive cooperative binding behavior for phosphate anions under physiological conditions.588 Proton NMR spectroscopic titrations carried out in HEPES buffer at pH 7.4 allowed binding constants of 3.90 × 104 for Na[H2PO4], 5.10 × 104 for K[H2PO4], and 3.13 × 104 M-1 for Li[H2PO4] to be calculated. The affinities of these phosphate salts were found to correlate well with the known affinities of the respective cations for the crown ether moiety.
In later studies, Bencini and Bianchi demonstrated that the zinc(II) phenanthroline complexes 542:Zn and 543:Zn2 interacted with bis(4-nitrophenyl)phosphate (BNPP) in CD3OD as inferred from 31P NMR spectroscopic studies.589 Significantly stronger binding was observed with the ditopic receptor 543:Zn2, presumably due to the chelation ability of the dinuclear complex. Phosphorous and 1H NMR spectroscopic titrations provided support for the proposed formation of 1:1 complexes as well as the presence of π-surface interactions between the phenanthroline groups and the p-nitrophenyl moieties of BNPP. These complexes were also found to effect phosphoester hydrolysis, with ditopic 543:Zn2 displaying a rate six-fold higher than that of the monotopic receptor 542:Zn.
The bis-(cobalt(II) dipicolylamine) complex 544a was originally developed by J. Chin and co-workers for use as a phosphate hydrolysis catalyst.590 Using the same organic frame, but with coordinated Zn(II) cations instead of Co(II) (complex 544b), D. H. Kim and Han were able to develop a colorimetric indicator displacement assay for inorganic phosphate in aqueous media.591 The displacement of pyrocatechol violet from receptor 544b in HEPES buffered water at pH 7.0 with inorganic phosphate led to significant spectroscopic changes. A drastic visible color change, from blue to yellow, was also observed. An association constant of 1.1 × 105 M-1 was measured for receptor 544b and inorganic phosphate through ITC experiments. Interestingly, little or no response was observed with other anions, including acetate, carbonate, nitrate, azide, perchlorate, sulfate, fluoride, chloride, and bromide. Receptor 544b was later found to bind AMP but not 3′,5′-cAMP.592 Based on this selectivity, the researchers were able to develop a visual assay of cyclic nucleotide phosphodiesterase (PDE) activity. In this assay, a solution of 3′,5′-cAMP and receptor 544b was added to a solution of PDE. PDE then effected the conversion of 3′,5′-cAMP to AMP, and the presence of receptor 544b allowed for real-time monitoring of the increasing AMP concentration. More recently, R. Smith and co-workers developed a pyrophosphate-selective indicator displacement assay based on receptor 544b.593 Eleven commercially available indicators were screened for spectroscopic changes upon the addition of 544b in a solution of aqueous HEPES buffer at pH 7.4. Indicators with binding affinities that fell between the affinities for inorganic phosphate and pyrophosphate were identified. When complexed to receptor 544b, these indicators (bromo pyrogallol red, mordant blue 9, and zincon) displayed a selective response to the addition of pyrophosphate over inorganic phosphate. Binding constants were not reported using this method.
Hamachi and co-workers have extensively pursued zinc(II) dipicolylamine (Zn(Dpa)) as a useful molecular recognition motif for phosphates, particularly phosphorylated proteins and peptides. The underlying host:guest interactions presumably derive from metal-ligand coordination chemistry. The Zn(Dpa) unit has also been studied as a selective phosphate binding motif. For example, receptor 545 was found to interact selectively with inorganic phosphate and methylphosphate over carbonate, acetate, nitrate, sulfate, azide, and chloride in aqueous media at pH 7.2.594 Furthermore, the addition of ATP to receptor 545 resulted in fluorescence enhancement. An affinity constant of 2.2 × 106 M-1 was measured for the 545:ATP complex. Significantly weaker binding behavior was observed with AMP and ADP, and no binding behavior was observed for 3′,5′-cAMP. The selectively of receptor 545 for ATP was attributed to the higher charge of the triphosphate. Phosphorous NMR spectroscopic studies provided support for the conclusion that receptor 545 bound the two terminal phosphate units of ATP. Reasonable selectivity for ATP over other nucleoside triphosphates was also observed.
A more application driven study by the same researchers relied on the use of receptor 545 in an assay of glycosyltransferase activity.595 These studies first confirmed the selectivity of receptor 545 for phosphate monoesters over phosphate diesters. This selectivity permitted for the formation of 5′-uridine diphosphate (UDP) to be monitored in the presence of the starting glycosylated nucleotide. This allowed the glycosylation reaction to be monitored without having to modify the enzyme or its substrates.
Analysis of the anion binding behavior of receptor 546 revealed a similar selectivity trend as that previously observed with receptor 545.596 Binding constants on the order of 104-105 M-1 were measured for receptor 546 with inorganic phosphate, adenosine nucleotides, and a variety of phosphate monoesters. The binding interactions were again monitored via fluorescence titration. No change in the emission spectrum of receptor 546 was observed upon additional of 3′,5′-cAMP or dimethylphosphate. Furthermore, a mono-Zn(Dpa) anthracene (547) did not undergo a change in fluorescence in the presence of these phosphate species. Displacement experiments with this mono-Zn(Dpa) receptor led to the determination of binding constants 1-2 orders of magnitude lower than those observed with the bis-Zn(Dpa) analogues 545 and 546. Proton NMR spectroscopic studies and single crystal X-ray diffraction analysis of receptor 546 and phenylphosphate supported equal participation of the two Zn(Dpa) units, with each metal center interacting with a phosphate oxygen atom (Figure 9). The fluorescent enhancement observed upon phosphate anion addition to these receptors was attributed to an increase in the extent to which the second zinc atom was complexed to the Dpa ligand in the presence of phosphate. It was proposed that this increased metal-ligand interaction serves to reduce the inherent PET quenching of the anthracene fluorescence by the Dpa amine ligand.
Figure 9.
View of the 546:phenylphosphate complex. Drawing generated from X-ray diffraction data originally published in reference 594. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
In further application-based studies by these researchers, receptors 545 and 546 were found to bind phosphorylated peptides over non-phosphorylated peptides under neutral aqueous conditions.597 Binding constants for the interactions between 545 and 546 with 548a and d were determined through fluorescence spectroscopy and were found to range from 104 - 107 M-1. In contrast, no binding behavior was observed for the non-phosphorylated sequence 548g or for a phosphorylated sequence bearing positively charged arginine residues (548c). The observed charge selectivity was attributed to charge repulsion between the cationic zinc centers and the arginine side chains. When the phosphorylated peptides 548a and d were added to 9-Zn(Dpa)-anthracene (547), the observed binding constants were two to three orders of magnitude lower than what was observed for the ditopic receptors. The reduced binding affinities seen for the mono-Zn(Dpa) complex were consistent with the conclusion that ditopic chelation played a critical role in the binding abilities of receptors 545 and 546. Further binding analyses with a larger set of phosphorylated peptides (548a-f) supported the conclusion that the affinity was strongly dependent on the overall charge of the peptide, with the strongest affinities being recorded for negatively charged sequences 548a,b (K ≈ 106 – 107 M-1).596 More detailed ITC studies revealed that the binding behavior was entropy driven, presumably due to solvation effects. Taking advantage of the receptor:peptide interactions, these researchers were able to show that receptor 546 could be used to monitor phosphatase activity via changes in the fluorescence emission intensity in the case of various peptide substrates. Other applications of this class of receptors included fluorescent staining of phosphoproteins on an SDS-PAGE gel598 and the disruption of phosphoprotein-protein surface interactions.599 Many of the above studies have been reviewed by Ojida and Hamachi.600,601
Hamachi and co-workers also investigated the utility of bipyridine bis(ZnDpa) receptors 549 and 550 as cross-linkers in solutions of hyperphosphorylated peptide sequences.602 These interactions were attributed to the stabilization of helix formation as a result of the simultaneous binding of multiple phosphate residues enforced by the complexes. This stabilization was monitored using circular dichroism (CD) spectroscopy. Fluorescence quenching of the receptors in the presence of the peptides was also observed. No significant conformational change was observed when receptors 549 and 550 were presented with monophosphorylated peptide sequences, or when mono-Dpa analogues were presented with the hyperphosphorylated sequences. These results provided support for the proposed cross-linking interactions.
Other fluorophore scaffolds for the Zn(Dpa) subunit were investigated later in the context of this work. For example, the acridine-based receptors 551 and 552 were synthesized. These systems displayed a reduction in emission intensity and a blue-shift in the emission wavelength in the presence of various nucleotides in HEPES buffer at pH 7.2.603 Binding affinities in the range of 106 -107 M-1 were measured for tri- and diphosphates in this aqueous medium, while much lower affinities were observed for nucleotide monophosphates, cyclic monophosphates, and monohydrogenphosphate. Little to no response was observed with non-phosphate anions. The spectroscopic changes seen in the presence of phosphates were attributed to a change in the coordination mode of zinc upon the addition of the phosphate anions. Hamachi and co-workers proposed that in the absence of anions the zinc coordinates to the nitrogen atom of the acridine as well as to the nitrogen atoms of the Dpa units. These interactions led to an increase in the emission intensity and a red-shift in the acridine fluorescence spectrum. This effect was reversed in the presence of the di- or triphosphates. Presumably, these species coordinate to the Dpa units and reduce coordination to the acridine nitrogen atom. These sensors were then used to monitor ATP and ADP hydrolysis as well as glycotransferase activity.
A xanthone-based bis-ZnDpa scaffold (553) was also investigated.604 Under similar conditions as used for the acridine-based studies, receptor 553 was found to interact with nucleotide tri- and diphosphates. Slightly lower binding affinities (105 – 106 M-1) were measured with receptor 553 as compared to receptors 551 and 552. Similar selectivities were observed. The interaction of receptor 553 with ATP was also studied by ITC. These studies revealed that the binding process was both enthalpically and entropically driven. Spectroscopically, emission changes were observed at three separate wavelengths upon addition of phosphate anions to receptor 553. Again, a rearrangement of the zinc coordination environment was proposed as the source of the observed spectral changes. More recently, a xanthene-based scaffold (554) was explored as part of continued efforts to improve the sensitivity of this class of receptor.605 Receptor 554 was found to exhibit unexpectedly low fluorescence in aqueous solution. Single crystal X-ray diffraction analysis of receptor 554 revealed a structure in which a water molecule coordinated to both zinc centers had added into the xanthene chromophore. High-resolution fast atom bombardment (FAB) mass spectrometry of the receptor also supported this addition reaction. The resulting disruption of the conjugation was proposed as the cause of the low emission intensity. This reaction was found to be reversible through pH titrations. A strong increase in fluorescence was observed upon the addition of ATP to receptor 554 at pH 7.4 (50 mM HEPES, 10 mM NaCl, 1 mM MgCl2). It was suggested that the coordination of ATP by the zinc centers shifted the equilibrium away from the water adduct. A strong binding constant (K = 1.3 × 106 M-1) and high sensitivity (< 10-6 M ATP) were observed in further fluorescence experiments. Job plot analyses were consistent with a 1:1 host:guest stoichiometry for ATP. Similar binding constants were observed with other nucleotides (ADP, GTP, CTP, UDP) and IP3. A significantly higher binding constant was measured for the pyrophosphate complex (K = 4.0 × 107 M-1), presumably due to the higher charge density of this anion. Little to no change in the emission intensity was observed upon the addition of monophosphate species (HPO42-, 3′,5′-cGMP, 3′,5′-cAMP) and other small inorganic anions (AcO-, SO42-, NO3-, HCO3-). This receptor was also applied as a stain for ATP in human cells.
Hamachi and co-workers have also studied the ability of this class of receptors to sense phosphate when incorporated in a hydrogel. In preliminary studies, the incorporation of di-Zn(Dpa) receptor 545 into a semi-wet hydrogel was investigated.606 The hydrogel material was based on a glycosylated amino acid-type hydrogelator, which was transparent enough to allow supramolecular binding events to be monitored in the gel matrix. For example, a binding constant of 1.1 × 105 M-1 for receptor 545 and inorganic phosphate was inferred from fluorescence titrations in a hydrogel matrix. This value was three-fold lower than the binding constant reported for 545 and inorganic phosphate in aqueous solution. Fluorescence enhancements were also observed upon addition of phenylphosphate and phosphotyrosine to the 545-hydrogel. Addition of sulfate, however, did not lead to a change in the emission spectrum. Further studies examined the response of receptors 545, 555, and 556 in hydrogel materials to a wider variety of anions.607 Receptor 545 displayed a binding constant of greater than 106 M-1 for ATP when studied through fluorescence titrations in the hydrogel matrix. No changes in the emission spectrum were observed for sulfate, nitrate, acetate, azide, bromide, chloride, or fluoride. Receptor 555, containing an environmentally sensitive dansyl fluorophore, displayed differing responses to phosphate anions when in the hydrogel. For example, a blue-shift in emission maxima and an increase in emission intensity was observed upon addition of phenylphosphate. On the other hand, addition of ATP, inorganic phosphate, and phosphotyrosine led to a red-shift in emission maxima and a decrease in emission intensity. The binding constants for receptor 555 with ATP and phenylphosphate were measured to be 1.8 × 105 M-1 and 7.2 × 103 M-1, respectively. Interestingly, receptor 555 did not display a response to anions in aqueous solution or when immobilized in an agarose gel. Using confocal microscopy, fluorescent receptors 555 and 556 were observed to travel from the aqueous solution to hydrophobic fibers in the gel matrix in the absence of anions. However, the addition of ATP or phenylphosphate altered the hydrophobicity of these fluorescent receptors, causing the complexes to move in the opposite direction.
Hong and co-workers utilized Zn(Dpa) units to create the azophenol receptor 557 that was designed for the visual sensing of pyrophosphate.608 While monovalent anions caused no change in the absorption spectrum, the addition of pyrophosphate led to a strong bathochromic shift. This change could be observed visually by a change in the solution color from yellow to red over the pH range 6.5-8.3. A strong binding affinity was observed, with an association constant of 6.6 × 108 M-1 being recorded in HEPES buffer at pH 7.4. Single crystal X-ray diffraction analysis of the complex led to the conclusion that the anion bridges the two zinc centers in the solid phase. The visual color change was attributed to the decrease in the association between the phenolic OH and the zinc centers that takes place upon anion coordination. A red absorbance shift and strong increase in emission intensity were observed for receptor 558 in the presence of pyrophosphate.609 A binding constant of 2.9 × 108 M-1 was determined under conditions similar to those used to analyze receptor 557 (HEPES buffer, pH 7.4). Much smaller spectroscopic changes were observed with ATP, ADP, and AMP as compared to what was seen with pyrophosphate. Based on this selectivity, it was proposed that this sensor could be useful in bioanalytical assays, particularly ones that require the measurement of small amounts of pyrophosphate in the presence of ATP.
In efforts to improve the binding affinity of these receptors, amide substituents were included in the design of receptor 559.610 The interaction of this receptor with pyrophosphate was followed through an indicator displacement assay using pyrocatechol violet in 10 mM HEPES buffer at pH 7.4. An extremely high binding affinity was inferred, and the binding constant could only be determined through competitive binding experiments with receptor 558 (K = 5.39 × 1010 M-1). Job plot analysis confirmed a 1:1 host:guest stoichiometry. No change in the absorption spectrum was observed upon the addition of inorganic phosphate, fluoride, chloride, bromide, acetate, bicarbonate, sulfate, azide, nitrate, perchlorate, or citrate anions. A single crystal X-ray diffraction analysis of the 559:pyrophosphate complex revealed chelation of the anion by both metal centers and the formation of hydrogen bonds with all four amide units. Notably, a 100-fold increase in binding affinity in aqueous media was achieved by the incorporation of four hydrogen bond donor units.
A FRET-based assay was achieved by attaching one unit of a fluorophore pair to the bis(Zn(DPA)) host and phosphate guest, respectively (560, 561).611 In this case, methyl red was appended to the recognition unit (560) while fluorescein was appended to phosphate (561). Fluorophore 561 then served as the indicator in a displacement assay with host 560, in which decomplexation of the indicator decreased the FRET efficiency of the system. This signal could be used to detect a number of anions, in the order pyrophosphate > ATP > ADP > AMP ≈ inorganic phosphate > acetate ≈ fluoride in 10 mM HEPES buffer at pH 7.4. A binding constant of 108 M-1 was estimated for the 560:pyrophosphate complex using this method.
While little activity was seen for monomers of other Zn(Dpa) complexes, the pyrene-based receptor 562 was found to form dimers with pyrophosphate and ATP.612 This dimerization could be observed through excimer formation in aqueous solution (HEPES buffer, pH 7.4). The addition of monovalent anions, including hydrogenphosphate and AMP, did not lead to spectroscopic changes. While no binding constants were reported, a significantly more intense excimer emission was observed for pyrophosphate than for ATP. More recently, these researchers reported the binding of FAD by simple receptor 563.613 This receptor served to increase the innate fluorescence of FAD, and this emission increase was used to determined a binding constant of 1.11 × 105 M-1 in water (10 mM HEPES, pH 7.4). Little change was observed in the emission of FMN upon addition of this receptors. In efforts toward a biological application, receptor 563 was used to treat human white blood cells, which are known to contain high levels of FAD. This allowed for imaging of these cells through flow cytometry.
Smith and co-workers analyzed Zn(Dpa) sensors for biological applications. In preliminary studies, receptor 545 (described previously) was found to bind phosphatidylserine (PS), an anionic phospholipid, on cell surfaces.614 The binding behavior was monitored through an increase in fluorescence intensity. No change in the emission spectrum was observed when receptor 545 was treated with zwitterionic phospholipids or when a mono-Zn(Dpa) sensor (547) was treated with PS. Receptor 545 was then used to detect apoptotic cells, which are commonly identified by the presence of PS on the cell surfaces. No cell internalization or cytotoxicity was detected. A library of functionalized Zn(Dpa) sensors were then analyzed in hopes of obtaining a red-shifted excitation wavelength; this was done via the incorporation of a 7-nitrobenz-2-oxa-1,3-diaza-4-yl (NBD) fluorophore. 615 Among the several sensors containing this latter moiety, the bis-Zn(Dpa) system 564 proved to be the most effective for the detection of PS-containing vesicles. Interestingly, fluorescence enhancement was only observed for PS located on vesicle surfaces. Again, no vesicle internalization or increased permeability was observed. Receptor 564 was able to detect vesicles containing as little as 5% PS. The sensing mechanism was attributed to the sensitivity of the NBD fluorophore to environmental polarity.
Fluorescent indicator displacement assays were also analyzed for use in the detection of PS.616 Two Zn(Dpa) receptors described previously (i.e. 544b and 563) were incorporated into displacement assays that were based on the use of a coumarin (565) as the indicator in aqueous media at pH 7.4. The bis-Zn(Dpa) receptors again displayed selectivity for hydrogenphosphate and pyrophosphate over other anions. Receptor 563 was found to bind pyrophosphate more strongly than it bound hydrogenphosphate. The association constants for these two substrates were 1.5 × 107- M-1 and 7.3 × 105 M-1, respectively. Interestingly, receptor 563 was found to bind both phosphate species more strongly than receptor 544b. This increased binding affinity was attributed to the lower net positive charge of receptor 544b as a result of the phenolate anion. Analysis of the para-substituted derivate of 563 did not reveal any binding interactions with phosphate derivatives, again emphasizing the importance of the bridging coordination of the two zinc centers in these systems. Such displacement assays were then applied to vesicles containing PS.617 In studies with pyrocatechol violet as the indicator, only receptor 544b proved responsive to low (5%) concentrations of PS in the vesicles. Receptor 563, however, proved to be the most responsive receptor when using fluorescence indicator 565; this resulted in detection limits as low was 5% PS. No displacement was observed with monodisperse PS.
A macrocyclic peptide was employed as a scaffold for a bis-Zn(Dpa) pyrophosphate sensor by Jolliffe and co-workers.618 This gave rise to receptor 566. In combination with this receptor, indicator 565 was used to achieve fluorescence sensing in aqueous media (HEPES buffer, pH 7.2). It was proposed that the larger spacing between the two zinc centers in this macrocycle-containing framework would lead to greater selectivity for pyrophosphate than had been observed with the previously described bis-Zn(Dpa) receptors. A high association constant (log K = 8.0) was indeed obtained for receptor 566 and pyrophosphate. The selectivity for pyrophosphate was good, with pyrophosphate being bound approximately two orders of magnitude more tightly than ATP, ADP, or citrate. No other anions appeared to bind this receptor. The observed selectivities were similar to those observed with the previously investigated bis-Zn(Dpa) systems. The larger scaffold, therefore, increased the association constant for pyrophosphate but did not significantly affect the overall selectivity.
In addition to their work with receptor 515 described earlier, the Yoon group investigated different scaffolds for preparing bis-Zn(Dpa) systems. Simple acridine receptor 567 displayed differing spectral responses for inorganic phosphate and pyrophosphate, respectively, in aqueous solution (HEPES buffer, pH 7.4).619 Specifically, emission enhancement was seen upon addition of inorganic phosphate to receptor 567. In contrast, quenching was observed when exposed to pyrophosphate. Addition of either anion led to a bathochromic shift in the emission maximum. The quenching mechanism with respect to pyrophosphate addition was assumed to be consistent with that previously observed for related receptors 551 and 552, in which the anions coordinate to the Dpa units and reduce coordination to the acridine nitrogen atom. On the other hand, the emission increase seen upon the addition of inorganic phosphate was attributed to the formation of an additional hydrogen bond between the acridine nitrogen and the bound phosphate. Binding constants of 4.85 × 107 M-1 and 9.36 × 104 M-1 were determined for receptor 567 with pyrophosphate and inorganic phosphate, respectively. No spectral change was observed upon the addition of a variety of other anions.
Sensor 568 was also prepared by Yoon and co-workers. This system proved to be reasonably selective for pyrophosphate based on the formation of 2 + 2 complexes that induced excimer emission.620 An association constant of 4.1 × 105 M-1 was determined in aqueous solution (HEPES buffer, pH 7.4). The only response among the other anions tested was a slight increase in emission upon addition of ATP. Despite this response, pyrophosphate could be reliably detected in the presence of 10 molar equiv. of ATP.
Das and co-workers analyzed the interaction of the mono-Zn(Dpa) receptor 569 with phosphate derivatives.621 Receptor 569 was found to bind selectively dihydrogenphosphate among other monovalent anions in acetonitrile (K = 5.62 × 105 M-1). A striking yellow to red color change was also observed upon addition of dihydrogenphosphate. The binding behavior of inorganic phosphate and other phosphate derivates was then tested in aqueous solution (HEPES buffer, pH 7.2). Under these conditions, responses were only observed upon the addition of ATP, ADP, and CTP, as inferred from red-shifted absorbance bands. Association constants for these three anions were determined to be 1.13 × 103 M-1, 2.50 × 102 M-1, and 7.72 × 102 M-1, respectively. The largest shift (ca. 20 nm) was observed for ATP. ATP was also the only anion to produce a visual color change from yellow to pink. The addition of AMP, pyrophosphate, and inorganic phosphate did not give rise to any appreciable spectral changes. The stronger binding of ATP as compared to CTP was attributed to the weaker electron donating character of CTP as a result of its constituent pyrimidone moiety. This receptor was later utilized as a non-toxic stain against ATP production in yeast and bacterial cells.622
In 2009, Guo, He, and co-workers reported triphosphate and pyrophosphate sensor 570.623 This receptor incorporated a 4-amino-7-aminosulfonyl-2,1,3-benzoxidiazole fluorophore that served as a visible light excited reporter unit. A five -six-fold emission enhancement was observed upon the addition of one equivalent of triphosphate or pyrophosphate to receptor 570 in aqueous media (10 mM HEPES, 1% DMSO, pH 7.2). The strong change in emission intensity was attributed to a chelation effect in which the polyanionic guests were bound by both Zn(Dpa) centers. No change in emission was observed in the presence of inorganic phosphate, acetate, nitrate, chloride, carbonate, sulfate, perchlorate, or fluoride. The importance of the second Zn(Dpa) binding site was supported by the lack of response of receptor 571 to any of the studied anions under the same conditions. The anion binding properties of receptor 572, in which the Zn(Dpa) units are directly conjugated to the fluorophore) were also investigated. This receptor did not respond to the addition of any anions, presumably due to a reduced zinc-binding ability of the Dpa as a result of the electron-withdrawing sulfonamide substituent. No binding constants were reported in this study.
Qian, Tang, and co-workers studied a bis(2-benzimidiazolylmethyl)amino group (see 573) as a replacement for the Dpa unit of receptor 544b in hopes that steric interactions between the benzimidazole units would increase the metal-metal distance.624 This increase was expected to facilitate selective pyrophosphate recognition in aqueous media. Following the synthesis of receptor 573, an indicator displacement assay was developed using fluorescein as a fluorescent indicator. Addition of pyrophosphate to the 573:fluorescein ensemble led to displacement of the indicator as inferred from an increase in fluorescence intensity (10 mM HEPES buffer, pH 7.4). No change in fluorescence was observed upon the addition of oxalate, inorganic phosphate, acetate, hydrogensulfate, fluoride, chloride, iodide, or bromide to the ensemble. Titrations with pyrophosphate led to the determination of a stability constant of 1.3 × 105 M-1 and a stoichiometry of 1:1 through Job plot analysis. It was thus concluded that receptor 573 is reasonably selective for pyrophosphate.
Receptors containing ammonium-expanded analogues of the Dpa systems were reported by Pina, García-España, and co-workers.625 Among these is the anthracene-based receptor 574. This system displayed a significant decrease in emission intensity in the presence of triphosphate, pyrophosphate, iodide, citrate, and cyanurate (pH 8, 0.15 M NaCl). Each of these anions were bound with similar affinity (log K 5.2 - 5.8), and very little change in fluorescence was observed upon the addition of inorganic phosphate, fluoride, or D, L-isocitrate. At the same time, addition of triphosphate, pyrophosphate, citrate, and D,L-isocitrate to dansyl-based receptor 575 led to an overall increase in emission intensity (pH 7.5, 0.15 M NaCl). No change in emission was observed with the other anions tested. This receptor displayed more selective binding than receptor 574. Significantly higher values were reported for triphosphate and pyrophosphate (log K = 4.9 and 4.1, respectively) than citrate and D,L-isocitrate (log K = 2.7 and 2.1, respectively). This study highlights the importance of the reporter moiety in these systems.
In 2007, Guo, Wang, and co-workers described the dihydrogenphosphate sensing abilities of a tetranuclear zinc(II) complex based on a cresolic ligand (576).626 A single crystal X-ray diffraction analysis of complex 576 revealed a 4:2 metal:ligand structure in which all of the ligand heteroatoms are bound to pentacoordinate zinc(II) centers. This receptor was found to be strongly fluorescent in unbuffered methanol. Addition of NaH2PO4 led to a dramatic decrease in fluorescence intensity while the addition of Na2CO3, NaHCO3, NaNO3, NaNO2, NaF, NaCl, NaBr, NI, or Na2SO4 led to rather small changes in the emission spectrum. Proton NMR spectroscopic experiments supported a binding mechanism in which the dihydrogenphosphate anion displaced the central phenol oxygen from the zinc(II) center. It was proposed that this displacement disrupted the ligand-to-metal charge transfer in the native complex, resulting in decreased emission. Evidence for both 1:1 and 1:2 host:guest binding stoichiometries was found, and an overall association constant was estimated to be 1.0 × 105 M-1.
Polyamine-phenol-based zinc complexes (577 and 578) were developed as anion receptors by Fusi, Micheloni, and co-workers in 2009.627 Potentiometric studies in aqueous solution (0.15 M NaCl) revealed the formation of 1:1 complexes with the receptors and both inorganic phosphate and pyrophosphate. At near-neutral pH, a strong preference was observed for pyrophosphate by 577 and for inorganic phosphate by 578. This selectivity was attributed to the respective Zn-Zn distances in each receptor. The longer distance in receptor 577 was thought to better accommodate pyrophosphate while the more compact 578 system could chelate inorganic phosphate more effectively. This trend was further supported by 1H NMR spectroscopic experiments in D2O (HEPES buffer, pH 7.4). Fluorescence studies with receptor 577 revealed a quenching effect upon addition of inorganic phosphate but an increase in emission intensity upon addition of pyrophosphate (HEPES buffer, pH 7.4). Little change in the emission spectrum was observed upon the addition of anions to receptor 578. The development of an indicator displacement assay based on pyrocatechol violet allowed for the determination of binding constants with inorganic phosphate, pyrophosphate, ATP, and glucose-6-phosphate guests in the HEPES buffer system at pH 7.4. In line with the potentiometric and 1H NMR spectroscopic studies, the strongest complexes were formed between receptor 577 and pyrophosphate and receptor 578 and inorganic phosphate, respectively (log K ≈ 4.4 for both complexes). In addition, receptor 577 was found to bind ATP strongly (log K = 3.8) while receptor 578 was found to bind glucose-6-phosphate strongly (log K = 3.5). The selectivity observed for these latter guests was again attributed to the specific inter-Zn distances of the two receptors. No interactions were observed with chloride, bromide, perchlorate, nitrate, or sulfate in these systems.
A unique bimetallic zinc receptor (579) was developed by Churchill and co-workers.628 The chiral, lysine-based ligand allowed for the detection of anions through fluorescence, absorption, and CD spectroscopy in aqueous media (10 mM HEPES, pH 7.4). A bathochromic shift in the absorption peak was observed upon the addition of pyrophosphate (40 nm), ATP (36 nm), and ADP (1 nm). No significant changes were found for fluoride, bromide, iodide, acetate, bicarbonate, inorganic phosphate, AMP, or 3′,5′-cAMP. The use of pyrocatechol violet indicator in a displacement assay allowed for the visual detection of pyrophosphate, ATP, and ADP. Fluorescence quenching was also observed for these three anions (pyrophosphate, ATP, ADP), and binding constants were calculated using this method. Pyrophosphate and ATP exhibited similar binding affinity (K ≈ 4 × 105 M-1) while ADP bound with significantly lower affinity (K = 9.14 × 103 M-1). The stronger binding interactions with pyrophosphate and ATP were attributed to the greater electronic charge density of these anions relative to ADP. Phosphorous NMR of the three respective complexes displayed large shifts of the bound guests compared to the free anions. Only the signal for the α-P center (the phosphorous atom closest to the sugar unit) of ATP did not shift, which led to the conclusion that the β- and γ-P centers were more involved in complex formation. Importantly, receptor 579 allowed for the selective detection of pyrophosphate in the presence of excess chloride and inorganic phosphate.
In 2007, a zirconium-based anion receptor (580) was developed by Wang, Wu, and co-workers.629 In this receptor design, anion binding to the zirconium center would displace the indicator (4′-N,N-dimethylamino-6-methyl-3-hydroxylflavone) and thus cause a change in the emission intensity of the indicator. In accord with such expectations, an increase in emission intensity of approximately 1.5-fold was indeed observed upon the addition of dihydrogenphosphate, acetate, and chloride, albeit a larger increase (6-fold) was observed upon the addition of fluoride. These experiments were conducted in a HEPES buffer at pH 7.6, and anions were added as their TBA salts. No binding constants were reported.
Molybdenum has also been incorporated into synthetic phosphate receptors by Pérez and co-workers.630 A specific combination of amide functionality and molybdenum metal-carbonyl fragments was used to produce receptors 581. These systems are of interest because they might allow anion binding to be monitored via IR spectroscopy. In this report, however, dihydrogenphosphate was only analyzed through 1H NMR spectroscopy in DMSO-d6. Nevertheless, these experiments allowed for the measurement of binding affinities. Binding constants for receptor 581a and 581b with dihydrogenphosphate were measured to be 856 M-1 and 257 M-1, respectively. Little selectivity was observed among the anions analyzed.
Steed and co-workers have utilized ruthenium(II) as both a scaffold and a recognition site in receptor 582.631 In this receptor, an amino-ferrocene cleft was assembled through ruthenium(II)-pyridine coordination. Anion binding studies were performed using 1H NMR spectroscopy with NO3-, AcO-, ReO4-, CF3SO3-, H2PO4-, Cl-, and HSO4- as the TBA salts in CDCl3. Receptor 582 was found to bind H2PO4- with a binding constant of 550 M-1for the 1:1 complex. However, selectivity in this system was observed for NO3- and HSO4-. Addition of Cl- and AcO- anions led to displacement of the pyridine ligands from the ruthenium(II) center.
Palladium and platinum metals have also found a place in phosphate anion recognition. For example, Vilar and co-workers reported the anion binding ability of dipalladium thiolate complexes functionalized with urea units.632,633 Proton NMR spectroscopic titrations in DMSO-d6 of receptor 583a with TBA anion salts revealed the following trend in binding strength: dihydrogenphosphate > phenylphosphate > bromide > chloride > hydrogensulfate. While the strongest binding was observed for dihydrogenphosphate (log K = 3.5), little selectivity was recorded between this anion, phenylphosphate, and bromide. Replacing the phenyl substituents with ethyl groups (583b) did not improve the selectivity. The anion binding ability of these receptors was attributed to a combination of hydrogen bonding interactions with the urea moieties and electrostatic interactions between the anion and the metal center. It was also proposed that the dipalladium thiolate backbone preorganized the receptor cavity. These receptors were also observed to decompose in solution over time. As result, platinum derivatives (584) were investigated in efforts to increase stability.634 A slight change in the trend in binding affinity was observed (dihydrogenphosphate ≈ phenylphosphate > acetate > hydrogensulfate > halides ≈ nitrate) through 1H NMR spectroscopic studies in DMSO-d6. The highest selectivity for dihydrogenphosphate was reported for receptor 584a, and the highest binding affinity for this anion was reported with receptor 584c (log K = 3.7). No decomposition was observed for receptors 584. A qualitative indicator displacement assay in CH3CN:CH2Cl2 solution was also developed by first coordinating the indicator methyl red to receptor 584c. Significant displacement was observed upon addition of dihydrogenphosphate and acetate to this complex through UV-Vis spectroscopy. This displacement was visible to the naked eye as an orange-to-yellow color change.
Platinum was utilized by Loeb and co-workers to organize urea-functionalized ligands for anion binding as in receptor 585.635 Proton NMR spectroscopy in DMSO-d6 led to the conclusion that the recognition units in 585 existed in the 1,2-alternate, 1,3-alternate, or cone conformations depending on the anion added (all as the corresponding TBA salts). While the halides (Cl-, Br-, I-) were complexed in a 1,2- or 1,3-alternate conformations, sulfate and dihydrogenphosphate were complexed in a cone conformation. The binding constants for the oxoanion complexes were found to be higher than could be determined using this method (K > 105 M-1), even in the highly competitive solvent DMSO. Single crystal X-ray diffraction analysis of the 585:SO4- complex led to the suggestion the observed strong binding was due to a combination of metal chelation and hydrogen bonding interactions between the anions and all eight NH groups of the complex. It was proposed that these interactions served to anchor the guest within the host cavity.
Kikuchi and co-workers analyzed the phosphate recognition of receptor 586, a system designed to combine the sensitivity of coumarin fluorescence with a Cd(II)-azamacrocycle.636 In neutral aqueous solution, Cd(II) was coordinated by the four nitrogen atoms of the macrocycle and the aromatic amino group of the coumarin subunit. While Cu(II) and Zn(II) complexes were also investigated, only the Cd(II) complex was found to quench appreciably the fluorescence of the indicator. Potentiometric titrations supported the conclusion that Zn(II) cation did not coordinate to the coumarin indicator while the Cd(II) center did. Addition of anions to the Cd(II) complex 586 led to a shift in the excitation spectrum. Presumably, this shift is due to displacement of the coordinated coumarin group by the anion. The spectroscopic shifts were used to determine binding affinities. Both pyrophosphate and citrate displayed dissociation constants in the 10-5 M range in HEPES buffer at pH 7.4. Monoanions (iodide, bromide, chloride, and inorganic phosphate), however, were characterized by much weaker interactions. Nucleotides also bound with strong to moderate affinities, although the various cyclic nucleotides that were tested were found to bind weakly if at all. The latter selectivity allowed receptor 586 to be used as the reporter group in a PDE assay, in a similar manner to the 544b-based assay previously described.
The Anslyn group designed and synthesized a cadmium(II) boronic acid-trispyridyl receptor (587) with the goal of binding carboxy- and phosphosugars.637 UV-Vis displacement experiments in methanol:water (3:1) allowed for the measurement of binding affinities. Association constants of log K = 3.95 and 4.76 were reported for AMP and for ribose 5-phosphate, respectively. The strongest affinity, however, was observed for carboxylated sugars. The binding interactions were proposed to derive from a combination of boronic acid-sugar bonding and metal-anion complexation.
Tin, particularly in the guise of organotin compounds, has a long history of use in the area of phosphate-selective polymer electrodes. In one of the first successful studies, Arnold and Glazier incorporated bis(p-chlorobenzyl)tin dichloride into a PVC membrane.108,638 This electrode proved selective for dibasic phosphate at pH 7.0 over acetate, sulfate, chloride, bromide, nitrate, and iodide. The detection limit was found to be 3.3 × 10-5 M with a linear response range from 2.2 × 10-4 M to 1.2 × 10-2 M. The electrode lifetime was reported to be at least 28 days. Further studies revealed that electrodes containing bis(p-methylbenzyl)tin and dibenzyltin derivatives lost much of the previously observed selectivity for dibasic phosphate seen in the case of the bis(p-chlorobenzyl)tin systems. Importantly, however, these electrodes remained selective for dibasic phosphate over chloride, which is generally considered as the most competitive contaminant in real world systems due in part to its high concentration.639 The observed selectivity trend led to the suggestion that electron-withdrawing substituents on the benzyl ring increased the selectivity for dibasic phosphate over other anions, presumably through an increase in tin-carbon hyperconjugation. The interaction of bis(p-chlorobenzyl)tin dichloride with several other anions was also studied. For example, bis(p-chlorobenzyl)tin dichloride electrodes were found to be as selective for thiocyanate as they were for dibasic phosphate. This chlorobenzyl derivative was also observed to interact with adenosine nucleotide derivatives in the order ATP > ADP > AMP > 3′,5′-cAMP. However, a slight selectivity for dibasic phosphate over these nucleotides was maintained. On the other hand, this electrode was found to be more selective for benzoate, salicylate, and pyrophosphate than dibasic phosphate while less selective for arsenate and fluoride.
Arnold and co-workers analyzed the response of a bis(p-fluorobenzyl)tin dichloride membrane with the hope of attaining an even greater phosphate selectivity.640 As expected, the resulting electrode proved selective for dibasic phosphate over thiocyanate, iodide, bromide, nitrate, acetate, and chloride. A strong correlation between the detection limit and the Hammet constant of this and the previously studied benzene substituents (p-methylbenzyl, benzyl, and p-chlorobenzyl) was observed. Both the fluoro derivative and the chloro derivative, however, were found to have an even stronger response for tribasic citrate than dibasic phosphate.
Simon and co-workers further probed the interaction of anions with trialkyl and dialkyl tin complexes via 119Sn NMR spectroscopy in CDCl3.641 For example, tributyltin chloride underwent a greater chemical shift upon addition of dihydrogenphosphate than upon addition of hydrogensulfate (TBA salts). These studies supported the proposal that tetracoordinate tin complexes are formed through ion exchange. Dioctyltin dichloride, however, was observed to form 1:2 complexes with dihydrogenphosphate (Kryptofix® 222/potassium salt) at low concentrations of anion. The host became oligomeric and/or polymeric upon the addition of more than 0.5 equiv. of phosphate. The chemical shifts of this tin derivative were consistent with the formation of a pentacoordinate tin species.
The multivalent tin receptor 588 was investigated by Chaniotakis and co-workers.642 When incorporated into a PVC membrane, this receptor displayed a selective response for inorganic phosphate over a variety of other anions, including salicylate, perchlorate, and thiocyanate. The observed selectivity correlated more closely to the metal coordination ability of the different anions than with the Hofmeister series. The lifetime of these electrodes, however, was less than 24 hours. A series of ditopic tin receptors (589a-i) were also studied in an effort to optimize the substitution pattern and fine tune the distance between the metal centers.643 In general, the best phosphate selectivity was observed for receptors with tin centers containing one electron withdrawing organic substituent and two halide substituents, presumably due to the increased Lewis acidity at the tin atom. In addition, linkers containing an odd number of carbon atoms appeared to complex phosphate more successfully. The binding behavior of receptors 589a, 589g, 589j, 589k, and 589l were analyzed by both 119Sn NMR spectroscopy and potentiometric methods.111 On the basis of the potentiometric studies, it was concluded that organic substituents that were electron-withdrawing in nature (Ph, 589a, k, l) displayed much better responses to inorganic phosphate than those lacking such substituents. On the other hand, a receptor that was more lipophilic (octyl, 589j) also displayed an improved response. The latter effect was attributed to improved partitioning of the carrier into the membrane. Both the inorganic phosphate response and the selectivity were found to decrease with increasing carbon spacer length (589a > 589k > 589l). Tin-119 NMR spectroscopic studies in CH2Cl2 confirmed that the phosphate selectivity of the receptor 589a based electrode, the best of the systems analyzed, was likely due to an increase in the stability constant of the receptor:phosphate complex. However, the lifetime of these electrodes was found to be on the order of 3 to 20 days, too short for repeated, long-term use.
Yu, Liu, and co-workers investigated divalent tin systems, such as bis(tribenzyltin) oxide, as ion selective electrode sensor elements.644,645 The corresponding electrode proved selective for dibasic phosphate over nitrate, chloride, acetate, and sulfate. Similar responses were observed, however, for iodide and bromide. An electrode lifetime of approximately one month was reported.
More recently, Suzuki and co-workers examined the effects of anionic additives on trialkyl/aryltin chloride electrodes.646 In preliminary studies, tributyltin chloride was incorporated into a PVC membrane containing sodium tetrakis[3,5-bis(trifluoromethyl)-phenyl]borate (NaTFPB) as an additive. The presence of the additive led to a high phosphate response at pH 7.0. The percentage of additive as well as the tin substitution (butyl, octyl, or phenyl) was then optimized. The most successful combination in terms of both selectivity and response proved to be tributyltin chloride mixed with NaTFPB at 25 mol % relative to tin ionophore. This result was tentatively attributed to facilitated chloride release arising from coordination of the additive to the tin center, and an increase in the Lewis acidity of the metal.
In addition to transition metals and main group cations, lanthanides (Ln) have also proven successful in the detection and complexation of phosphate species. Lanthanide-based receptors typically rely on the change in emissive properties of the cation that arises when anions displace water molecules from the metal center. For example, addition of anions to the chiral Eu and Tb complexes 590 (Ln = Eu(III), Tb(III)) results in changes in the circularly polarized luminescence (CPL) of the receptor in aqueous media buffered to pH 7.4.647,648 Apparent binding affinities for monohydrogenphosphate were estimated at log K = 4.15 or greater for all four complexes (590a,b:Eu and 590a,b:Tb). The phosphate affinities were significantly higher than those measured for bicarbonate, acetate, and lactate. Control experiments provided support for the suggestion that the dianionic ligand is bound to the metal center in a monodentate fashion. In general, the methylated ligand complexes (590b) proved to interact with anions more strongly than complexes of receptor 590a. Presumably, this reflects differences in the coordination environment of the two ligands. In addition, the terbium complexes of general structure 590 were found to respond to anions more effectively than the corresponding europium complexes. This trend was postulated to be due to differences in the pKa values of the metal centers. The closely related complex 591 was later bound to gold nanoparticles for improved luminescent sensing.649 Addition of diketone antenna compound 592 to these particles led to a strongly luminescent complex in HEPES buffer at pH 7.4. Displacement of the antenna by a variety of anions led to emission quenching. However, only addition of FMN resulted in almost complete quenching of the luminescence, allowing selective detection of this anion over AMP, ADP, ATP, 3′,5′-cAMP, NADP, inorganic phosphate, and a number of carboxylates.
The Anslyn group designed and synthesized the multifunctional receptor 593 that contains an N-oxide bipyridine europium complex and an ammonium “pinwheel” functionality.650 Host 593 was found to form a 1:1 complex with 2,3-BPG in 50% methanol/acetonitrile. The complex was thought to be stabilized through a europium-phosphate interaction acting in combination with ammonium-phosphate and ammonium-carboxylate interactions. The europium:2,3-BPG interaction was the one considered responsible for the fluorescence quenching. From the change in emission, a binding constant of 6.7 × 105 M-1was calculated for this solvent system. Binding studies in pure methanol supported the formation of 2:1 593:2,3-BPG complexes. This solvent-dependent stoichiometry was attributed to the complexation of each of the two phosphate groups by the europium center of a different receptor as a result of a reduction in the ammonium-anion binding interaction. The proposed model was supported by several subsequent experiments. For example, the removal of the ammonium centers in receptor 593 also led to a 2:1 binding stoichiometry, even in the original methanol/acetonitrile solvent system. Compounds containing one phosphate group and one carboxylate group also demonstrated 2:1 binding behavior. Furthermore, phenylphosphate was found to have a slightly reduced binding affinity of 2.0 × 105 M-1 relative to 2,3-BPG, a finding that was attributed to coordination of the monoanion at only the europium center. These results provided support for the conclusion that the three anionic groups of 2,3-BPG were indeed bound to the three cationic centers of host 593 in the methanol/acetonitrile solvent mixture. However, the addition of water to the solvent system led to a quenching of the fluorescence, preventing the use of receptor 593 in aqueous media.
Ziessel and co-workers found that europium and terbium complexes of receptors 594 and 595 displayed emission changes upon the addition of inorganic phosphate and ATP in aqueous media. In preliminary studies, both the absorption and emission of complexes 594 were observed to undergo changes upon the addition of inorganic phosphate or ATP in aqueous media at pH 7.0 (Tris/HClO4 buffer).651 No spectral changes were observed upon the addition of AMP or ADP. Consistent with what was seen with system 593 above, spectral analysis supported the displacement of water from the inner coordination sphere of the lanthanide. In addition, a peak corresponding to the ternary 594:Eu:ATP complex was observed in the ESI-mass spectrum. The nature of the anion interactions in 594:Eu were further probed using optical spectroscopy; these interactions were also analyzed using computational studies based on density functional theory.652 In the computational studies, mono-, di-, and triphosphate anions were generally found to be bound to the europium center in a multi-dentate fashion. Addition of water to the theoretical monohydrogenphosphate complex led to monovalent complexation. Upon binding, these anions displaced one or more of the ligand nitrogen atoms depending on the binding affinity of the guest. Based on these computational studies, it was proposed that increasing the lanthanide complexation ability of the ligand would enhance the selectivity of the system. To test these predictions, the alkyl nitrogen moiety was replaced by a phenylphosphine group and the methylene spacers were removed yielding receptor 595. With this new system, a 20-fold increase in binding affinity of the ligand to the europium metal was observed relative to 594. The binding behavior of anions toward 595:Eu was analyzed through UV-Vis, emission and luminescence lifetime measurements in a Tris/HCl buffer at pH 7.0. No spectral changes were observed with the addition of nitrate or AMP. However, ATP, ADP, and monohydrogenphosphate were found to bind. Similar association constants (log K ≈ 5.5) were recorded for all three anions, although the spectral changes were more pronounced with ATP and monohydrogenphosphate than with ADP. The increase in luminescence lifetimes for these complexes was attributed to a decrease in the nonradiative decay pathways available with coordinated water molecules in the absence of the anion. While stronger binding affinity was observed with 595:Eu relative to 594:Eu, little change in selectivity was observed.
While many lanthanide-based anion receptors have focused on differences in emission intensity, the Eu(III) complex 596 displayed differential UV-Vis spectral shifts when exposed to phosphate anions in aqueous media at pH 7.4.653 In preliminary studies, a pink to blue color shift was observed upon the addition of monohydrogenphosphate or pyrophosphate but not upon the addition of a variety of other inorganic anions. The detection limit for monohydrogenphosphate, even in the presence of other anions, was determined to be 6.0 × 10-7 M at this pH. A 2:1 anion:receptor binding stoichiometry was determined for monohydrogenphosphate and receptor 596, as was the second association constant (3.2 × 105 M-1). The association constant for the 1:1 complex with pyrophosphate was determined to be 5.9 × 103 M-1. When nucleotides were analyzed, moderate responses were observed for AMP and ADP, while a strong new absorption band was observed upon addition of ATP (K = 2.2 × 105 M-1). These results highlight the sensing power of lanthanide cations. In appropriately designed complexes, these metals will give rise to distinct spectral responses to different substrates that are thought to reflect slight differences in the overall coordination environment.
A number of diketone antibiotics have also been used as ligands in europium(III) sensing complexes. For example, C. Jiang and co-workers reported that the addition of ATP to a europium(III):doxycycline complex (597a) led to a strong increase in the luminescence intensity.654 This change was particularly apparent in aqueous ammonium chloride at pH 10. The emission intensity was found to correlate linearly with the ATP concentration in the range of 1.00 × 10-7 - 2.00 × 10-6 M-1. ATP detection was found to be independent of several possible intereferents found in bodily fluids and could be applied to commercial ATP samples. An analogous complex with oxytetracycline (597b) displayed similar spectroscopic changes but an increased linear concentration range (8.00 × 10-8 - 1.50 × 10-6 M).655 These researchers also reported the detection of NADP using a tetracycline ligand (597c) at concentrations as low as 6.9 × 10-8 M at pH 7.6. 656 Duerkop and co-workers studied the effect of inorganic phosphate on the luminescence of the europium(III) tetracycline complex (597c).657 Quenching was observed to be linear with phosphate concentration over the range of 5 × 10-6 - 7.50 × 10-4 M at neutral pH. This signal was also subject to little interference by common cations and anions. Schäferling and Wolfbeis studied the effect of several other phosphate derivatives on the luminescence of complex 597c.658 In this case, quenching was found to be strongest with ATP and GTP, followed by pyrophosphate > AMP > ADP ≈ 5′-guanosine diphosphate (GDP) ≈ 3′,5′-cAMP. This selectivity allowed for the use of complex 597c to monitor kinase assays in real time.
C. Jiang and co-workers also developed a terbium(III)-based luminescence sensor for ATP by using a norfloxacin ligand (598).659 Coordination of the norfloxacin ligand led to an emissive complex in aqueous solution, presumably due to energy transfer processes from the ligand to the terbium(III) center. Addition of ATP to the Tb3+:L complex in Tris-HCl buffered aqueous solution at pH 7.4 further increased the emission intensity. It was proposed that multi-dentate coordination of ATP to the terbium(III) center displaced coordinated water molecules, which are known to quench lanthanide luminescence. The change in signal intensity was found to vary linearly with the concentration of ATP over the 1.0 × 10-6 M - 1.60 × 10-5 M range, and a detection limit of 4 × 10-8 M was reported. Less than 10% interference was measured for a range of metal ions, proteins, and metabolites expected to be found in biological fluids. This system was further used to quantify the levels of ATP in commercial drug samples.
A different lanthanide-based system was reported by Yang and co-workers, who combined ytterbium and pyrocatechol violet in a 2:1 ratio to form receptor 599.660,661 This complex proved successful in the detection of phosphate-derivatives in aqueous media (HEPES buffer, pH 7.0). In preliminary studies, the addition of ATP or monohydrogenphosphate led to the decomplexation of pyrocatechol violet and concurrent formation of 1:1 Yb:anion complexes. The resultant optical changes were observed both visually and spectrophotometrically. While several other lanthanide and transition metals were analyzed, only ytterbium proved successful in these assays. Little to no interference was observed in the presence of other anions, even when the latter were added at a 1000-fold excess relative to the two test phosphate anions. Other nucleotides resulted in a much lower response. Spectrophotometric titrations with ATP and monohydrogenphosphate resulted in conditional binding constants of 5.85 × 1015 M-1 and 2.0 × 1015 M-1 for the interaction of these two anions to this ytterbium complex. The sensitivity of this system was found to lie in the 10-5 M range in HEPES buffer.
The uranyl cation has also been extensively exploited to produce phosphate receptors, particularly by Reinhoudt and co-workers. In early studies involving complexes of this high valent actinide cation, receptors 600a-c and 601a-b were made and tested; these receptors were found to be selective for dihydrogenphosphate in acetonitrile / 1% DMSO as inferred from conductometric studies.662 The highest association constants were observed with receptors 600b and 600c (K = 2.5 × 106 M-1 and 5.0 × 106 M-1, respectively). The efficiency of these receptors was attributed to the presence of anion-binding amide groups and increased preorganization. These complexes were also observed by 31P NMR spectroscopy and FAB mass spectrometry. In addition, high selectivities over chloride, hydrogensulfate, nitrite, and thiocyanate were reported. Further studies focused on receptors 601a-d.663 Single crystal X-ray diffraction analysis of receptor 601b and H2PO4- was performed. The resulting structure revealed the expected two apical oxygen bonds on the uranyl cation, as well as coordination to the four positions of the salophene ligand. An additional coordination site at the uranium cation was seen to be filled by an oxygen atom of the phosphate. Hydrogen bonds were also observed between the phosphate oxygen atoms and the methoxy substituents of the ligand. Single crystal X-ray diffraction analysis of receptor 601d and H2PO4- revealed similar uranyl coordination and clear hydrogen bonding interactions between the dihydrogenphosphate and the amide units. Several trends in the anion binding properties of these compounds were further analyzed using conductometry. The incorporation of the nitro groups in receptor 601c, for example, increased the binding affinity for chloride and nitrite but not dihydrogenphosphate. While receptors 602 and 603 contained additional hydrogen bonding units as compared to receptor 601a-c, weaker complexation constants were observed with dihydrogenphosphate. This effect was attributed to the proposed smaller cavity size of these latter receptors. Receptors 600a, 600b, 600f, and 604, however, displayed an increase in complex stability due to the presence of additional amide units. Complexation studies in DMSO/water mixtures revealed weak binding interactions.
Lipophilic receptors 605 were incorporated into PVC membranes cast on chemically modified field effect transistors (CHEM-FETs).664 These FETs were found to sense dihydrogenphosphate with selectivity observed relative to nitrate, bromide, chloride, and sulfate. The lowest detection limit (1.6 × 10-4 M) was observed with receptor 605b, which may form hydrogen bonds between the methoxy groups and the phosphate. A reduced selectivity for dihydrogenphosphate over nitrate was observed with receptors 605b and 605c as compared to receptor 605a. Presumably, this reflects the electron-donating nature of the substituents on the former systems. An investigation of these compounds in ion-selective electrodes at varying pH led to the suggestion that the hydrogen bonding available in receptor 605b led to an increased selectivity over the hydroxide ion as compared to receptors 605a and 605c.665 More detailed studies compared the response of receptors 605a and 605b to receptors 601a and 601b in phosphate selective electrodes.666 The latter set displayed reduced phosphate selectivity, presumably due to reduced solubility in the PVC membrane. The best selectivity and response in these electrodes was observed with receptor 605a. An analysis of ion selective electrode durability was also carried out. It was revealed that receptor 601e had a selectivity that was comparable to that of receptor 605a, but with a lifetime that was improved by up to two months.667 Similar trends but lower lifetimes (2 weeks) were observed with the CHEMFETs.668
Recently, Mandolini and Rissanen analyzed receptor 601f, which possesses bulky tert-butyl substituents, and macrocyclic receptor 606.669 As inferred from UV-Vis titrations in DMSO, receptor 601f is characterized by significant fluoride selectivity. In addition, the interaction of receptor 601f with hydrogenphosphate (log K = 2.32) was found to be significantly weaker than that found with receptor 601a (log K = 4.00). The reduced binding of receptor 601f was attributed to steric hindrance due to the tert-butyl substituents, which inhibits complexation. The small cavity of the receptor 606 led a to selectively toward fluoride over other anions. All anions were studied as the TBA salts.
Reinhoudt and co-workers also analyzed several uranyl receptors for their ability to function as ion pair receptors and transporters. Ditopic receptor 607d was designed to extract an ion pair, in this case KH2PO4, from an aqueous layer into apolar media.670 Proton NMR spectroscopic experiments carried out in DMSO allowed for the determination of a complexation constant of 1.1 × 103 M-1 for the interaction of receptor 607d with H2PO4-. These experiments also provided support for the presence of hydrogen bonding interactions between the anion and the amide moiety of the receptor. A similar association constant for the anion was determined through cyclic voltammetry. This latter method also permitted the determination of an association constant for potassium of 1.0 × 102 M-1. Complexation of the cation:anion pair by receptor 607d was observed through FAB mass spectrometry, thus providing direct evidence of effective ion pair extraction. The calixarene-salen compounds 608 were also found to function as ditopic receptors.671 These receptors were found to bind dihydrogenphosphate, with association constants of 3.5 × 102 M-1 and 3.9 × 102 M-1 being recorded for receptors 608a and 608b, respectively. Peaks corresponding to the ion pair NaH2PO4:608b complex were also seen in the FAB mass spectrum.
The ability of these receptors to transport ion pairs across membranes was also analyzed. Receptors 607 were tested for their ability to transport KH2PO4 but were found to only be effective when used in combination with calixarene 609.672 Receptors 607e-g, 610, and 611 were studied for their ability to transport NPr4H2PO4 over NPr4Cl through a supported liquid membrane.673 Receptors 610 and 611 provided for the selective co-transport of chloride over phosphate. The receptors 607e and 607f, however, allowed for the selective co-transport of dihydrogenphosphate, as predicted. A 2:1 carrier:anion ratio was observed for the latter dihydrogenphosphate complexes, presumably due to the presence of the sulfamido moieties.
Receptors 612 and 613 incorporate thymine bases, a moiety included in order to promote specific ditopic binding of AMP.674 Proton NMR spectroscopic experiments in DMSO supported a ditopic binding mode. Binding constants for the AMP complexes of receptors 612 and 613 were determined to be 75 M-1 and 1.2 × 103 M-1, respectively, in this solvent. The relatively low binding interaction observed with receptor 612 was attributed to steric crowding as well as to the presence of only one open coordination site. Both host:guest complexes were also observed through FAB mass spectrometry.
4.3.2 Metal Cations as Non-coordinating Reporter Groups
As seen in many of the previously discussed examples, metal cations have played an important role in anion complexation. In this section, metal ions that act largely as non-coordinating reporter sites will be reviewed, particularly metallocene as an electrochemical reporter and [Ru(bpy)3]2+ as a fluorescent reporter.
Metallocene as an electrochemical reporter
Reversible electrochemical behavior is generally seen for metallocene (Mc)/metallocenium (Mc+) redox processes. These couples can be affected by the presence of anions. Generally, anion binding interactions result in cathodic shifts of both the oxidation and reduction peaks of the Mc/Mc+. These shifts are attributed to the interaction of the neutral receptor with the anion as well as the stabilization of the metallocenium species by coordination of the anion.675 The magnitude of the shift is correlated with several factors, including the strength of the receptor-anion interaction as well as the distance between the bound anion and the metallocene unit. Insolubility of the metallocenium:anion ion pairs in organic media, however, can lead to adsorption of the receptors onto the electrode. This adsorption leads to irreversible and/or poorly characterized electrochemical spectra. In addition, irreversible ligand oxidation can also interfere with this sensing method.
In 1993 Beer and co-workers began an investigation of the binding behavior of ferrocene and cobaltocene-appended phosphate receptors. Many of the systems developed by this group relied on the phosphate binding functionalities discussed in previous sections. The key difference was the use of a metallocene subunit as an electrochemical reporter group. In early studies, the amide-based receptor 614, bearing two kinds of metallocenes, was found to form 1:1 complexes with dihydrogenphosphate by 1H NMR spectroscopy in acetonitrile.676 In cyclic voltammetric (CV) experiments, both the ferrocenyl and cobaltocenium centers were observed to undergo cathodic shifts upon addition of dihydrogenphosphate, chloride, and hydrogensulfate (TBA salts) in acetonitrile. Similar results were obtained in studies with receptors 615a, 616, 617, and 618. Among this set of receptors, the tripodal receptor 617 displayed the largest cathodic perturbation in the presence of dihydrogenphosphate. Interestingly, no perturbation of the dihydrogenphosphate signal was observed in the presence of a ten-fold excess of hydrogensulfate or chloride anions for receptors 616, 617, and 618. Further investigation of the phosphate binding behavior of receptors 615a and 616 revealed a discrepancy in the selectivity observed through CV and 1H NMR spectroscopic methods. Based on 1H NMR spectroscopic analyses, a significantly stronger interaction with hydrogensulfate than dihydrogenphosphate in acetonitrile was inferred.677 In contrast, a stronger electrochemical response for dihydrogenphosphate over hydrogensulfate was inferred through CV studies. This disparity was attributed to an increased participation of the ferrocene moiety in the electrochemical experiments as compared to the NMR spectroscopic studies. The researchers concluded that complex stabilities were not a reliable indicator of electrochemical response.
The simple cobaltocene amide receptors 619 also displayed a selective electrochemical response towards dihydrogenphosphate (TBA salt) in acetonitrile.678 The association constant corresponding to the interaction between dihydrogenphosphate and receptor 619b was measured to be 3.2 × 102 M-1 in DMSO-d6 via 1H NMR spectroscopy.
The binding behavior of various analogues that contain several other recognition moieties was analyzed in order to probe further the inherent anion selectivity of this class of receptors.679 The amine- and amide-based receptors 615a, 615b, 620, and 621 displayed low dihydrogenphosphate binding affinities and selectivities as inferred from both 1H NMR spectroscopic and CV measurements carried out in chloroform and acetonitrile using several test TBA anion salts. In separate electrochemical studies, the amidopyridine receptors 616 and 622 displayed the strongest response to dihydrogenphosphate as compared to other anions; however, these receptors did not produce reversible redox waves.
Interestingly, 1H NMR spectroscopic analyses provided support for significantly different binding modes for hydrogensulfate and dihydrogenphosphate in their interactions with receptor 622. Hydrogensulfate was considered to interact more significantly with the amine side than the ferrocene unit of the receptor, presumably due to its ability to act as a proton donor to the terminal amine. The opposite binding mode was observed with the more basic dihydrogenphosphate anion. In addition, only the addition of dihydrogenphosphate resulted in a bathochromic shift of the d-d transition absorption band. Hydrogensulfate, however, was able to inhibit the UV-Vis spectral response to dihydrogenphosphate binding. The mono-functionalized receptor 616 did not display any anion-based UV-Visible spectroscopic response.
The anion binding behavior of urea receptors 623 and 624 was also analyzed via 1H NMR spectroscopic studies carried out in acetonitrile-d3 with TBA anion salts.680 Selectivity for chloride and dihydrogenphosphate was observed, presumably based on steric effects. For example, receptors with bulky tert-butyl ester substituents (623b and 624b) displayed chloride selectivity while their hexyl-substituted counterparts displayed the more common dihydrogenphosphate selectivity. Furthermore, stronger binding constants were observed with difunctionalized receptors 624 over monofunctionalized receptor 623. Reversible CV responses were reported for both classes of receptors in the presence of dihydrogenphosphate, acetate, and chloride.
Efforts were also made by Beer and co-workers to achieve phosphate complexation in competitive solvents such as DMSO, methanol, and water. For example, the polyammonium receptors 625 and 626 displayed cathodic responses to monohydrogenphosphate and ATP in aqueous media near pH 7.681,682 The thiourea receptor 627a was able to bind dihydrogenphosphate (TBA salt) in DMSO (K = 2.6 × 102 M-1). At the same time, guanidinium receptor 627b was observed to complex pyrophosphate (TBA salt) in a 2:1 receptor:anion ratio in 1:1 MeOH:H2O (K = 4.6 × 103 M-2).683 While binding constants were determined by 1H NMR spectroscopy, nearly concordant results were obtained using CV measurements.
Martínez-Máñez and co-workers also prepared a series of polyammonium-functionalized ferrocenes 628-639 and investigated the ability of these receptors to recognize phosphate anions in aqueous media. The largest responses were observed at lower pH when the protonation state of the amines was maximized. Low selectivities were generally observed. For instance, receptors 628 displayed a redox response to ATP, inorganic phosphate, sulfate, and nitrate in aqueous media.684 Among the series of receptors 628, the strongest response was observed with receptor 628b. This receptor also proved somewhat selective for ATP over inorganic phosphate and sulfate at a pH of 4.9 in 7:3 THF:water. Macrocyclic receptor 629 displayed a low response under these conditions, which was attributed to the steric bulk of the ferrocene units when compared to the unfunctionalized azamacrocycle. More detailed studies of receptor 628b were performed through both potentiometric and CV methods.685 Potentiometric studies in 7:3 THF:water confirmed that receptor 628b forms complexes with ATP and inorganic phosphate over a wide pH range. Using this method, the concentration of ATP could be determined in the presence of inorganic phosphate or sulfate. On the other hand, a preference for hydrogensulfate over dihydrogenphosphate was observed through CV studies with receptor 628b in dry acetonitrile with TBA anion salts.
A comparison of the binding behavior of receptors 625a, 628a, 630, 631a, and 631b revealed differing selectivities in aqueous media.686,687 Potentiometric studies supported the conclusion that for most species the sulfate complex dominated at lower pH ranges while the inorganic phosphate complex dominated at neutral and basic pH. This trend was also reflected in CV studies. Indeed, receptor 631b could be used to detect inorganic phosphate quantitatively at pH 7, even in the presence of competing sulfate or nitrate ions. Little inorganic phosphate interaction was observed with receptor 628a, supporting the suggestion that the cyclic receptors may bind the anion more closely to the ferrocene moieties. Receptors 631a and 631b displayed a strong response to ATP and ADP, as well as a weaker response to AMP.
The effect of polyoxo ligands as well as the number of appended ferrocene units was studied by comparing several receptors in water (633 - 635) and in a dioxane-water mixture (7:3, 629 and 632).688 Under these conditions, receptor 629 displayed a stronger electrochemical response for inorganic phosphate (pH 6-7) than receptor 632, presumably due to the increased number of nitrogen atoms in receptor 629. While receptors 633 and 634 each contain four nitrogen atoms, a higher binding affinity for ATP was observed for 634 in potentiometric studies. Molecular modeling led to the suggestion that receptor 634 could adopt a conformation allowing all four nitrogen atoms to interact with the phosphate. Complexes with receptor 633 were presumed to be stabilized by interaction with only two nitrogen atoms. Similar trends were observed in the case of ATP complexation; however, these receptors displayed modest electrochemical responses to inorganic phosphate and ATP. The binding behavior of receptors 629 and 632 was then analyzed in acetonitrile using TBA anion salts. In this solvent, a selective response to dihydrogenphosphate over hydrogensulfate was observed.
Further studies compared the binding behavior of receptors 628b, 629, 632, and 636-638 in acetonitrile using TBA anion salts.689 In these studies, no electrochemical shifts were observed upon addition of chloride or bromide anions. Only receptor 628b displayed a change in the ferrocene oxidation potential upon addition of hydrogensulfate. Dihydrogenphosphate addition resulted in more pronounced shifts in both the oxidation and reduction waves. Receptor 629 displayed the strongest interaction with dihydrogenphosphate. In general, greater responses were observed with those receptors containing more amine groups, presumably due to increased proton transfer from the anion. On the other hand, the number of ferrocene centers did not appear to impact the CV spectrum directly. Selectivity studies revealed that the dihydrogenphosphate responses of receptors 629, 636, and 637 were not affected by the presence of a ten-fold excess of chloride, bromide, or hydrogensulfate anions.
Receptor 639 was designed to provide both an electrochemical and fluorescent response to anions. However, it was found to be insensitive to the presence of anions in acetonitrile except as its copper(II) complex (639:Cu).690 This latter complex displayed an electrochemical response to both fluoride and dihydrogenphosphate, as well as a fluorescence response to dihydrogenphosphate, nitrate and fluoride (all anions added as TBA salts). Log K values determined from fluorescent titrations were reported as 4.2-4.5 for these three anions, leading to the conclusion that this receptor is not particularly selective. It is important to note that many of the receptors in these studies displayed electrochemical signals in response to transition metal cations as well as to anions.
Pyrrolic ferrocene receptors were first reported by Sessler and co-workers in 1998.691,692 Preliminary studies compared the anion binding properties of 640a to that of its acyclic counterpart 641. This was done using both 1H NMR spectroscopy and electrochemical analyses (both carried out in acetonitrile using various TBA anion salts). The two pyrrolic systems were found to bind chloride and dihydrogenphosphate in preference to bromide and hydrogensulfate. In addition, a slight preference for dihydrogenphosphate over chloride was observed with receptor 640a. Fluoride was bound too strongly for an association constant to be determined by NMR spectroscopic methods, but Job plots supported a 2:1 guest:host stoichiometry. All other anions were found to form 1:1 complexes. The stronger binding of 640a was attributed to the macrocyclic effect of preorganization. Both receptors produced electrochemical signals, with the strongest response reported for dihydrogenphosphate followed by fluoride and chloride.
Further studies examined derivatives 640b-d, although solubility problems required the use of dichloromethane / 2% DMSO as the solvent. This precluded a direct comparison with 640a. The highest binding affinity was observed for receptor 640d, a finding that provides support for the conclusion that the additional oxygen atoms present in this system participate in hydrogen bonding with the anion, thereby increasing the stability of the complex. A similar trend was inferred from the electrochemical studies, although little difference was observed in the responses of 640c and 640d. This latter result led to the suggestion that additional factors, such as the proximity of the bound anion to the ferrocene unit, greatly affected the electrochemical response.
Gale and co-workers found that that cleft-like pyrrole-ferrocene compounds 642-645 also function as phosphate sensors under conditions of electrochemical analysis.693,694 The association constants for these receptors with a variety of anions (fluoride, chloride, bromide, dihydrogenphosphate, hydrogensulfate, and benzoate as their TBA salts) were first determined via 1H NMR spectroscopy titrations carried out in dichloromethane. Based on these analyses, it was concluded that the conjugated receptors 642 and 643 bind the targeted anions more strongly than receptors 644 and 645. Selectivity for fluoride among other anions was observed with all four receptors. The highest association constant for dihydrogenphosphate was measured with receptor 643 (K = 2.96 × 102 M-1). Distortion of the redox wave was observed for receptors 642 and 644 in the presence of dihydrogenphosphate. CV studies with receptors 643 and 645, however, led to the observation of strong, measurable electrochemical responses for dihydrogenphosphate.
Gale and co-workers also investigated a small library of amide and urea ferrocene receptors (646-651).695 In this case, 1H NMR spectroscopic titrations carried out in DMSO-d6 / H2O (0.5%) with TBA anion salts proved consistent with the presence of dihydrogenphosphate-receptor interactions. However, as a rule, receptors 646-651 displayed a preference for carboxylate anions over dihydrogenphosphate. Electrochemical studies supported the conclusions drawn from the NMR spectroscopic studies. Receptors 648-651 were found to produce the highest electrochemical response upon anion addition.
Moutet and co-workers also investigated the interactions of dihydrogenphosphate and ATP with a variety of amide and cationic ferrocene receptors. In preliminary studies, the amide-bipyridine receptor 652a was observed to bind dihydrogenphosphate in dichloromethane as inferred from both 1H NMR spectroscopic and CV experiments using TBA anion salts.696 The NMR spectroscopic studies provided support for strong anion-amide interactions. This conclusion was further confirmed by the finding that weak or essentially non-existent binding was seen in the case of receptors 652b-d. No interaction with the bipyridine unit was observed by 1H NMR spectroscopy. While evidence of fluoride, chloride and hydrogensulfate complexation was also obtained from CV experiments, the strongest and most well-characterized response was observed for dihydrogenphosphate. The bipyridine-free amide receptors 653-657 displayed electrochemical responses to dihydrogenphosphate and ATP.697 The binding behavior of receptors 653-656 with dihydrogenphosphate was analyzed in dichloromethane (653, 654 and 656) or acetonitrile (655) through 1H NMR spectroscopic titrations. A strong perturbation of the chemical shift of the amide protons provided support for the conclusion that hydrogen bonding at these sites was the primary binding force leading to complex formation. Interestingly, both types of amide protons in receptor 656 were found to shift upon treatment with dihydrogenphosphate. Monitoring these shifts allowed a binding constant of 89 M-1 to be determined for dihydrogenphosphate. Similar binding constants were observed with fluoride anion. However, in this case the iron center was considered to be more involved in binding than the amide groups. In dichloromethane, receptor 654 was found to bind dihydrogenphosphate and fluoride more strongly than receptor 653. This finding serves to highlight the importance of the amide substituents. Receptor 654 was also found to have a greater affinity for the dihydrogenphosphate and fluoride anions than receptor 655, an observation that was attributed to macrocycle 655 being too small and rigid to accommodate well the phosphate anion. It should be noted, however, that receptor 655 was studied in acetonitrile while receptor 654 was studied in dichloromethane. Receptor 655 also displayed a low electrochemical response. Interestingly, both 656 and 657 produced well-characterized electrochemical responses when exposed to dihydrogenphosphate and ATP. However, receptor 657 did not display evidence of binding as inferred from 1H NMR spectroscopic analyses. Nevertheless, taken in concert these results led to the suggestion that the cyclotriveratrylene scaffold and additional ferrocene units in receptors 656 and 657 create phosphate binding sites appropriate for amperometric sensing. Little electrochemical response was observed for these receptors in the presence of hydrogensulfate, and fluoride produced irreversible waves.
Well-characterized electrochemical behavior with dihydrogenphosphate in dichloromethane was seen for the simple ammonium receptor 658.698 However, little response was observed in methanol. Presumably, this is due to the counteracting effects of ion pair solubilization and solvation of the anion. In these experiments, all anions were studied as the TBA salts. The cationic viologen receptors (659, 660 and poly-661) were observed to be efficient electrochemical sensors for ATP, even in aqueous media.699 While the redox activity of the viologen was not well behaved, monitoring the ferrocene activity in unbuffered water revealed a strong interaction with ATP and a smaller interaction with hydrogensulfate, dithionite, and phenylphosphate. Interestingly, no response was observed with dihydrogenphosphate, sulfate, hydrogenphosphate, trifluoroacetate, fluoride or chloride. Receptor 660 displayed the greatest and most selective ATP response. This finding was attributed to the presence of additional interactions between the viologen and adenine moieties, as well as to an increased level of preorganization. Interestingly, films of poly-661 could be deposited on electrode surfaces and the resulting systems were found to display a selective response for ATP that could be measured quantitatively.
Molina and co-workers studied several urea substituted ferrocene compounds, including the ditopic receptors (662) and the cyclophane-type receptors (663-664). The ditopic receptor 662 was designed to bind concurrently both anions and cations through interactions with the urea moieties and the crown ether moieties, respectively.700 Indeed, both dihydrogenphosphate (K = 1.5 × 104 M-1) and fluoride (K = 1.5 × 103 M-1) were found to interact with the urea groups as judged from 1H NMR spectroscopic titrations carried out in chloroform using TBA anion salts. Potassium cation was then able to bind the resulting 662:H2PO4- complex with an association constant of 7.3 × 103 M-1 in this solvent. A strong, selective electrochemical response was observed for dihydrogenphosphate in dichloromethane (a different solvent than the chloroform used in the NMR spectroscopic studies). A lower response, however, was observed in the presence of the potassium cation. The authors attributed this variation to a conformational change that occurs upon the binding of potassium, a movement that was thought to bring the two crown ether arms closer together, thereby making the urea groups less accessible to the anion. The cyclic receptors 663 and 664 were both found to bind dihydrogenphosphate with association constants of 7.2 × 102 M-1 (chloroform) and 4.5 × 102 M-1 (DMSO), respectively, as inferred from 1H NMR spectroscopic studies (studied as the TBA salts).701 While fluoride was also found to interact with these receptors, chloride, nitrate, and hydrogensulfate did not produce any spectral changes. Although the binding behavior of receptor 664 in a much polar solvent supported stronger complexation, electrochemical studies revealed the formation of an unidentified new species after oxidation in the absence of anion, presumably due to an irreversible chemical reaction following electron transfer (EC mechanism). Receptor 663, however, gave a well-behaved cathodic shift and demonstrated a strong selectivity for dihydrogenphosphate over other anions. Molecular modeling of the two receptors led to the conclusion that intramolecular hydrogen bonding interactions served to stabilize cavities that are appropriate for dihydrogenphosphate.
Later studies analyzed the anion binding of the urea receptors 665-669.702 This series of receptors allowed for fluoride and dihydrogenphosphate detection using 1H NMR spectroscopy and electrochemical methods. Furthermore, the incorporation of fluorescent moieties allowed for the use of receptor 670 as a fluorescent sensor. This receptor was able to recognize hydrogenpyrophosphate, ATP and ADP, and signal their presence via fluorescence and UV-Vis spectroscopies as well as electrochemical methods.703 In the case of ATP and ADP, hydrogen bonding interactions were considered to be most important in terms of stabilizing complex formations. However, in the case of hydrogenpyrophosphate, the induced spectroscopic and electrochemical changes were thought to reflect simultaneous hydrogen bonding and deprotonation events. Fluorescence titrations with receptor 670 in acetonitrile allowed binding constants of 4.5 × 105 M-1 and 1.4 × 104 M-1, to be determined for ATP and ADP, respectively. An apparent association constant for hydrogenpyrophosphate of ≈104 M-1 was also calculated. Naked-eye detection was achieved via the incorporation of colorimetric units into the guanidinium receptor 673 as part of a larger study including receptors 671-674.704 Electrochemical studies revealed the ability of each these receptors to detect fluoride, acetate, hydrogensulfate, dihydrogenphosphate, and hydrogenpyrophosphate. ITC studies in DMSO allowed for the determination of association constants on the order of 104 M-1 for dihydrogenphosphate in the case of receptors 671, 672, and 674. Furthermore, naked-eye detection proved possible with receptors 671-674 upon addition of the fluoride, acetate, dihydrogenphosphate and hydrogenpyrophosphate. Anions were studied as the TBA salts in these studies.
Recently Torroba and Riant have prepared and studied the N,N-diethylthiobarbituric receptors 675-679.705 In this case, UV-Vis and 1H NMR spectroscopic studies carried out in acetonitrile supported the conclusion that the dihydrogenphosphate, benzoate, acetate, fluoride and cyanide anions (TBA salts) interacted with both the thiobarbituric unit and the ferrocene moiety of the receptors. However, dihydrogenphosphate was found to bind to a much lower extent than the other studied anions. While no binding constant could be measured for dihydrogenphosphate, naked-eye detection proved possible with receptor 675 at anion concentrations of 10-5 M. These receptors were also found to complex transition metal cations.
Several other more specialized ferrocene receptors have also been reported. For example, the binding behavior of the phosphorous containing macrocycle 680 was analyzed by Mathieu, Delavaux-Nicot, and co-workers.706 In this case, a strong cathodic shift was observed by CV in acetonitrile upon the addition of dihydrogenphosphate (TBA salt). Smaller shifts were observed for hydrogensulfate and chloride. The addition of up to two equivalents of the latter anions did not affect the phosphate-induced signal. Molecular modeling supported the conclusion that the cavity of receptor 680 was compatible with the dihydrogenphosphate anion in terms of size and shape.
Wang and co-workers recently reported a bis-amidoferrocene receptor containing an ethylene glycol linkage (681).707 Electrochemical analyses were conducted in dichloromethane with 0.1 M [TBA-BF4]. CV curves revealed the growth of a new oxidation peak after the addition of 0.5 equivalents of dihydrogenphosphate (presumably added as the TBA salt) relative to the ferrocene unit. Above two equivalents of dihydrogenphosphate, no change in the oxidation peak was observed. This behavior was attributed to the formation of a 2:1 host:guest complex below 0.5 equivalents of anion, and these complexes did not exhibit dramatically altered electrochemical activities. Between 0.5 and 2 equivalents of anion, 1:1 host:guest complexes were formed, and these complexes led to the formation of a new oxidation peak. After 2 equivalents of anion were added, it was proposed that 1:2 host:guest complexes predominated in the solution.
Many examples of metallocene-based phosphate receptors derived from a calix[4]arene core have been reported by Beer and co-workers. In early studies, receptor 682 was found to complex dihydrogenphosphate in DMSO-d6 as judged from 1H NMR spectroscopic titrations (K = 2.8 × 103 M-1).708 Chloride and bromide, however, were bound even more strongly. Precipitation of the host:guest complexes prevented an analysis of these systems by electrochemical methods. Increasing the bulkiness of the lower rim substituents (683 and 684) led to similar association constants but produced better selectivities for dihydrogenphosphate in the case of receptor 684.709 The bridged receptor 685 was found to bind dihydrogenphosphate slightly more strongly than 684 (K = 6.38 × 103 M-1in DMSO-d6).710,711 Receptors 683-685 were also observed to interact with a variety of carboxylate anions. While receptor 683 displayed the lowest binding affinity of the series, it also displayed the only high selectivity for dihydrogenphosphate. Reversible cathodic shifts were observed for receptors 683-685 in acetonitrile with the magnitude of the response in line with the observed stability constants recorded in DMSO-d6. As with receptor 618 described earlier, receptors 686 and 687, derived from calixarenes substituted on the lower rim, gave rise to strong cathodic shifts in the presence of dihydrogenphosphate (TBA salt), at least in organic media.712 Overall, calixarenes of this type proved useful as scaffolds for creating amide-metallocene anion receptors since, by and large, these compounds displayed both high stability constants and good electrochemical responses. Oddly, receptor 688 did not display a response to anions, perhaps due to the presence of competing steric or intramolecular hydrogen bonding interactions. Thus, the specifics of the design are seen to be important in terms of receptor performance.
More recently, Beer and co-workers reported calixarenes functionalized at the upper rim with amido- and ureidoferrocene units (689 - 691).713 These receptors were found to bind benzoate, chloride, and dihydrogenphosphate through 1H NMR spectroscopic studies in 1:1 deuterated acetonitrile:DMSO. The spectra obtained were consistent with the presence of hydrogen bonding interactions with the urea and amide units, respectively, at the upper rim of the macrocycle. Significantly stronger binding constants were observed with tetraurea receptor 690 than di-substituted receptor 689, presumably due to increased hydrogen bonding interactions with the additional recognition moieties. Tetraurea receptor 690 and tetraamido receptor 691 displayed similar association constants with dihydrogenphosphate (K = 150 M-1 and 120 M-1, respectively); however, receptor 690 displayed a greater selectivity for dihydrogenphosphate over benzoate and chloride. Cyclic and square-wave voltammetry studies revealed the presence of a new peak upon addition of dihydrogenphosphate to all three receptors while only peak shifts were observed with benzoate and chloride (1:1 CH2Cl2:CH3CN). As a result, the authors suggested that the new peak resulted from the kinetically slow binding of dihydrogenphosphate relative to the cyclic and square-wave voltammetry time scale. All anions were studied as the TBA salts.
In 2008, Lemaire and Saint-Aman synthesized receptors 692-693 in which the ferrocene moieties were connected to calixarene units through urea or amide groups.714,715 Electrochemical analyses, as well as 1H NMR spectroscopic studies, carried out in dichloromethane led to the conclusion that receptors 692-693 interact with dihydrogenphosphate (TBA salt); however, the resulting binding constants were rather low (e.g., K = 36 M-1 for receptor 692a). In a unique variation on the calixarene ferrocene paradigm, Guo and co-workers developed the thiacalix[4]arene-based receptor 694.716 In this case, electrochemical studies carried out in dichloromethane:acetonitrile revealed a cathodic shift that was correlated with formation of a dihydrogenphosphate (TBA salt) complex.
The effect of organizing electrochemically active receptors on surfaces was also investigated by the Beer group.717 Solution studies of the free receptor 695 and its surface bound analogue 696, carried out via 1H NMR spectroscopy in acetonitrile/dichloromethane with TBA anion salts, led to the suggestion that these receptors formed much stronger anion complexes than the previous systems. Interestingly, electrochemical analyses revealed an irreversible phenomenon in the case of receptor 695 but well-behaved redox behavior for receptor 696. This latter system was found to display selectivity for dihydrogenphosphate. Good selectivity was also observed when receptors 696 and 697 were absorbed onto gold monolayers. An increase in the cathodic shift was observed for the surface-bound version of 696 relative to what was observed for this same receptor in acetonitrile/dichloromethane solution. Such an observation provides support for the conclusion that absorption to the surface amplified the signal, presumably by preorganizing the system. Both the surface and solution based forms of receptor 696 were found to be free of interference for dihydrogenphosphate recognition at up to 100 equivalents of halide.
Amidoferrocene moieties have also been attached to dendrimers for electrochemical anion sensing, particularly by Astruc and co-workers. Early studies in 1997 investigated the electrochemical behavior of dendrimers bearing 3, 9, 18, and 36 amidoferrocene units, of which two are shown (698a (9 units) and 698b (18 units)).718 These studies revealed a clear “dendritic effect” in which an increase in the number of arms, and therefore amidoferrocene units, led to an increase in anion association as inferred from 1H NMR spectroscopic and CV studies with TBA anion salts. With high generation dendrimers (e.g., 36 units), however, saturation was reached. Presumably, this reflects the fact that many of the ferrocene units were no longer available to the anion. In the case of the amino dendrimers analyzed in this study, the highest anion response was observed with the system bearing 18-amidoferrocene units. For this receptor, a new wave was observed in the presence of dihydrogenphosphate in dichloromethane. Apparent association constants with values > 105 M-1 were calculated for the interaction of dendrimers 698a and 698b with this anion. In addition, strong selectivity for dihydrogenphosphate over hydrogensulfate, chloride, and nitrate was observed.
Further studies compared the incorporation of Cp (699a) vs. Cp* (699b) substituted amidoferrocene ligands attached to a polyamine core.719 Dendrimers of generation 1-5 were studied, with generation 3 (G3) shown here (699). Interestingly, the anion response of the Cp* derivatives (e.g., 699b) proved to be longer lived and more reversible than that of the unsubstituted Cp congeners (e.g., 699a). The improved behavior of the Cp* receptors was attributed to the enhanced stability and increased lipophilicity that are characteristic of Cp* ligands. Furthermore, appreciable phosphate selectivity was observed, with little interference from chloride or hydrogensulfate. The strongest apparent association constant for dihydrogenphosphate interactions was estimated to be 7 × 104 M-1 (DMF) with the fourth generation (G4) Cp* functionalized ferrocenium dendrimer. While no dendritic effect was observed for the Cp* substituted dendrimers in dichloromethane, a modest dendritic effect was reported for analyses in DMF. Anions were studied as the TBA anion salts.
In 2003, Astruc and co-workers reported a series of polyamine dendrimers that were functionalized through phenol-N hydrogen bonds with silicon linkages to the amidoferrocene.720,721 Receptor 700 represents the first generation dendrimer (G1), which bears 12 amidoferrocene groups. Electrochemical studies in dichloromethane based on G1-G4 dendrimers revealed a positive dendritic effect for dihydrogenphosphate (studied as its TBA salt). No dendritic effect was reported for ATP, however, possibly due to the increased steric restraints of anion complexation. Addition of dihydrogenphosphate or ATP produced new waves in the cyclic voltammogram of these dendrimers in dichloromethane.
Dendrons similar to those above have also been attached to gold nanoparticles in an effort to create unique dihydrogenphosphate electrochemical sensors.722-725 With these surface-supported systems, a new redox wave was again observed upon addition of dihydrogenphosphate. Electrochemical titrations supported a 1:1 ferrocene: dihydrogenphosphate stoichiometry. Interestingly, a 2:1 ferrocene: anion ratio was observed with ATP. The ferrocene signals associated with this response proved to be independent of the concentrations of the ferrocene ligand on the gold surface. The response was also independent of the length of the carbon spacer in the case of the linear amidoferrocene ligands. An increased response was seen when electron-withdrawing groups were present on the Cp rings. This result provides support for the presence of amide-anion hydrogen bonding interactions. A decreased response was observed when the Cp rings were substituted with electron-donating groups. A small interference from hydrogensulfate and chloride was observed in these systems. All studies were performed in dichloromethane with TBA anion salts.
Astruc and co-workers also showed that the derivatized nanoparticles could be adsorbed to a Pt electrode surface to produce a phosphate responsive electrode. These electrodes were able to sense dihydrogenphosphate and ATP over hydrogensulfate and chloride in dichloromethane. The facile synthesis of these nanoparticle systems and the similarity in their response to the free dendrimer systems lends support to the conclusion that this multi-faceted approach to electrochemical sensor development could prove to be of general utility.
Recently, Astruc and co-workers prepared the organometallic cluster dendrimers 701 and 702 and showed that these macromolecules could be used to recognize ATP with selectivity over dihydrogenphosphate.726 In CV studies carried out in dichloromethane, dendrimer 701 was found to display larger cathodic shifts in the presence of ATP than did dendrimer 702. This difference in dendrimer response was attributed to the shorter distance between the metallic fragments in dendrimer 701. The incorporation of dendrimer 702 onto a Pt electrode also allowed for selective ATP detection.
Other researchers have also analyzed dendritic systems. For example, Losada, Cuadrado, and co-workers developed silicon-amine based dendrimers incorporating simple (non-amido) ferrocene units (e.g., 703).727 These dendrimers displayed an electrochemical response to the presence of anions (TBA salts) in both dichloromethane solution and when immobilized on an electrode. Strong but irreversible electrochemical responses were observed for dihydrogenphosphate, even in the presence of other anions. Dendrimers without the inner amine groups displayed a much smaller response to anions. As a result, the inner amine groups of the dendrimer were considered to be a crucial determinant of the selective recognition shown by dendrimers based on 703, presumably as the result of favorable hydrogen bonding interactions with the anion.
In another example, Cuadrado and co-workers reported a strong response for dihydrogenphosphate in DMSO when G1-G4 polyamine dendrimers appended with urea-ferrocene units were analyzed.728 Receptor 704 represents the fourth generation dendrimer containing 32 urea-ferrocene units. Despite the high polarity of the solvent, submillimolar concentrations of dihydrogenphosphate could be selectively detected in the presence of chloride and hydrogensulfate when the latter anions were present at equal concentrations to dihydrogenphosphate (all anions studied as the TBA salts).
[Ru(bpy)3]2+ as fluorescence and electrochemical reporter
The unique [Ru(bpy)3]2+ unit can serve as both an electrochemical and a luminescent reporter group. When functionalized with amide or other hydrogen bonding moieties, an increase in emission of the MLCT band is generally observed in the presence of anions. These effects are not observed with unfunctionalized [Ru(bpy)3]2+. It has been proposed that the observed increase in emission is a consequence of the bound anion serving to rigidify the receptor and thus inhibiting vibrational and rotational relaxation modes of nonradiative decay. As observed with the ferrocene systems, the binding affinities can be tuned quite dramatically through an appropriate choice of the spacer/bridging moieties. Similar effects have been observed in many case where Re(bpy)(CO)3Cl units were used to replace the [Ru(bpy)3]2+ moieties. However, these latter systems will not be discussed in detail here.
Beer and co-workers were among the first to report applications of this unique signaling unit. The researchers focused first on bipyridyl cores bearing amine and amide substituents. Many of these systems displayed good selectivities for phosphate containing analytes. For instance, in the case of receptor 705a 1H NMR spectroscopy and CV studies carried out in acetonitrile revealed a selective response to dihydrogenphosphate, even in the presence of hydrogensulfate and chloride.729 It was proposed that anion coordination primarily involves interactions with the amide-bipyridyl functionality. A similar selectivity was observed with the calixarene-based receptor 706.709 This latter system displayed a binding constant of 4.4 × 103 M-1 with dihydrogenphosphate in DMSO-d6 as determined from 1H NMR spectroscopic analyses. This binding affinity was similar to that reported for the cobaltocene analogue 685, described earlier in the section. The response of receptor 705a to dihydrogenphosphate, as well as that of receptors 687b and 707 - 710, was further investigated through 1H NMR spectroscopic, electrochemical and optical techniques.730 All receptors in this study displayed a dihydrogenphosphate association constant that was at least 10-fold higher than the corresponding association constant for chloride anion as inferred from 1H NMR spectroscopic analyses carried out in DMSO-d6. Receptor 707b displayed a much stronger binding response than receptor 708, a finding that provided further support for the importance of amide hydrogen bonding interactions in moderating the anion recognition process. Increased binding affinities were also seen as the degree of macrocyclic preorganization increased. Specifically, binding affinities were found to follow the sequence 707c < 709 and 710 < 705a for dihydrogenphosphate in DMSO-d6.
Phosphate selectivity was also observed through electrochemical experiments carried out in acetonitrile with receptors 705a, 707c, 709, and 710. Here, a strong selectivity for dihydrogenphosphate over hydrogensulfate, chloride, and bromide was observed for each of the four studied receptors. Notably, the response of receptor 705a to dihydrogenphosphate was not affected by the addition of up to 10 equivalents of hydrogensulfate and chloride anions. Optical studies with receptors 705a, 707c, and 710 revealed an increase in the intensity of the MLCT absorption band in the presence of dihydrogenphosphate. In acetonitrile solution, a bathochromic shift in the emission band as well as an increase in the quantum yield was observed upon the addition of dihydrogenphosphate. These changes in emission spectral features were attributed to the increased structural rigidity of the anion:receptor complex as compared to the free receptor. All anions were studied as their TBA salts.
The hybrid ferrocene receptor 711 was found to stabilize a strong 2:1 guest:host complex when exposed to dihydrogenphosphate (TBA salt) as inferred from both 1H NMR and UV-Vis spectroscopic studies carried out in acetonitrile.94 While the presence of the ferrocene moiety served to quench the emission of the [Ru(bpy)3]2+ subunit in the free receptor, a strong emission intensity was observed in the presence of dihydrogenphosphate. Addition of chloride or hydrogensulfate did not affect the luminescence, presumably due to weaker host-guest interactions with these two anions.
Dimeric derivatives 712 were also found to be selective for dihydrogenphosphate over chloride and bromide.731 The anion binding ability of these receptors was studied by 1H NMR spectroscopy in DMSO-d6 using TBA anion salts. Anion binding affinity was found to be highly dependent on the nature of the linker, the bipyridyl substituent, and the metal center. For example, the meta-substituted 712a bound dihydrogenphosphate two orders of magnitude more strongly than para-substituted 712b, and the ethylene-substituted 712c displayed very low anion binding affinity. In addition, ester-containing receptor 712d was found to bind dihydrogenphosphate more strongly than methyl-derivative 712f. The different metal centers studied bound dihydrogenphosphate in the order Os > Ru > Re. Of the combinations studied, the highest binding affinity was found for receptor 712h (K > 3.0 × 105 M-1).
Macrocyclic derivatives of receptor 707c (713) were found to interact more strongly with dihydrogenphosphate (TBA salt) than the linear analogues (K > 105 M-1 vs. 1.3 × 103 M-1 for 713 and 707c, respectively, in DMSO-d6).732 Smaller macrocycles such as receptor 714, however, proved too sterically hindered to allow for appropriate hydrogen bonding interactions. These receptors displayed only weak affinities (K < 103 M-1 for H2PO4- in DMSO-d6).
The use of the polyammonium receptors 715 allowed these kinds of analyses to be extended into aqueous media. These systems were found to interact strongly with dihydrogenphosphate and ATP over the pH range 2-10, as observed through potentiometric titrations.733 Interestingly, luminescence quenching was observed upon the addition of dihydrogenphosphate and ATP in these systems (pH = 6). The mechanism of this response was not determined. In a mixed acetonitrile:water (9:1) medium, the charged imidazolium receptors 716 and 717 were found to display a significant luminescence enhancement in the presence of ATP. 734 Association constants could not be determined for this anion. On the other hand, receptor 717 displayed an association constant of 1.4 × 103 M-1 with dihydrogenphosphate as measured by emission titrations in 9:1 acetonitrile:water. However, it is to be noted that under these conditions, much stronger binding affinities were observed for the chloride, bromide, and acetate anions.
The anion binding properties of receptor 718, a quinone derivative, and receptor 719 were analyzed by 1H NMR spectroscopy in DMSO-d6.735 The dihydrogenphosphate binding affinity was found to be similar for both receptors (K ≈ 2 × 102 M-1) under these conditions. Notably, both receptors proved selective for acetate and chloride over dihydrogenphosphate. All anions studied as the TBA salts.
Ditopic receptors, such as compound 720, were also investigated by Beer and co-workers. Receptor 720 was found to bind TBA dihydrogenphosphate (K = 9.0 × 102 M-1 in DMSO).736 In addition, a clear ability to complex K+ into the crown ether moieties was observed using 1H NMR spectroscopy. The presence of K+ reduced the affinity for dihydrogenphosphate, presumably due to a conformational change that precluded the formation of optimal anion-receptor hydrogen bonds. The anion binding behavior of this receptor could also be observed through an increase in the Ru(bpy)3 luminescence intensity.
The Watanabe group analyzed the anion complexation of metalloreceptor 721. This system contains two bis(acylaminoimidazoline) subunits in addition to a Ru(bpy)3 core.737 The guanidinium units of this receptor displayed largely reduced basicities (pKa ≈ 2, 4) as compared to traditional guanidinium groups. Thus, in contrast to what was true for many other systems, the guanidinium units were initially neutral in these studies. On the other hand, 1H NMR spectroscopic studies revealed that protonation of 721, as opposed to complexation, takes place when treated with diphenylhydrogenphosphate (DPHP) or dibenzyl hydrogenphosphate (DBHP) in acetonitrile. In contrast, when acetone was used as the solvent the formation of a 1:1 inner-cleft complex was observed in the presence of DBHP. Complex formation was observed in both solvents in the presence of anionic diphenylphosphate (DPP). The nature of these interactions was further probed using absorbance and luminescence spectroscopy in acetone. Since no responses were observed during control experiments with Ru(bpy)32+, the changes when receptor 721 was exposed to this set of phosphate anions were attributed to specific chemical differences (complex formation, deprotonation) and not counter anion exchange or solvent polarity differences. Addition of DBHP produced a red-shift and decreased the intensity of the MLCT band as well as the luminescence of receptor 721. These responses were attributed to the increase in the positive charge at the guanidinium moiety, presumably through proton transfer, that occurs upon complex formation. Addition of DPP produced a slight blue-shift and served to increase intensity of the MLCT band and the overall luminescence. The enhancement of the emission response was attributed to an increase in the rigidity of the receptor. From the observed anion-induced UV-Vis absorption changes, binding constants of 3.3 × 104 M-1 and 4.8 × 103 M-1 were calculated for DPP and DBHP, respectively. The decreased affinity seen for DBHP was attributed to electrostatic repulsion between the cationic ruthenium center and the incipient positive charge produced on the guanidinium binding site of receptor 721 during complexation. Notably, strong spectral responses were observed in relatively polar solvents even in the absence of a positive charge on the guanidinium moiety. In these studies, the anions were added as the tetraethylammonium salts.
Several other research groups have also used the Ru(bpy)3 reporter unit to probe phosphate binding interactions. For example, Smith and co-workers developed the ditopic receptor 722, which was designed to bind phosphorylated sugars through anion-amide and diol-boronic acid interactions.738 Strong binding (log K ≈ 3) was observed for fructose-6-phosphate, glucose-1-phosphate and galactose-6-phosphate in water as inferred through the enhancement in the emission spectrum. Interestingly, the binding behavior of fructose alone was found to be greatly increased when sodium phosphate was used as the buffer. A cooperative binding model was proposed in which the phosphate anion formed hydrogen bonds with both the amide units and the alcohols of the saccharide.
Another set of Ru(bpy)3-based phosphate anion receptors was introduced by Vos and co-workers. These researchers compared the emission behavior of receptors 707c, 723, and 724 in the presence of dihydrogenphosphate (TBA salt) in acetonitrile.739 Emission intensity and lifetime increases were observed for all three receptors up to the point where 2.5 equivalents of dihydrogenphosphate had been added. When greater quantities of anion were added, receptor 724 displayed an unexpected decrease in the emission intensity. Similar binding was, to a first approximation, observed for both the bipyridine receptor 707c and the phenanthroline receptor 723. An extreme sensitivity to water was also observed.
Lin and co-workers also explored the use of phenanthroline ligands. These researchers developed colorimetric sensor 725, which contains a functionalized phenanthroline core with nitrophenylhydrazine substituents as the L groups.740 Naked-eye and UV-Vis studies led to the suggestion that receptor 725 formed a complex with dihydrogenphosphate (TBA salt) in DMSO. However, a stronger binding behavior was seen with acetate.
The dithiocarbamate ruthenium receptors 726-727 were found to function as anion redox sensors.741 In this case, the addition of TBA dihydrogenphosphate produced a cathodic shift in the ruthenium-centered redox couple in acetonitrile. Proton NMR spectroscopic titrations in DMSO-d6:CD3CN (1:1) provided support for the electrochemical results. Binding constants of 70 M-1 and 45 M-1 for receptors 727a and 727b, respectively, were reported.
Recently, Sun and co-workers studied the quinoxaline(sulfonamide) receptors 728-729 and related metalloreceptors 730-731.742 Changes in the acidity of the N-H sulfonamide proton appeared to be the largest determinant of anion binding behavior within the limited set of receptors 728 and 729. Metal coordination to receptors 728 and 729 to yield 730 and 731, respectively, increased the association constants by one to two orders of magnitude for the studied anions (fluoride, cyanide, acetate, dihydrogenphosphate, and chloride as the corresponding TBA salts). UV-Vis and 1H NMR spectroscopic studies carried out in different solvents provided support for the conclusion that all of the receptors subject to study were bound to dihydrogenphosphate through hydrogen bonding interactions.
Metal dipyrrolyl-bipyridine receptors 732-733 were prepared by Sessler and co-workers.743,744 Relative to other test anions studied, the ruthenium receptor 732a was found to display the highest affinity toward dihydrogenphosphate (as the TBA salt). A binding constant of 1.04 × 105 M-1 was obtained through fluorescence titrations carried out in DMSO.
The anion binding abilities of similar systems were studied by Lin and co-workers in 2009.745 UV-Vis titrations in dry DMSO were carried out in the case of receptors 734 - 735 by adding the TBA salts of fluoride, chloride, bromide, iodide, acetate, hydroxide, and dihydrogenphosphate to solutions of the receptor. Association constants in the range of 102 - 104 M-1 were reported for binding to dihydrogenphosphate, although a preference was observed for fluoride and acetate. Little to no spectroscopic changes were observed upon the addition of halide ions. In general, the benzene-substituted compounds 734b and 735b bound anions more strongly than did the toluene derivatives 734a and 735a. The relatively lower binding affinity of the toluene-functionalized receptors was attributed to the electron donating ability of the methyl group relative to the hydrogen atom of the benzene derivatives.
Dimetallic systems were analyzed as possible sensors for phosphate by Martínez-Máñez and co-workers. In preliminary studies, receptor 736 was found to serve as a selective fluorescent sensor for ATP in 7:3 acetonitrile:water at slightly acidic pH values (4-6).746 No changes in the emission spectrum were observed upon the addition of chloride, sulfate, or inorganic phosphate. Further studies examined the dimetallic complex produced upon coordination of Cu2+ to the cyclam unit of ligand 736.747 For example, in acetonitrile:water (7:3), the stability constant between [736:Cu)]4+ and ATP was determined to be log K = 6.10 through potentiometric titrations. CV studies supported the conclusion that non-coordinating anions (ATP, ADP, GMP, sulfate, and inorganic phosphate) interacted with the 736:Cu receptor through electrostatic (Coulombic) interactions rather than metal-anion coordination. A larger shift was observed in the CV spectrum of coordinating anions such as chloride, bromide, and hydroxide.
4.4. Macrocycles
Macrocycles offer several advantages as phosphate receptors. One advantage derives from the effective preorganization of the constituent binding moieties. A variety of macrocycle classes have been analyzed, including calixarene, calixpyrrole, porphyrin, and other polypyrrolic macrocycles. Each class exhibits different geometrical organization and in some cases metal-coordination propensities.
Calixarenes can be functionalized at either or both the upper and lower rim with a variety of substituents. Bulky functionalities on either rim are often used to lock the structures in the cone or other conformations. In an early example, Reinhoudt and co-workers functionalized the upper rim with sulfonamide groups to produce receptors such as 737.748 While receptors of this type were found to display selectivity for hydrogensulfate, receptor 737 was also found to interact with dihydrogenphosphate. An association constant of 3.5 × 102 M-1 in chloroform was determined through 1H NMR spectroscopy. Loeb and Cameron examined amide functionalized calixarene 738, which displayed 1:1 binding with a variety of anions as inferred from 1H NMR spectroscopic analyses carried out in CDCl3.749 A preference was observed for Y-shaped anions over tetrahedral anions such as dihydrogenphosphate, for which an association constant of 22 M-1 was calculated. Both studies relied on TBA anion salts.
Schneider and co-workers later introduced trimethyl ammonium groups onto the upper rim and demonstrated that various functionalized calix[4]arene (e.g., 739, 740), calix[6]arene (e.g., 741a) and calix[8]arene (e.g., 741b) derivatives interact with nucleotides in water as inferred from 1H NMR spectroscopic studies.750 The calculated binding constants for receptors 739 and 740 were similar (e.g., 11 × 103 M-1 vs. 7 × 103 M-1, respectively, with ATP), but the larger macrocycles (741a, 741b) displayed significantly higher binding affinities (ca. 7 × 104 M-1 with ATP). These studies, along with molecular modeling, revealed several interesting trends. In accord with studies previously discussed, each ion pair interaction was calculated to contribute approximately 5 kJ/mol of binding energy. The authors concluded that these ion pairing interactions, however, disrupted the insertion of the nucleobase into the calixarene cavity. The increased binding affinity of larger macrocycles was thus attributed to more complete insertion of the nucleobase into the macrocyclic cavity. Little selectivity was observed among the different nucleobases. These receptors were also found to interact with double stranded DNA.
A comparison of cone and 1,3-alternate binding behavior was conducted by Lhoták, Stibor, and co-workers using the urea receptors 742a and 743a.751 While receptor 742a was predicted to interact with two anions, a 1:1 binding stoichiometry was observed for most anions as deduced from 1H NMR spectroscopic titrations carried out in deuterated chloroform/acetonitrile (4:1). The associated affinity constants proved similar to those determined in the case of receptor 743a. Association constants for dihydrogenphosphate were found to be approximately 2 × 103 M-1 for both receptors (i.e. 742a and 743a), and little anion selectivity was observed. The putative lack of binding to the second site of receptor 742a was attributed to a negative allosteric effect in which widening of one side of the receptor caused the other side to be compressed. It was proposed that compression of the second site rendered it unsuitable for anion binding.
An extensive 1H NMR spectroscopic study was used to analyze receptors 742-747.752 These studies led to the conclusion that neither cone nor partial cone conformations displayed a preference for dihydrogenphosphate over other anions in deuterated chloroform:acetonitrile (4:1). At the same time, receptors 742b and 742d with 1,3-alternate conformations were found to exhibit slightly greater affinities for dihydrogenphosphate as compared to other anions under these conditions (e.g., K(H2PO4-) = 3.2 × 103 M-1 and K(PhCO2-) = 2.1 × 103 M-1 for receptor 742b). All anions were studied as their TBA salts.
The amide-containing receptor 748 was synthesized by Chen and Huang. This system is characterized by the use of a functionalized anthracene to strap the upper rim of a calix[4]arene with a fluorescent substituent.753 Fluorescence quenching was observed upon the addition of dihydrogenphosphate (TBA salt) in acetonitrile. A higher degree of quenching, however, was observed upon addition of acetate. Thus, this system, although interesting, is not selective for phosphate.
Chawla developed the upper rim functionalized neutral semicarbazone calixarene 749.754 UV-Vis and NMR spectroscopic studies confirmed that receptor 749 is highly selective for dihydrogenphosphate (TBA salt). A binding constant of 1890 M-1 was measured in DMSO.
A number of studies have also focused on the analysis of lower rim-functionalized receptors. Nam and co-workers examined the urea-functionalized receptors 750 and 751. The three calix[6]arenes (receptors 750) were found to bind dihydrogenphosphate (TBA salt), albeit with different affinities. The strongest binding interaction was observed with receptor 750a, which displayed a binding constant of 8.3 × 102 M-1 in deuterated dichloromethane as deduced from a 1H NMR spectroscopic titration.755 This receptor, however, displayed a much stronger affinity for chloride than dihydrogenphosphate. Methylation of the remaining alcohol groups yielded receptor 750b. This substitution pattern was found to result in lower binding constants, presumably due to reduced hydrogen bonding interactions. The incorporation of thioureas (750c) also served to reduce the phosphate binding affinity relative to 750a. The weaker observed binding behavior was attributed to increased intermolecular interactions as a result of the enhanced acidity. The same kinds of relative enhancements were seen in the case of the calix[4]arene derivatives (751); again, the urea derivatives proved to be better receptors. On the other hand, with these smaller macrocycles, an increased binding affinity was seen for the methylated receptor 751b relative to alcohol substituted receptor 751a.756 This effect was attributed to increased intramolecular hydrogen bonding of the hydroxyl groups in the smaller macrocycles in the absence of anions. No selectivity for dihydrogenphosphate was observed. Nam, Jeon, and co-workers also developed an electrochemical calix[4]arene derivative (752) containing quinone moieties as well as urea functionalities.757 Proton NMR spectroscopic studies in CDCl3 displayed an increased binding affinity for dihydrogenphosphate (K = 1.0 × 103 M-1) relative to the more traditional calixarene receptor 751b (K = 2.5 × 102 M-1). This increased binding affinity was attributed to an additional hydrogen bonding interaction between the hydrogen of the anion and the oxygen of the quinone moiety of receptor 752. Receptor 752 bound anions in the order of hydrogensulfate > dihydrogenphosphate > acetate > chloride > bromide > perchlorate. Cathodic shifts were observed through CV upon addition of these anions to a solution of receptor 752 in acetonitrile. The magnitude of the shift followed a similar binding trend as that observed in the NMR spectroscopic studies. All anions were studied as their TBA salts.
In another example, Beer and co-workers functionalized the lower rim of calix[4]arene to produce the positively charged pyridinium-containing receptor 753. This system was found to form 2:1 anion:receptor complexes in DMSO with TBA anion salts.758 On the basis of 1H NMR spectroscopic titration studies, a binding constant of 4.5 × 104 M-2 for the interaction with dihydrogenphosphate was calculated. Selectivity for dihydrogenphosphate over chloride, bromide, and hydrogensulfate was also observed. Furthermore, receptor 753 was found to give rise to an electrochemical response to dihydrogenphosphate.
Moderate dihydrogenphosphate selectivity was also observed with receptor 754, which contains positively charged phosphonium groups.759 Using 1H NMR spectroscopy, Nam, Vicens and co-workers were able to measure a binding constant of 7.5 × 102 M-1 in chloroform using the TBA salt.
Highly selective dihydrogenphosphate binding in acetonitrile was achieved with the fluorescent calixarene-based receptor 755 developed by Chen and Chen.760 A strong increase in the emission intensity, along with a small blue-shift in the fluorescence maximum, was observed upon the addition of dihydrogenphosphate. This spectral change was attributed to inhibition of PET quenching arising either from an increase in the hydrogen bonding interactions or enhancements in the rigidity of the receptor. On the basis of fluorescence titrations, a 1:2 receptor:anion stoichiometry for dihydrogenphosphate and a binding constant of 5.4 × 109 M-2 was inferred. Strong selectivity over a variety of other anions was observed, including a 2700-fold greater binding affinity for dihydrogenphosphate as compared to fluoride. Proton NMR spectroscopic studies in chloroform/DMSO led to the suggestion that phosphate binding derived from multiple hydrogen bonding interactions, including those with the amides, sulfonamides, hydroxyl groups, and various CH protons. These multi-point interactions were proposed to be the most likely source of the observed selectivity. The same researchers later examined the calixarene-based receptors 756, which were functionalized with chromogenic azophenol groups as sensing units at the upper rim and with hydrogen bonding units on the lower rim.761 These receptors were found to bind dihydrogenphosphate, as well as fluoride and acetate, in acetonitrile. A color change of light yellow to blue was observed upon anion addition. The absorption spectrum of receptor 756b was found to shift to different wavelengths upon the addition of dihydrogenphosphate, fluoride, and acetate, thus allowing selective detection of all three anions. Binding constants with dihydrogenphosphate were found to be on the order of 104 M-1 for all three receptors. Interestingly, the carboxylate groups at the lower rim were thought to play a critical role in dihydrogenphosphate binding, but not for the recognition of acetate or fluoride. These studies were carried out using the TBA salts of the anions in question.
An alternative approach to the colorimetric detection of dihydrogenphosphate was introduced by Gunnlaugsson and Matthews.762 These researchers utilized receptor 757, which incorporates two amidourea substituents at the lower rim. UV-Vis titrations in DMSO with TBA anion salts led to the conclusion that this receptor interacts with both dihydrogenphosphate and hydrogenpyrophosphate (log β = 4.86 and 5.72 for these two substrate, respectively). A higher affinity, however, was observed for fluoride. Color changes from yellow to purple were observed upon the addition of fluoride or hydrogenpyrophosphate TBA salts.
The use of receptor 758, which incorporates four amido urea units, led to increased binding constants.763 For example, in the case of receptor 758a log K values near 5 for were observed for acetate, fluoride, dihydrogenphosphate, and pyrophosphate (TBA salts) were recorded in DMSO as deduced from UV-Vis spectroscopic titrations. Fluoride or pyrophosphate addition resulted in a strong clear-to-red color change in solutions of receptor 758b. On the other hand, acetate or dihydrogenphosphate addition resulted in weaker, yellow color changes. However, a 1:1 binding stoichiometry was seen only in the case of pyrophosphate. Proton NMR spectroscopic studies of receptor 758b confirmed that the amidourea moieties participated in anion binding.
Beer and co-workers prepared calixarenes 759 and 760 as potential ditopic anion-cation receptors. Receptor 759 was designed to interact with anions at the amide functionality of the lower rim while complexing cations within the crown ether unit.764 After formation, the potassium and ammonium complexes of receptor 759 were found to bind a variety of anions (add as the TBA salts) as revealed by 1H NMR spectroscopic studies carried out in acetonitrile. With this system, the strongest interaction was seen with dihydrogenphosphate. The binding affinity was too high to be determined using NMR spectroscopic methods but was estimated to be greater than 104 M-1. The free base receptor 759, on the other hand, did not bind anions. In the case of receptor 760, it was the calixarene moieties that were designed to function as the cation binding sites.765 The interaction of 760 and control compound 761 with various anions was analyzed using 1H NMR spectroscopy in DMSO-d6. Compound 760 displayed a preference for dihydrogenphosphate over acetate, benzoate, and chloride. The association constant corresponding to the interaction between receptor 760 and dihydrogenphosphate was measured to be ca. 700 M-1 in this solvent. Interestingly, acetate was found to interact with receptor 761 more strongly than did dihydrogenphosphate. This led these researchers to suggest that the calixarene moiety contributes to phosphate binding even in the absence of a cation. Studies of receptor 760 as a potential ditopic binding agent were only carried out using halide anion salts.
More application-driven studies of calixarene receptors have also been preformed. For example, Roundhill and co-workers compared the dihydrogenphosphate extraction ability of the amide and amine calix[4]arene derivatives 762 and 763.766 These researchers found that the amine derivatives 763 were more efficient at extracting phosphate from a neutral aqueous phase into an organic layer (chloroform). This effect was attributed to the enhanced interactions between the phosphate and the charged amines present in the protonated form of 763 as compared to the neutral amide receptor 762.
In a different study, Gupta and co-workers developed a phosphate selective electrode based on the calix[6]arene-derived ionophore 764. The resulting ISEs displayed a working concentration range of 1.77 × 10-5 to 0.1 M for monohydrogenphosphate with a response time of 20 seconds.767 Good selectivity was achieved over a variety of anions. A slight interference was observed for ClO4-. The lifetime was found to be approximately one month. Furthermore, phosphate measurements made using this calixarene-based electrode in field samples were comparable to values obtained with a traditional molybdenum assay. In another example, receptors 765 were analyzed by Arrigan and co-workers after incorporation into a PVC-based ISE.768 The resulting electrodes were found to be selective for monohydrogenphosphate in aqueous media over a concentration range of 5.0 × 10-5 to 1.0 × 10-1 M.
Alterations made to the core structure of calixarenes, such as varying the linkages between the phenyl rings, have also been explored. For example, Anslyn and co-workers designed the expanded azacalixarene 766. This receptor is characterized by several hydrogen bonding sites, as well as a cationic and hydrophobic core.769 Through a fluorescent indicator (HPTS) displacement assay, receptor 766 was observed to interact with IP3, fructose-1,6-diphosphate, and gluconic acid in aqueous media buffered to pH 7.2 with HEPES. The highest binding affinity was measured with IP3 (2.4 × 104 M-1). Proton NMR spectroscopic studies with fructose-1,6-diphosphate led to the conclusion that the most important binding interactions involve the secondary amines of the core, rather than the primary amines at the upper rim.
Akkaya and co-workers achieved a cationic core by replacing the phenyl rings of a traditional calixarene with pyridinium units (767).770 As in the previous Anslyn system, HPTS was used as the fluorescent indicator in a displacement assay for the sensing of polyanions. The anion binding interactions of receptor 767 were studied through fluorescence titration in a 3-(N-morpholino)propanesulfonic acid (MOPS) buffer at pH 7.5. This assay was found to be selective for ATP (log K = 2.87) over ADP, AMP, NADP, NAD, pyrophosphate, citrate, succinate, and fumarate (log K = 0.03 - 0.51). The observed selectivity was attributed to the increased charge of ATP relative to the other tested anions.
Receptor 768, a functionalized cavitand reported by Diederich and Sebo, was also found to be an effective anion receptor.771 For instance, 1H NMR spectroscopic titrations revealed that receptor 768 interacted with a variety of nucleotides at pH 8.3 in D2O. An increase in the binding affinity was observed with increasing anion charge (AMP < ADP < ATP). Furthermore, a slight selectivity for AMP (K = 1.0 × 104 M-1) over other nucleotide monophosphates was found. Analysis using both the NMR spectroscopic chemical shifts and molecular modeling led to the conclusion that ion pairing and hydrogen bonding interactions were largely responsible for the observed binding. Selectivity was attributed to hydrophobic interactions such as inclusion of the nucleotide base within the receptor cavity.
The fluorescent cavitand 769 was investigated later by Yoon, Lee, and co-workers.772 In this case, relatively minor fluorescence changes were observed in the presence of pyrophosphate and dihydrogenphosphate in 40% aqueous DMSO. However, significant fluorescence quenching was observed upon the addition of GTP (K = 7.38 × 104 M-1). Much lower binding affinities were observed for CTP and ATP. Phosphorous NMR spectroscopic studies led to the suggestion that the receptor interacts most strongly with the phosphate sites of the nucleotide.
In another study involving the use of a modified calixarene core, Lhoták and co-workers designed the ureido-thiacalixarenes 770 and 771.773 In this case, 1H NMR spectroscopic studies carried out in a chloroform : acetonitrile mixture (4 : 1) revealed hydrogen bonding interactions between various TBA anion salts (e.g., chloride, bromide, iodide, cyanide, benzoate, nitrate, dihydrogenphosphate, and hydrogensulfate) and the urea moieties. Stronger interactions were observed with the bis-urea receptor 771 than receptor 770. However, in neither case was significant selectivity observed for dihydrogenphosphate, although this analyte did display an association constant of 1.5 × 104 M-1 with receptor 770. Attempts to measure the binding constants for receptor 771 were hampered by the formation of species with mixed anion:receptor stoichiometries.
Cyclodextrins (CyD's) are also species that contain cone-shaped cavities and surfaces that can be functionalized to promote selective anion binding behavior. As a general rule, the resulting scaffolds are water-soluble while still providing a hydrophobic inner cavity. As early as 1970, Hoffman and Bock studied the complexation of nucleotides by unsubstituted cyclodextrins through UV-Vis spectroscopy in a buffered aqueous solution (0.01 M Tris, 0.1 M NaCl, pH 7.6).774 Under these conditions, significant spectral shifts were observed upon the addition of a variety of adenine derivatives as well as inosine monophosphate (IMP) to cycloheptaamylose (β-CyD). Little change was observed upon the addition of these guests to cyclohexaamylose (α-CyD) or upon the addition of CMP, GMP, UMP, or 2′-deoxy-5′-thymidine monophosphate (dTMP) to either CyD. Spectral changes of adenine derivatives with β-CyD increased in the order adenine < adenosine < 5′-ADP < 5′-ATP < 5′-AMP. Among a number of adenosine monophosphates (3′-AMP, 5′-AMP, 2′,3′-cAMP, 2′-AMP), a significantly stronger signal was observed upon addition of 3′-AMP. An equilibrium constant of 22 M-1 was measured for the 3′-AMP:β-CyD complex. Formoso reported similar binding trends with β-CyD through the use of circular dichroism (CD) spectroscopy in a sodium phosphate buffered solution at pH 7.775,776 Using this method, a 1:1 binding constant of 41 M-1 was measured for the complex of β-CyD and 5′-AMP at 25°C. Studies at various temperatures allowed for the calculation of thermodynamic parameters. Interestingly, the measurement of a favorable enthalpy (ΔH° = -4.9 kcal/mol) and an unfavorable entropy (ΔS° = -8.7 e.u.) was inconsistent with the dominance of hydrophobic host:guest interactions, which would be expected to display favorable entropy and opposing or negligible enthalpy.
In 1979, Knowles and co-workers functionalized O-methylated α-CyD with ammonium groups (772) and demonstrated the selective binding of benzylphosphate in aqueous media.777 Binding affinities were measured through the competitive displacement of nitrophenol as monitored by UV-Vis spectroscopy. A dissociation constant of 3.1 × 10-2 M was reported for benzylphosphate. It was suggested that the benzyl group binds inside the cavity while the ammonium groups, all charged at pH 7, would interact with the phosphate oxygen atoms. The proposed binding model was supported by the observation of strong selectivity for benzylphosphate over benzyl alcohol and inorganic phosphate. Furthermore, substantially lower binding affinities were observed with a monoammonium derivative analogous to 772 (i.e., 773). While the binding behavior of receptor 772 and benzylphosphate at pH 5.5 and 9.5 was also analyzed, the strongest binding interaction between these two compounds was observed at pH 7.
Schneider and Eliseev later investigated the binding of amino-β-CyD's 774 and 775 with a variety of nucleotides.778-780 In this case, 1H NMR spectroscopic experiments carried out in water at pD 6.0 revealed binding interactions with inorganic phosphate, ATP, ribophosphates, and a variety of nucleotide monophosphates. The strongest binding behavior, as predicted based on electrostatic interactions, was observed between receptor 775 and ATP (K = 3.24 × 106 M-1). Chemical shift analysis and 2-D NMR spectroscopic experiments led to the suggestion that the nucleobase of AMP was partially included within the CyD cavity along with the ribose unit. The ribose unit was also proposed to participate in hydrogen bonding interactions with the CyD. These interactions, along with the approximately 4 salt bridges that were expected to be formed with the ammonium groups of this receptor, were suggested to be largely responsible for the observed binding affinity.
Thatcher and co-workers examined the binding of a variety of 4-isopropylphenylphosphate esters (776) to the ammonium-functionalized CyD's 777a, 777b, and 778a using UV-Vis spectroscopy.781 In the case of compound 776d, 31P NMR spectroscopy was used. A number of trends were observed in aqueous media (pH 7.3, Tris buffer). For instance, when compared to unsubstituted β-CyD, receptor 777a displayed a significantly higher binding affinity only for dianions 776c and 776d. As this increase was not observed for various monoanions, these researchers suggested that the interactions of monoanions 776a and 776b remained largely hydrophobic in nature. In addition, a lower affinity for the binding of the phenylphosphate derivatives 776a-c to receptor 777b relative to the larger receptor 777a was observed. Proton NMR spectroscopic studies confirmed the inclusion of the phenyl ring within the CyD cavity of both receptors. Interestingly, the free energy of the complexation proved to be 1-1.5 kcal/mol weaker for both receptors than what would be expected based on a combination of the presumed hydrophobic and electrostatic interactions. This disparity was attributed to the exclusion of the propyl group of the guest into the aqueous media on the other side of the ring as a result of the strong electrostatic interactions between the phosphate and ammonium groups. Molecular modeling and a slow observed dissociation rate supported this conclusion.
Darcy and co-workers studied the binding behavior of the CyD derivative 778a with a variety of phosphates via potentiometric titrations and 1H NMR spectroscopic measurements.782 Log association constants as high as 9.57 were measured with receptor 778a and ATP. A comparison of equally charged anions revealed that stronger complexes were formed with guests containing a hydrophobic moiety for cavity inclusion (AMP and p-nitrophenylphosphate) than those that lacked such a feature (e.g., ribose-5-phosphate). Proton NMR spectroscopic experiments supported this conclusion and led to the suggestion that the ribose unit was not included within the cavity unless it was part of a cavity-bound nucleotide.
Seto and co-workers reported that receptor 778a inhibited phosphatase activity by binding arylphosphate esters.783 In later studies, these researchers analyzed the effect of buffers on the binding behavior of both receptors 778a and 778b in aqueous media at pH 7.0 using both 1H and 31P NMR spectroscopy.784 Negatively charged buffers were found to compete with arylphosphate esters by binding to the receptors, a phenomenon that reduced the apparent association constant. On the other hand, positively charged buffers served to enhance the binding affinity, sometimes up to 4 orders of magnitude over negatively charged buffers. Similar association constants were observed with both receptors 778a and 778b for p-nitrophenylphosphate and phosphotyrosine. In contrast, an appreciable difference was observed with N-acetylphosphotyrosine methyl ester. This ester was found to be bound to receptor 778a with an association constant of 6.1 × 105 M-1 and to receptor 778b with an association constant of 1.5 × 105 M-1 in Tris buffer. This difference in affinities was attributed to the larger size of the N-acetylphosphotyrosine methyl ester anion, which makes it a better match for the larger cavity in CyD 778a. Dynamic NMR spectroscopic experiments revealed that the buffer identity had a strong effect on the on rate, leading to complex formation but little effect on the corresponding dissociation rate.
Smith and co-workers later used β-CyD's as scaffolds to probe the thermodynamics associated with the binding of aryl phosphate monoesters to receptors bearing guanidinium (779b, 780b), as opposed to ammonium (779a, 780a), substituents.785,786 The thermodynamic parameters were determined in water at pH 7.00 using ITC. Free energies of association of approximately 3-4 kcal/mol were generally observed but significant differences were observed in the enthalpic and entropic contributions. Ammonium compounds were observed to be characterized by greater entropic contributions, while enthalpic contributions were more important in the case of the guanidinium species. This led to the conclusion that guanidiniums provided stronger interactions but only when properly oriented.
Binding events were also probed in the case of phosphotyrosine using indicator displacement assays and fluorescence titrations in aqueous media at pH 7. These studies revealed that monosubstituted β-CyD derivative 781 did not bind phosphotyrosine. However, the more highly substituted CyD's 780a and 780b, were found to bind phosphotyrosine with binding affinities of 350 M-1 and 310 M-1, respectively. Inclusion of the aromatic ring within the macrocyclic core of these latter two receptors was inferred based on an observed increase in the quantum yield of phosphotyrosine in the presence of receptor 780a or 780b.
Yuan, Fujita, and co-workers recently reported that ATP is complexed well by the guanidinium substituted receptor 782.787 For instance, at a pD of 5 in D2O, the formation of the ATP:782 complex was found to be irreversible under conditions of NMR spectroscopic analysis. This irreversibility prevented the determination of an association constant. Proton and 31P NMR spectroscopic analysis, however, did lead to the conclusion that the ATP was most likely bound through all three phosphate groups, as well as via partial inclusion of the adenine moiety within the core. Unfortunately, ITC analyses of the interaction between ATP and receptor 782 were complicated by the formation of other weak complexes. Nevertheless, these researchers suggested that binding would be driven in part by a strong entropic driving force, which was thought to reflect the extensive desolvation that would be expected upon analyte binding. Weaker ATP binding was observed in the case of the amine receptor 777a.
Calixpyrroles have also been investigated for their anion binding abilities. These species form complexes with Lewis basic anions in organic media. Anion complexation is believed to occur primarily through hydrogen bonding interactions between the anion and the pyrrolic NH units. Early efforts to develop calixpyrroles as anion receptors were spearheaded by Sessler and co-workers who synthesized a variety of derivatives in order to tune the selectivity of the basic calixpyrrole skeleton. In the original reports, the octamethyl calixpyrrole 783 was found to display a selectivity for fluoride anion (TBA salt) in dichloromethane (K > 104 M-1) as judged from 1H NMR spectroscopic titrations. A relatively low binding affinity for dihydrogenphosphate (K = 97 M-1) was measured in this solvent under analogous conditions.788 Bromide substitution at the pyrrole β positions (784a) increased the binding affinity to 6.5 × 102 M-1 but did not alter the previously observed selectivity.789 The binding affinity of dihydrogenphosphate in acetonitrile (0.5% water) was found to be slightly higher for receptor 783 (K = 1.3 × 103 M-1) than that observed in dichloromethane.465 Under the same conditions, an increase in the binding affinity was observed for the octafluoro-substituted system 784b (K = 9.1 × 103 M-1). However, the use of different solvents precluded a direct comparison of the binding behavior of receptors 784a and 784b.
The Sessler group also examined the binding behavior of various fluorinated calix[n]pyrroles (784b and 785) by means of ITC analyses carried out in DMSO with TBA anion salts. In general, higher binding affinities for phosphate anions (1.7 × 104 M-1 for 784b, 9.6 × 103 M-1 for 785a, and 1.5 × 104 M-1 for 785b) were found relative to what was seen for the unsubstituted receptor 783 (5.1 × 103 M-1).790 Substitution with aryl groups at the meso positions (786), on the other hand, did not increase binding affinities or change the selectivity relative to what was seen for the parent calix[4]pyrrole receptor 783.791,792
The anion affinity of calixpyrrole 783 was also investigated by Schmidtchen and co-workers using ITC.793 Although both the counter cation (e.g., K+-cryptand vs. TBA) and solvent (CH3CN vs. CD2Cl2 in the original study) were changed, several differences in the anion binding behavior were noted relative to what was reported originally by Sessler and co-workers based on NMR spectroscopic studies. For example, no preference in selectivity for fluoride over chloride was observed, and the binding affinities for Cl- and H2PO4- under these disparate conditions proved higher than recorded previously. Specifically, a binding constant of 1.5 × 104 M-1 was determined with dihydrogenphosphate by Schmidtchen using ITC. Quite recently, a partial reconciliation of these results was made, with the conclusion being drawn that non-first order effects, including solvation, ion pairing, and solvent dependence are more important than originally assumed.331,794,795
The use of receptor 783 to create a rudimentary fluoride anion sensor using a displacement assay with p-nitrophenolate and UV-Vis readout was reported by Gale, Sessler, and Twyman.796 Recently, Machado and co-workers exploited calix[4]pyrrole in displacement assays for naked-eye anion detection with Brooker's merocyanine dye in acetonitrile.797 The dye solution, which is violet in color, becomes orange upon the addition of receptor 783. Using this approach, fluoride, and to lesser extent chloride and dihydrogenphosphate, could be visually detected via a reversal of this color change (anions added as the TBA salts).
In work that was seminal in its time, Miyaji, et al. exploited calixpyrrole to create the first naked-eye detectable chemosensor for anions. This was done by linking anthraquinone chromophores to a calixpyrrole core to create receptors 787a and 787b. These systems permitted the visual detection of fluoride, chloride and dihydrogenphosphate in dichloromethane.798 Receptor 787c was observed to bind anions by 1H NMR spectroscopy but did not undergo a colorimetric change upon the addition of anions. An alternate substitution with anthracene at the β positions (788) allowed sensing through anion-induced fluorescence quenching.799 Interestingly, a slight selectivity for dihydrogenphosphate over chloride was observed in acetonitrile for receptors 788. Binding affinities were found to be stronger than those previously observed (log K = 4.20 and 3.56 for 788a and 788b, respectively). The higher binding constant for receptor 788a was attributed to electronic communication between the substituent and the pyrrole nitrogen. The fluoride anion was found to be bound with the greatest affinity among the test set of anions explored, whereas no binding behavior was observed for hydrogensulfate or bromide (all anions studied as their TBA salts).
In an effort to improve the phosphate anion selectivity, fluorophores were attached through sulfonamide and thiourea groups; this led to receptors 789.800 It was expected that the additional hydrogen bonding sites would also participate in anion binding interactions. Indeed, support for this model was obtained through 1H NMR spectroscopic analyses. The additional binding interactions led to selectivity for dihydrogenphosphate and pyrophosphate over chloride in acetonitrile/water mixtures. Selectivity over fluoride was not observed. The presence of water was found to be necessary to achieve appreciable anion binding, presumably as a consequence of preventing receptor aggregation. In these studies, 0.01% water was used for the studies involving receptors 789a and 789b, and 4% water was used for those involving receptor 789c. At this latter concentration of water, receptor 789c was found to bind pyrophosphate with a binding constant of ≥ 106 M-1 as inferred from fluorescence quenching experiments. This receptor was able to operate as a fluorescent sensor in acetonitrile solutions containing up to 20% water. These studies used TBA anion salts.
Anion detection by supramolecular self-assembly was explored by Shao and co-workers in the context of calixpyrroles.801 The addition of any one of the studied receptors (783, 790-792) to a solution of chloranil in chloroform led to the formation of a supramolecular structure with charge-transfer character, resulting in a blue colored solution. The resulting systems could then be employed in naked-eye detection of anions, with addition of TBA fluoride and TBA dihydrogenphosphate producing a visual and reversible blue-to-orange color change. UV-Vis spectroscopic studies revealed that the absorption spectrum of the receptor-chloranil-anion solutions were distinct from that of chloranil alone. This result led to the suggestion that the system was more complicated than a simple displacement assay.
Sessler, Lee and co-workers further developed a chromogenic calix[4]pyrrole system in which a dipyrrolylquinoxaline moiety was incorporated as a “strap” across the macrocycle core (793).802 Significant changes in the absorption spectrum were observed upon the addition of various anions as their TBA salts in a solution of 97:3 CH3CN:DMSO. Binding affinities increased with decreasing anion size (fluoride > chloride > bromide > acetate > dihydrogenphosphate. ITC experiments were in good agreement with the absorbance-based studies. Anion-dependent visual color changes were also observed. While dihydrogenphosphate displayed the lowest binding affinity (K = 1.13 × 103 M-1), addition of this anion displayed the largest visual color change. This large color change was attributed to a different binding mode for dihydrogenphosphate complexation relative to that of other anions.
Several other calixpyrrole derivatives were explored by Sessler and co-workers in the context of pursuing various applications of anion recognition. For instance, several functionalized calix[4]pyrrole derivatives were prepared and used to create the calixpyrrole modified silica gels 794b and 795b, which were employed in the separation of dihydrogenphosphate from a variety of other anions by HPLC.803 ATP, ADP, and AMP could also be separated. It was suggested that the modest anion binding behavior of the parent calixpyrroles 794a and 795a facilitated the separation of anions. The low binding affinity was expected to lead to more rapid exchange as compared to a receptor with high affinity.
The membrane transport ability of receptor 783 was measured through incorporation into a PVC-based ISE.804 For reference, the transport was compared to that of pyridine derivatives 796 and 797. An ISE based on receptor 783 was found to be selective for inorganic phosphate over halides from pH 3.5-9.0. In addition, a low response to fluoride was measured. The selectivity trend observed for this ISE was thus the reverse of what was in organic media. This result was attributed to the high hydration energy, and therefore slow kinetics, of fluoride in such systems. ISE's incorporating receptors 796 and 797 also displayed selectivity for inorganic phosphate. The response was most intense at low pH, presumably due to protonation of the pyridine moieties. A modification of this system was developed by Shishkanova and co-workers.805 In these studies, PVC membranes derived from receptor 783 were coated with a conducting polymer, polyaniline (PANI). These membranes demonstrated an improvement in anionic sensitivity, particularly towards highly hydrated guests, such as fluoride and dihydrogenphosphate. The improved response was attributed to an increase in the transfer of anions from the sample into the PVC matrix as a result of the conducting polymer.
Sessler and co-workers also analyzed the membrane transport of receptors 798 and 799 using both aqueous-organic-aqueous transport type systems and ISE's. In these receptors, the attached cytosine group was expected to provide nucleotide selectivity for GMP.806 The proposed selectivity was based upon a combination of nucleobase pairing interactions and hydrogen bonding interactions at the pyrrole units. In the membrane studies, the expected selectivity for GMP was observed for meso-linked receptor 799 but not for receptor 798. The success of receptor 799 was attributed to the increased spacing and flexibility between the two recognition moieties of this receptor. When incorporated into ISE's, selectivity for CMP was observed, presumably due to the innate affinity of unsubstituted calixpyrrole 783 for CMP.
In another application-driven study, Sessler, Gale and co-workers reported that the ferrocene receptor 800 displayed an electrochemical response when exposed to anions (TBA salts) in acetonitrile/DMSO mixtures.807 The strongest signal was observed for dihydrogenphosphate. Later studies by Radecka and co-workers in aqueous solution demonstrated that the anion recognition process was highly dependent on the nature of the matrix in which the receptor was immobilized.808 When receptor 800 was incorporated into carbon paste electrodes, the largest shift was induced by dihydrogenphosphate at pH 4.0. ISE's containing this receptor in a liquid membrane displayed responses in line with the Hofmeister sensitivity sequence. As a result, the response for dihydrogenphosphate was actually the lowest among a series of other anions (bromide, chloride, and fluoride) at the same pH.
More elaborate analogues of receptor 800, namely receptors 801, were investigated by Cheng and co-workers.809 These latter receptors featured an aminophenyl linkage between the calix[4]pyrrole and the ferrocene units. Electrochemical studies in acetonitrile demonstrated that addition of TBA dihydrogenphosphate resulted in the largest cathodic shift among the various anions tested (i.e. fluoride, chloride, bromide, acetate, and hydrogensulfate). On the other hand, based on 1H NMR spectroscopic titrations carried out in CD3CN, receptor 801 was observed to have a strong preference for the fluoride and acetate anions rather than dihydrogenphosphate. The disparity in these results was attributed to the fact that increased stabilizing interactions between the bound anion and the ferrocene subunits and phenyl groups to which these groups were connected were possible in the case of dihydrogenphosphate, as compared to fluoride and acetate. These enhanced interactions with dihydrogenphosphate were expected to give rise to a greater perturbation of the electrochemical signal.
Gale and co-workers have also investigated the anion binding behavior of various calixpyrrole derivatives. For example, receptor 802 containing a tribromo-pyrrole ring was found to have stronger affinity toward anions than receptor 783.810 In this case, 1H NMR spectroscopic titrations carried out in dichloromethane led to the conclusion that the NH group of the tribromo-pyrrole interacted with the anions (TBA salts). This additional interaction (relative to that provided by 783) was used to explain the observed selectivity towards carboxylate anions over most halide anions, as well as over hydrogensulfate and dihydrogenphosphate. Addition of fluoride led to peak broadening in the 1H NMR spectrum, precluding the determination of a binding constant.
Recently, Anzenbacher and co-workers developed various anion receptors based on modified versions of octamethylcalix[4]pyrrole. These systems included receptors 803, which contain electron-withdrawing dye moieties and which were designed to permit the naked-eye detection of fluoride, acetate, pyrophosphate and dihydrogenphosphate.811 UV-Vis studies carried out in DMSO / 0.5% water, revealed a higher anion affinity for receptors 803 than receptor 783. As a class, receptors 803 were found to bind fluoride, acetate, and pyrophosphate more strongly than chloride and dihydrogenphosphate (all anions studied as TBA salts). For example, association constants with receptor 803a were found to be 5.8 × 105 M-1 for pyrophosphate and 5.2 × 103 M-1 for dihydrogenphosphate in this solvent mixture. These studies were expanded to include various 1,3-indane-based calixpyrroles (804).812 A change in the color of the receptor solution was observed upon the addition of fluoride, acetate and, to a lesser extent, dihydrogenphosphate. Additional UV-Vis experiments revealed similar binding trends as those observed with receptor 803. An association constant for receptor 804d with dihydrogenphosphate of 1.6 × 105 M-1 was determined in DMSO / 0.5% water.
Danil de Namor and co-workers have carried out thermodynamic studies involving several calix[4]pyrrole compounds and various phosphate anion guests. In preliminary 1H NMR spectroscopic studies, these researchers reported a 1:1 binding stoichiometry between receptor 783 and dihydrogenphosphate or hydrogenpyrophosphate (TBA salts) in both acetonitrile and DMF.813 Conductance measurements confirmed the stoichiometry of the dihydrogenphosphate complexes in acetonitrile. Hydrogenpyrophosphate, however, displayed a 2:1 host:guest binding stoichiometry in acetonitrile, as inferred from analogous studies. Extraction experiments involving a partitioning between water-DCM with receptor 783 and hydrogenpyrophosphate provided support for the proposed 2:1 host:guest binding stoichiometry. On the basis of separate ITC measurements, an enthalpy-entropy compensation effect was inferred to be operative upon changing the solvent from acetonitrile to DMF. Thus, these studies are broadly supportive of those of Sessler, Schmidtchen, and Gale that led to the conclusion that the binding behavior of calix[4]pyrrole 783 is strongly dependent on the conditions, including choice of solvent.
Danil de Namor and co-workers also analyzed the anion binding interactions of the isomeric receptors 805.814 Interestingly, a 1:2 host:guest binding stoichiometry was observed for these receptors in the case of dihydrogenphosphate (TBA salt) in acetonitrile. Isomer 805b displayed a higher overall affinity for dihydrogenphosphate (log K = 9.46) than receptor 805a (log K= 6.1), as inferred from 1H NMR spectroscopic studies. On the other hand, fluoride displayed a 1:1 stoichiometry with receptor 805a (log K = 3.08) but a 1:2 stoichiometry with receptor 805b (log K = 9.72) in acetonitrile.
These researchers also studied receptor 806. In general, this latter system gave rise to weaker anion interactions than receptor 783.815 The influence of the medium was also examined. While 806 displayed a preference for the fluoride anion in acetonitrile, in DMF dihydrogenphosphate was bound more strongly than F- (log K = 4.81 vs. 4.23, respectively). This change in selectivity was attributed to solvation effects. All anions were studied as the TBA salts.
The effect of altering the calixarene core was analyzed by the Danil de Namor group by replacing one of the pyrrole units with a thiophene unit (807).816 This receptor was found to form a 2:1 (host:anion) complex with dihydrogenphosphate (TBA salt) in acetonitrile. Different binding behavior, however, was observed using propylene carbonate (PC) as the solvent. In this case, a 1:1 binding stoichiometry was observed. The change in stoichiometry was attributed to enhanced solvation of the dihydrogenphosphate anion in PC. A log K value of 4.22 was measured through ITC studies.
A modified sulphonium calixpyrrole 808 was reported by Schmidtchen and co-workers.817 This system was used to examine the effect of installing positive charges at the meso-positions of a calix[4]pyrrole core. Higher anion affinities were observed with this receptor, presumably due to a combination of hydrogen bonds and electrostatic effects. ITC titrations carried out in DMSO yielded a binding constant for the first interaction with dihydrogenphosphate (TBA salt) of 3.3 × 105 M-1. A second binding constant, attributed to non-specific electrostatic interactions, was reported to be 6.5 × 103 M-1. In addition, receptor 808 displayed a strong preference for dianions, such as 1,3-benzenedicarboxylate, over monoanions, such as benzoate (triethylammonium salts).
In a different modification of the core, Sessler and co-workers examined the anion binding ability of calix[4]pyrrole[2]carbazole receptor 809.818 It was proposed that replacing two of the four bridging methylene units of calixpyrrole 783 with carbazole groups would enhance ion affinity through increased hydrogen bonding interactions and through expansion of the macrocycle core. In addition, the carbazole unit was expected to serve as a fluorescence signaling moiety. Emission studies in dichloromethane led to the detection of strong 1:1 complexes of receptor 809 with acetate, benzoate, pyrophosphate, and dihydrogenphosphate. Significantly lower affinities were observed for the complexation of oxalate, succinate, and chloride while no appreciable binding was observed for the bromide, nitrate, and hydrogensulfate anions. All anions were studied as the TBA salts. Binding constants for dihydrogenphosphate and pyrophosphate were recorded at 7.2 × 104 M-1 and 6.4 × 104 M-1, respectively; however, a preference was observed for carboxylate anions.
In 2005, Sessler and Lee prepared the expanded bipyrrole-containing calixpyrroles 810.819 In this case, 1H NMR spectroscopic titrations carried out in acetonitrile revealed binding constants for dihydrogenphosphate (TBA salt) that varied considerably. For instance, a K > 240 M-1 was recorded for receptor 810a, and a K >104 M-1 was found for receptor 810b. These differences in binding affinity were attributed in large measure to the different macrocycle geometries resulting from the greater bipyrrole-bipyrrole distance caused by the thiophene linker in 810 relative to 810a. The highest affinity, however, was observed with carboxylate anions.
Sessler and co-workers have also explored the behavior of calix[n]bispyrrolylbenzenes (811) as anion receptors.820 While these receptors exhibited a preference for halide anions, association constants for dihydrogenphosphate were determined by 1H NMR spectroscopic methods and found to be 6.3 × 103 M-1 and 1.7 × 103 M-1 in dichloromethane-d2 for 811a and 811b, respectively (all anions studied as their TBA salts). The improved affinities as compared to receptor 783 (K = 97 M-1 in CD2Cl2, see above) were attributed to the increased donor ability of the bispyrrolylbenzene subunit in 811 as compared to the dimethyldipyrromethane moiety present in 783. Geometrical effects may also play a role in accounting for these observed differences in affinity.
The anion binding behavior of N-confused calix[4]pyrroles (NCCPs) has also been explored. In these calixpyrrole isomers, the inverted nature of one of the pyrroles precludes formation of an ideal cone conformation.821 On the other hand, these species do bind anions. The binding of negatively charged analytes generally occurs as the result of interactions involving the β-CH of the inverted pyrrole group as well as the three N-H's of the non-inverted pyrroles. An extensive study of N-confused calix[4]pyrroles (812) as anion hosts was conducted by Anzenbacher and co-workers.822 NMR spectroscopic titrations carried out in DMSO-d6 with TBA anion salts led to the conclusion that receptor 812 bound spherical anions more weakly than calixpyrrole 783. At the same time, the chromogenic receptors 812b and 812c displayed a high inherent affinity for hydrogenpyrophosphate (5.6 × 103 M-1 for receptor 812b) and dihydrogenphosphate (8.1 × 105 M-1 for receptor 812c), as inferred from UV-Vis titrations. A preference for acetate was also observed for both receptors.
Other examples of N-confused calix[4]pyrroles (e.g., 813) were reported by Dehaen and co-workers.823 UV-Vis studies of receptors 813, which include a chromophoric azo group, were conducted in acetonitrile with TBA anion salts. No clear trend in binding affinity was observed among the different substituents, with log β values ranging from 3.32 to 3.87 for dihydrogenphosphate. The most stable dihydrogenphosphate complex was found with receptor 813e (log β = 3.87). When similar studies were performed in dichloromethane, however, the substituents were observed to influence the measured binding affinities. The binding affinity increased as a function of the electron-withdrawing nature of the azo group (log β = 3.10 for receptor 813d and log β = 4.45 for receptor 813e). In a further extension of this theme, Dehaen and Gale reported the synthesis and binding behavior of receptors 814.824 Here, 1H NMR spectroscopic titrations carried out in DMSO-d6 / 0.5% water with TBA salts led to the suggestion that receptors 814 are selective for carboxylate anions. However, a relatively low affinity for dihydrogenphosphate (38-197 M-1) was also observed. This selectivity was rationalized in terms of the basicity of the analytes.
Several phosphate receptors based on porphyrin macrocycles have also been developed.825 While free base porphyrins are generally poor anion receptors, the incorporation of peripheral substituents can be used to improve their anion recognition capacity and, in many cases, allowed for the selective complexation of anionic guests. For example, porphyrin 815a, prepared by Aoyama and Ogoshi, contained protonated pyridyl substituents as well as a protonated porphyrin core.826 It was predicted that this receptor would interact with anions through a combination of hydrogen bonds and electrostatic interactions. Indeed, discrimination between monoalkylphosphate and dialkylphosphate anions was observed via 1H and 31P NMR spectroscopic analyses carried out in chloroform. The phenyl-substituted porphyrin 815b displayed a lower anion affinity, presumably due to the loss of the pyridinium-anion interactions.
Král and Schmidtchen investigated a different charged receptor unit, namely the tetraguanidinium porphyrin 816.827 These compounds were found to form ordered aggregates in water. Circular dichroism (CD) studies led to the conclusion that receptor 816 bound dihydrogenphosphate, which disturbed the aggregates. The largest spectroscopic change was observed for acetate. Král and co-workers also prepared the tetrabrucin-porphyrin 817.828 UV-Vis titrations, carried out in MeOH/HEPES buffer (1:1), revealed that receptor 817 interacts more strongly with ATP (K = 6.4 × 104 M-1) than with ADP or AMP. The binding interactions were predicted to originate from electrostatic interactions between the ammonium groups and the phosphate moieties, as well as π-surface interactions involving the aromatic elements of both compounds.
The neutral β-functionalized porphyrin 818 with an appended disulfonamine chromophore was developed by Starnes and co-workers.829 In this case, UV-Vis-based binding studies carried out in dichloromethane with TBA anion salts led to the conclusion that the anion selectivity of receptor 818 correlated with the basicity of the analytes. The strongest interactions were observed for fluoride, followed by dihydrogenphosphate (K = 7 × 10-4 M-1).
Another strategy pursued by several groups has involved the incorporation of metal into the porphyrin structure. The presence of a metal center allows the resulting porphyrin complexes to act as anion sensors via axial ligation. In early work by Pasternack, Gibbs, and Gaudemer, the interaction of pyridyl porphyrin 819 and its metal complexes with nucleosides and nucleotides was reported.830 In this case, 1H NMR spectroscopic studies carried out in neutral aqueous solution provided support for the notion that more stable complexes are formed with purine nucleotides than the corresponding pyrimidine derivatives. Binding constants were determined to be on the order of 103 M-1. Detailed spectrophotometric experiments established that only metal complexes with zero or one axial ligand (such as the Cu(II) or Zn(II) complexes) interacted appreciably with the nucleotides. These results led to the suggestion that an open axial position was necessary to obtain a measurable binding affinity. Indeed, species with two axial ligands (such as the complexes Mn(III)) displayed no spectroscopic changes upon nucleotide addition.
The cobalt(III) porphyrin complex 820 was prepared by Jyo and co-workers.831 This metalloporphyrin was then incorporated into the membrane of an ion-selective electrode. While it was demonstrated that receptor 820 interacts with dihydrogenphosphate, the highest affinity was observed with SCN-. Later, Kuroda and Ogoshi studied the binding behavior of porphyrin 821, a Rh(III) complex that bears two ammonium “tails” off the macroyclic periphery.832 In this case, UV-Vis studies carried out in aqueous solution demonstrated that receptor 821 binds most nucleotides well (i.e., K(dAMP)) = 860 M-1), except for UMP. Further studies involved analysis of dicationic metal porphyrin 822.833 This receptor was used to extract AMP from aqueous solution into a chloroform phase. Under these conditions, receptor 822 displayed selectivity for AMP over other nucleotide monophosphates, such as UMP, GMP, and CMP.
A tetracationic cobaltocenium porphyrin receptor (823) was prepared by Beer and co-workers.834 This system, which relies on peripheral metal substitution, was found to interact with anionic guests (TBA salts) in acetonitrile. Both hydrogen bonds with the amide groups and electrostatic interactions with the positively charged cobalt centers were thought to account for the observed binding. As such, an electrochemical response was expected in the presence of anions. In fact, as determined via CV analyses, the largest cathodic shift was seen upon the addition of dihydrogenphosphate. Other amide systems developed by Beer and co-workers are the zinc porphyrins 824a and 824b.835 UV-Vis titrations revealed that both receptors displayed a preference for tetrahedral anions (TBA salts). Receptor 824b displayed selectivity for hydrogensulfate over dihydrogenphosphate (log K = 4.8) in DMSO. In contrast, dihydrogenphosphate (log K = 5.4) proved to be the anion bound by receptor 824a in acetonitrile. Square-wave and cyclic voltammetry analyses provided further support for these conclusions. Notably, receptor 824a displayed the most dramatic cathodic shift in the presence of dihydrogenphosphate relative to hydrogensulfate, chloride, nitrate, and perchlorate in acetonitrile. Beer and co-workers also analyzed the binding behavior of other amide-functionalized metalloporphyrins of general structure 825.836 In this case, UV-Vis titrations in different solvents with TBA anion salts led to the conclusion that the identity of the metal influenced both the affinity and the selectivity of the anion recognition process. For instance, in DMSO receptors 825f and 825g were found to interact strongly with dihydrogenphosphate, displaying log K values of 4.4 and 3.6, respectively. In general, halide anions were found to interact more strongly than dihydrogenphosphate with these cadmium(II) and mercury(II) receptors. However, the other metal receptors did not bind any of the tested anions in this competitive solvent.
Metal and free-base complexes of the tetraurea porphyrins (826) were studied by Burns and Scheidt.837 As inferred from 1H NMR spectroscopic studies carried out in DMSO-d6, these receptors display a strong preference for halide anions over dihydrogenphosphate. A binding constant of 1.4 × 103 M-1 was measured for the interaction of dihydrogenphosphate with receptor 826c, while an association constant of greater than 105 M-1 was estimated for chloride in this solvent. In the case of dihydrogenphosphate, the binding affinity was found to increase when more electron-withdrawing substituents were present on the receptor. The metal complexes of receptor 826 displayed weaker anion binding affinity than the free base forms. In later studies, Burns reported that receptor 826c interacts more strongly with dihydrogenphosphate than chloride in dichloromethane.838,839 This result stands in contrast to what was observed in DMSO. On the other hand, in the case of the diurea receptor 827 similar studies revealed a higher selectivity for dihydrogenphosphate in both solvents. The halide preference found for receptor 826c in DMSO was attributed to the presence of a single solvent molecule within the receptor-anion complex. It was proposed that this solvent molecule provided an extra charge-dipole interaction that helped stabilize the complex. Such ancillary solvent effects were not thought to operative in the case of dihydrogenphosphate and receptor 826c, or across the board for the diurea receptor 827. These findings were supported by single crystal X-ray diffraction analyses (826c:dihydrogenphosphate complex shown in Figure 10). All studies relied on the use of TBA anion salts.
Figure 10.
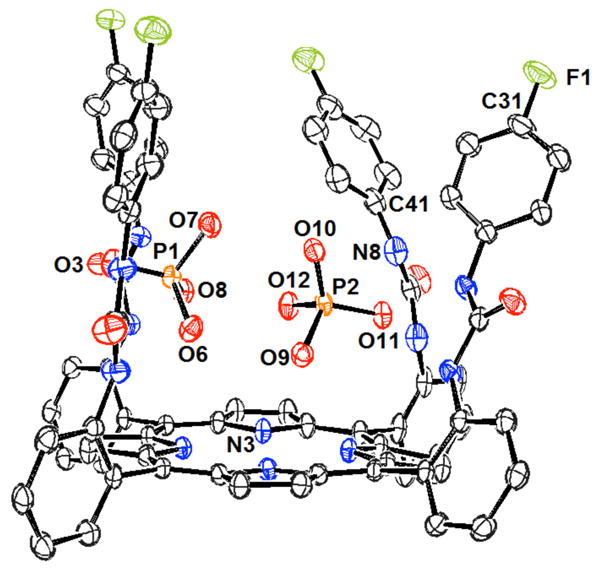
View of the 826c:(H2PO4)2 complex. Drawing generated from X-ray diffraction data originally published in reference 836. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
Hong and co-workers later investigated the anion binding behavior of the free-base and zinc(II) complexes of the functionalized tetrabenzylurea porphyrins 828.840 Anion interactions were analyzed using UV-Vis and 1H NMR spectroscopic titrations carried out in DMSO. Receptors 828a and 828b, which contain phenol groups as substituents, were found to display a preference for acetate and dihydrogenphosphate over other anions. The association constants corresponding to the binding of dihydrogenphosphate (TBA salt) to receptors 828a and 828b were reported to be 4.5 × 104 and 2.0 × 104 M-1, respectively. The incorporation of colorimetric units, giving analogues 828c and 828d, permitted the naked-eye detection of anions. UV-Vis studies with receptors 828c and 828d revealed interactions with fluoride, acetate, and dihydrogenphosphate. The anion selectivity was found to correlate with basicity.
Recently, Li and co-workers prepared the double strapped porphyrin 829.841 While the free-base form (porphyrin 829a) did not display significant anion binding affinities through UV-Vis titrations carried out in chloroform:DMSO (50:1), the corresponding zinc complex, 829b, bound dihydrogenphosphate (TBA salt) under these conditions. A binding affinity of 8.43 × 105 M-1 for dihydrogenphosphate and 829b was reported. A preference for this latter anion over other test anions, such as fluoride, acetate, chloride, bromide, iodide, nitrate, and perchlorate, was reported. This strong affinity and selectivity for H2PO4- was attributed to a combination of hydrogen bond interactions involving the urea groups and coordination to the Lewis acidic metal center.
Boronic acid substituted porphyrins of general structure 830 were introduced by Shinkai and co-workers as glucose phosphate receptors.842,843 It was found that the zinc complex, receptor 830b, could discriminate between glucose-1-phosphate (dipotassium salt) and glucose-6-phosphate (disodium salt) as inferred from 31P NMR spectroscopic analyses carried out in DMSO-d6. Proton NMR spectroscopic titrations led to measured binding constants of 250 M-1 and 1500 M-1 for glucose-1-phosphate and glucose-6-phophate, respectively. CD studies also supported this selectivity. For instance, a strong Cotton effect was observed upon the addition of glucose-6-phosphate to receptor 830b. In contrast, a weak Cotton effect was observed upon the addition of glucose-1-phosphate. The stronger interaction with glucose-6-phosphate was attributed to a judicious combination of interactions between the boronic acid and the 1,2-diol and complexation of the phosphate group to the centrally bound zinc center. This study was expanded to include other analytes, such as 3,4-dihydroxyphenylalanine (DOPA) and its derivatives.
A large number of so-called expanded porphyrins have been reported to date. These larger analogues of porphyrins have often been found to bind anions in their protonated forms. Sapphyrins are the oldest and the best studied of the expanded porphyrins in terms of their phosphate recognition properties. In contrast to porphyrins, sapphyrins possess a large and basic inner cavity.844,845 This cavity, when protonated, has been found to be well suited for interacting with phosphate anions. Sapphyrin and many of its derivatives are positively charged at neutral pH. Further, these macrocycles have been found to interact well with phosphate anions via a combination of electrostatic interactions and hydrogen bonds. This was established inter alia by X-ray diffraction analyses. For example, single crystal X-ray diffraction analysis revealed that receptor 837a (discussed in detail below) formed 1:1 complexes with hydrogenphosphate and dihydrogenphosphate in the solid phase (Figure 11).846 Interestingly, the oxygen atoms in dihydrogenphosphate were observed to be bound by five nitrogen-based hydrogen bonds, with N-O bond distances in the range of 2.8-2.9 Å. On the other hand, complexes of 2:1 guest:host stoichiometry were found with diphenylphosphate and monobasic phenylphosphate. With these anions, the molecule “above” and the molecule “below” the macrocyclic plane were held in place by three and two hydrogen bonds, respectively, with N-O bond lengths in the range of 2.8-3.0 Å. In addition, the phenyl group of monobasic phenylphosphate was found to lie in the plane of the macrocycle, at distances (3.5-3.6 Å) in the range of van der Waals contact interactions.
Figure 11.

View of the 837a:dihydrogenphosphate complex. Drawing generated from X-ray diffraction data originally published in reference 843. In this representation, solvent molecules and most hydrogen atoms have been omitted for clarity.
Early studies by Sessler and co-workers demonstrated that sapphyrin 831 could serve as a carrier in the transport of nucleotides and nucleotide analogues using a U-tube membrane model.847 This model consisted of two aqueous phases (Aq. 1 and Aq. 2) separated by an organic layer. Transport was observed when the protonated sapphyrin was dissolved in CH2Cl2 and GMP was dissolved in the first aqueous phase (Aq. 1) at pH 2.5. GMP was rather quickly thereafter observed to build up in Aq. 2. The attachment of cytosine units onto the periphery of this expanded porphyrin (receptor 832a) allowed for the transport of nucleotides at, or near, neutral pH.848 Receptor 832a displayed selectivity for GMP over AMP and CMP. However, the incorporation of an additional cytosine unit (receptor 833a) led to a loss in selectivity, perhaps as the result of permitting other, less selective, hydrogen bonding interactions. Further studies were carried out to confirm that the transport selectivity was indeed determined by the interaction with the complementary nucleobase.849,850 For this purpose, receptors 832b and 833b bearing guanine units were prepared. As predicted, higher selectivities were found toward cytidine derivatives.
In a different study, sapphyrin 831 was included in a PVC liquid membrane and used to create an ion-selective electrode.173 This ISE was found to give a response toward AMP that was slightly more intense than that for GMP at pH 6.6. This selectivity was attributed to increased π-surface interactions with the adenine base.
The anion recognition features of sapphyrins were further studied by Sessler and co-workers in the context of other possible applications. For example, the silica-bound sapphyrin 832c was prepared and used as a solid support for chromatographs. It was found to allow the HPLC-based separation of monomeric and small oligomeric nucleotides at pH 7.851 Separation was observed between mono-, di-, and triphosphates. Separation between nucleobases, however, was not achieved with this receptor-modified surface. By incorporating a cytosine group within the support system, producing functionalized 833c, separation of nucleotide monophosphates could be achieved.852 On the other hand, peak broadening was seen when this support was used for mixtures of di- and triphosphates. This broadening was attributed to kinetically slow associations and/or dissociations for these anions resulting from relatively enhanced binding affinities. Sapphyrin 832c also interacted with inorganic anions, such as arsenate, phosphate, sulfate, nitrate, chloride, bromide, and iodide, as inferred from the fact that decreased elution times for AMP were observed in the presence of these anions.853 The greatest inhibition was observed with arsenate and phosphate anions. On this basis, it was inferred that these tetrahedral anions interacted most strongly with sapphyrin under the conditions of the experiment (aqueous media, pH 6-8).
Sessler and co-workers also reported that oligosapphyrins 834-836 could serve as carriers for nucleotide di- and triphosphate anions at neutral pH.854 Specifically, sapphyrin trimers 834 and 835 were found to be efficient carriers for nucleotide diphosphates. The tetramer 836 was reported to allow for the transport of all the phosphorylated species included in the study. However, it did display a preference for adenosine-derived nucleotides.
The phosphate binding behavior of more water-soluble sapphyrins has also been analyzed. For example, Sessler and co-workers examined interactions between sapphyrins 837a-b and inorganic phosphate using NMR spectroscopy and UV-Vis analyses carried out in highly competitive media.846,855 Association constants on the order of 104 M-1 were measured in methanol. Likewise, association constants on the order of 102 M-1 were recorded in 10 mM aqueous bis-Tris at pH 6.1. Sessler and co-workers also exploited the aggregation tendencies of certain sapphyrins, such as 838, to effect phosphate detection in aqueous media.856 Interactions were analyzed by monitoring changes in the absorbance and fluorescence spectra as a function of phosphate anion concentration in aqueous media (25 mM PIPES buffer, pH = 7.0). It was found that phosphate helped stabilize the monomeric form of sapphyrins 838. Since the monomeric form was far more fluorescent than various aggregated forms, phosphate addition led to an increase in fluorescence intensity. Effective association constants ranging from approximately 6 to 19 M-1 were reported. In later studies, Sessler and co-workers prepared receptors 837c-f and 839.857 These water solubilized sapphyrins were found to act as catalysts for the hydrolysis of bis(4-nitrophenyl)phosphate (BNPP) near neutral pH. The interactions between receptor 837d and BNPP were studied using UV-Vis spectroscopy. On the basis of these analyses, a binding constant of 400 M-1 was determined at pH 7.5 (0.01 M HEPES/0.1 M NaNO3).
Charvatova and co-workers prepared capillaries coated with sapphyrin 837a for use in open-tubular capillary electrochromatography (CE).858,859 These capillaries were found to separate nucleotide monophosphates selectively as a function of the nucleobase. Furthermore, significant separation was observed between mono-, di-, and triphosphates. Moderate resolution was observed in the case of di- and triphosphates and was found to be correlated with the nature of the nucleobase. In the course of the study it was found that phosphoserine and phosphothreonine (added as the phenylthiohydantoin derivatives) could not be eluted readily from the column. Presumably, this reflects a strong interaction with the sapphyrin-coated capillary. These researchers suggested that the nature of the organic moiety present in the analytes had a significant effect on the nature and strength of the interactions with the solid phase.
Other expanded porphyrins have also been employed as phosphate receptors. For example, Sessler and co-workers analyzed the interactions between rubyrin 840, a hexapyrrolic expanded porphyrin and several nucleotides. This was done using a triphasic U-tube transport system operating at neutral pH, in analogy to what was used in the case of sapphyrins.860 Upon the addition of triisopropylsilyl-protected cytidine (C-Tips), the transport of GMP was found to be 30-fold faster in the case of rubyrin than with sapphyrin. This increase was attributed to the formation of a cooperative supramolecular structure. Separate from this, the binding behavior of meso-functionalized smaragdyrin 841 was analyzed by Chandrashekar and co-workers.861 In this case, UV-Vis studies conducted in methanol led to the conclusion that receptor 841 bound AMP with a binding constant of 2.6 × 105 M-1. However, the affinity for fluoride (K = 7.6 × 105 M-1) was found to be higher than that for AMP.
Expanded pyrrolic systems containing diamidodipyrromethane units were developed by Sessler and Ustynyuk and tested as anion receptors.862 One of these, receptor 842, displayed a preference for tetrahedral anions as inferred from UV-Vis titrations in acetonitrile (anions studied as TBA salts).863 The strongest anion interaction was observed for dihydrogenphosphate. A 2:1 (guest:host) binding stoichiometry was inferred on the basis of Job plots and fits to the binding curves. These latter analyses allowed first and second association constants of 3.4 × 105 and 2.6 × 104 M-1, respectively, to be calculated. Further studies demonstrated that relatively simple structural modifications could give rise to different anion binding selectivities. For example, receptor 843 was found to interact strongly with hydrogensulfate, (K = 1.1 × 105 M-1), while receptor 844 proved selective for chloride (K = 1.2 × 105 M-1) in acetonitrile. The association constants for dihydrogenphosphate were measured to be 2.9 × 104 and 1.5 × 104 M-1, respectively, in this same solvent.
Following these studies, several additional macrocycles were prepared via anion templation. After removal of the anion, compounds 845-847 were obtained.864,865 All three systems were found to act as anion receptors, displaying selectivity toward dihydrogenphosphate and hydrogensulfate (TBA salts) as inferred from UV-Vis titrations carried out in acetonitrile. Receptor 847 displayed a 3:1 guest:host stoichiometry with dihydrogenphosphate, and the first binding constant was found to be log K = 6.7.
Recently, a new series of pyrrolic macrocycles with amido-imine functionalities, namely receptors 848-850, was developed by Sessler and Katayev.866 The seemingly minor changes within this set were reflected in demonstrably different anion binding properties. For example, in acetonitrile, receptor 848 was found to interact strongly with the hydrogensulfate anion (TBA salt). In contrast, the acetate anion was bound in preference over other anions by receptor 849. Furthermore, the more rigid receptor 850 displayed a high affinity toward chloride. In general these receptors also displayed a strong affinity toward dihydrogenphosphate. Binding constants on the order of 105 M-1 were reported for this latter anion in acetonitrile. In related studies, the bipyrrole-derived macrocycles 851-854 were synthesized by Katayev and co-workers.867 Competitive 1H NMR spectroscopic titrations were performed in the presence of acetate in DMSO / 0.5% water. These experiments revealed that receptor 852 displayed selectivity toward dihydrogenphosphate (TBA salt). An association constant of greater than 104 M-1 was inferred from these studies.
Sessler and Furuta developed the first example of an expanded pyrrole macrocyclic containing dipyrrolylquinoxaline units.868 This system, receptor 855, displayed a high affinity toward fluoride and dihydrogenphosphate (TBA salts) in dichloromethane as determined from UV-Vis spectroscopic titrations. A log K of 3.8 was reported for the binding of dihydrogenphosphate. The presence of the quinoxaline subunits allowed for the naked-eye detection of both anions in this apolar solvent.
Perhaps the most complicated pyrrolic anion receptor reported to date is the bipyrrole-based [2]catenane 856 prepared by Sessler, Vögtle and co-workers.869 In this case, 1H NMR spectroscopic titrations carried out in 1,1,2,2-tetrachloroethane-d2 confirmed that this multi-dimensional, catenated system interacts strongly with several anions (TBA salts). A high selectivity toward dihydrogenphosphate was reported. The binding constant for this anion was found to be greater than 107 M-1. It was proposed that receptor 856 contained a tetrahedral cavity between the rings. This geometry would be expected to result in a strong and selective receptor for dihydrogenphosphate.
5. Conclusions
The goal of obtaining strong and selective binding agents for phosphate and phosphorylated compounds has attracted the attention of many research groups, each with a slightly different approach. This has resulted in what is now an extensive set of artificial receptors. While many of the determinants and underlying binding affinities and selectivities are common to all supramolecular interactions, several factors are particularly important in phosphate binding. For example, phosphate selectivity is often determined by the geometry of the receptor binding cavities. Further, the arrangement of various hydrogen bond donors and acceptors can be used to maximize interactions with the shape and polarity of this tetrahedral anion. These hydrogen bonding interactions play an important role in nearly all the successful binding systems reported in this review, including those wherein electrostatic interactions appear to dominate the binding affinity. As a general rule, receptors with C3v symmetry appear to maximize these various stabilizing interactions, although favorable orientations have also been achieved through the use of well-designed macrocyclic and cleft receptors. While challenging to design and prepare, appropriately sized receptors with an ability to form inclusion complexes with phosphate anions have also proven to be highly effective.
Polyammonium-based compounds, with which this review began, bind anions mainly via electrostatic interactions. These systems provide some of the strongest phosphate interactions in aqueous media at neutral pH. These interactions depend strongly on the distribution of the charge within the receptor, both in terms of charge density and size/shape complementarity. Increasing the charge density of alkyl polyammonium receptors requires a careful balance of the spacing between the nitrogen atoms, the size of the macrocyclic ring and the methylation pattern. At the same time, hydrogen bonding interactions, even in the simplest polyammonium systems, can be critical contributors to the phosphate binding affinity. However, the overwhelming influence of electrostatic interactions in polyammonium systems can “swamp” the more subtle selectivities that would be expected to be provided by other recognition motifs.
Guanidinium receptors are also positively charged near neutral pH. As a general rule, these systems give rise to phosphate complexes that are less stable than those produced with polyammonium systems. Presumably, this reflects the lower charge density of the guanidinium group in water. The high pKa of these units, however, allows guanidinium-based receptors to stabilize electrostatic interactions with phosphate anions over a much wider pH range than ammonium receptors. The use of guanidiniums can also allow for the placing of charged groups in much closer proximity to the targeted anions than do ammonium motifs. Binding studies via ITC are also simplified, as protonation and deprotonation events do not need to be accounted for in the data analysis. Perhaps the most attractive feature of guanidinium subunits is that these units stabilize complexes characterized by the presence of two parallel hydrogen bonds. This geometry generally imparts a strong preference for oxoanions over other anions of like charge. As a consequence, guanidinium units are found in some of the most successful phosphate receptors produced to date.
The large number of neutral receptors based on hydrogen bond donor subunits has allowed insight into the design criteria needed to achieve selective phosphate recognition. In general the most effective hydrogen bond donors for phosphate recognition are thiourea motifs that are both relatively acidic and which stabilize parallel hydrogen bonding geometries. With these and other motifs, factors that increase the effective acidity serve to increase the hydrogen bond donating ability. As a given rule, this enhances binding. However, a balance must be maintained in order to avoid deprotonation by the anion of interest. The use of receptors with multi-point binding moieties preorganized for interactions with tetrahedral anions can overcome the innate selectivity arising from differences in anion basicity, something that often favors fluoride and acetate over dihydrogenphosphate. This approach has been exploited successfully through the design of clefts that are too large for other competing species or inaccessible to spherical anions.
Another caveat that must be considered is that certain phosphate anions have a propensity to dimerize in less polar media. This can complicate binding studies carried out in organic solvents. On the other hand, this phenomenon may be exploited to produce highly selective receptors that target the dimeric form. The organic solubility of many neutral hydrogen-bond receptors has been used to create effective phosphate transport systems. At the same time, other applications may require the use of systems that function in aqueous environments.
The incorporation of metal cations into appropriately designed frameworks can give highly effective phosphate anion receptors. For example, metal complexes of polyammonium macrocycles were found to bind phosphate anions two to three-fold more strongly than their diprotonated counterparts. This is thought to reflect the presence of favorable Lewis acid-base interactions. Metal coordination has also been used to preorganize the surrounding ligand structure and to generate cavities optimized for phosphate binding. Separate from this, combining metallic units with electrochemical or luminescent properties with other types of receptor motifs can provide for useful signaling. This approach can also be used to introduce ancillary electrostatic binding interactions. Metal coordination-based strategies also offer advantages for phosphate binding and sensing in aqueous media, including increased solubility of the receptor and a source of consistent Lewis acid-base interactions.
Finally, macrocyclic receptors, in particular pyrrolic receptors, have been extensively studied as phosphate anion receptors. In many cases, high anion binding constants were observed, which is ascribed to the inherent structural preorganization. In the case of sapphyrins, systems that are charged at neutral pH, significant selectivity for nucleotides was observed based in the case of systems bearing complementary nucleobase functionality. In the case of neutral, non-aromatic pyrrole macrocycles, several specific geometries were noted that gave rise to high phosphate anion selectivities. However, this optimization remains difficult to achieve and, in fact, low phosphate selectivities are generally observed with most simple pyrrolic receptors, including the easily synthesized calixpyrroles.
As noted throughout this review, considerable progress has been made in the area of phosphate and phosphorylated substrate recognition. On the other hand, there is clearly room for improvement. Although many receptors have been reported, few display appreciable selectivity for phosphate over other anions. Discrimination between phosphate guests has been even more rarely achieved. Achieving this selectivity in a routine, predictable manner is thus a clear challenge for the future. Another challenge is generating receptors, particularly neutral systems, that function well in aqueous media. While much has been learned through the study of binding interactions in organic media, applications involving biological phosphate targets will require the use of selective receptors whose function is not impaired when exposed to water. While the use of highly charged systems, such as those based on guanidinium subunits, is one way to address this problem, many of the most attractive applications, including those associated with through-membrane transport and selective phosphorylated substrate extraction, will likely require the use of neutral receptor systems or those of low charge density that function in the presence of water and salts. The need to produce such systems, as well as a desire to understand non-first order effects, such as ion pairing, aggregation, and solvation, underscores the benefits that could accrue from further work in the phosphate recognition area. It is hoped that by summarizing the current state-of-the-art, this review will help advance efforts in this all-important area and facilitate the development of receptors that can be exploited for use in various real-world applications.
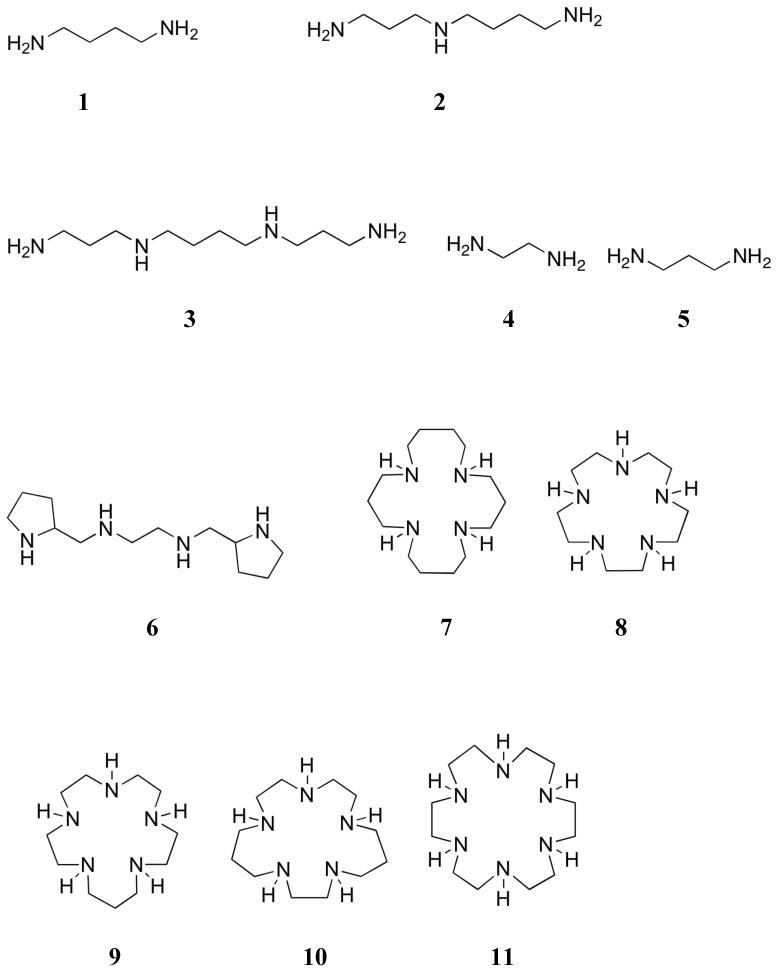

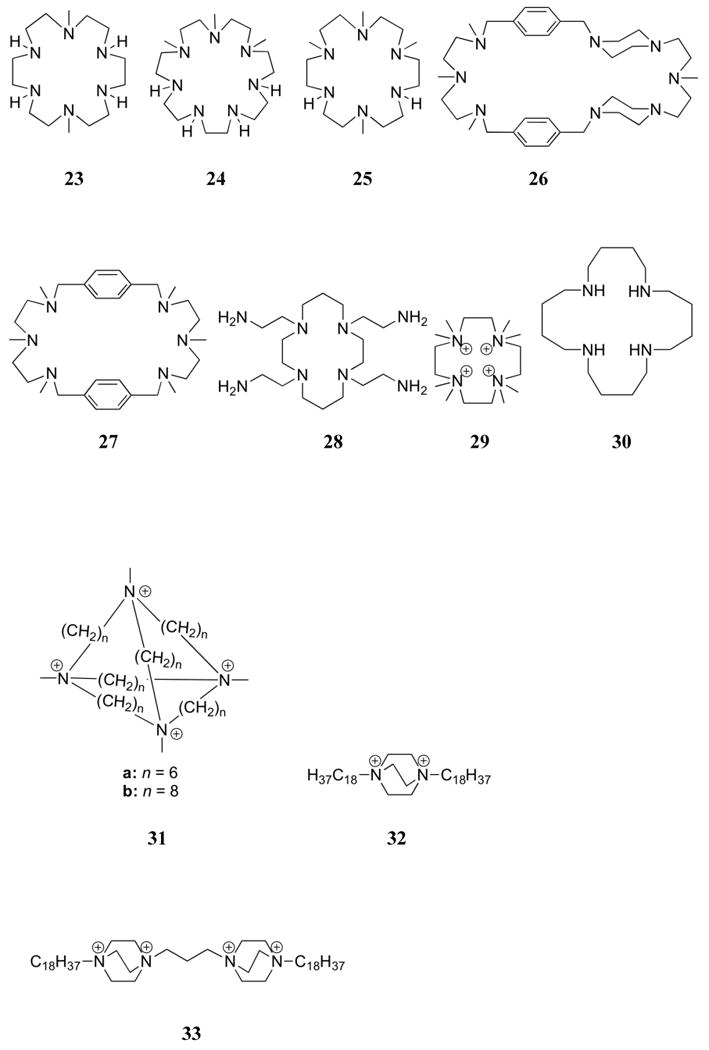
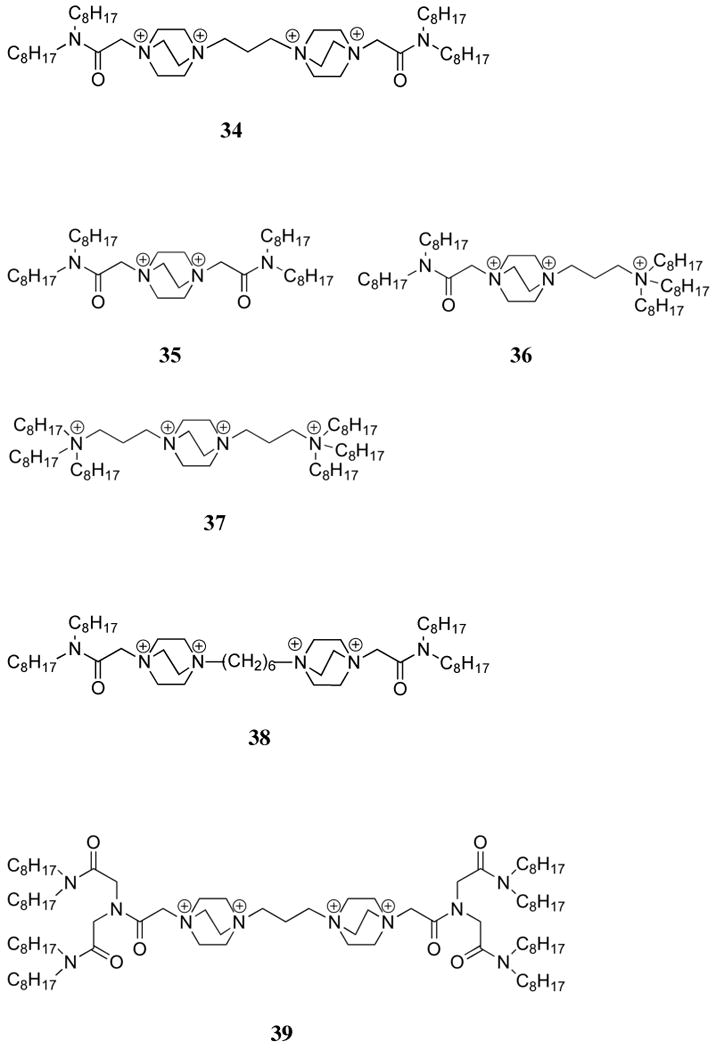
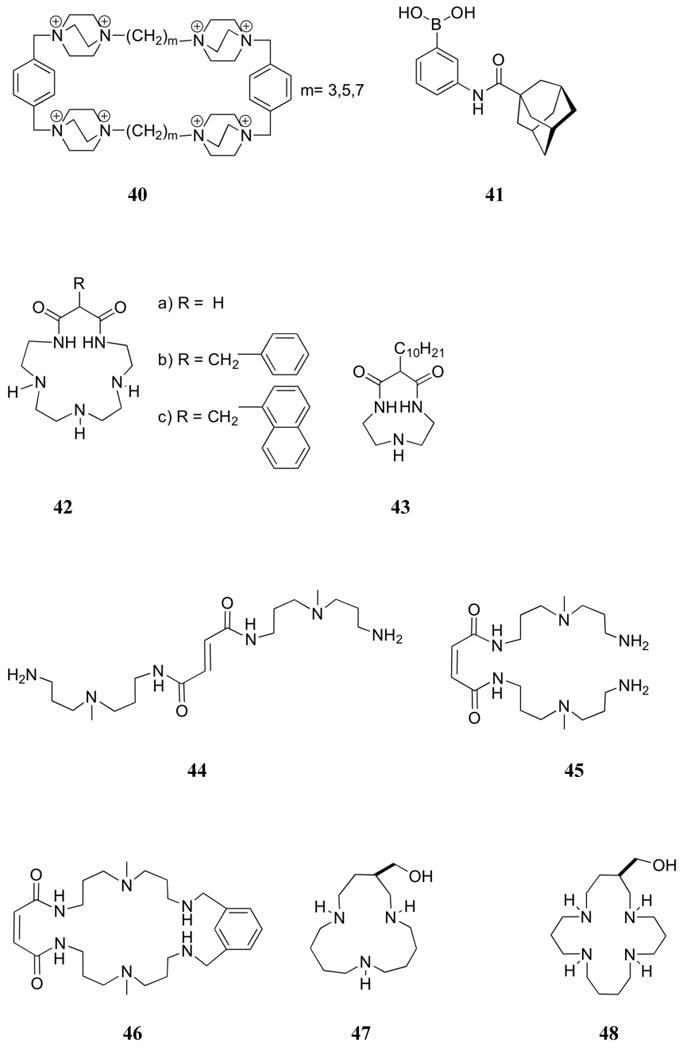
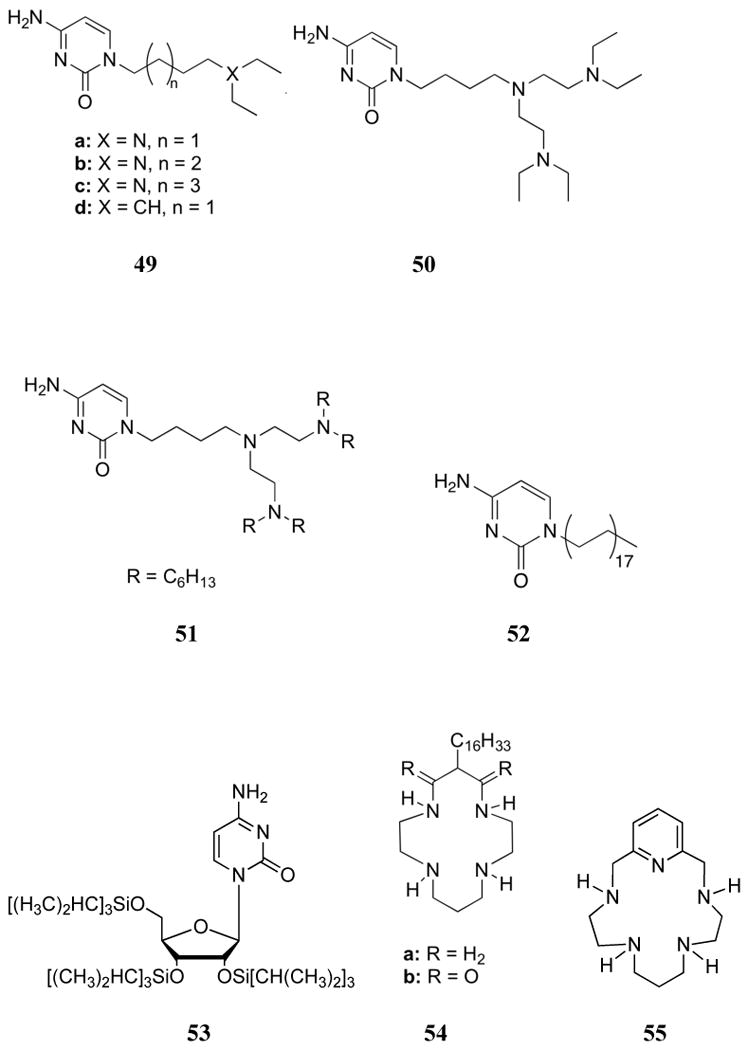
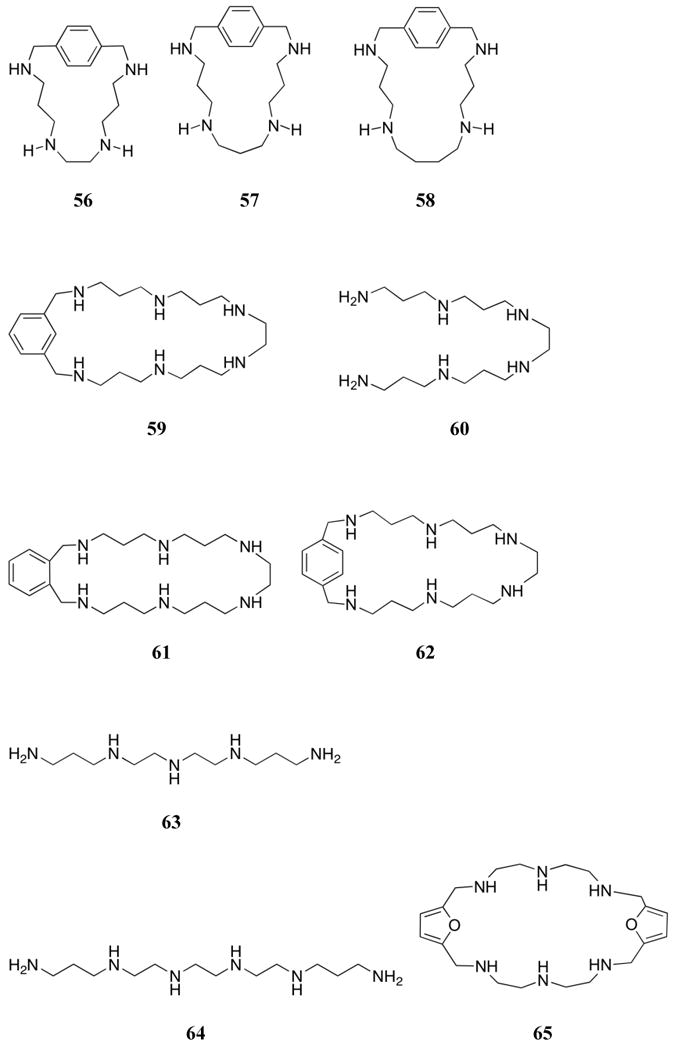
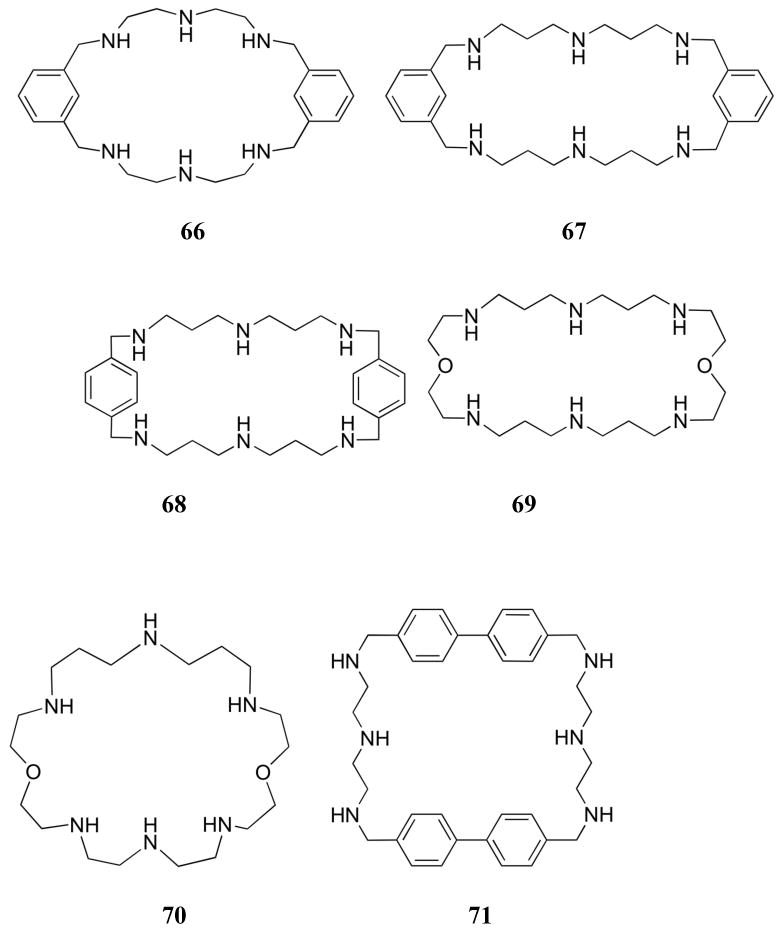
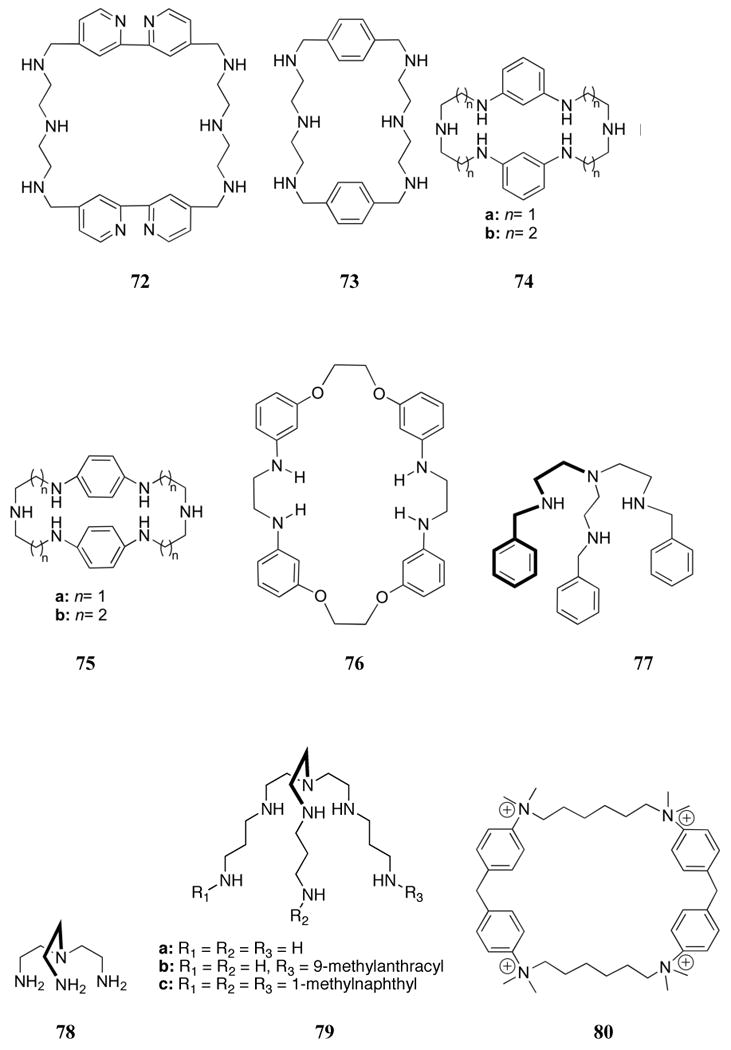
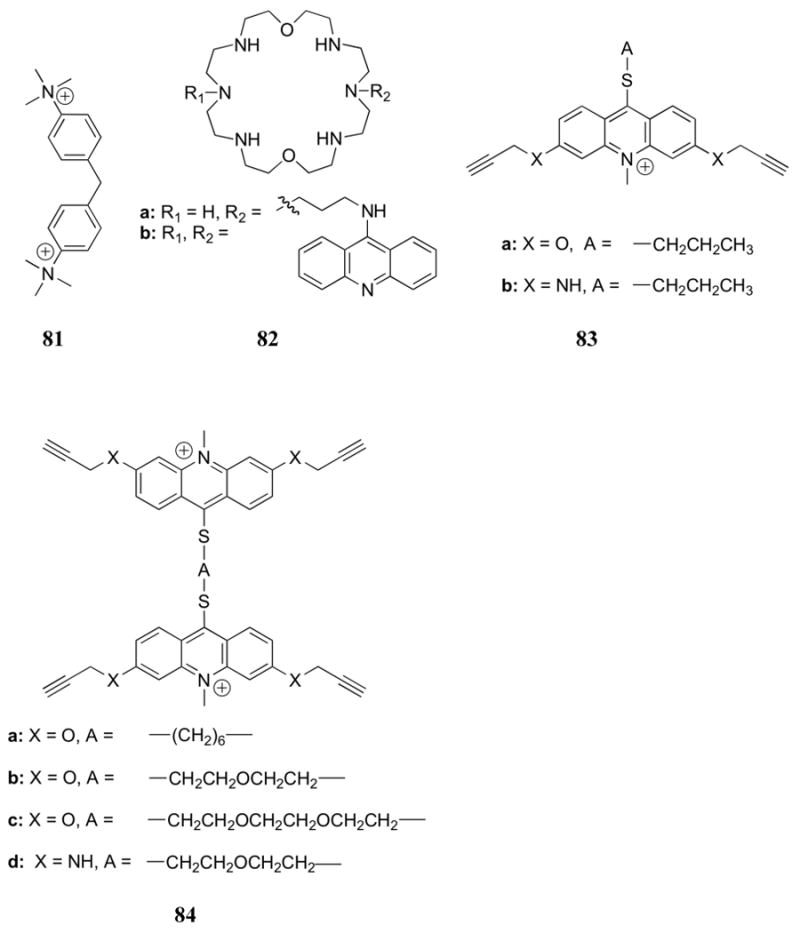
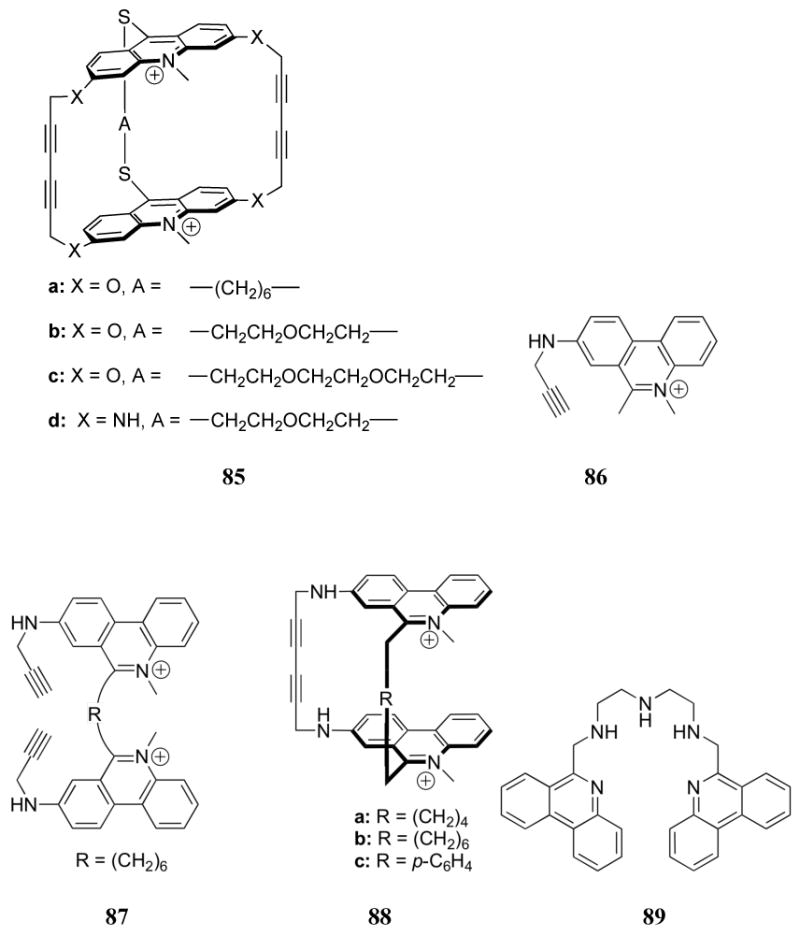
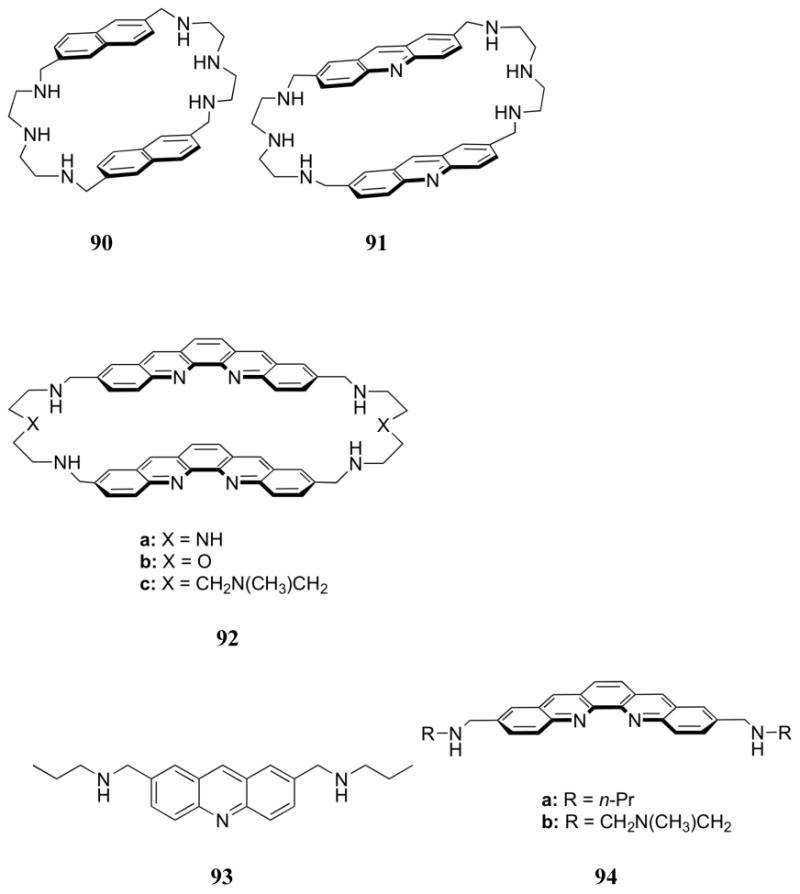
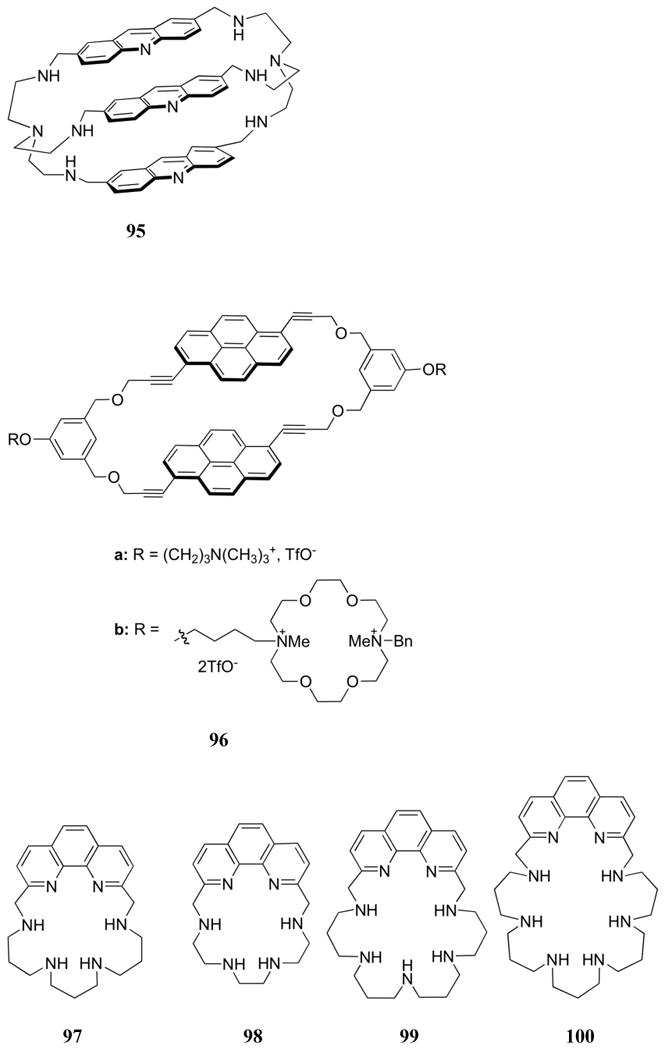

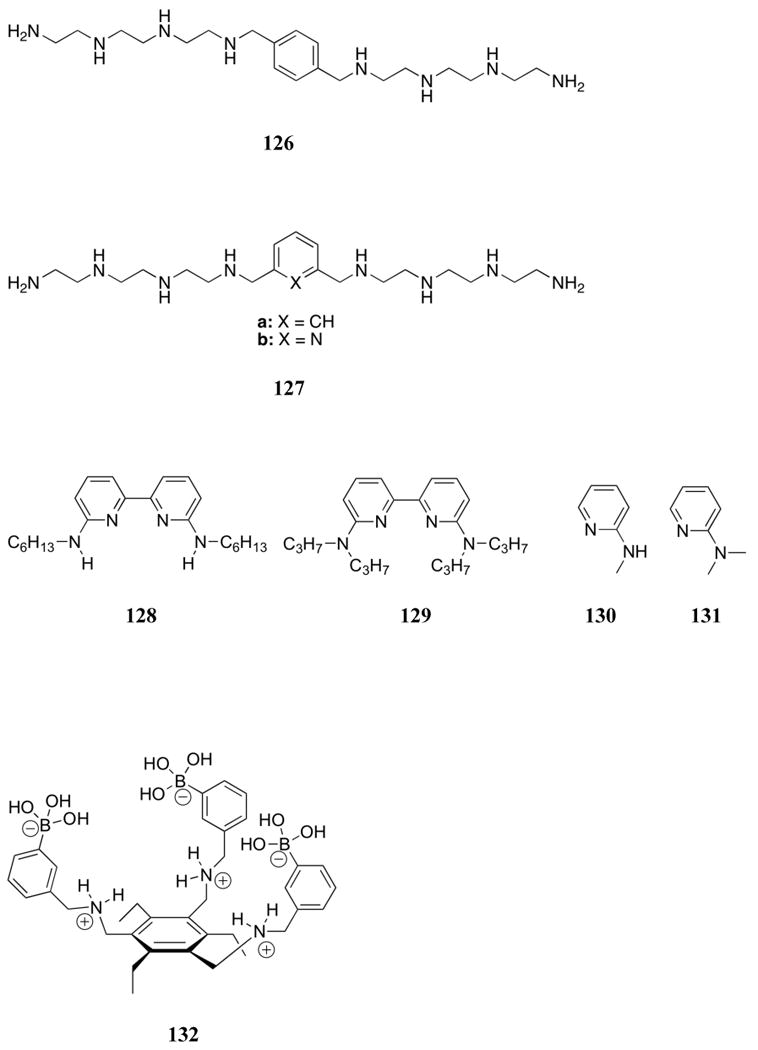



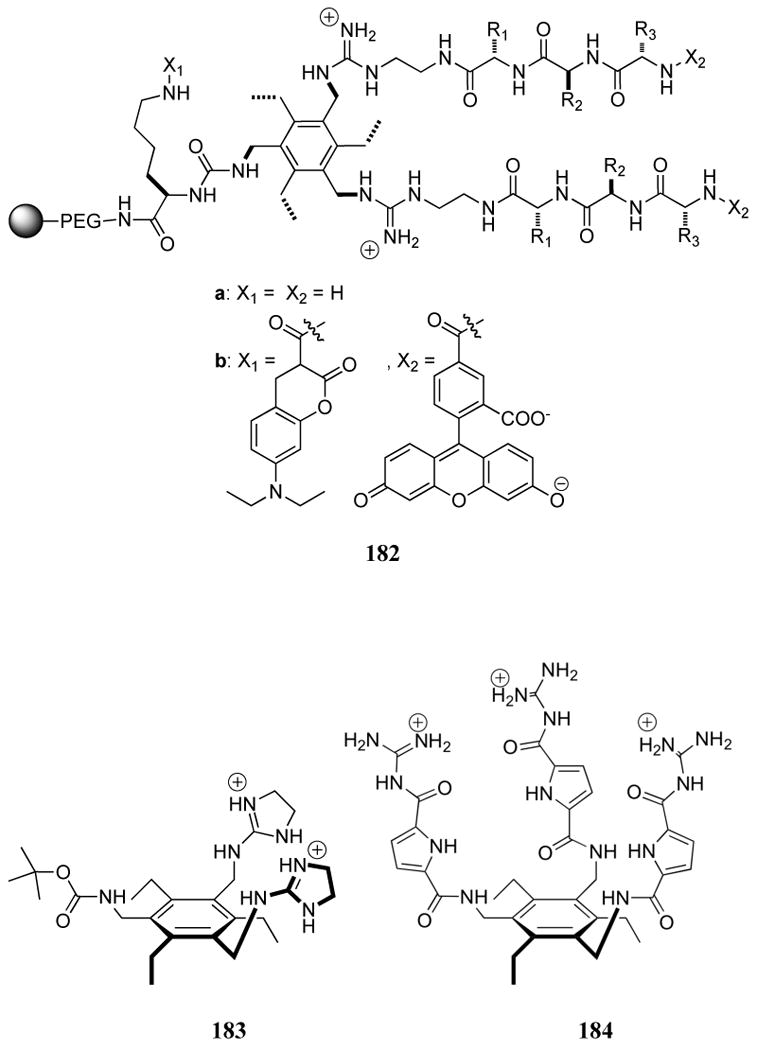
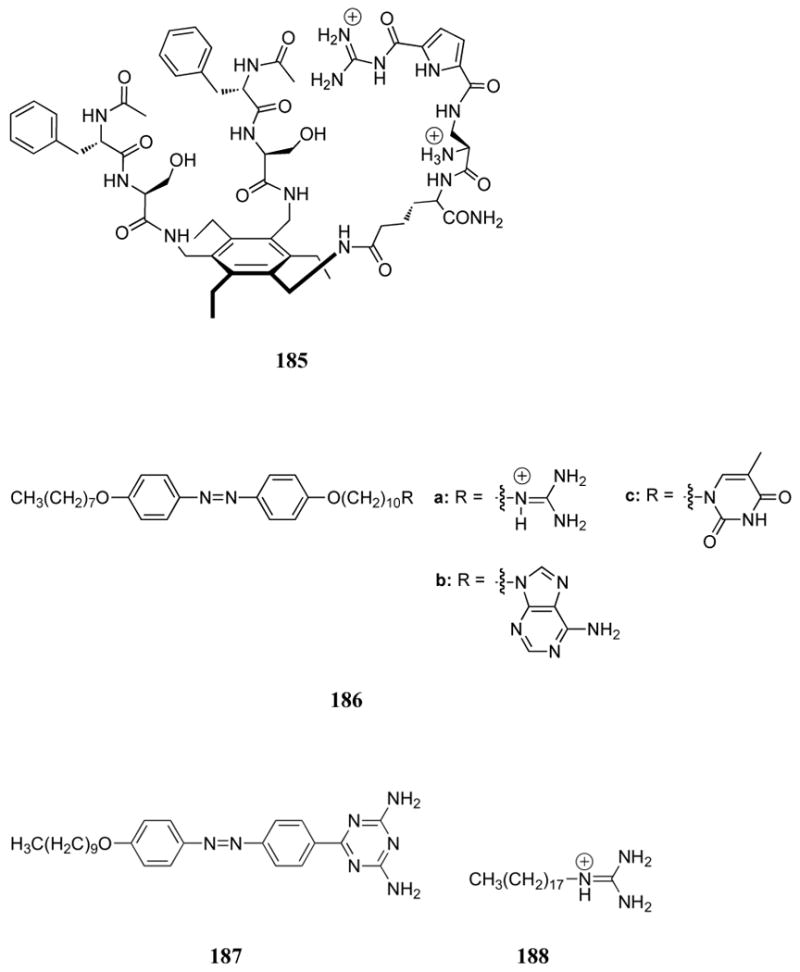

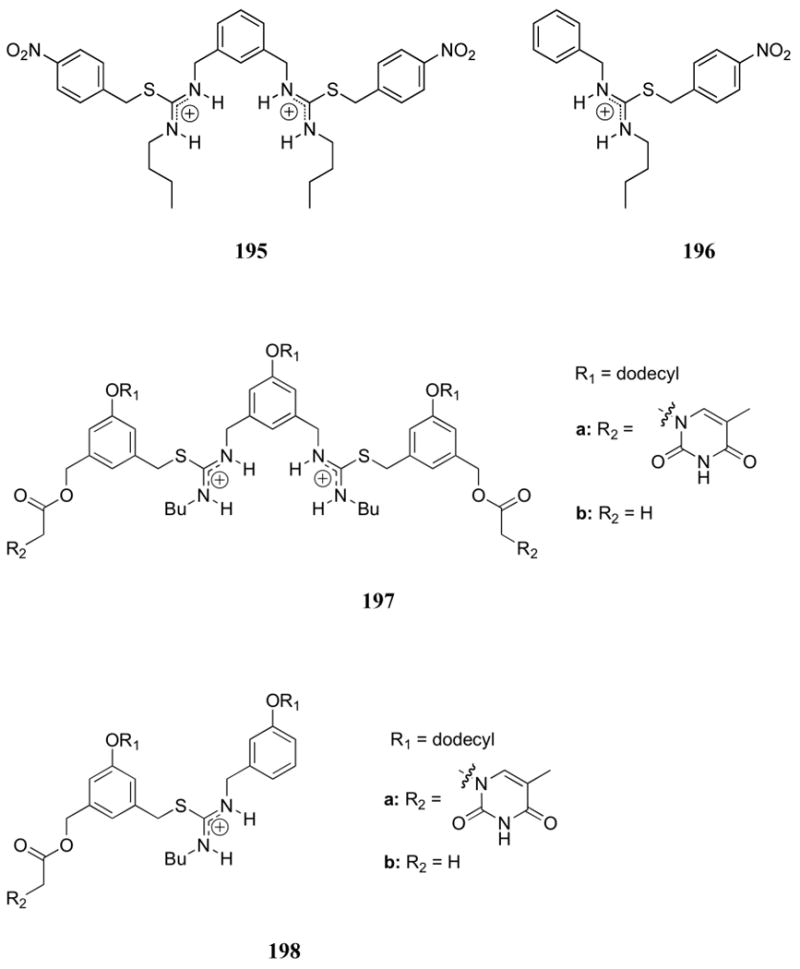

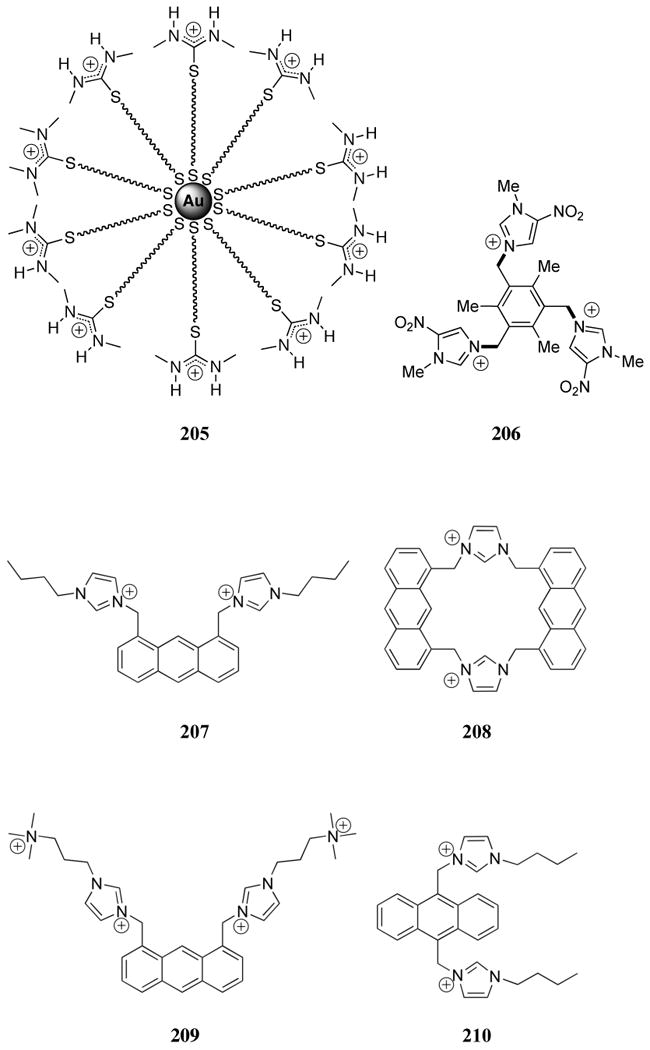
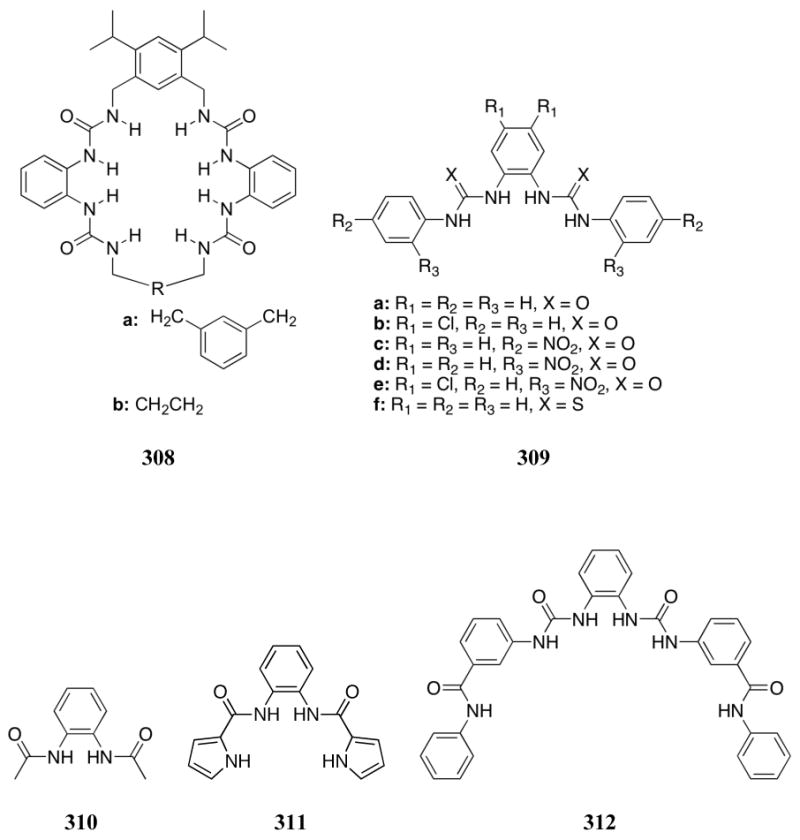
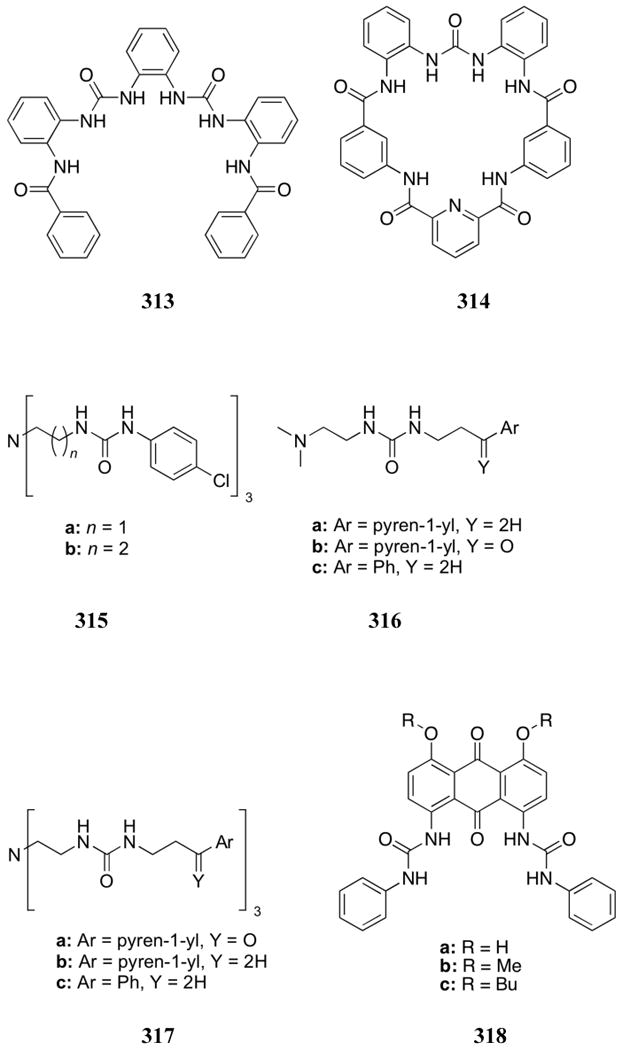
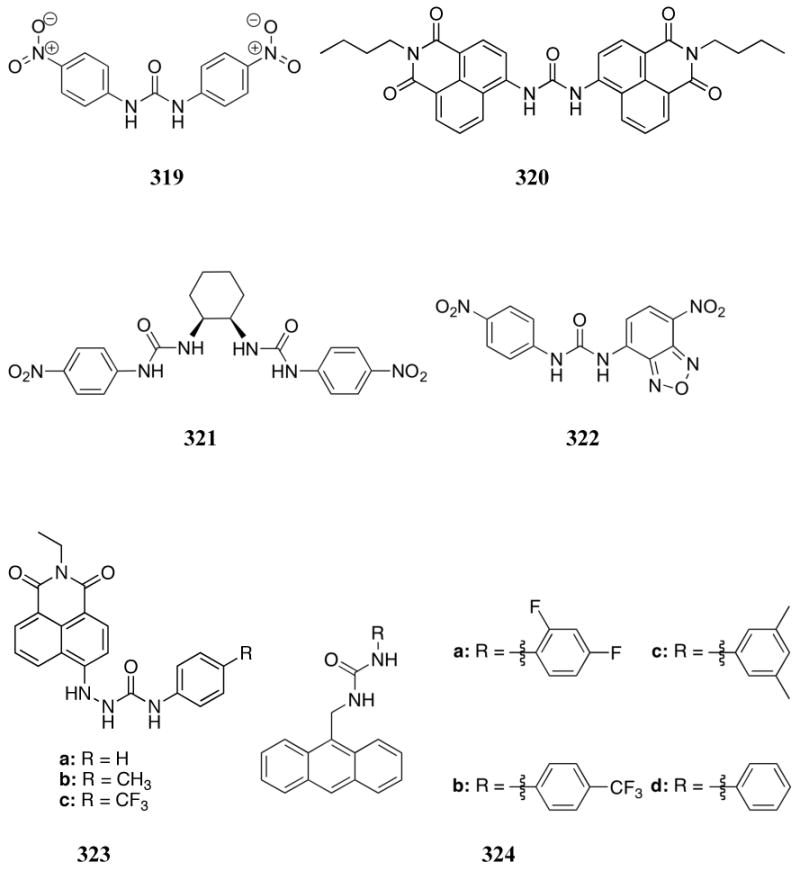
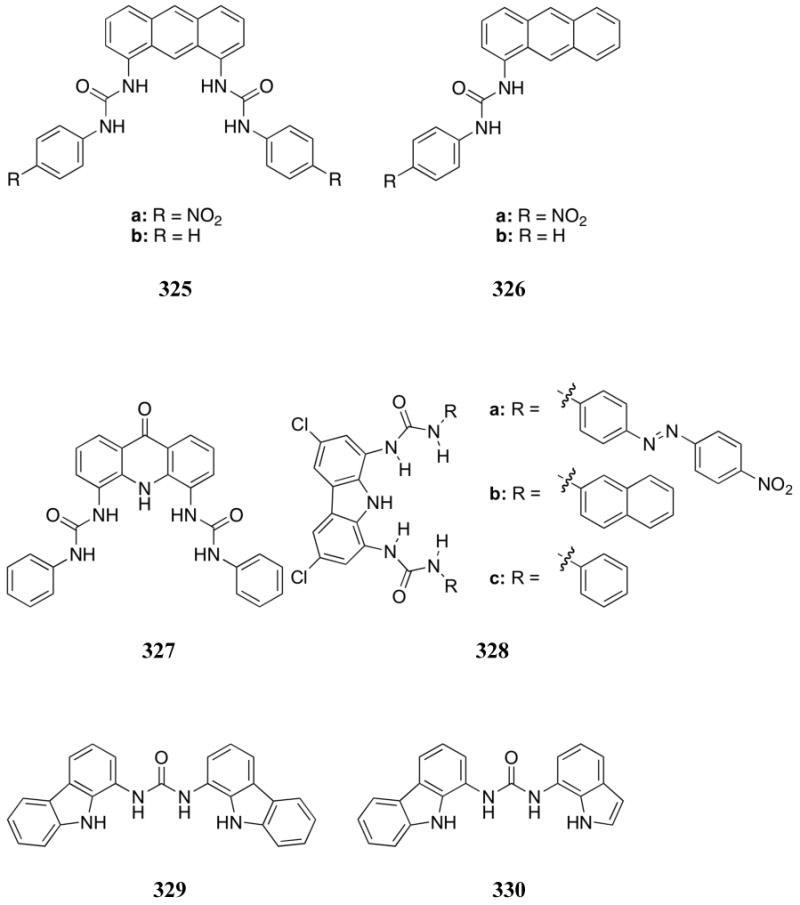

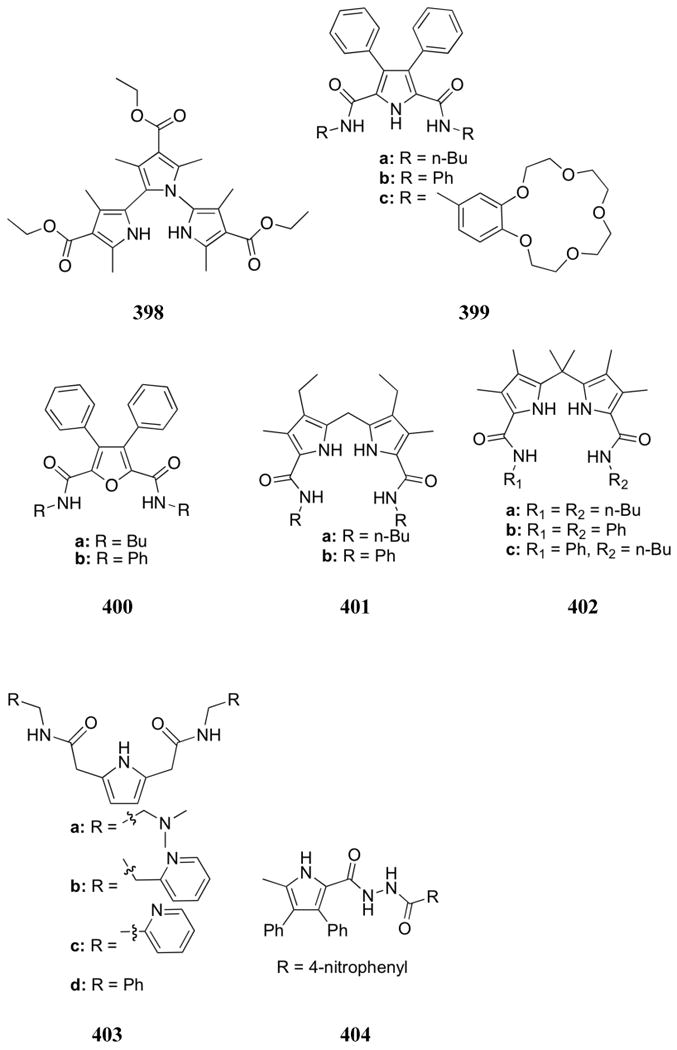
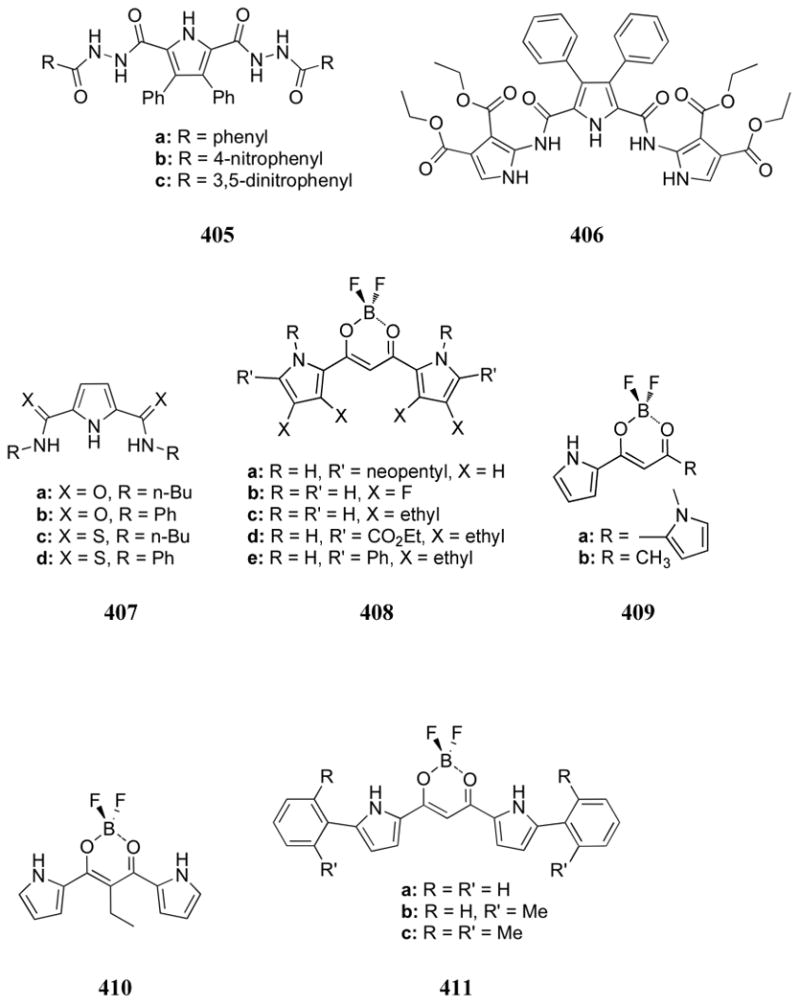
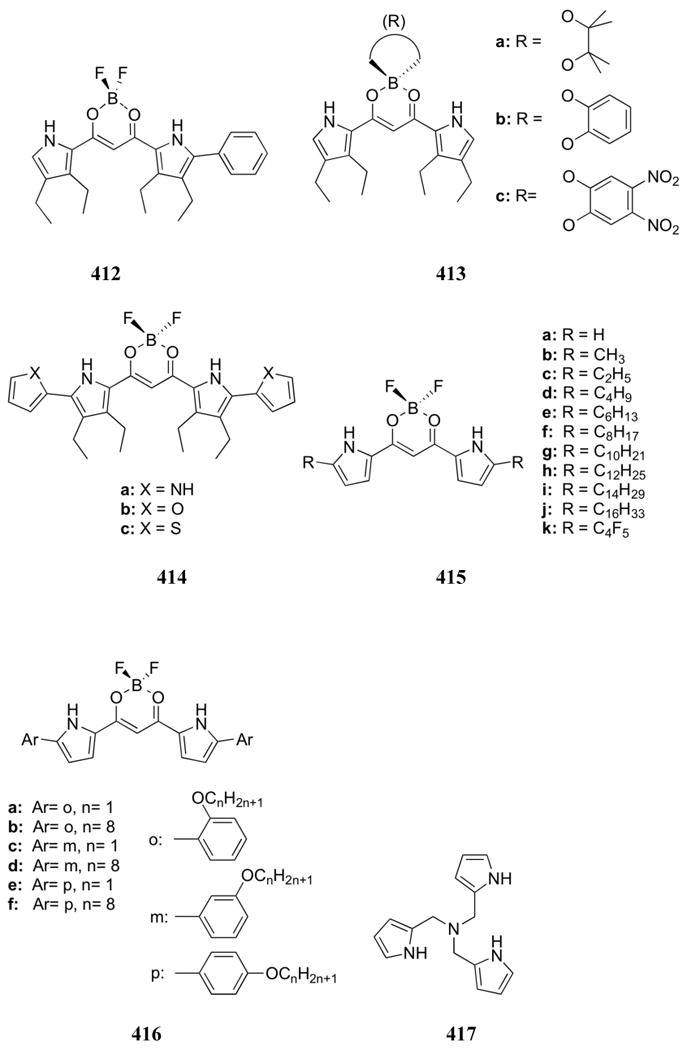
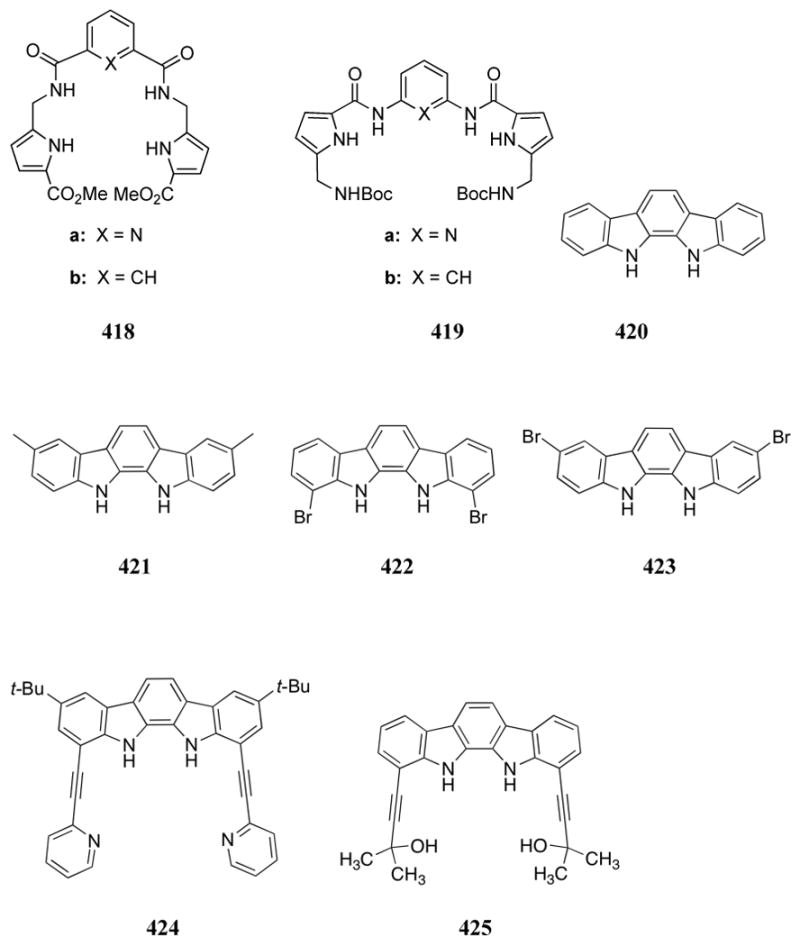





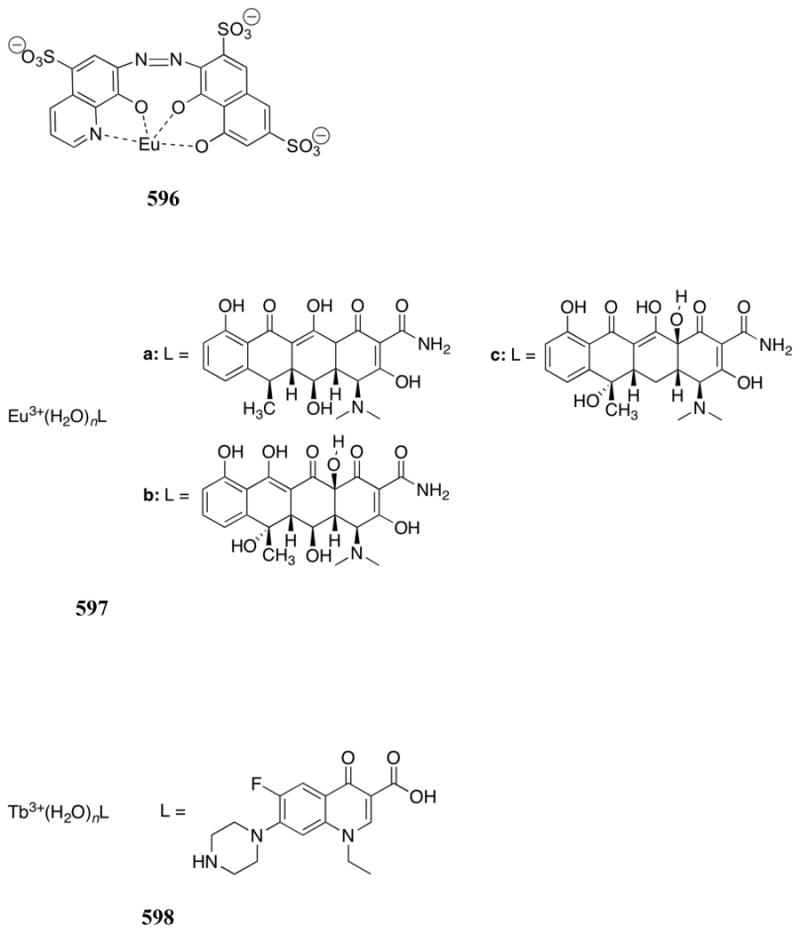
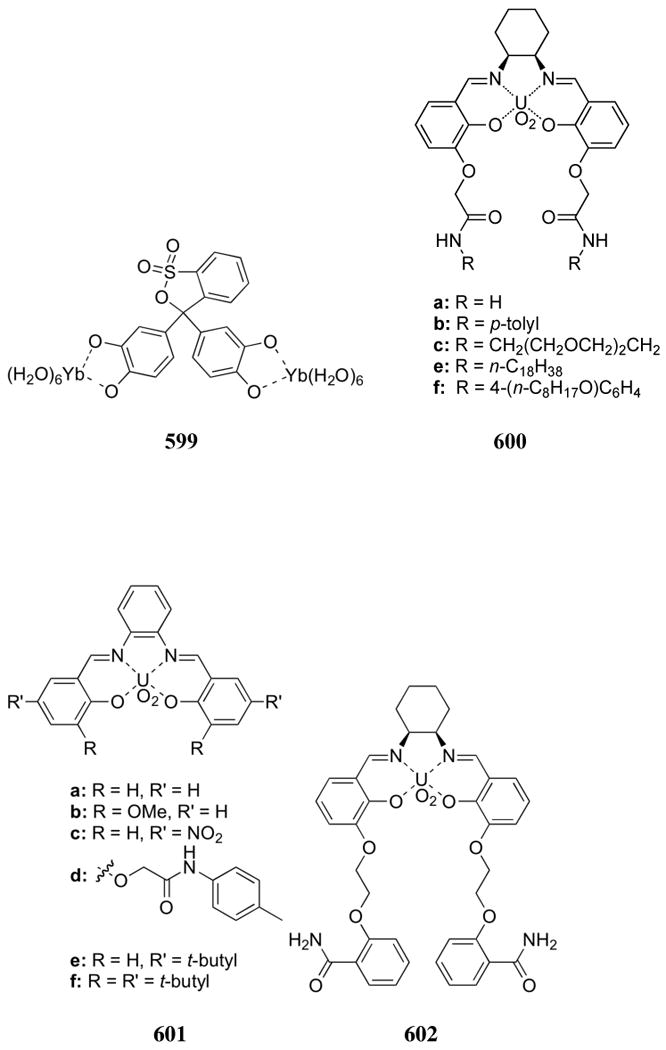
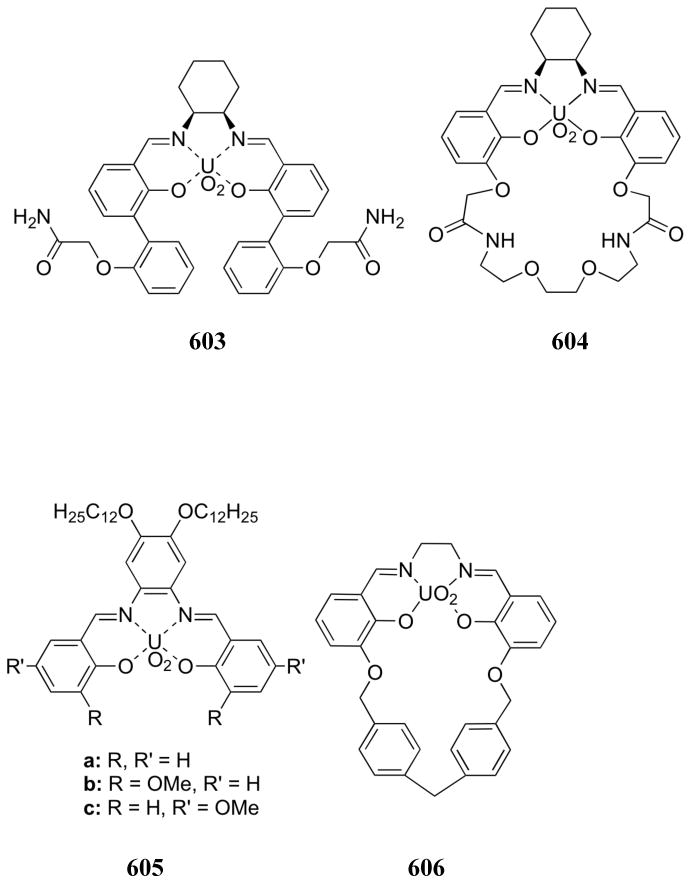
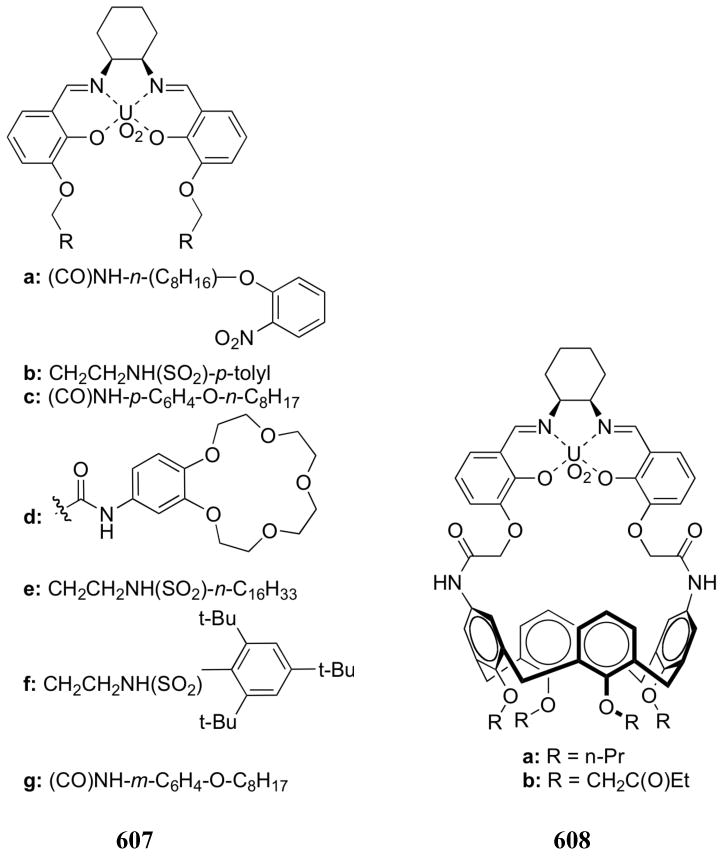
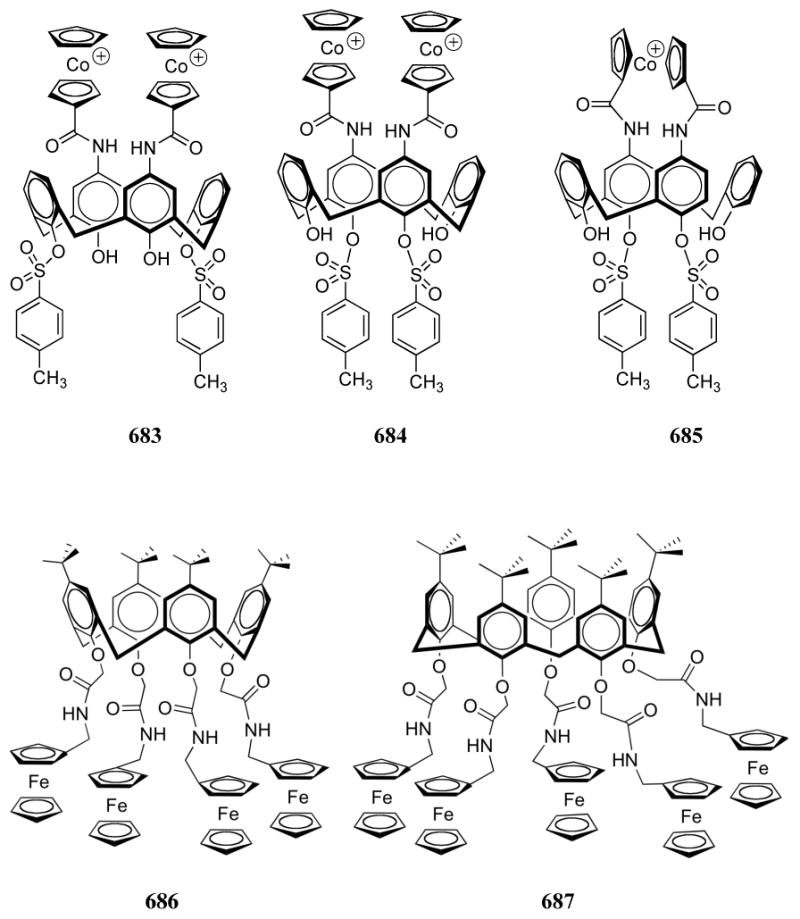
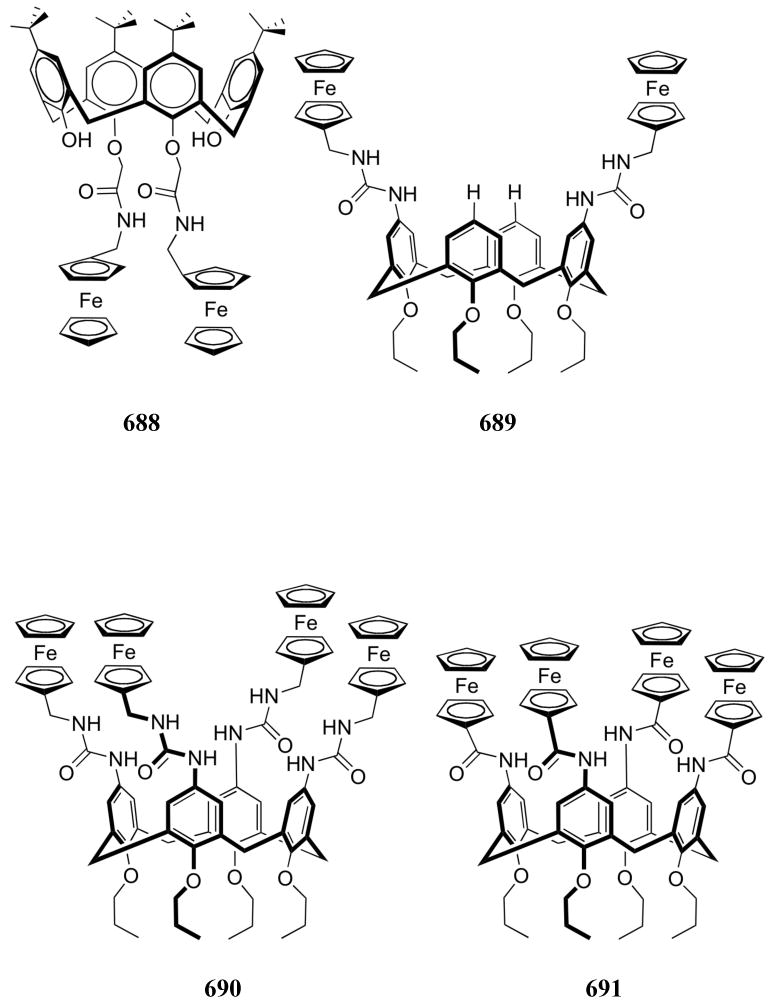
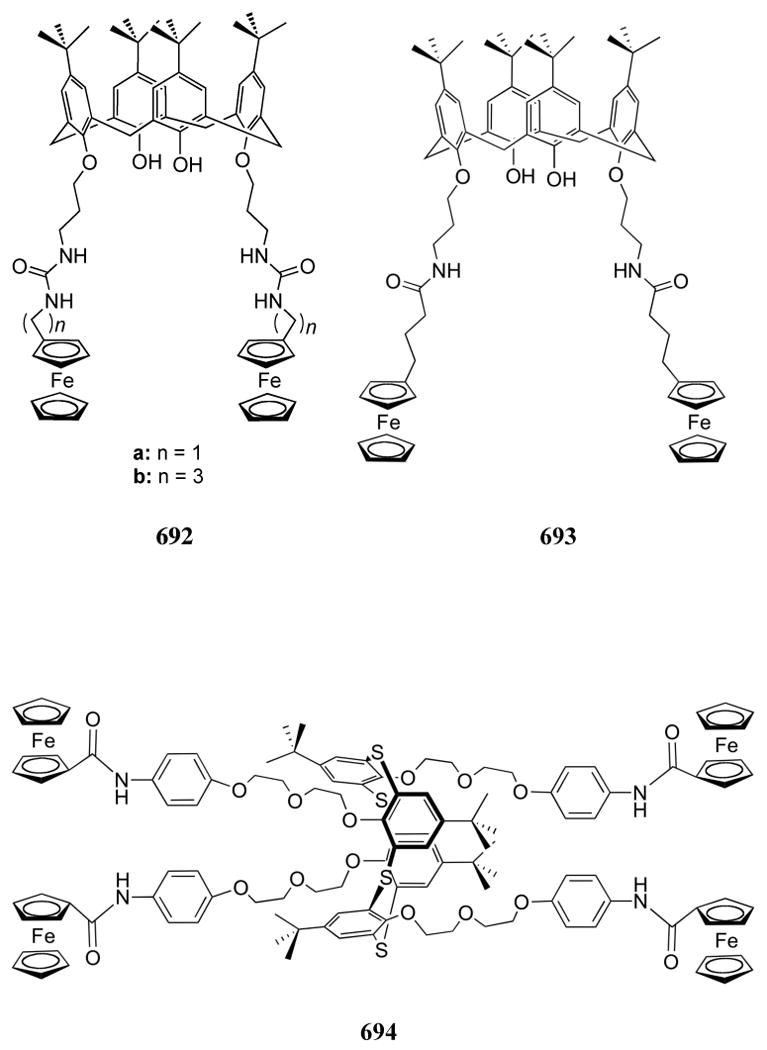
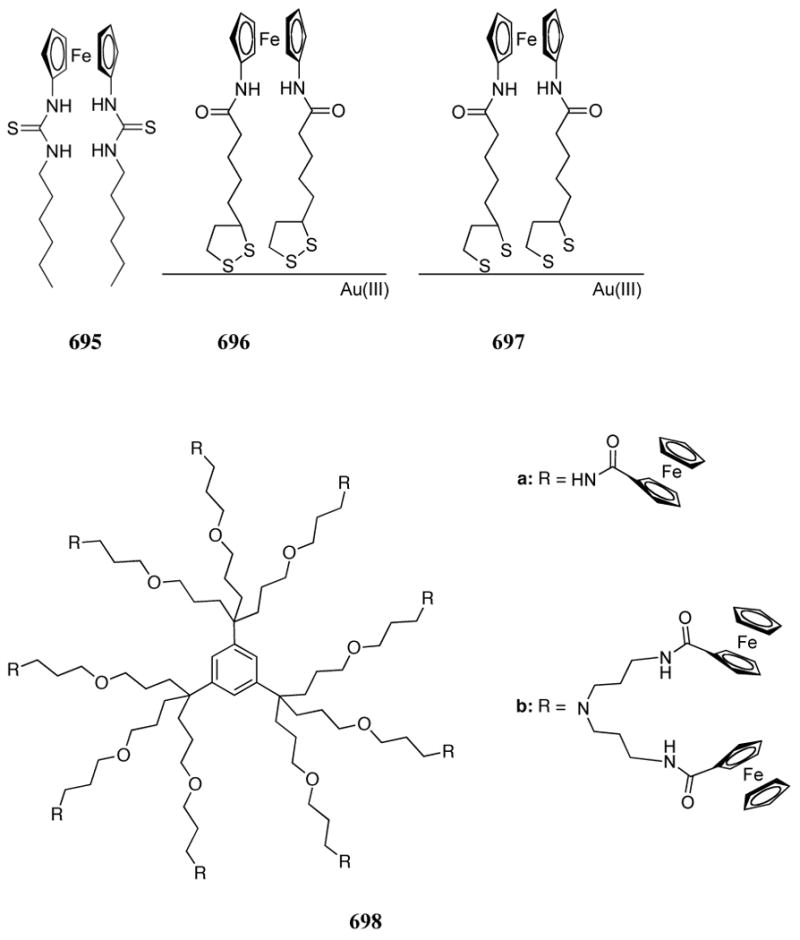

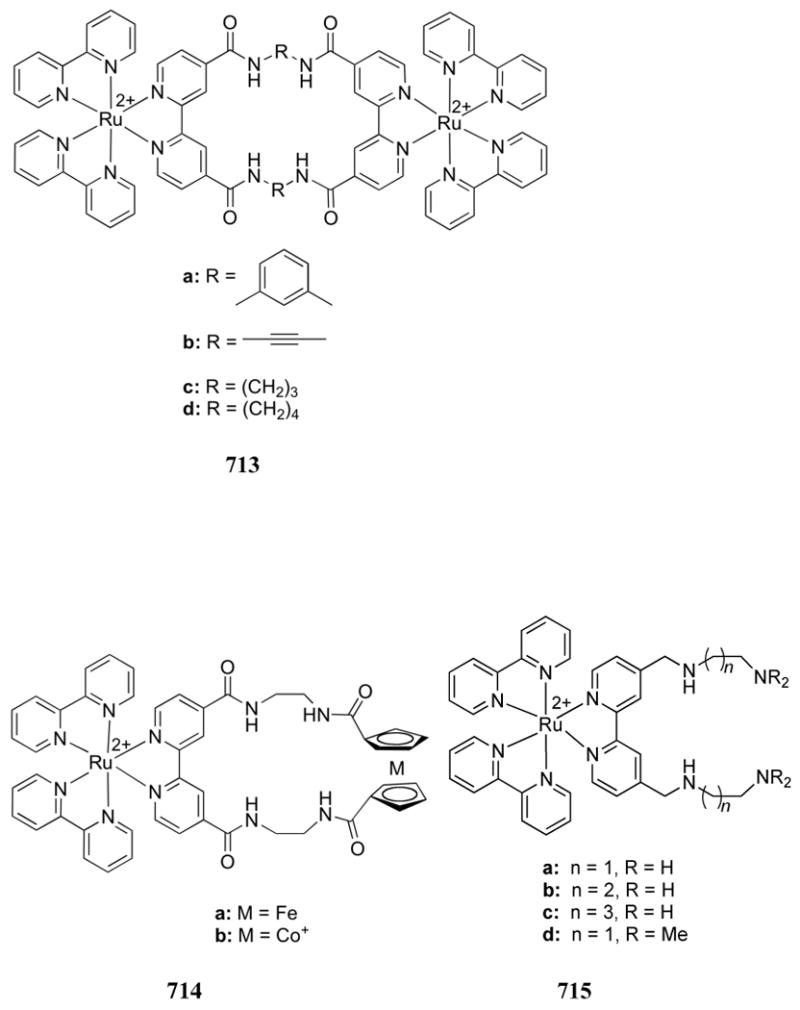
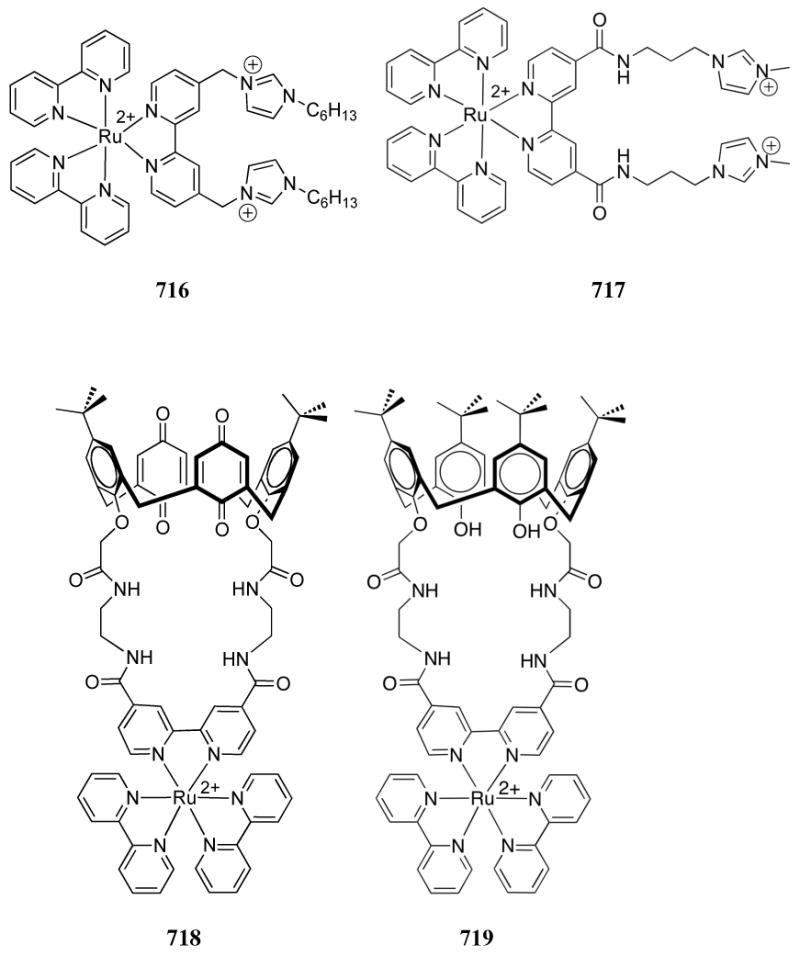
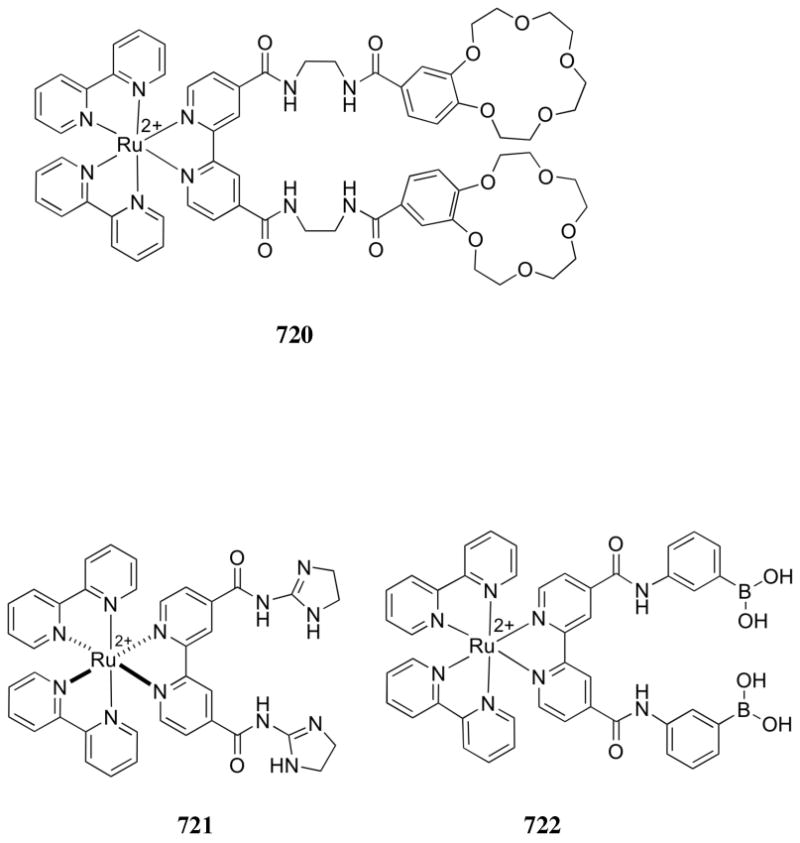
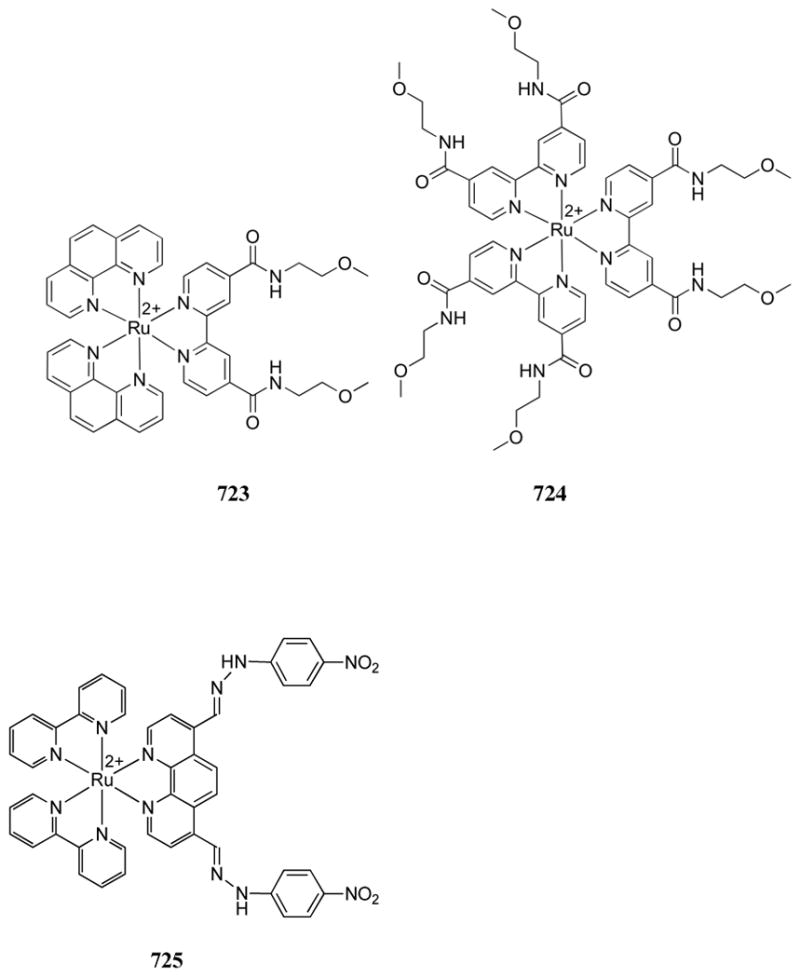
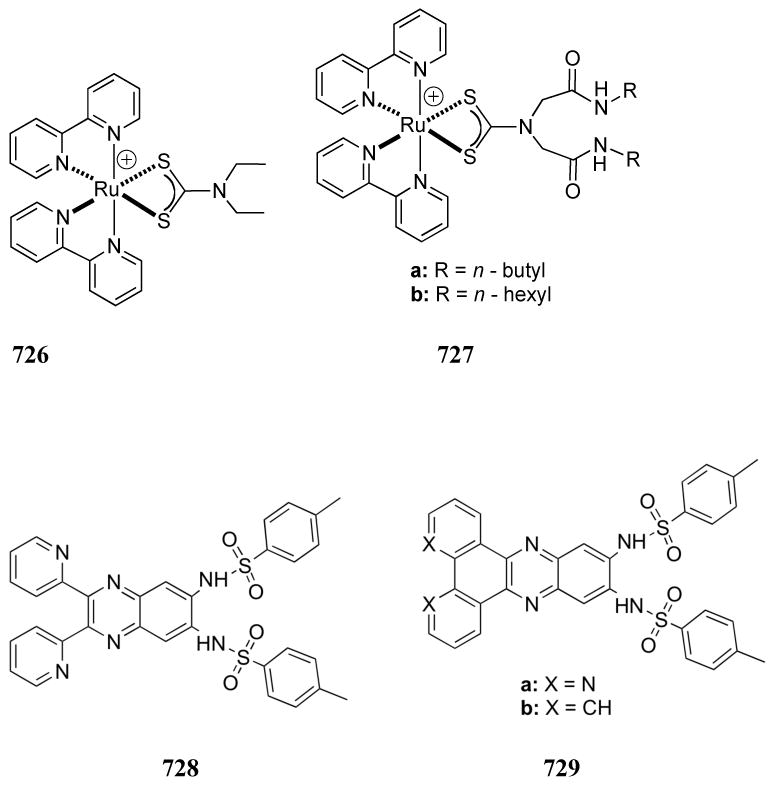
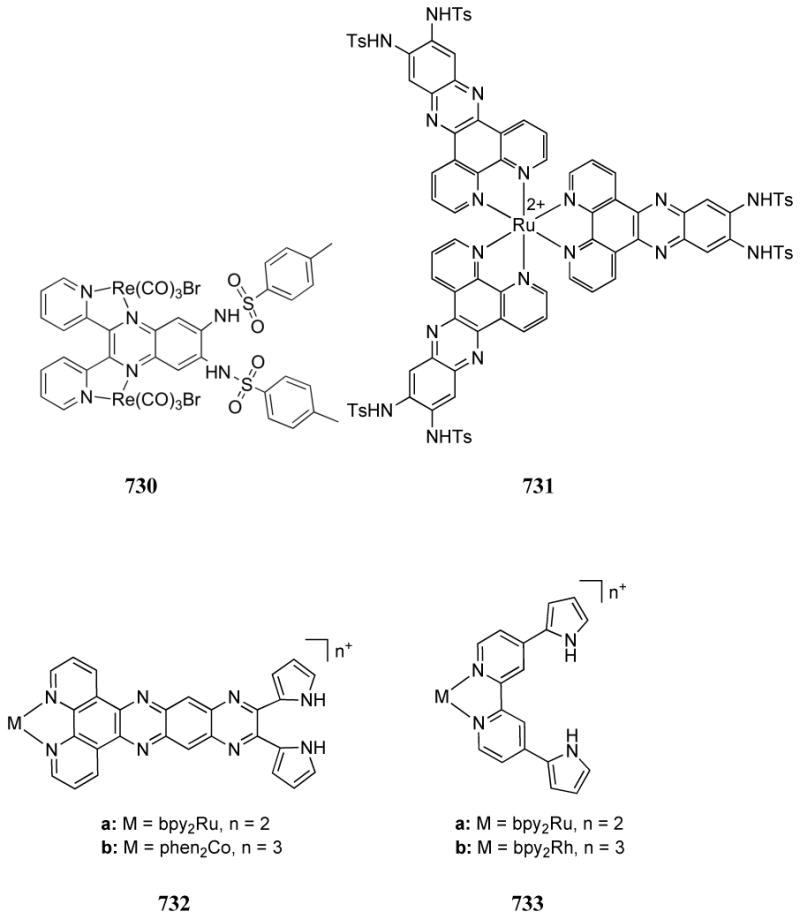
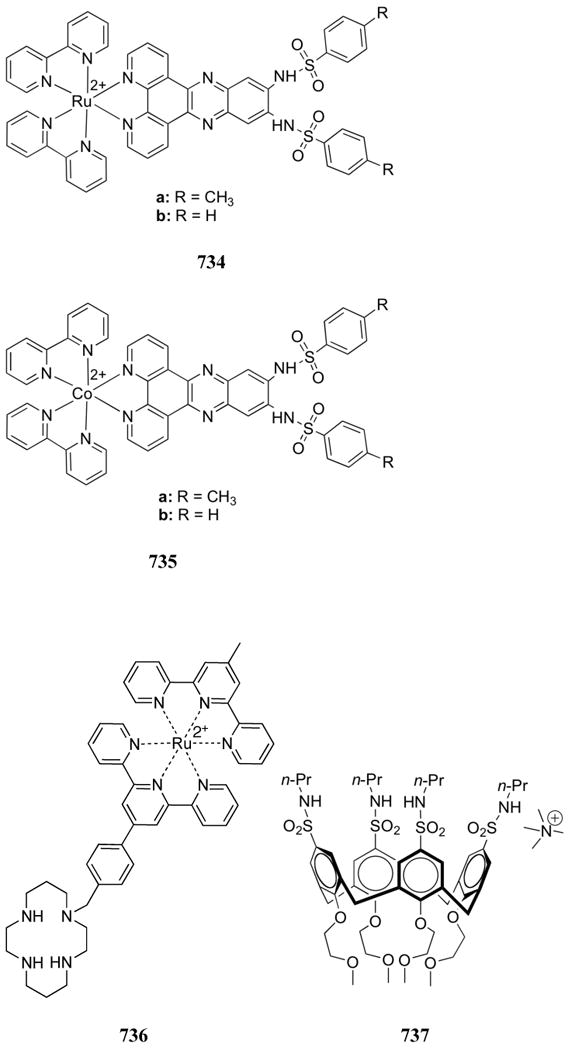
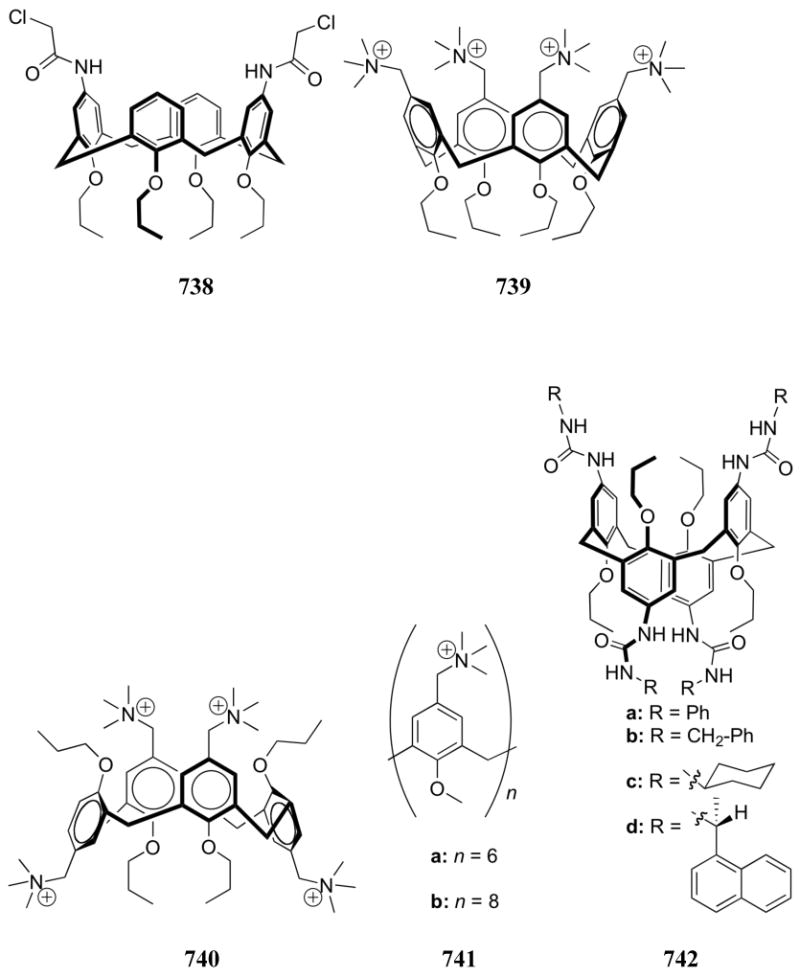
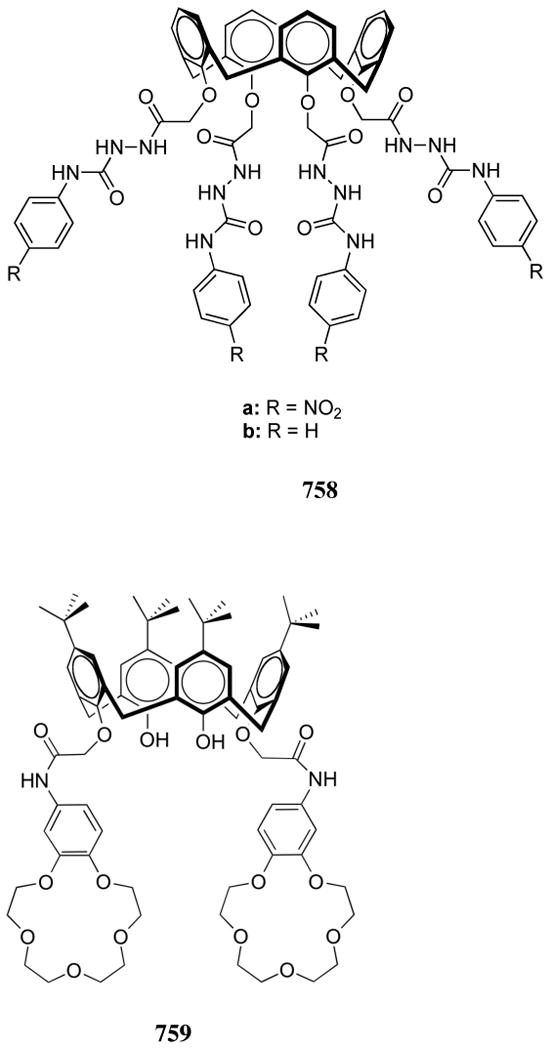
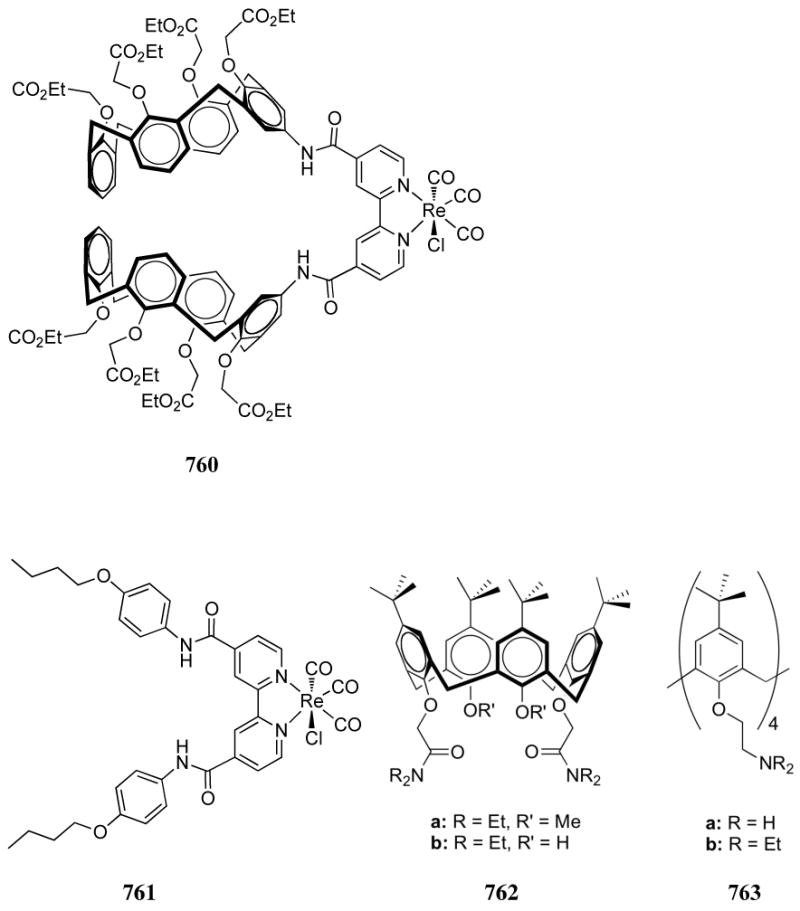
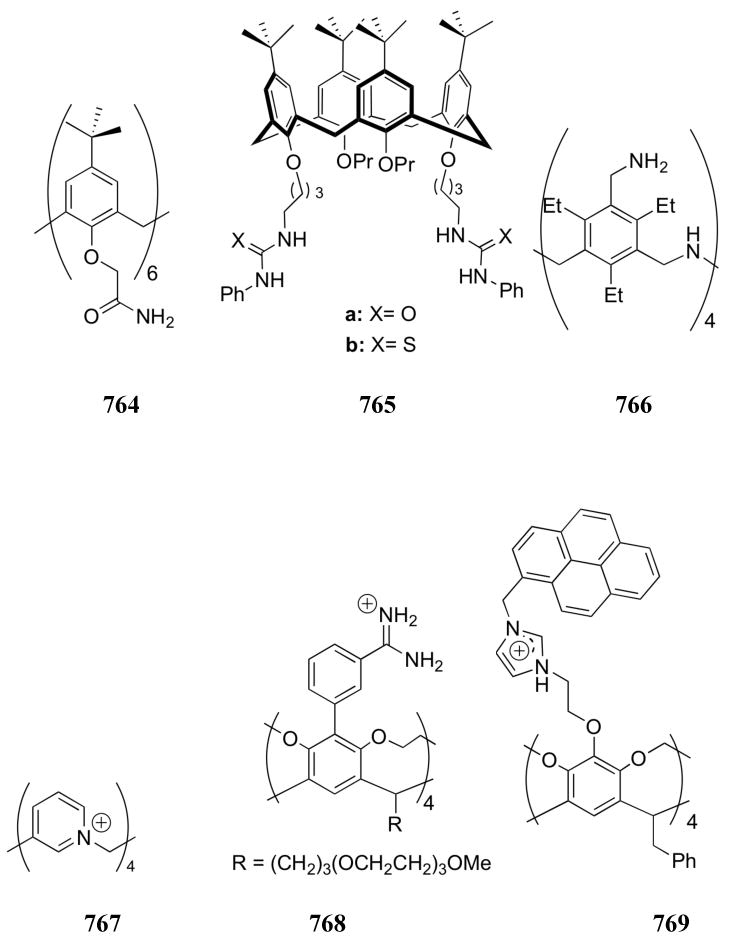
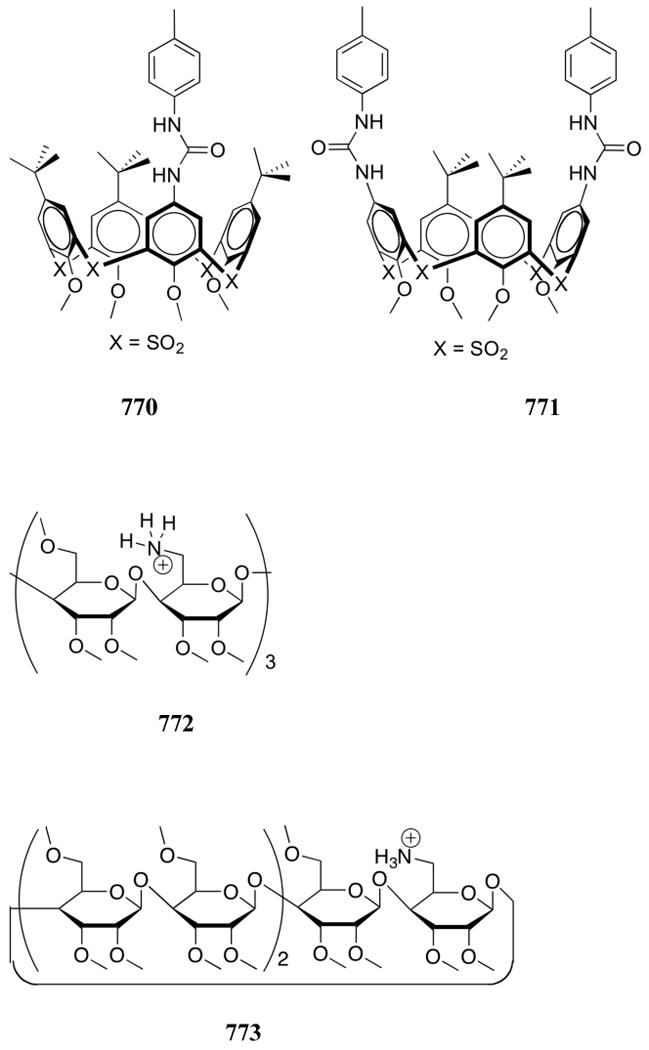
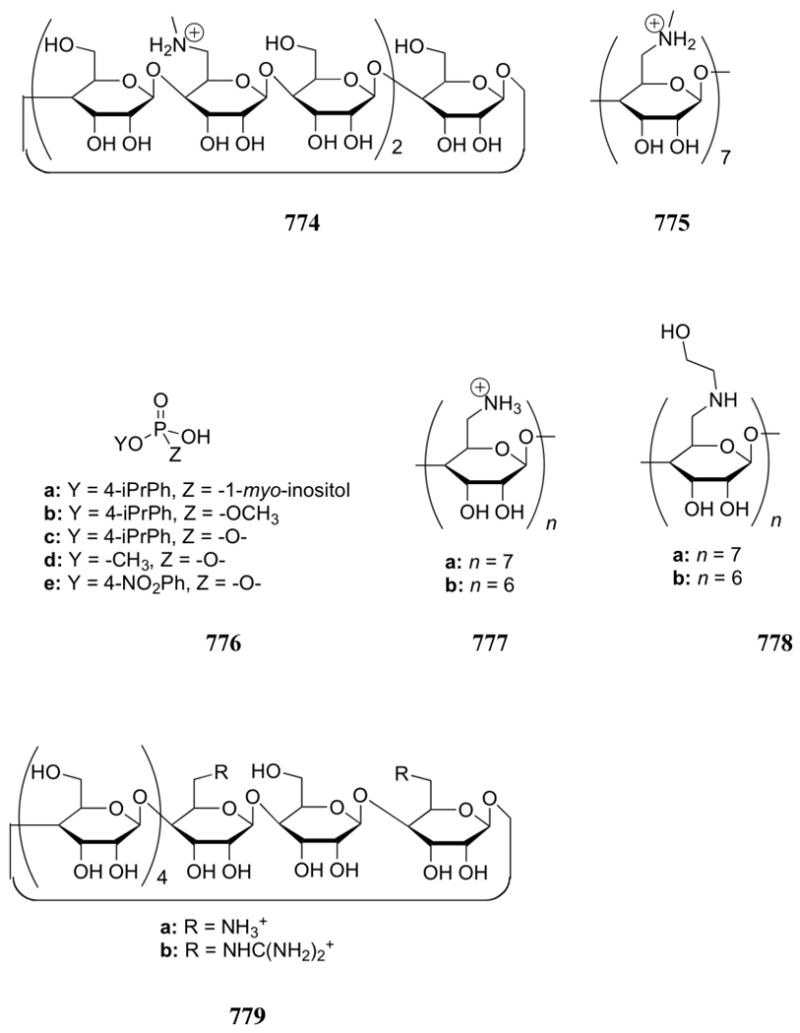
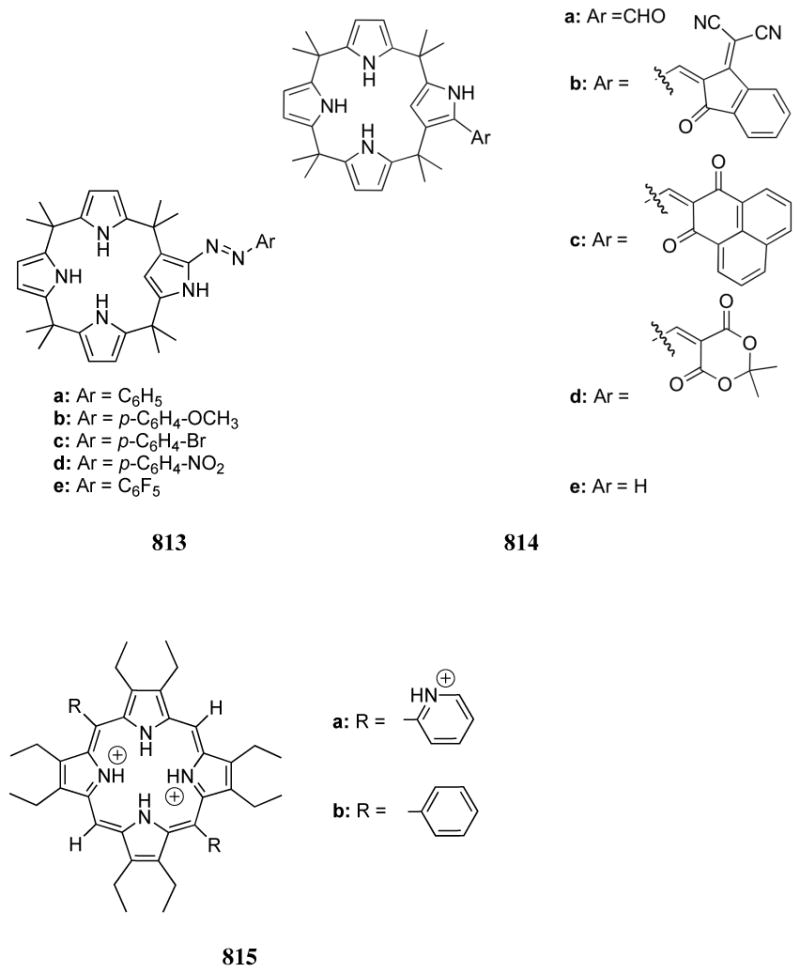

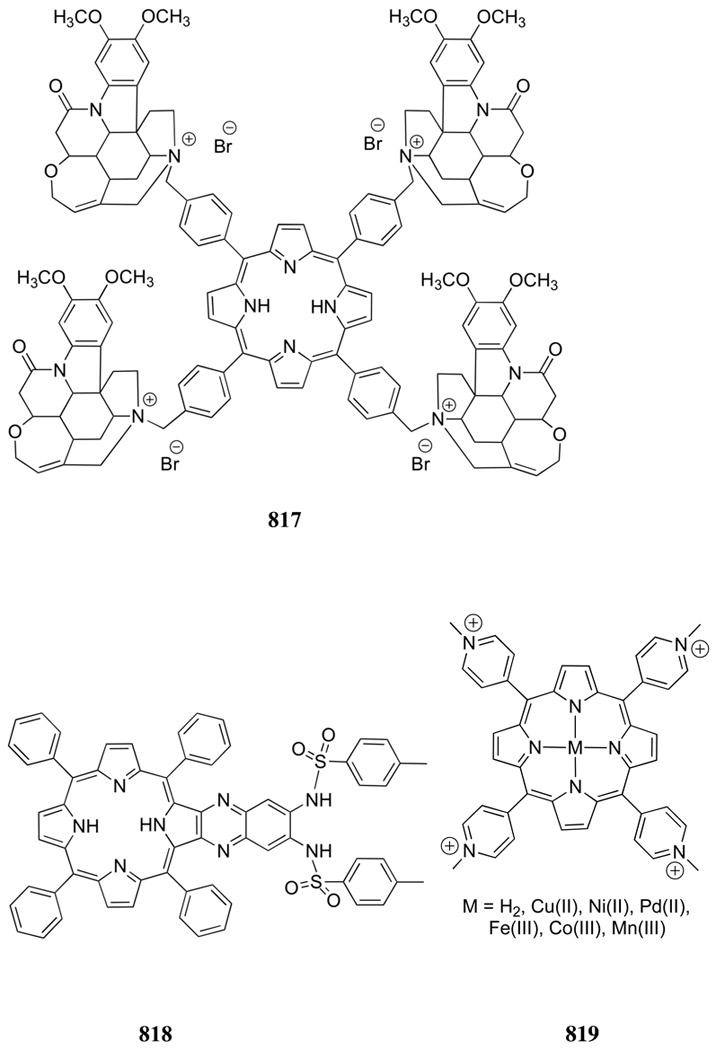
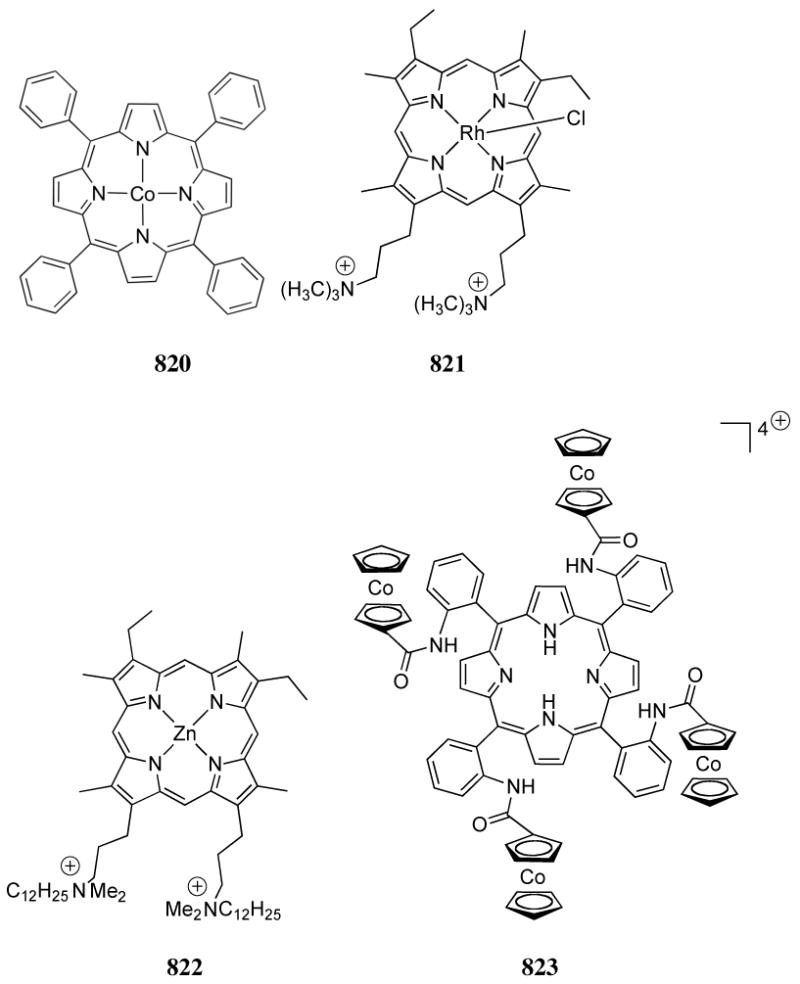


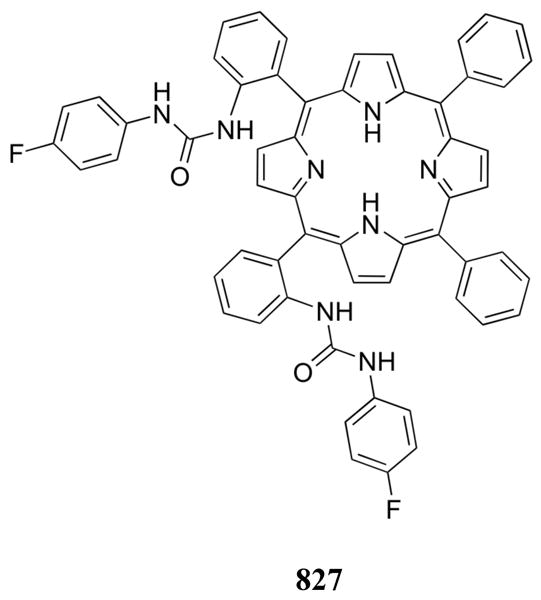

Acknowledgments
The authors gratefully acknowledge the contributions of Ms. Taylor Beaver, Professor Marco Bonizzoni, Dr. Chance Rainwater, Mr. Vladimir Roznyatovskiy, Dr. Natalie Barkey, and Dr. Sung Ok Kang. Professor Sessler thanks the National Institutes of Health (GM 58907) and the Robert A. Welch Foundation (F-1018). Support under the WCU (World Class University) program (R32-2010-10217-0) is also acknowledged. Professor Anslyn thanks the NIH, NSF, and Welch Foundation for support over his 22 year career.
Biographies

Dr. Hargrove received a B.S. in Chemistry and Spanish from Trinity University, San Antonio, Texas in 2004. She earned a Ph.D. in organic chemistry from the University of Texas at Austin in 2010 under the guidance of Dr. Eric Anslyn and Dr. Jonathan Sessler. Her dissertation work focused on the development of selective receptors for biologically relevant analytes through the combination of organic chemistry and molecular biology. Her studies also included an internship with Dr. Jean-Pierre Sauvage at Université Louis Pasteur, Strasbourg, France. Her honors include a National Science Foundation Integrated Graduate Education and Research Training (NSF-IGERT) fellowship. She is currently a TRDRP postdoctoral fellow at the California Institute of Technology with Dr. Peter Dervan.

Dr. Sonia Nieto was born in Oviedo, Spain, on September 13, 1980. She received her PhD in Inorganic Chemistry in 2006 at the University of Oviedo (Supervisors: Professor Julio Perez and Dr. Lucia Riera). In August 2006 she joined Eric V. Anslyn's group at the University of Texas at Austin enjoying a MEC postdoctoral fellowship, where she worked in the development of new protocols for the enantiomeric excess determination of amines. In April 2009 she moved to Zaragoza, Spain, and joined Esteban Urriolabeitia's group as a JAE-doc researcher at the Instituto de Ciencia de Materiales de Aragon (CSIC). Her current research is focused in the enantioselective synthesis of ortho-functionalized α-amino acids employing palladacycles.

Tianzhi Zhang is a Senior Research Chemist with the Henkel Corporation in Rocky Hill, CT. She received her B.S. degree in Physical Chemistry in 1996 from Jilin University, P. R. China, and her Ph.D. degree in Organic Chemistry in 2007 from the University of Texas at Austin under the supervision of Professor Eric V. Anslyn. Her doctoral studies focused on the design and synthesis of differential receptors for phosphorylated compounds. At Henkel, her research interests are in the controlled synthesis of functional polymeric materials.

Professor Jonathan L. Sessler received a B.S. degree in chemistry in 1977 from the University of California, Berkeley. He obtained a Ph.D. from Stanford University in 1982 (supervisor: Professor James P. Collman). He was an NSF-CNRS and NSF-NATO Postdoctoral Fellow with Professor Jean-Marie Lehn at L'Université Louis Pasteur de Strasbourg, France. He was then a JSPS Visiting Scientist in Professor Iwao Tabushi's group in Kyoto, Japan. In 1984 he accepted a position as an Assistant Professor of Chemistry at the University of Texas at Austin, where he is currently the Roland K. Pettit Centennial Chair. Dr Sessler is a co-founder of two companies, Pharmacyclics, Inc. and Anionics, Inc.

Eric V. Anslyn received his Ph.D. in Chemistry from the California Institute of Technology in 1987 under the direction of Robert H. Grubbs. After two years at Columbia University as a National Science Foundation Post-Doctoral Fellow with Ronald Breslow, he joined the chemistry faculty at the University of Texas at Austin in 1989. He was named a Sloan Scholar, Dreyfus Teacher Scholar, Searle Scholar, and Presidential Young Investigator and then promoted to Associate Professor in 1994. In 1997 he was named Full Professor, and in 2004 he was awarded the Norman Hackerman Professorship in Chemistry and Biochemistry. He is also a 2005 Cope Scholar Awardee. In 2006 Professor Anslyn was elected as a fellow to the American Association for the Advancement of Science and in 2007 was inducted as an Honorary Professor at East China University of Science and Technology. Currently, he sits as an Associate Editor for the Journal of the American Chemical Society and serves on the editorial board of Supramolecular Chemistry. His research encompasses physical organic and bioorganic chemistry and generally explores the use of synthetic receptors for sensing and catalysis applications.
References
- 1.Adams RLP, Knowler JT, Leader DP. The Biochemistry of the Nucleic Acids. 10th. Chapman and Hall; New York: 1986. [Google Scholar]
- 2.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 3.Mason CF. Biology of Freshwater Pollution. Longman; New York: 1991. [Google Scholar]
- 4.Tiessen H, editor. Phosphorus in the Global Environment : Transfers, Cycles, and Management. Wiley; New York: 1995. [Google Scholar]
- 5.Charra B, Calemard E, Ruffet M, Chazot C, Terrat JC, Vanel T, Laurent G. Kidney Int. 1992;41:1286. doi: 10.1038/ki.1992.191. [DOI] [PubMed] [Google Scholar]
- 6.Gutzwiller JP, Schneditz D, Huber AR, Schindler C, Gutzwiller F, Zehnder CE. Nephrol, Dial, Transplant. 2002;17:1037. doi: 10.1093/ndt/17.6.1037. [DOI] [PubMed] [Google Scholar]
- 7.Margolis HC, Duckworth JH, Moreno EC. J Dent Res. 1988;67:1468. doi: 10.1177/00220345880670120601. [DOI] [PubMed] [Google Scholar]
- 8.Delmez JA, Slatopolsky E. Am J Kidney Dis. 1992;19:303. doi: 10.1016/s0272-6386(12)80446-x. [DOI] [PubMed] [Google Scholar]
- 9.Hruska KA, Teitelbaum SL. N Engl J Med. 1995;333:166. doi: 10.1056/NEJM199507203330307. [DOI] [PubMed] [Google Scholar]
- 10.Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Am J Kidney Dis. 1998;31:607. doi: 10.1053/ajkd.1998.v31.pm9531176. [DOI] [PubMed] [Google Scholar]
- 11.Block GA, Port FK. Am J Kidney Dis. 2000;35:1226. doi: 10.1016/s0272-6386(00)70064-3. [DOI] [PubMed] [Google Scholar]
- 12.Robinson C. Dent Dig. 2000;1:1. [Google Scholar]
- 13.Minutolo R, Bellizzi V, Cioffi M, Iodice C, Giannattasio P, Andreucci M, Terracciano V, Di Iorio BR, Conte G, De Nicola L. J Am Soc Nephrol. 2002;13:1046. doi: 10.1681/ASN.V1341046. [DOI] [PubMed] [Google Scholar]
- 14.Timms AE, Zhang Y, Russell RGG, Brown MA. Rheumatology. 2002;41:725. doi: 10.1093/rheumatology/41.7.725. [DOI] [PubMed] [Google Scholar]
- 15.Albaaj F, Hutchison A. Drugs. 2003;63:577. doi: 10.2165/00003495-200363060-00005. [DOI] [PubMed] [Google Scholar]
- 16.Block G, Port FK. Sem Dialysis. 2003;16:140. doi: 10.1046/j.1525-139x.2003.160301.x. [DOI] [PubMed] [Google Scholar]
- 17.Young EW, Albert JM, Satayathum S, Goodkin DA, Pisoni RL, Akiba T, Akizawa T, Kurokawa K, Bommer J, Piera L, Port FK. Kidney Int. 2005;67:1179. doi: 10.1111/j.1523-1755.2005.00185.x. [DOI] [PubMed] [Google Scholar]
- 18.Dhingra R, Sullivan LM, Fox CS, Wang TJ, D'Agostino RB, Gaziano JM, Vasan RS. Arch Intern Med. 2007;167:879. doi: 10.1001/archinte.167.9.879. [DOI] [PubMed] [Google Scholar]
- 19.Taylor AE, Miller CW. J Biol Chem. 1914;18:215. [Google Scholar]
- 20.Fiske CH, Subbarow Y. J Biol Chem. 1925;66:375. [Google Scholar]
- 21.Martin JB, Doty DM. Anal Chem. 1949;21:965. [Google Scholar]
- 22.Gee A, Deitz VR. Anal Chem. 1953;9:1320. [Google Scholar]
- 23.Taussky HH, Shorr E, Kurzmann G. J Biol Chem. 1953;202:675. [PubMed] [Google Scholar]
- 24.Duff EJ, Stuart JL. Analyst. 1971;96:802. [Google Scholar]
- 25.Baadenhuijsen H, Seuren-Jacobs HEH, Jansen AP. Clin Chem. 1977;23:1275. [PubMed] [Google Scholar]
- 26.Matsuo T, Shida J, Kurihara W. Anal Chim Acta. 1977;91:385. [Google Scholar]
- 27.Shida J, Matsuo T. Bull Chem Soc Jpn. 1980;53:2868. [Google Scholar]
- 28.Mirzoyan FV, Tarayan VM, Petrosyan AA. Ukr Khim Zh (Russ Ed) 1980;46:995. [Google Scholar]
- 29.Motomizu S, Wakimoto T, Toei K. Anal Chim Acta. 1982;138:329. [Google Scholar]
- 30.Garber CC, Miller RC. Clin Chem. 1983;29:184. [PubMed] [Google Scholar]
- 31.Muñoz MA, Balón M, Fernandez C. Clin Chem. 1983;29:372. [PubMed] [Google Scholar]
- 32.Munoz A, Mas Torres F, Estela JM, Cerda V. Anal Chim Acta. 1997;350:21. [Google Scholar]
- 33.Katewa SD, Katyare SS. Anal Biochem. 2003;323:180. doi: 10.1016/j.ab.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 34.Worsfold PJ, Gimbert LJ, Mankasingh U, Omaka ON, Hanrahan G, Gardolinski PCFC, Haygarth PM, Turner BL, Keith-Roach MJ, McKelvie ID. Talanta. 2005;66:273. doi: 10.1016/j.talanta.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Vogel GL, Chow LC, Brown WE. Caries Res. 1983;17:23. doi: 10.1159/000260645. [DOI] [PubMed] [Google Scholar]
- 36.Diacu E, Ioannou PC, Polydorou CK, Efstathiou CF. Analyst. 1995;120:2613. doi: 10.1039/an9952002115. [DOI] [PubMed] [Google Scholar]
- 37.Carey CM, Vogel GL. J Res Natl Inst Stand Technol. 2000;105:267. doi: 10.6028/jres.105.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engblom SO. Biosens Bioelectron. 1998;13:981. doi: 10.1016/s0956-5663(98)00001-3. [DOI] [PubMed] [Google Scholar]
- 39.Estela JM, Cerda V. Talanta. 2005;66:307. doi: 10.1016/j.talanta.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 40.Hanrahan G, Salmassi TM, Khachikian CS, Foster KL. Talanta. 2005;66:435. doi: 10.1016/j.talanta.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 41.Pawar GB, Todai NK, Jaffar MB. Clin Chem. 1978;24:1847. [PubMed] [Google Scholar]
- 42.Kallner A. Clin Chim Acta. 1975;59:35. doi: 10.1016/0009-8981(75)90215-6. [DOI] [PubMed] [Google Scholar]
- 43.Motomizu S, Wakimoto T, Toei K. Talanta. 1983;30:333. doi: 10.1016/0039-9140(83)80076-9. [DOI] [PubMed] [Google Scholar]
- 44.Shkinev VM, Spivakov BY, Vorob'eva GA, Zolotov YA. Anal Chim Acta. 1985;167:145. [Google Scholar]
- 45.Guilbault GG, Nanjo M. Anal Chim Acta. 1975;78:69. doi: 10.1016/S0003-2670(01)84753-X. [DOI] [PubMed] [Google Scholar]
- 46.Gajovic N, Habermuller K, Warsinke A, Schuhmann W, Scheller FW. Electroanalysis. 1999;11:1377. [Google Scholar]
- 47.Kubo I, Inagawa M, Sugawara T, Arikawa Y, Karube I. Anal Lett. 1991;24:1711. [Google Scholar]
- 48.Amine A, Palleschi G. Anal Lett. 2004;37:1. [Google Scholar]
- 49.Ghosh A, Ronner P, Cheong E, Khalid P, Matschinsky FM. J Biol Chem. 1991;266:22887. [PubMed] [Google Scholar]
- 50.Luque de Castro MD, Quiles R, Fernández-Romero JM, Fernández E. Clin Chem. 1995;41:99. [PubMed] [Google Scholar]
- 51.Schulz DW, Passonneau JV, Lowry OH. Anal Biochem. 1967;19:300. doi: 10.1016/0003-2697(67)90166-2. [DOI] [PubMed] [Google Scholar]
- 52.Hofmeister F. Arch Exp Pathol Pharmakol. 1888;24:247. [Google Scholar]
- 53.Marcus Y. Biophys Chem. 1994;51:111. [Google Scholar]
- 54.Katayev EA, Ustynyuk YA, Sessler JL. Coord Chem Rev. 2006;250:3004. [Google Scholar]
- 55.Flatt LS, Lynch V, Anslyn EV. Tetrahedron Lett. 1992;33:2785. [Google Scholar]
- 56.Chu F, Flatt LS, Anslyn EV. J Am Chem Soc. 1994;116:4194. [Google Scholar]
- 57.Gale PA, Hiscock JR, Moore SJ, Caltagirone C, Hursthouse MB, Light ME. Chem--Asian J. 2010;5:555. doi: 10.1002/asia.200900230. [DOI] [PubMed] [Google Scholar]
- 58.Antonisse MMG, Reinhoudt DN. Chem Commun. 1998:443. [Google Scholar]
- 59.Snowden TS, Anslyn EV. Curr Opin Chem Biol. 1999;3:740. doi: 10.1016/s1367-5931(99)00034-4. [DOI] [PubMed] [Google Scholar]
- 60.Beer PD, Gale PA. Angew Chem, Int Ed. 2001;40:486. [PubMed] [Google Scholar]
- 61.Aoki S, Kimura E. Rev Mol Biotechnol. 2002;90:129. doi: 10.1016/s1389-0352(01)00070-8. [DOI] [PubMed] [Google Scholar]
- 62.Hartley JH, James TD, Ward CJ. J Chem Soc, Perkin Trans 1. 2000:3155. [Google Scholar]
- 63.Sessler JL, Gale PA, Cho WS. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]
- 64.Stibor I, editor. Anion Sensing. Vol. 255 Springer-Verlag; New York, NY: 2005. [Google Scholar]
- 65.Bowman-James K. Acc Chem Res. 2005;38:671. doi: 10.1021/ar040071t. [DOI] [PubMed] [Google Scholar]
- 66.Amendola V, Esteban-Gómez D, Fabbrizzi L, Licchelli M. Acc Chem Res. 2006;39:343. doi: 10.1021/ar050195l. [DOI] [PubMed] [Google Scholar]
- 67.Gale PA. Acc Chem Res. 2006;39:465. doi: 10.1021/ar040237q. [DOI] [PubMed] [Google Scholar]
- 68.Yoon J, Kim SK, Singh NJ, Kim KS. Chem Soc Rev. 2006;35:355. doi: 10.1039/b513733k. [DOI] [PubMed] [Google Scholar]
- 69.Amendola V, Bonizzoni M, Esteban-Gómez D, Fabbrizzi L, Licchelli M, Sancenón F, Taglietti A. Coord Chem Rev. 2006;250:1451. [Google Scholar]
- 70.Davis AP. Coord Chem Rev. 2006;250:2939. [Google Scholar]
- 71.Gale PA, Quesada R. Coord Chem Rev. 2006;250:3219. [Google Scholar]
- 72.García-España E, Díaz P, Llinares JM, Bianchi A. Coord Chem Rev. 2006;250:2952. [Google Scholar]
- 73.Gunnlaugsson T, Glynn M, Tocci GM, Kruger PE, Pfeffer FM. Coord Chem Rev. 2006;250:3094. [Google Scholar]
- 74.Kang SO, Hossain MA, Bowman-James K. Coord Chem Rev. 2006;250:3038. [Google Scholar]
- 75.Rice CR. Coord Chem Rev. 2006;250:3190. [Google Scholar]
- 76.Schmidtchen FP. Coord Chem Rev. 2006;250:2918. [Google Scholar]
- 77.Wichmann K, Antonioli B, Soehnel T, Wenzel M, Gloe K, Gloe K, Price JR, Lindoy LF, Blake AJ, Schroeder M. Coord Chem Rev. 2006;250:2987. [Google Scholar]
- 78.Gamez P, Mooibroek TJ, Teat SJ, Reedijk J. Acc Chem Res. 2007;40:435. doi: 10.1021/ar7000099. [DOI] [PubMed] [Google Scholar]
- 79.Berlicki L, Rudzinska E, Mlynarz P, Kafarski P. Curr Org Chem. 2006;10:2285. [Google Scholar]
- 80.Hirsch AKH, Fischer FR, Diederich F. Angew Chem, Int Ed. 2007;46:338. doi: 10.1002/anie.200603420. [DOI] [PubMed] [Google Scholar]
- 81.Tamaru Si, Hamachi I. Struct Bond. 2008;129:95. [Google Scholar]
- 82.Martell AE, Motekaitis RJ, Lu Q, Nation DA. Polyhedron. 1999;18:3203. [Google Scholar]
- 83.Hynes MJ. J Chem Soc, Dalton Trans. 1993:311. [Google Scholar]
- 84.Wiskur SL, Ait-Haddou H, Lavigne JJ, Anslyn EV. Acc Chem Res. 2001;34:963. doi: 10.1021/ar9600796. [DOI] [PubMed] [Google Scholar]
- 85.Martínez-Máñez R, Sancenón F. Chem Rev. 2003;103:4419. doi: 10.1021/cr010421e. [DOI] [PubMed] [Google Scholar]
- 86.Nguyen BT, Anslyn EV. Coord Chem Rev. 2006;250:3118. [Google Scholar]
- 87.de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem Rev. 1997;97:1515. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 88.Czarnik AW. ACS Symp Ser. 1993;538:104. [Google Scholar]
- 89.Bissell RA, Prasanna de Silva A, Gunaratne HQN, Lynch PLM, Maguire GEM, McCoy CP, Sandanayake KRAS. Top Curr Chem. 1993;168:223. [Google Scholar]
- 90.de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Adv Supramol Chem. 1997;4:1. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 91.Gunnlaugsson T, Ali HDP, Glynn M, Kruger PE, Hussey GM, Pfeffer FM, Santos CMG, Tierney J. J Fluoresc. 2005;15:287. doi: 10.1007/s10895-005-2627-y. [DOI] [PubMed] [Google Scholar]
- 92.Sauvage JP, editor. Transition Metals in Supramolecular Chemistry. Wiley; New York: 1999. [Google Scholar]
- 93.Beer PD. Chem Commun. 1996:689. [Google Scholar]
- 94.Beer PD, Graydon AR, Sutton LR. Polyhedron. 1996;15:2457. [Google Scholar]
- 95.Nishizawa S, Kaneda H, Uchida T, Teramae N. J Chem Soc, Perkin Trans 2. 1998:2325. [Google Scholar]
- 96.Shionoya M, Furuta H, Lynch V, Harriman A, Sessler JL. J Am Chem Soc. 1992;114:5714. [Google Scholar]
- 97.Choi K, Hamilton AD. Angew Chem, Int Ed. 2001;40:3912. doi: 10.1002/1521-3773(20011015)40:20<3912::AID-ANIE3912>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 98.Zhang X, Guo L, Wu FY, Jiang YB. Org Lett. 2003;5:2667. doi: 10.1021/ol034846u. [DOI] [PubMed] [Google Scholar]
- 99.Beer PD, Bayly SR. Top Curr Chem. 2005;255:125. [Google Scholar]
- 100.Bakker E, Buhlmann P, Pretsch E. Electroanalysis. 1999;11:915. [Google Scholar]
- 101.Bakker E, Meyerhoff ME. Anal Chim Acta. 2000;416:121. [Google Scholar]
- 102.Bakker E, Pretsch E. TrAC, Trends Anal Chem. 2001;20:11. [Google Scholar]
- 103.Sokalski T, Ceresa A, Zwickl T, Pretsch E. J Am Chem Soc. 1997;119:11347. [Google Scholar]
- 104.Mathison S, Bakker E. Anal Chem. 1998;70:303. doi: 10.1021/ac990387s. [DOI] [PubMed] [Google Scholar]
- 105.Buehlmann P, Pretsch E, Bakker E. Chem Rev. 1998;98:1593. doi: 10.1021/cr970113+. [DOI] [PubMed] [Google Scholar]
- 106.Grabner EW, Vermes I, König KH. J Electroanal Chem Interfacial Electrochem. 1986;214:135. [Google Scholar]
- 107.Goediker W, Cammann K. Anal Lett. 1989;22:1237. [Google Scholar]
- 108.Glazier SA, Arnold MA. Anal Lett. 1989;22:1075. [Google Scholar]
- 109.Liu J, Masuda Y, Sekido E, Wakida S, Hiiro K. Anal Chim Acta. 1989;224:145. [Google Scholar]
- 110.Liu J, Masuda Y, Sekido E. J Electroanal Chem Interfacial Electrochem. 1990;291:67. [Google Scholar]
- 111.Tsagkatakis I, Chaniotakis N, Altmann R, Jurkschat K, Willem R, Martins JC, Qin Y, Bakker E. Helv Chim Acta. 2001;84:1952. [Google Scholar]
- 112.Kubena BD, Luecke H, Rosenberg H, Quiocho FA. J Biol Chem. 1986;261:7995. [PubMed] [Google Scholar]
- 113.Luecke H, Quiocho FA. Nature. 1990;347:402. doi: 10.1038/347402a0. [DOI] [PubMed] [Google Scholar]
- 114.Quiocho FA. Kidney Int. 1996;49:943. doi: 10.1038/ki.1996.132. [DOI] [PubMed] [Google Scholar]
- 115.Wang Z, Luecke H, Yao N, Quiocho FA. Nat Struct Biol. 1997;4:519. doi: 10.1038/nsb0797-519. [DOI] [PubMed] [Google Scholar]
- 116.Yao N, Ledvina PS, Choudhary A, Quiocho FA. Biochemistry. 1996;35:2079. doi: 10.1021/bi952686r. [DOI] [PubMed] [Google Scholar]
- 117.Ledvina PS, Tsai AL, Wang Z, Koehl E, Quiocho FA. Protein Sci. 1998;7:2550. doi: 10.1002/pro.5560071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perrault DM, Chen X, Anslyn EV. Tetrahedron. 1995;51:353. [Google Scholar]
- 119.Nakai C, Glinsmann W. Biochemistry. 1977;16:5636. doi: 10.1021/bi00644a039. [DOI] [PubMed] [Google Scholar]
- 120.Bunce S, Kong ESW. Biophys Chem. 1978;8:357. doi: 10.1016/0301-4622(78)80017-9. [DOI] [PubMed] [Google Scholar]
- 121.Lomozik L, Gasowska A. J Inorg Biochem. 1998;72:37. [Google Scholar]
- 122.Gasowska A. J Inorg Biochem. 2003;96:346. doi: 10.1016/s0162-0134(03)00150-8. [DOI] [PubMed] [Google Scholar]
- 123.Lomozik L, Jastrzab R, Gasowska A. Polyhedron. 2000;19:1145. [Google Scholar]
- 124.Yip LC, Balis ME. Biochemistry. 1980;19:1849. doi: 10.1021/bi00550a019. [DOI] [PubMed] [Google Scholar]
- 125.Lahti R, Hannukainen R, Lonnberg H. Biochem J. 1989;259:55. doi: 10.1042/bj2590055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Labadi I, Jenei E, Lahti R, Lonnberg H. Acta Chem Scand. 1991;45:1055. doi: 10.3891/acta.chem.scand.45-1055. [DOI] [PubMed] [Google Scholar]
- 127.Labadi I, Sillanpaa R, Lonnberg H. J Chem Soc, Dalton Trans. 1992:765. [Google Scholar]
- 128.Alves da Silva J, Felcman J, Merce ALR, Mangrich AS, Lopes RSC, Lopes CC. Inorg Chim Acta. 2003;356:155. [Google Scholar]
- 129.Kimura E, Kodama M, Yatsunami T. J Am Chem Soc. 1982;104:3182. [Google Scholar]
- 130.Dietrich B, Hosseini MW, Lehn JM, Sessions RB. J Am Chem Soc. 1981;103:1282. [Google Scholar]
- 131.Kimura E, Watanabe A, Nihira H. Chem Pharm Bull. 1983;31:3264. doi: 10.1248/cpb.31.3264. [DOI] [PubMed] [Google Scholar]
- 132.Kimura E, Fujioka H, Yatsunami A, Nihira H, Kodama M. Chem Pharm Bull. 1985;33:655. doi: 10.1248/cpb.33.655. [DOI] [PubMed] [Google Scholar]
- 133.Umezawa Y, Kataoka M, Takami W, Kimura E, Koike T, Nada H. Anal Chem. 1988;60:2392. doi: 10.1021/ac00172a014. [DOI] [PubMed] [Google Scholar]
- 134.Hosseini MW, Lehn JM, Mertes MP. Helv Chim Acta. 1983;66:2454. [Google Scholar]
- 135.Hosseini MW, Lehn JM. Helv Chim Acta. 1987;70:1312. [Google Scholar]
- 136.Hosseini MW, Lehn JM, Mertes MP. Helv Chim Acta. 1985;68:818. [Google Scholar]
- 137.Cordier D, Hosseini MW. New J Chem. 1990;14:611. [Google Scholar]
- 138.Domenech A, García-España E, Ramirez JA, Celda B, Martinez MC, Monleon D, Tejero R, Bencini A, Bianchi A. J Chem Soc, Perkin Trans 2. 1999:23. [Google Scholar]
- 139.Wilson HR, Williams RJP. J Chem Soc, Faraday Trans 1. 1987;83:1885. [Google Scholar]
- 140.Dietrich B, Hosseini MW, Lehn JM, Sessions RB. Helv Chim Acta. 1983;66:1262. [Google Scholar]
- 141.Kimura E. Top Curr Chem. 1985;128:113. [Google Scholar]
- 142.Bianchi A, Micheloni M, Paoletti P. Coord Chem Rev. 1991;110:17. [Google Scholar]
- 143.Bartolini M, Bianchi A, Micheloni M, Paoletti P. J Chem Soc, Perkin Trans 2. 1982:1345. [Google Scholar]
- 144.Bencini A, Bianchi A, García-España E, Scott EC, Morales L, Wang B, Deffo T, Takusagawa F, Mertes MP, Mertes KB, Paoletti P. Bioorg Chem. 1992;20:8. [Google Scholar]
- 145.Bencini A, Bianchi A, García-España E, Fusi V, Micheloni M, Paoletti P, Ramirez JA, Rodriguez A, Valtancoli B. J Chem Soc, Perkin Trans 2. 1992:1059. [Google Scholar]
- 146.Andres A, Bazzicalupi C, Bencini A, Bianchi A, Fusi V, García-España E, Giorgi C, Nardi N, Paoletti P, Ramirez JA, Valtancoli B. J Chem Soc, Perkin Trans 2. 1994:2367. [Google Scholar]
- 147.Bazzicalupi C, Bencini A, Bianchi A, Cecchi M, Escuder B, Fusi V, García-España E, Giorgi C, Luis SV, Maccagni G, Marcelino V, Paoletti P, Valtancoli B. J Am Chem Soc. 1999;121:6807. [Google Scholar]
- 148.Andres A, Arago J, Bencini A, Bianchi A, Domenech A, Fusi V, García-España E, Paoletti P, Ramirez JA. Inorg Chem. 1993;32:3418. [Google Scholar]
- 149.Bencini A, Bianchi A, Giorgi C, Paoletti P, Valtancoli B, Fusi V, García-España E, Llinares JM, Ramirez JA. Inorg Chem. 1996;35:1114. doi: 10.1021/ic9506926. [DOI] [PubMed] [Google Scholar]
- 150.Bazzicalupi C, Bencini A, Bianchi A, Fusi V, Giorgi C, Paoletti P, Stefani A, Valtancoli B. J Chem Soc, Perkin Trans 2. 1995:275. [Google Scholar]
- 151.Bazzicalupi C, Bencini A, Bianchi A, Fusi V, Giorgi C, Granchi A, Paoletti P, Valtancoli B. J Chem Soc, Perkin Trans 2. 1997:775. [Google Scholar]
- 152.Bencini A, Bianchi A, Burguete MI, Domenech A, García-España E, Luis SV, Nino MA, Ramirez JA. J Chem Soc, Perkin Trans 2. 1991:1445. [Google Scholar]
- 153.Bianchi A, Micheloni M, Paoletti P. Inorg Chim Acta. 1988;151:269. [Google Scholar]
- 154.Schmidtchen FP. Angew Chem. 1977;89:751. [Google Scholar]
- 155.Schmidtchen FP. Angew Chem, Int Ed. 1977;16:720. [Google Scholar]
- 156.Schmidtchen FP. Chem Ber. 1981;114:597. [Google Scholar]
- 157.Schmidtchen FP. Top Curr Chem. 1986;132:101. [Google Scholar]
- 158.Tabushi I, Imuta J, Seko N, Kobuke Y. J Am Chem Soc. 1978;100:6287. [Google Scholar]
- 159.Tabushi I, Kobuke Y, Imuta J. J Am Chem Soc. 1980;102:1744. [Google Scholar]
- 160.Tabushi I, Kobuke Y, Imuta J. J Am Chem Soc. 1981;103:6152. [Google Scholar]
- 161.Li T, Diederich F. J Org Chem. 1992;57:3449. [Google Scholar]
- 162.Li T, Krasne SJ, Persson B, Kaback HR, Diederich F. J Org Chem. 1993;58:380. [Google Scholar]
- 163.Menger FM, Catlin KK. Angew Chem. 1995;107:2330. [Google Scholar]
- 164.Menger FM, Catlin KK. Angew Chem, Int Ed. 1995;34:2147. [Google Scholar]
- 165.Riggs JA, Hossler KA, Smith BD, Karpa MJ, Griffin G, Duggan PJ. Tetrahedron Lett. 1996;37:6303. [Google Scholar]
- 166.Carey CM, Riggan WB., Jr Anal Chem. 1994;66:3587. doi: 10.1021/ac00093a009. [DOI] [PubMed] [Google Scholar]
- 167.Le Goff T, Braven J, Ebdon L, Scholefield D. Anal Chim Acta. 2004;510:175. [Google Scholar]
- 168.Nelissen HFM, Smith DK. Chem Commun. 2007:3039. doi: 10.1039/b706227c. [DOI] [PubMed] [Google Scholar]
- 169.Marecek JF, Burrows CJ. Tetrahedron Lett. 1986;27:5943. [Google Scholar]
- 170.Marecek JF, Fischer PA, Burrows CJ. Tetrahedron Lett. 1988;29:6231. [Google Scholar]
- 171.Furuta H, Magda D, Sessler JL. J Am Chem Soc. 1991;113:978. [Google Scholar]
- 172.Tohda K, Tange M, Odashima K, Umezawa Y, Furuta H, Sessler JL. Anal Chem. 1992;64:960. doi: 10.1021/ac00032a023. [DOI] [PubMed] [Google Scholar]
- 173.Tohda K, Naganawa R, Xiao ML, Tange M, Umezawa K, Odashima K, Umezawa Y, Furuta H, Sessler JL. Sens Actuators, B. 1993;14:669. [Google Scholar]
- 174.Andres A, Burguete MI, García-España E, Luis SV, Miravet JF, Soriano C. J Chem Soc, Perkin Trans 2. 1993:749. [Google Scholar]
- 175.Aguilar JA, García-España E, Guerrero JA, Luis SV, Llinares JM, Miravet JF, Ramirez JA, Soriano C. J Chem Soc, Chem Commun. 1995:2237. [Google Scholar]
- 176.Aguilar JA, García-España E, Guerrero JA, Luis SV, Llinares JM, Ramirez JA, Soriano C. Inorg Chim Acta. 1996;246:287. [Google Scholar]
- 177.Aguilar JA, Celda B, Fusi V, García-España E, Luis SV, Martinez MC, Ramirez JA, Soriano C, Tejero R. J Chem Soc, Perkin Trans 2. 2000:1323. [Google Scholar]
- 178.Aguilar J, Díaz P, Escarti F, García-España E, Gil L, Soriano C, Verdejo B. Inorg Chim Acta. 2002;339:307. [Google Scholar]
- 179.Lu Q, Motekaitis RJ, Reibenspies JJ, Martell AE. Inorg Chem. 1995;34:4958. doi: 10.1021/ic951384a. [DOI] [PubMed] [Google Scholar]
- 180.Nation DA, Reibenspies J, Martell AE. Inorg Chem. 1996;35:4597. [Google Scholar]
- 181.Lu Q, Martell AE, Motekaitis RJ. Inorg Chim Acta. 1996;251:365. [Google Scholar]
- 182.Nation DA, Lu Q, Martell AE. Inorg Chim Acta. 1997;263:209. [Google Scholar]
- 183.Anda C, Llobet A, Salvado V, Reibenspies J, Motekaitis RJ, Martell AE. Inorg Chem. 2000;39:2986. doi: 10.1021/ic990818p. [DOI] [PubMed] [Google Scholar]
- 184.Anda C, Llobet A, Martell AE, Donnadieu B, Parella T. Inorg Chem. 2003;42:8545. doi: 10.1021/ic034205v. [DOI] [PubMed] [Google Scholar]
- 185.Anda C, Llobet A, Salvado V, Martell AE, Motekaitis RJ. Inorg Chem. 2000;39:3000. doi: 10.1021/ic9908468. [DOI] [PubMed] [Google Scholar]
- 186.Ragunathan KG, Schneider HJ. J Chem Soc, Perkin Trans 2. 1996:2597. [Google Scholar]
- 187.Anda C, Martinez MA, Llobet A. Supramol Chem. 2005;17:257. [Google Scholar]
- 188.Kumar A, Mehtab S, Singh UP, Aggarwal V, Singh J. Electroanalysis. 2008;20:1186. [Google Scholar]
- 189.Hossain MA, Liljegren JA, Powell D, Bowman-James K. Inorg Chem. 2004;43:3751. doi: 10.1021/ic049762b. [DOI] [PubMed] [Google Scholar]
- 190.Albelda MT, García-España E, Jimenez HR, Llinares JM, Soriano C, Sornosa-Ten A, Verdejo B. Dalton Trans. 2006:4474. doi: 10.1039/b605639c. [DOI] [PubMed] [Google Scholar]
- 191.Schneider HJ, Theis I. Angew Chem. 1989;101:757. [Google Scholar]
- 192.Schneider HJ, Theis I. Angew Chem, Int Ed. 1989;28:753. [Google Scholar]
- 193.Schneider HJ, Blatter T, Palm B, Pfingstag U, Ruediger V, Theis I. J Am Chem Soc. 1992;114:7704. [Google Scholar]
- 194.Hosseini MW, Blacker AJ, Lehn JM. J Chem Soc, Chem Commun. 1988:596. [Google Scholar]
- 195.Hosseini MW, Blacker AJ, Lehn JM. J Am Chem Soc. 1990;112:3896. [Google Scholar]
- 196.Fenniri H, Hosseini MW, Lehn JM. Helv Chim Acta. 1997;80:786. [Google Scholar]
- 197.Claude S, Lehn JM, Schmidt F, Vigneron JP. J Chem Soc, Chem Commun. 1991:1182. [Google Scholar]
- 198.Claude S, Lehn JM, Schmidt F, Vigneron JP. J Chem Soc, Chem Commun. 1991:1656. [Google Scholar]
- 199.Cudic P, Zinic M, Tomisic V, Simeon V, Vigneron JP, Lehn JM. J Chem Soc, Chem Commun. 1995:1073. [Google Scholar]
- 200.Malojcic G, Piantanida I, Marinic M, Zinic M, Marjanovic M, Kralj M, Pavelic K, Schneider HJ. Org Biomol Chem. 2005;3:4373. doi: 10.1039/b509094f. [DOI] [PubMed] [Google Scholar]
- 201.Dhaenens M, Lehn JM, Vigneron JP. J Chem Soc, Perkin Trans 2. 1993:1379. [Google Scholar]
- 202.Teulade-Fichou MP, Vigneron JP, Lehn JM. Supramol Chem. 1995;5:139. [Google Scholar]
- 203.Baudoin O, Gonnet F, Teulade-Fichou MP, Vigneron JP, Tabet JC, Lehn JM. Chem--Eur J. 1999;5:2762. [Google Scholar]
- 204.Teulade-Fichou MP, Vigneron JP, Lehn JM. J Chem Soc, Perkin Trans 2. 1996:2169. [Google Scholar]
- 205.Inouye M, Fujimoto K, Furusyo M, Nakazumi H. J Am Chem Soc. 1999;121:1452. [Google Scholar]
- 206.Abe H, Mawatari Y, Teraoka H, Fujimoto K, Inouye M. J Org Chem. 2004;69:495. doi: 10.1021/jo035188u. [DOI] [PubMed] [Google Scholar]
- 207.Bazzicalupi C, Beconcini A, Bencini A, Fusi V, Giorgi C, Masotti A, Valtancoli B. J Chem Soc, Perkin Trans 2. 1999:1675. [Google Scholar]
- 208.Bazzicalupi C, Biagini S, Bencini A, Faggi E, Giorgi C, Matera I, Valtancoli B. Chem Commun. 2006:4087. doi: 10.1039/b611031b. [DOI] [PubMed] [Google Scholar]
- 209.Bazzicalupi C, Bencini A, Biagini S, Faggi E, Meini S, Giorgi C, Spepi A, Valtancoli B. J Org Chem. 2009;74:7349. doi: 10.1021/jo901423m. [DOI] [PubMed] [Google Scholar]
- 210.Bazzicalupi C, Bencini A, Berni E, Bianchi A, Fornasari P, Giorgi C, Masotti A, Paoletti P, Valtancoli B. J Phys Org Chem. 2001;14:432. [Google Scholar]
- 211.Anda C, Bazzicalupi C, Bencini A, Berni E, Bianchi A, Fornasari P, Llobet A, Giorgi C, Paoletti P, Valtancoli B. Inorg Chim Acta. 2003;356:167. [Google Scholar]
- 212.Huston ME, Akkaya EU, Czarnik AW. J Am Chem Soc. 1989;111:8735. [Google Scholar]
- 213.Vance DH, Czarnik AW. J Am Chem Soc. 1994;116:9397. [Google Scholar]
- 214.Albelda MT, Bernardo MA, García-España E, Godino-Salido ML, Luis SV, Melo MJ, Pina F, Soriano C. J Chem Soc, Perkin Trans 2. 1999:2545. [Google Scholar]
- 215.Albelda MT, Aguilar J, Alves S, Aucejo R, Díaz P, Lodeiro C, Lima JC, García-España E, Pina F, Soriano C. Helv Chim Acta. 2003;86:3118. [Google Scholar]
- 216.Sancenón F, Benito A, Lloris JM, Martínez-Máñez R, Pardo T, Soto J. Helv Chim Acta. 2002;85:1505. [Google Scholar]
- 217.Descalzo AB, Jimenez D, Marcos MD, Martínez-Máñez R, Soto J, El Haskouri J, Guillém C, Beltrán D, Amorós P, Borrachero MV. Adv Mater. 2002;14:966. [Google Scholar]
- 218.Descalzo AB, Marcos MD, Martínez-Máñez R, Soto J, Beltrán D, Amorós P. J Mater Chem. 2005;15:2721. [Google Scholar]
- 219.Aucejo R, Díaz P, García-España E, Alarcon J, Delgado-Pinar E, Torres F, Soriano C, Guillém MC. New J Chem. 2007;31:44. [Google Scholar]
- 220.Develay S, Tripier R, Le Baccon M, Patinec V, Serratrice G, Handel H. Dalton Trans. 2005:3016. doi: 10.1039/b507819a. [DOI] [PubMed] [Google Scholar]
- 221.Develay S, Tripier R, Le Baccon M, Patinec V, Serratrice G, Handel H. Dalton Trans. 2006:3418. doi: 10.1039/b517695f. [DOI] [PubMed] [Google Scholar]
- 222.Develay S, Tripier R, Bernier N, Le Baccon M, Patinec V, Serratrice G, Handel H. Dalton Trans. 2007:1038. doi: 10.1039/b616862k. [DOI] [PubMed] [Google Scholar]
- 223.Le Bris N, Bernard H, Tripier R, Handel H. Inorg Chim Acta. 2007;360:3026. [Google Scholar]
- 224.Delepine AS, Tripier R, Handel H. Org Biomol Chem. 2008;6:1743. doi: 10.1039/b719514a. [DOI] [PubMed] [Google Scholar]
- 225.Delepine AS, Tripier R, Le Bris N, Bernard H, Honraedt A, Handel H. Inorg Chim Acta. 2009;362:3829. [Google Scholar]
- 226.Mutai T, Abe Y, Araki K. J Chem Soc, Perkin Trans 2. 1997:1805. [Google Scholar]
- 227.Cabell LA, Monahan MK, Anslyn EV. Tetrahedron Lett. 1999;40:7753. [Google Scholar]
- 228.Lara KO, Godoy-Alcantar C, Rivera IL, Eliseev AV, Yatsimirsky AK. J Phys Org Chem. 2001;14:453. [Google Scholar]
- 229.Gerasimchuk OA, Mason S, Llinares JM, Song M, Alcock NW, Bowman-James K. Inorg Chem. 2000;39:1371. doi: 10.1021/ic9911116. [DOI] [PubMed] [Google Scholar]
- 230.Kimura E, Kuramoto Y, Koike T, Fujioka H, Kodama M. J Org Chem. 1990;55:42. [Google Scholar]
- 231.Arturoni E, Bazzicalupi C, Bencini A, Caltagirone C, Danesi A, Garau A, Giorgi C, Lippolis V, Valtancoli B. Inorg Chem. 2008;47:6551. doi: 10.1021/ic800549e. [DOI] [PubMed] [Google Scholar]
- 232.Bencini A, Biagini S, Giorgi C, Handel H, Le Baccon M, Mariani P, Paoletti P, Paoli P, Rossi P, Tripier R, Valtancoli B. Eur J Org Chem. 2009:5610. [Google Scholar]
- 233.Dietrich B, Guilhem J, Lehn JM, Pascard C, Sonveaux E. Helv Chim Acta. 1984;67:91. [Google Scholar]
- 234.Grell D, Grell E, Bugnon P, Dietrich B, Lehn JM. J Therm Anal Calorim. 2004;77:483. [Google Scholar]
- 235.Bazzicalupi C, Bencini A, Bianchi A, Danesi A, Giorgi C, Valtancoli B. Inorg Chem. 2009;48:2391. doi: 10.1021/ic8013128. [DOI] [PubMed] [Google Scholar]
- 236.Olivier C, Grote Z, Solari E, Scopelliti R, Severin K. Chem Commun. 2007:4000. doi: 10.1039/b711425g. [DOI] [PubMed] [Google Scholar]
- 237.Mateus P, Delgado R, Brandao P, Carvalho S, Felix V. Org Biomol Chem. 2009;7:4661. doi: 10.1039/b912940e. [DOI] [PubMed] [Google Scholar]
- 238.Mateus P, Delgado R, Brandao P, Felix V. J Org Chem. 2009;74:8638. doi: 10.1021/jo901742d. [DOI] [PubMed] [Google Scholar]
- 239.Perreault DM, Cabell LA, Anslyn EV. Bioorg Med Chem. 1997;5:1209. doi: 10.1016/s0968-0896(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 240.Best MD, Tobey SL, Anslyn EV. Coord Chem Rev. 2003;240:3. [Google Scholar]
- 241.Schug KA, Lindner W. Chem Rev. 2005;105:67. doi: 10.1021/cr040603j. [DOI] [PubMed] [Google Scholar]
- 242.Blondeau P, Segura M, Pérez-Fernández R, de Mendoza J. Chem Soc Rev. 2007;36:198. doi: 10.1039/b603089k. [DOI] [PubMed] [Google Scholar]
- 243.Houk RJT, Tobey SL, Anslyn EV. Top Curr Chem. 2005;255:199. [Google Scholar]
- 244.Watters JI, Matsumoto S. J Am Chem Soc. 1964;86:3961. [Google Scholar]
- 245.Cotton FA, Day VW, Hazen EE, Jr, Larsen S. J Am Chem Soc. 1973;95:4834. doi: 10.1021/ja00796a012. [DOI] [PubMed] [Google Scholar]
- 246.Cotton FA, Day VW, Hazen EE, Jr, Larsen S, Wong STK. J Am Chem Soc. 1974;96:4471. doi: 10.1021/ja00821a020. [DOI] [PubMed] [Google Scholar]
- 247.Springs B, Haake P. Bioorg Chem. 1977;6:181. [Google Scholar]
- 248.Dietrich B, Fyles TM, Lehn JM, Pease LG, Fyles DL. J Chem Soc, Chem Commun. 1978:934. [Google Scholar]
- 249.Dietrich B, Fyles DL, Fyles TM, Lehn JM. Helv Chim Acta. 1979;62:2763. [Google Scholar]
- 250.Gross R, Duerner G, Goebel MW. Liebigs Ann Chem. 1994:49. [Google Scholar]
- 251.Gross R, Bats JW, Goebel MW. Liebigs Ann Chem. 1994:205. [Google Scholar]
- 252.Nishizawa S, Kato Y, Teramae N. J Am Chem Soc. 1999;121:9463. [Google Scholar]
- 253.Schmidtchen FP. Tetrahedron Lett. 1989;30:4493. [Google Scholar]
- 254.Schiessl P, Schmidtchen FP. J Org Chem. 1994;59:509. [Google Scholar]
- 255.Berger M, Schmidtchen FP. J Am Chem Soc. 1996;118:8947. [Google Scholar]
- 256.Berger M, Schmidtchen FP. J Am Chem Soc. 1999;121:9986. [Google Scholar]
- 257.Fibbioli M, Berger M, Schmidtchen FP, Pretsch E. Anal Chem. 2000;72:156. doi: 10.1021/ac990696i. [DOI] [PubMed] [Google Scholar]
- 258.Stephan H, Gloe K, Schiessl P, Schmidtchen FP. Supramol Chem. 1995;5:273. [Google Scholar]
- 259.Jadhav VD, Schmidtchen FP. Org Lett. 2005;7:3311. doi: 10.1021/ol051109k. [DOI] [PubMed] [Google Scholar]
- 260.Galan A, Pueyo E, Salmeron A, de Mendoza J. Tetrahedron Lett. 1991;32:1827. [Google Scholar]
- 261.Alcazar V, Segura M, Prados P, de Mendoza J. Tetrahedron Lett. 1998;39:1033. [Google Scholar]
- 262.Galan A, de Mendoza J, Toiron C, Bruix M, Deslongchamps G, Rebek J., Jr J Am Chem Soc. 1991;113:9424. [Google Scholar]
- 263.Kato Y, Conn MM, Rebek J., Jr J Am Chem Soc. 1994;116:3279. [Google Scholar]
- 264.Deslongchamps G, Galan A, de Mendoza J, Rebek J., Jr Angew Chem, Int Ed. 1992;31:61. [Google Scholar]
- 265.Deslongchamps G, Galan A, de Mendoza J, Rebek J., Jr Angew Chem. 1992;104:58. [Google Scholar]
- 266.Andreu C, Galan A, Kobiro K, de Mendoza J, Park TK, Rebek J, Jr, Salmeron A, Usman N. J Am Chem Soc. 1994;116:5501. [Google Scholar]
- 267.Magrans JO, Ortiz AR, Molins MA, Lebouille PHP, Sanchez-Quesada J, Prados P, Pons M, Gago F, de Mendoza J. Angew Chem, Int Ed. 1996;35:1712. [Google Scholar]
- 268.Magrans JO, Ortiz AR, Molins MA, Lebouille PHP, Sanchez-Quesada J, Prados P, Pons M, Gago F, de Mendoza J. Angew Chem. 1996;108:1816. [Google Scholar]
- 269.Cuevas F, Di Stefano S, Magrans JO, Prados P, Mandolini L, de Mendoza J. Chem--Eur J. 2000;6:3228. doi: 10.1002/1521-3765(20000901)6:17<3228::aid-chem3228>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 270.Dixon RP, Geib SJ, Hamilton AD. J Am Chem Soc. 1992;114:365. [Google Scholar]
- 271.Jubian V, Veronese A, Dixon RP, Hamilton AD. Angew Chem, Int Ed. 1995;34:1237. [Google Scholar]
- 272.Ariga K, Anslyn EV. J Org Chem. 1992;57:417. [Google Scholar]
- 273.Kneeland DM, Ariga K, Lynch VM, Huang CY, Anslyn EV. J Am Chem Soc. 1993;115:10042. [Google Scholar]
- 274.Metzger A, Lynch VM, Anslyn EV. Angew Chem, Int Ed. 1997;36:862. [Google Scholar]
- 275.Niikura K, Metzger A, Anslyn EV. J Am Chem Soc. 1998;120:8533. [Google Scholar]
- 276.Niikura K, Anslyn EV. J Org Chem. 2003;68:10156. doi: 10.1021/jo030263m. [DOI] [PubMed] [Google Scholar]
- 277.Schneider SE, O'Neil SN, Anslyn EV. J Am Chem Soc. 2000;122:542. [Google Scholar]
- 278.McCleskey SC, Griffin MJ, Schneider SE, McDevitt JT, Anslyn EV. J Am Chem Soc. 2003;125:1114. doi: 10.1021/ja021230b. [DOI] [PubMed] [Google Scholar]
- 279.Schmuck C, Schwegmann M. Org Lett. 2005;7:3517. doi: 10.1021/ol051230r. [DOI] [PubMed] [Google Scholar]
- 280.Schmuck C, Heller M. Org Biomol Chem. 2007;5:787. doi: 10.1039/b613660e. [DOI] [PubMed] [Google Scholar]
- 281.Sasaki DY, Kurihara K, Kunitake T. J Am Chem Soc. 1991;113:9685. [Google Scholar]
- 282.Sasaki DY, Kurihara K, Kunitake T. J Am Chem Soc. 1992;114:10994. [Google Scholar]
- 283.Taguchi K, Ariga K, Kunitake T. Chem Lett. 1995:701. [Google Scholar]
- 284.Ariga K, Kamino A, Koyano H, Kunitake T. J Mater Chem. 1997;7:1155. [Google Scholar]
- 285.Jiménez Blanco JL, Bootello P, Benito JM, Ortiz Mellet C, García Fernández JM. J Org Chem. 2006;71:5136. doi: 10.1021/jo060360q. [DOI] [PubMed] [Google Scholar]
- 286.Goebel MW, Bats JW, Duermer G. Angew Chem. 1992;104:217. [Google Scholar]
- 287.Goebel MW, Bats JW, Duermer G. Angew Chem, Int Ed. 1992;104:207. [Google Scholar]
- 288.Yeo WS, Hong JI. Tetrahedron Lett. 1998;39:8137. [Google Scholar]
- 289.Yeo WS, Hong JI. Tetrahedron Lett. 1998;39:3769. [Google Scholar]
- 290.Kubo Y, Tsukahara M, Ishihara S, Tokita S. Chem Commun. 2000:653. [Google Scholar]
- 291.Kubo Y, Ishihara S, Tsukahara M, Tokita S. J Chem Soc, Perkin Trans 2. 2002:1455. [Google Scholar]
- 292.Kubo Y, Kato M, Misawa Y, Tokita S. Tetrahedron Lett. 2004;45:3769. [Google Scholar]
- 293.Nishizawa S, Cui YY, Minagawa M, Morita K, Kato Y, Taniguchi S, Kato R, Teramae N. J Chem Soc, Perkin Trans 2. 2002:866. [Google Scholar]
- 294.Seong HR, Kim DS, Kim SG, Choi HJ, Ahn KH. Tetrahedron Lett. 2004;45:723. [Google Scholar]
- 295.Misawa Y, Kubo Y, Tokita S, Ohkuma H, Nakahara H. Chem Lett. 2004;33:1118. [Google Scholar]
- 296.Kubo Y, Uchida S, Kemmochi Y, Okubo T. Tetrahedron Lett. 2005;46:4369. [Google Scholar]
- 297.Ihm H, Yun S, Kim HG, Kim JK, Kim KS. Org Lett. 2002;4:2897. doi: 10.1021/ol026373h. [DOI] [PubMed] [Google Scholar]
- 298.Kim SK, Singh NJ, Kim SJ, Kim HG, Kim JK, Lee JW, Kim KS, Yoon J. Org Lett. 2003;5:2083. doi: 10.1021/ol034498w. [DOI] [PubMed] [Google Scholar]
- 299.Yoon J, Kim SK, Singh NJ, Lee JW, Yang YJ, Chellappan K, Kim KS. J Org Chem. 2004;69:581. doi: 10.1021/jo035387d. [DOI] [PubMed] [Google Scholar]
- 300.Kwon JY, Singh NJ, Kim HN, Kim SK, Kim KS, Yoon J. J Am Chem Soc. 2004;126:8892. doi: 10.1021/ja0492440. [DOI] [PubMed] [Google Scholar]
- 301.Kim SK, Singh NJ, Kwon J, Hwang IC, Park SJ, Kim KS, Yoon J. Tetrahedron. 2006;62:6065. [Google Scholar]
- 302.Xu Z, Kim S, Lee KH, Yoon J. Tetrahedron Lett. 2007;48:3797. [Google Scholar]
- 303.Singh NJ, Jun EJ, Chellappan K, Thangadurai D, Chandran RP, Hwang IC, Yoon J, Kim KS. Org Lett. 2007;9:485. doi: 10.1021/ol062849b. [DOI] [PubMed] [Google Scholar]
- 304.Kim SK, Seo D, Han SJ, Son G, Lee IJ, Lee C, Lee KD, Yoon J. Tetrahedron. 2008;64:6402. [Google Scholar]
- 305.Lee HN, Singh NJ, Kim SK, Kwon JY, Kim YY, Kim KS, Yoon J. Tetrahedron Lett. 2007;48:169. [Google Scholar]
- 306.Xu Z, Singh NJ, Lim J, Pan J, Kim HN, Park S, Kim KS, Yoon J. J Am Chem Soc. 2009;131:15528. doi: 10.1021/ja906855a. [DOI] [PubMed] [Google Scholar]
- 307.Sato K, Onitake T, Arai S, Yamagishi T. Heterocycles. 2003;60:779. [Google Scholar]
- 308.Fahlbusch T, Frank M, Schatz J, Schmaderer H. Eur J Org Chem. 2006:1899. doi: 10.1021/jo052319d. [DOI] [PubMed] [Google Scholar]
- 309.Lu QS, Dong L, Zhang J, Li J, Jiang L, Huang Y, Qin S, Hu CW, Yu XQ. Org Lett. 2009;11:669. doi: 10.1021/ol8027303. [DOI] [PubMed] [Google Scholar]
- 310.Wang S, Chang YT. J Am Chem Soc. 2006;128:10380. doi: 10.1021/ja063733d. [DOI] [PubMed] [Google Scholar]
- 311.Ghosh K, Saha I. Tetrahedron Lett. 2008;49:4591. [Google Scholar]
- 312.Ghosh K, Saha I, Patra A. Tetrahedron Lett. 2009;50:2392. [Google Scholar]
- 313.Kumar A, Pandey PS. Org Lett. 2008;10:165. doi: 10.1021/ol702457w. [DOI] [PubMed] [Google Scholar]
- 314.Nguyen QPB, Le TN, Kim TH. Bull Korean Chem Soc. 2009;30:1743. [Google Scholar]
- 315.Amendola V, Boiocchi M, Fabbrizzi L, Palchetti A. Chem--Eur J. 2005;11:120. doi: 10.1002/chem.200400592. [DOI] [PubMed] [Google Scholar]
- 316.Turkewitsch P, Wandelt B, Ganju RR, Darling GD, Powell WS. Chem Phys Lett. 1996;260:142. [Google Scholar]
- 317.Turkewitsch P, Wandelt B, Darling GD, Powell WS. J Photochem Photobiol, A. 1998;117:199. doi: 10.1021/ac981558g. [DOI] [PubMed] [Google Scholar]
- 318.Belcher WJ, Fabre M, Farhan T, Steed JW. Org Biomol Chem. 2006;4:781. doi: 10.1039/b516027h. [DOI] [PubMed] [Google Scholar]
- 319.Turner DR, Paterson MJ, Steed JW. J Org Chem. 2006;71:1598. doi: 10.1021/jo052339f. [DOI] [PubMed] [Google Scholar]
- 320.Ghosh K, Sarkar AR, Masanta G. Tetrahedron Lett. 2007;48:8725. [Google Scholar]
- 321.Ghosh K, Sarkar AR, Patra A. Tetrahedron Lett. 2009;50:6557. [Google Scholar]
- 322.Neelakandan PP, Hariharan M, Ramaiah D. Org Lett. 2005;7:5765. doi: 10.1021/ol052246k. [DOI] [PubMed] [Google Scholar]
- 323.Neelakandan PP, Hariharan M, Ramaiah D. J Am Chem Soc. 2006;128:11334. doi: 10.1021/ja062651m. [DOI] [PubMed] [Google Scholar]
- 324.Vilozny B, Schiller A, Wessling RA, Singaram B. Anal Chim Acta. 2009;649:246. doi: 10.1016/j.aca.2009.07.032. [DOI] [PubMed] [Google Scholar]
- 325.Coudret C, Harriman A. J Chem Soc, Chem Commun. 1992:1755. [Google Scholar]
- 326.Jung YG, Yeo WS, Lee SB, Hong JI. Chem Commun. 1997:1061. [Google Scholar]
- 327.Ariga K, Kunitake T. Acc Chem Res. 1998;31:371. [Google Scholar]
- 328.Buhlmann P, Amemiya S, Nishizawa S, Xiao KP, Umezawa Y. J Inclusion Phenom. 1998;32:151. [Google Scholar]
- 329.Choi K, Hamilton AD. Coord Chem Rev. 2003;240:101. [Google Scholar]
- 330.Chmielewski MJ, Zielinski T, Jurczak J. Pure Appl Chem. 2007;79:1087. [Google Scholar]
- 331.Sessler JL, Gross DE, Cho WS, Lynch VM, Schmidtchen FP, Bates GW, Light ME, Gale PA. J Am Chem Soc. 2006;128:12281. doi: 10.1021/ja064012h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Alunni S, Pero A, Reichenbach G. J Chem Soc, Perkin Trans 2. 1998:1747. [Google Scholar]
- 333.Kang SO, Begum RA, Bowmna-James K. Angew Chem, Int Ed. 2006;45:7882. doi: 10.1002/anie.200602006. [DOI] [PubMed] [Google Scholar]
- 334.Werner F, Schneider HJ. Helv Chim Acta. 2000;83:465. [Google Scholar]
- 335.Raposo C, Perez N, Almaraz M, Mussons ML, Caballero MC, Moran JR. Tetrahedron Lett. 1995;36:3255. [Google Scholar]
- 336.Hirst SC, Tecilla P, Geib SJ, Fan E, Hamilton AD. Isr J Chem. 1992;32:105. [Google Scholar]
- 337.Choi K, Hamilton AD. J Am Chem Soc. 2001;123:2456. doi: 10.1021/ja005772+. [DOI] [PubMed] [Google Scholar]
- 338.Choi K, Hamilton AD. J Am Chem Soc. 2003;125:10241. doi: 10.1021/ja034563x. [DOI] [PubMed] [Google Scholar]
- 339.Camiolo S, Coles SJ, Gale PA, Hursthouse MB, Mayer TA, Paver MA. Chem Commun. 2000:275. [Google Scholar]
- 340.Kavallieratos K, de Gala SR, Austin DJ, Crabtree RH. J Am Chem Soc. 1997;119:2325. [Google Scholar]
- 341.Kavallieratos K, Bertao CM, Crabtree RH. J Org Chem. 1999;64:1675. doi: 10.1021/jo982382l. [DOI] [PubMed] [Google Scholar]
- 342.Brooks SJ, Evans LS, Gale PA, Hursthouse MB, Light ME. Chem Commun. 2005:734. doi: 10.1039/b413654c. [DOI] [PubMed] [Google Scholar]
- 343.Sun SS, Lees AJ. Chem Commun. 2000:1687. [Google Scholar]
- 344.Sun SS, Lees AJ, Zavalij PY. Inorg Chem. 2003;42:3445. doi: 10.1021/ic0206589. [DOI] [PubMed] [Google Scholar]
- 345.Liao JH, Chen CT, Fang JM. Org Lett. 2002;4:561. doi: 10.1021/ol0171564. [DOI] [PubMed] [Google Scholar]
- 346.Liao JH, Chen CT, Fang JM. J Chin Chem Soc. 2006;53:1439. [Google Scholar]
- 347.Chen KH, Liao JH, Chan HY, Fang JM. J Org Chem. 2009;74:895. doi: 10.1021/jo802173b. [DOI] [PubMed] [Google Scholar]
- 348.Kuo LJ, Liao JH, Chen CT, Huang CH, Chen CS, Fang JM. Org Lett. 2003;5:1821. doi: 10.1021/ol034364i. [DOI] [PubMed] [Google Scholar]
- 349.Kovalchuk A, Bricks JL, Reck G, Rurack K, Schulz B, Szumna A, Weisshoff H. Chem Commun. 2004:1946. doi: 10.1039/b405207b. [DOI] [PubMed] [Google Scholar]
- 350.Kondo SI, Hiraoka Y, Kurumatani N, Yano Y. Chem Commun. 2005:1720. doi: 10.1039/b417304j. [DOI] [PubMed] [Google Scholar]
- 351.Chmielewski MJ, Charon M, Jurczak J. Org Lett. 2004;6:3501. doi: 10.1021/ol048661e. [DOI] [PubMed] [Google Scholar]
- 352.Sessler JL, Rubin BL, Camiolo S, Cho WS, Pantos GD, Lynch VM. Supramol Chem. 2006;18:103. [Google Scholar]
- 353.Valiyaveettil S, Engbersen JFJ, Verboom W, Reinhoudt DN. Angew Chem. 1993;105:942. [Google Scholar]
- 354.Yoshida H, Saigo K, Hiratani K. Chem Lett. 2000:116. [Google Scholar]
- 355.Kameta N, Hiratani K. Chem Lett. 2006;35:536. [Google Scholar]
- 356.Causey CP, Allen WE. J Org Chem. 2002;67:5963. doi: 10.1021/jo020098v. [DOI] [PubMed] [Google Scholar]
- 357.Kang J, Jo JH, In S. Tetrahedron Lett. 2004;45:5225. [Google Scholar]
- 358.Kang J, Kim J. Tetrahedron Lett. 2005;46:1759. [Google Scholar]
- 359.Liu C, Qian X, Wang J, Li Z. Tetrahedron Lett. 2008;49:1087. [Google Scholar]
- 360.Hossain MA, Llinares JM, Powell D, Bowman-James K. Inorg Chem. 2001;40:2936. doi: 10.1021/ic015508x. [DOI] [PubMed] [Google Scholar]
- 361.Hossain MA, Kang SO, Llinares JM, Powell D, Bowman-James K. Inorg Chem. 2003;42:5043. doi: 10.1021/ic034735r. [DOI] [PubMed] [Google Scholar]
- 362.Kang SO, Llinares JM, Powell D, VanderVelde D, Bowman-James K. J Am Chem Soc. 2003;125:10152. doi: 10.1021/ja034969+. [DOI] [PubMed] [Google Scholar]
- 363.Hossain MA, Kang SO, Powell D, Bowman-James K. Inorg Chem. 2003;42:1397. doi: 10.1021/ic0263140. [DOI] [PubMed] [Google Scholar]
- 364.Kang SO, Powell D, Bowman-James K. J Am Chem Soc. 2005;127:13478. doi: 10.1021/ja054332l. [DOI] [PubMed] [Google Scholar]
- 365.Kang SO, Day VW, Bowman-James K. Org Lett. 2009;11:3654. doi: 10.1021/ol9014249. [DOI] [PubMed] [Google Scholar]
- 366.Szumna A, Jurczak J. Eur J Org Chem. 2001:4031. [Google Scholar]
- 367.Chmielewski M, Jurczak J. Tetrahedron Lett. 2004;45:6007. [Google Scholar]
- 368.Chmielewski MJ, Jurczak J. Chem--Eur J. 2005;11:6080. doi: 10.1002/chem.200500232. [DOI] [PubMed] [Google Scholar]
- 369.Chmielewski MJ, Szumna A, Jurczak J. Tetrahedron Lett. 2004;45:8699. [Google Scholar]
- 370.Chmielewski MJ, Jurczak J. Tetrahedron Lett. 2005;46:3085. [Google Scholar]
- 371.Piatek P, Jurczak J. Chem Commun. 2002:2450. doi: 10.1039/b207335h. [DOI] [PubMed] [Google Scholar]
- 372.Costero AM, Jose Banuls M, Jose Aurell M, Ward MD, Argent S. Tetrahedron. 2004;60:9471. [Google Scholar]
- 373.Zeng ZY, Xu KX, He YB, Liu SY, Wu JL, Wei LH, Meng LZ. Chin J Chem. 2004;22:1372. [Google Scholar]
- 374.Morey J, Orell M, Barcelo MA, Deya PM, Costa A, Ballester P. Tetrahedron Lett. 2004;45:1261. [Google Scholar]
- 375.Jain AK, Gupta VK, Raisoni JR. Talanta. 2006;69:1007. doi: 10.1016/j.talanta.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 376.Zhang Y, Yin Z, He J, Cheng JP. Tetrahedron Lett. 2007;48:6039. [Google Scholar]
- 377.Watanabe S, Sonobe M, Arai M, Tazume Y, Matsuo T, Nakamura T, Yoshida K. Chem Commun. 2002:2866. doi: 10.1039/b205751d. [DOI] [PubMed] [Google Scholar]
- 378.Nishizawa S, Buehlmann P, Iwao M, Umezawa Y. Tetrahedron Lett. 1995;36:6483. [Google Scholar]
- 379.Smith PJ, Reddington MV, Wilcox CS. Tetrahedron Lett. 1992;33:6085. [Google Scholar]
- 380.Kelly TR, Kim MH. J Am Chem Soc. 1994;116:7072. [Google Scholar]
- 381.Raposo C, Almaraz M, Martin M, Weinrich V, Mussons ML, Alcazar V, Caballero MC, Moran JR. Chem Lett. 1995:759. [Google Scholar]
- 382.Lakshminarayanan PS, Ravikumar I, Suresh E, Ghosh P. Chem Commun. 2007:5214. doi: 10.1039/b713365k. [DOI] [PubMed] [Google Scholar]
- 383.Ravikumar I, Lakshminarayanan PS, Arunachalam M, Suresh E, Ghosh P. Dalton Trans. 2009:4160. doi: 10.1039/b820322a. [DOI] [PubMed] [Google Scholar]
- 384.Jose DA, Kumar DK, Ganguly B, Das A. Inorg Chem. 2007;46:5817. doi: 10.1021/ic062466+. [DOI] [PubMed] [Google Scholar]
- 385.Xie H, Yi S, Yang X, Wu S. New J Chem. 1999;23:1105. [Google Scholar]
- 386.Choi HJ, Park YS, Yun SH, Kim HS, Cho CS, Ko K, Ahn KH. Org Lett. 2002;4:795. [PubMed] [Google Scholar]
- 387.Amemiya S, Buhlmann P, Umezawa Y. Chem Commun. 1997:1027. [Google Scholar]
- 388.Tohda K, Amemiya S, Ohki T, Nagahora S, Tanaka S, Buhlmann P, Umezawa Y. Isr J Chem. 1997;37:267. [Google Scholar]
- 389.Snellink-Ruel BHM, Antonisse MMG, Engbersen JFJ, Timmerman P, Reinhoudt DN. Eur J Org Chem. 2000:165. [Google Scholar]
- 390.Brooks SJ, Gale PA, Light ME. Chem Commun. 2005:4696. doi: 10.1039/b508144k. [DOI] [PubMed] [Google Scholar]
- 391.Yin Z, Li Z, Yu A, He J, Cheng JP. Tetrahedron Lett. 2004;45:6803. [Google Scholar]
- 392.Brooks SJ, Edwards PR, Gale PA, Light ME. New J Chem. 2006;30:65. [Google Scholar]
- 393.Brooks SJ, Gale PA, Light ME. Chem Commun. 2006:4344. doi: 10.1039/b610938a. [DOI] [PubMed] [Google Scholar]
- 394.Kang SO, Jeon S, Nam KC. Supramol Chem. 2002;14:405. [Google Scholar]
- 395.Boiocchi M, Del Boca L, Gomez DE, Fabbrizzi L, Licchelli M, Monzani E. J Am Chem Soc. 2004;126:16507. doi: 10.1021/ja045936c. [DOI] [PubMed] [Google Scholar]
- 396.Amendola V, Boiocchi M, Esteban-Gómez D, Fabbrizzi L, Monzani E. Org Biomol Chem. 2005;3:2632. doi: 10.1039/b504931h. [DOI] [PubMed] [Google Scholar]
- 397.Boiocchi M, Del Boca L, Esteban-Gómez D, Fabbrizzi L, Licchelli M, Monzani E. Chem--Eur J. 2005;11:3097. doi: 10.1002/chem.200401049. [DOI] [PubMed] [Google Scholar]
- 398.Ali HDP, Kruger PE, Gunnlaugsson T. New J Chem. 2008;32:1153. [Google Scholar]
- 399.Dos Santos CMG, Glynn M, McCabe T, Seixas de Melo JS, Burrows HD, Gunnlaugsson T. Supramol Chem. 2008;20:407. [Google Scholar]
- 400.Kwon JY, Jang YJ, Kim SK, Lee KH, Kim JS, Yoon J. J Org Chem. 2004;69:5155. doi: 10.1021/jo049281+. [DOI] [PubMed] [Google Scholar]
- 401.García-Garrido SE, Caltagirone C, Light ME, Gale PA. Chem Commun. 2007:1450. doi: 10.1039/b618072h. [DOI] [PubMed] [Google Scholar]
- 402.Thangadurai TD, Singh NJ, Hwang IC, Lee JW, Chandran RP, Kim KS. J Org Chem. 2007;72:5461. doi: 10.1021/jo070791o. [DOI] [PubMed] [Google Scholar]
- 403.Hiscock JR, Caltagirone C, Light ME, Hursthouse MB, Gale PA. Org Biomol Chem. 2009;7:1781. doi: 10.1039/b900178f. [DOI] [PubMed] [Google Scholar]
- 404.Wu Y, Peng X, Fan J, Gao S, Tian M, Zhao J, Sun S. J Org Chem. 2007;72:62. doi: 10.1021/jo061634c. [DOI] [PubMed] [Google Scholar]
- 405.Stephan H, Spies H, Johannsen B, Klein L, Vögtle F. Chem Commun. 1999:1875. [Google Scholar]
- 406.Buhlmann P, Nishizawa S, Xiao KP, Umezawa Y. Tetrahedron. 1997;53:1647. [Google Scholar]
- 407.Xiao KP, Buehlmann P, Umezawa Y. Anal Chem. 1999;71:1183. doi: 10.1021/ac980689e. [DOI] [PubMed] [Google Scholar]
- 408.Nishizawa S, Yokobori T, Shioya T, Teramae N. Chem Lett. 2001:1058. [Google Scholar]
- 409.Nishizawa S, Yokobori T, Kato R, Yoshimoto K, Kamaishi T, Teramae N. Analyst. 2003;128:663. doi: 10.1039/b301141k. [DOI] [PubMed] [Google Scholar]
- 410.Lee KH, Hong JI. Tetrahedron Lett. 2000;41:6083. [Google Scholar]
- 411.Nishizawa S, Kato R, Hayashita T, Teramae N. Anal Sci. 1998;14:595. [Google Scholar]
- 412.Hayashita T, Onodera T, Kato R, Nishizawa S, Teramae N. Chem Commun. 2000:755. [Google Scholar]
- 413.Nishizawa S, Teramae N. Anal Sci. 1997;13:485. [Google Scholar]
- 414.Nishizawa S, Shigemori K, Teramae N. Chem Lett. 1999:1185. [Google Scholar]
- 415.Shigemori K, Nishizawa S, Yokobori T, Shioya T, Teramae N. New J Chem. 2002;26:1102. [Google Scholar]
- 416.Kato R, Cui YY, Nishizawa S, Yokobori T, Teramae N. Tetrahedron Lett. 2004;45:4273. [Google Scholar]
- 417.Xie H, Yi S, Wu S. J Chem Soc, Perkin Trans 2. 1999:2751. [Google Scholar]
- 418.Tobe Y, Sasaki SI, Mizuno M, Naemura K. Chem Lett. 1998:835. [Google Scholar]
- 419.Sasaki Si, Mizuno M, Naemura K, Tobe Y. J Org Chem. 2000;65:275. doi: 10.1021/jo991237k. [DOI] [PubMed] [Google Scholar]
- 420.Herges R, Dikmans A, Jana U, Kohler F, Jones PG, Dix I, Fricke T, König B. Eur J Org Chem. 2002:3004. [Google Scholar]
- 421.Sasaki Si, Citterio D, Ozawa S, Suzuki K. J Chem Soc, Perkin Trans 2. 2001:2309. [Google Scholar]
- 422.Gunnlaugsson T, Davis AP, Glynn M. Chem Commun. 2001:2556. [Google Scholar]
- 423.Gunnlaugsson T, Davis AP, Hussey GM, Tierney J, Glynn M. Org Biomol Chem. 2004;2:1856. doi: 10.1039/b404706k. [DOI] [PubMed] [Google Scholar]
- 424.Ohshiro I, Ikegami M, Shinohara Y, Nishimura Y, Arai T. Bull Chem Soc Jpn. 2007;80:747. [Google Scholar]
- 425.Gunnlaugsson T, Davis AP, O'Brien JE, Glynn M. Org Lett. 2002;4:2449. doi: 10.1021/ol026004l. [DOI] [PubMed] [Google Scholar]
- 426.Gunnlaugsson T, Davis AP, O'Brien JE, Glynn M. Org Biomol Chem. 2005;3:48. doi: 10.1039/b409018g. [DOI] [PubMed] [Google Scholar]
- 427.Gunnlaugsson T, Kruger PE, Lee TC, Parkesh R, Pfeffer FM, Hussey GM. Tetrahedron Lett. 2003;44:6575. [Google Scholar]
- 428.Gunnlaugsson T, Kruger PE, Jensen P, Tierney J, Ali HDP, Hussey GM. J Org Chem. 2005;70:10875. doi: 10.1021/jo0520487. [DOI] [PubMed] [Google Scholar]
- 429.Pfeffer FM, Buschgens AM, Barnett NW, Gunnlaugsson T, Kruger PE. Tetrahedron Lett. 2005;46:6579. [Google Scholar]
- 430.Pfeffer FM, Seter M, Lewcenko N, Barnett NW. Tetrahedron Lett. 2006;47:5241. [Google Scholar]
- 431.Duke RM, O'Brien JE, McCabe T, Gunnlaugsson T. Org Biomol Chem. 2008;6:4089. doi: 10.1039/b807579d. [DOI] [PubMed] [Google Scholar]
- 432.Ali HDP, Quinn SJ, McCabe T, Kruger PE, Gunnlaugsson T. New J Chem. 2009;33:793. [Google Scholar]
- 433.Hennrich G, Sonnenschein H, Resch-Genger U. Tetrahedron Lett. 2001;42:2805. doi: 10.1021/ic000827u. [DOI] [PubMed] [Google Scholar]
- 434.Lee DH, Lee KH, Hong JI. Org Lett. 2001;3:5. doi: 10.1021/ol006690t. [DOI] [PubMed] [Google Scholar]
- 435.Lee DH, Lee HY, Lee KH, Hong JI. Chem Commun. 2001:1188. [Google Scholar]
- 436.Lee DH, Lee HY, Hong JI. Tetrahedron Lett. 2002;43:7273. [Google Scholar]
- 437.Wu FY, Li Z, Wen ZC, Zhou N, Zhao YF, Jiang YB. Org Lett. 2002;4:3203. doi: 10.1021/ol026357k. [DOI] [PubMed] [Google Scholar]
- 438.Wu FY, Li Z, Guo L, Wang X, Lin MH, Zhao YF, Jiang YB. Org Biomol Chem. 2006;4:624. doi: 10.1039/b513969d. [DOI] [PubMed] [Google Scholar]
- 439.Liu WX, Jiang YB. Org Biomol Chem. 2007;5:1771. doi: 10.1039/b703122j. [DOI] [PubMed] [Google Scholar]
- 440.Nie L, Li Z, Han J, Zhang X, Yang R, Liu WX, Wu FY, Xie JW, Zhao YF, Jiang YB. J Org Chem. 2004;69:6449. doi: 10.1021/jo049088f. [DOI] [PubMed] [Google Scholar]
- 441.Li Z, Liu Z, Liao QX, Wei ZB, Long LS, Jiang YB. Comptes Rendus Chimie. 2008;11:67. [Google Scholar]
- 442.Liu WX, Jiang YB. J Org Chem. 2008;73:1124. doi: 10.1021/jo702159r. [DOI] [PubMed] [Google Scholar]
- 443.Yang R, Liu WX, Shen H, Huang HH, Jiang YB. J Phys Chem B. 2008;112:5105. doi: 10.1021/jp711899h. [DOI] [PubMed] [Google Scholar]
- 444.Liu WX, Yang R, Li AF, Li Z, Gao YF, Luo XX, Ruan YB, Jiang YB. Org Biomol Chem. 2009;7:4021. doi: 10.1039/b910255h. [DOI] [PubMed] [Google Scholar]
- 445.Wei LH, He YB, Wu JL, Wu XJ, Meng LZ, Yang X. Supramol Chem. 2004;16:561. [Google Scholar]
- 446.Wu JL, He YB, Wei LH, Meng LZ, Yang TX, Liu X. Aust J Chem. 2005;58:53. [Google Scholar]
- 447.Wu Jl, Wei Lh, Zeng Zy, Liu Sy, Gong R, Meng Lz, He Yb. Chin J Chem. 2003;21:1553. [Google Scholar]
- 448.Zeng ZY, He YB, Wei LH, Wu JL, Huang YY, Meng LZ. Can J Chem. 2004;82:454. [Google Scholar]
- 449.Ros-Lis JV, Martínez-Máñez R, Sancenón F, Soto J, Rurack K, Weisshoff H. Eur J Org Chem. 2007:2449. [Google Scholar]
- 450.Gomez DE, Fabbrizzi L, Licchelli M, Monzani E. Org Biomol Chem. 2005;3:1495. doi: 10.1039/b500123d. [DOI] [PubMed] [Google Scholar]
- 451.Camiolo S, Gale PA, Hursthouse MB, Light ME. Org Biomol Chem. 2003;1:741. doi: 10.1039/b210848h. [DOI] [PubMed] [Google Scholar]
- 452.Evans LS, Gale PA, Light ME, Quesada R. Chem Commun. 2006:965. doi: 10.1039/b517308f. [DOI] [PubMed] [Google Scholar]
- 453.Caltagirone C, Bates GW, Gale PA, Light ME. Chem Commun. 2008:61. doi: 10.1039/b713431b. [DOI] [PubMed] [Google Scholar]
- 454.Jose DA, Kumar DK, Ganguly B, Das A. Tetrahedron Lett. 2005;46:5343. [Google Scholar]
- 455.Kim SK, Singh NJ, Kim SJ, Swamy KMK, Kim SH, Lee KH, Kim KS, Yoon J. Tetrahedron. 2005;61:4545. [Google Scholar]
- 456.Pfeffer FM, Gunnlaugsson T, Jensen P, Kruger PE. Org Lett. 2005;7:5357. doi: 10.1021/ol051497q. [DOI] [PubMed] [Google Scholar]
- 457.Pfeffer FM, Kruger PE, Gunnlaugsson T. Org Biomol Chem. 2007;5:1894. doi: 10.1039/b703208k. [DOI] [PubMed] [Google Scholar]
- 458.Lowe AJ, Dyson GA, Pfeffer FM. Org Biomol Chem. 2007;5:1343. doi: 10.1039/b703626b. [DOI] [PubMed] [Google Scholar]
- 459.Lowe AJ, Dyson GA, Pfeffer FM. Eur J Org Chem. 2008:1559. [Google Scholar]
- 460.Thiagarajan V, Ramamurthy P, Thirumalai D, Ramakrishnan VT. Org Lett. 2005;7:657. doi: 10.1021/ol047463k. [DOI] [PubMed] [Google Scholar]
- 461.Bonizzoni M, Fabbrizzi L, Taglietti A, Tiengo F. Eur J Org Chem. 2006:3567. [Google Scholar]
- 462.Zyryanov GV, Palacios MA, Anzenbacher P., Jr Angew Chem, Int Ed. 2007;46:7849. doi: 10.1002/anie.200702611. [DOI] [PubMed] [Google Scholar]
- 463.Lee GW, Singh N, Jang DO. Tetrahedron Lett. 2008;49:1952. [Google Scholar]
- 464.Black CB, Andrioletti B, Try AC, Ruiperez C, Sessler JL. J Am Chem Soc. 1999;121:10438. [Google Scholar]
- 465.Anzenbacher P, Jr, Try AC, Miyaji H, Jursikova K, Lynch VM, Marquez M, Sessler JL. J Am Chem Soc. 2000;122:10268. [Google Scholar]
- 466.Sessler JL, Maeda H, Mizuno T, Lynch VM, Furuta H. Chem Commun. 2002:862. doi: 10.1039/b111708d. [DOI] [PubMed] [Google Scholar]
- 467.Sessler JL, Pantos GD, Katayev E, Lynch VM. Org Lett. 2003;5:4141. doi: 10.1021/ol0355635. [DOI] [PubMed] [Google Scholar]
- 468.Aldakov D, Anzenbacher P., Jr Chem Commun. 2003:1394. [PubMed] [Google Scholar]
- 469.Aldakov D, Palacios MA, Anzenbacher P., Jr Chem Mater. 2005;17:5238. [Google Scholar]
- 470.Pohl R, Aldakov D, Kubat P, Jursikova K, Marquez M, Anzenbacher P., Jr Chem Commun. 2004:1282. doi: 10.1039/b315268e. [DOI] [PubMed] [Google Scholar]
- 471.Shevchuk SV, Lynch VM, Sessler JL. Tetrahedron. 2004;60:11283. [Google Scholar]
- 472.Gale PA, Camiolo S, Chapman CP, Light ME, Hursthouse MB. Tetrahedron Lett. 2001;42:5095. [Google Scholar]
- 473.Gale PA, Camiolo S, Tizzard GJ, Chapman CP, Light ME, Coles SJ, Hursthouse MB. J Org Chem. 2001;66:7849. doi: 10.1021/jo016020g. [DOI] [PubMed] [Google Scholar]
- 474.Camiolo S, Gale PA, Hursthouse MB, Light ME, Warriner CN. Tetrahedron Lett. 2003;44:1367. [Google Scholar]
- 475.Camiolo S, Coles SJ, Gale PA, Hursthouse MB, Tizzard GJ. Supramol Chem. 2003;15:231. [Google Scholar]
- 476.Vega IED, Camiolo S, Gale PA, Hursthouse MB, Light ME. Chem Commun. 2003:1686. [Google Scholar]
- 477.El Drubi Vega I, Gale PA, Hursthouse MB, Light ME. Org Biomol Chem. 2004;2:2935. doi: 10.1039/B409115A. [DOI] [PubMed] [Google Scholar]
- 478.Li R, Evans LS, Larsen DS, Gale PA, Brooker S. New J Chem. 2004;28:1340. [Google Scholar]
- 479.Evans LS, Gale PA, Light ME, Quesada R. New J Chem. 2006;30:1019. [Google Scholar]
- 480.Sessler JL, Pantos GD, Gale PA, Light ME. Org Lett. 2006;8:1593. doi: 10.1021/ol060193g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Zielinski T, Jurczak J. Tetrahedron. 2005;61:4081. [Google Scholar]
- 482.Maeda H, Kusunose Y. Chem--Eur J. 2005;11:5661. doi: 10.1002/chem.200500627. [DOI] [PubMed] [Google Scholar]
- 483.Maeda H, Ito Y. Inorg Chem. 2006;45:8205. doi: 10.1021/ic0608703. [DOI] [PubMed] [Google Scholar]
- 484.Fujimoto C, Kusunose Y, Maeda H. J Org Chem. 2006;71:2389. doi: 10.1021/jo052511f. [DOI] [PubMed] [Google Scholar]
- 485.Maeda H, Kusunose Y, Mihashi Y, Mizoguchi T. J Org Chem. 2007;72:2612. doi: 10.1021/jo062660d. [DOI] [PubMed] [Google Scholar]
- 486.Maeda HHY, Nakanishi T. J Am Chem Soc. 2007;129:13661. doi: 10.1021/ja074435z. [DOI] [PubMed] [Google Scholar]
- 487.Maeda H, Haketa Y. Org Biomol Chem. 2008;6:3091. doi: 10.1039/b806161k. [DOI] [PubMed] [Google Scholar]
- 488.Maeda H, Fujii Y, Mihashi Y. Chem Commun. 2008:4285. doi: 10.1039/b806361c. [DOI] [PubMed] [Google Scholar]
- 489.Maeda H, Mihashi Y, Haketa Y. Org Lett. 2008;10:3179. doi: 10.1021/ol801014z. [DOI] [PubMed] [Google Scholar]
- 490.Maeda H, Terasaki M, Haketa Y, Mihashi Y, Kusunose Y. Org Biomol Chem. 2008;6:433. doi: 10.1039/b718317h. [DOI] [PubMed] [Google Scholar]
- 491.Maeda H, Eifuku N. Chem Lett. 2009;38:208. [Google Scholar]
- 492.Yin Z, Zhang Y, He J, Cheng JP. Tetrahedron. 2006;62:765. [Google Scholar]
- 493.Yin Z, Zhang Y, He J, Cheng JP. Tetrahedron. 2006;62:2936. [Google Scholar]
- 494.Zhang Y, Yin Z, Li Z, He J, Cheng JP. Tetrahedron. 2007;63:7560. [Google Scholar]
- 495.Curiel D, Cowley A, Beer PD. Chem Commun. 2005:236. doi: 10.1039/b412363h. [DOI] [PubMed] [Google Scholar]
- 496.Kwon TH, Jeong KS. Tetrahedron Lett. 2006;47:8539. [Google Scholar]
- 497.Ju J, Park M, Suk Jm, Lah MS, Jeong KS. Chem Commun. 2008:3546. doi: 10.1039/b804284e. [DOI] [PubMed] [Google Scholar]
- 498.Chang KJ, Moon D, Lah MS, Jeong KS. Angew Chem, Int Ed. 2005;44:7926. doi: 10.1002/anie.200503121. [DOI] [PubMed] [Google Scholar]
- 499.Lee JY, Lee MH, Jeong KS. Supramol Chem. 2007;19:257. [Google Scholar]
- 500.Sessler JL, Cho DG, Lynch V. J Am Chem Soc. 2006;128:16518. doi: 10.1021/ja067720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Bates GW, Gale PA, Light ME. Chem Commun. 2007:2121. doi: 10.1039/b703905k. [DOI] [PubMed] [Google Scholar]
- 502.Bates GW, Triyanti, Light ME, Albrecht M, Gale PA. J Org Chem. 2007;72:8921. doi: 10.1021/jo701702p. [DOI] [PubMed] [Google Scholar]
- 503.Caltagirone C, Gale PA, Hiscock JR, Brooks SJ, Hursthouse MB, Light ME. Chem Commun. 2008:3007. doi: 10.1039/b806238b. [DOI] [PubMed] [Google Scholar]
- 504.Caltagirone C, Hiscock JR, Hursthouse MB, Light ME, Gale PA. Chem--Eur J. 2008;14:10236. doi: 10.1002/chem.200801639. [DOI] [PubMed] [Google Scholar]
- 505.Zielinski T, Dydio P, Jurczak J. Tetrahedron. 2008;64:568. [Google Scholar]
- 506.Dydio P, Zielinski T, Jurczak J. Chem Commun. 2009:4560. doi: 10.1039/b907164d. [DOI] [PubMed] [Google Scholar]
- 507.Pfeffer FM, Lim KF, Sedgwick KJ. Org Biomol Chem. 2007;5:1795. doi: 10.1039/b702804k. [DOI] [PubMed] [Google Scholar]
- 508.Wang Y, Lin H, Shao J, Cai ZS, Lin HK. Talanta. 2008;74:1122. doi: 10.1016/j.talanta.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 509.Caltagirone C, Mulas A, Isaia F, Lippolis V, Gale PA, Light ME. Chem Commun. 2009:6279. doi: 10.1039/b912942a. [DOI] [PubMed] [Google Scholar]
- 510.Caltagirone C, Gale PA, Hiscock JR, Hursthouse MB, Light ME, Tizzard GJ. Supramol Chem. 2009;21:125. [Google Scholar]
- 511.Dydio P, Zielinski T, Jurczak J. J Org Chem. 2009;74:1525. doi: 10.1021/jo802288u. [DOI] [PubMed] [Google Scholar]
- 512.Kim H, Kang J. Bull Korean Chem Soc. 2007;28:1531. [Google Scholar]
- 513.Peng X, Wu Y, Fan J, Tian M, Han K. J Org Chem. 2005;70:10524. doi: 10.1021/jo051766q. [DOI] [PubMed] [Google Scholar]
- 514.Kang J, Kim HS, Jang DO. Tetrahedron Lett. 2005;46:6079. [Google Scholar]
- 515.Moon KS, Singh N, Lee GW, Jang DO. Tetrahedron. 2007;63:9106. [Google Scholar]
- 516.Lee GW, Singh N, Jung HJ, Jang DO. Tetrahedron Lett. 2009;50:807. [Google Scholar]
- 517.Kang SO, Nguyen QPB, Kim TH. Bull Korean Chem Soc. 2009;30:2735. [Google Scholar]
- 518.Tong H, Zhou G, Wang L, Jing X, Wang F, Zhang J. Tetrahedron Lett. 2003;44:131. [Google Scholar]
- 519.Schnopp M, Ernst S, Haberhauer G. Eur J Org Chem. 2009:213. [Google Scholar]
- 520.Aoyama Y, Mizokami K, Toi H. Chem Lett. 1990:651. [Google Scholar]
- 521.Motomura T, Aoyama Y. J Org Chem. 1991;56:7224. [Google Scholar]
- 522.Manabe K, Okamura K, Date T, Koga K. J Am Chem Soc. 1992;114:6940. [Google Scholar]
- 523.Kruger PE, Mackie PR, Nieuwenhuyzen M. J Chem Soc, Perkin Trans 2. 2001:1079. [Google Scholar]
- 524.Lu H, Xu W, Zhang D, Zhu D. Chem Commun. 2005:4777. doi: 10.1039/b509133k. [DOI] [PubMed] [Google Scholar]
- 525.Miyaji H, Sessler JL. Angew Chem, Int Ed. 2001;40:154. doi: 10.1002/1521-3773(20010105)40:1<154::AID-ANIE154>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 526.Zhou LL, Zhang XH, Wu SK. Sens Actuators, B. 2005;B106:343. [Google Scholar]
- 527.Ou SJ, Zhang BG, Liao HP, Bai ZP. Wuji Huaxue Xuebao. 2006;22:817. [Google Scholar]
- 528.Shang XF, Xu XF. Biosystems. 2009;96:165. doi: 10.1016/j.biosystems.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 529.Li J, Lin H, Lin H. J Coord Chem. 2009;62:1921. [Google Scholar]
- 530.Neus Pina M, Rotger MC, Costa A, Ballester P, Deya PM. Tetrahedron Lett. 2004;45:3749. [Google Scholar]
- 531.Sancenón F, Descalzo AB, Martínez-Máñez R, Miranda MA, Soto J. Angew Chem, Int Ed. 2001;40:2640. doi: 10.1002/1521-3773(20010716)40:14<2640::AID-ANIE2640>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 532.Zhou LL, Sun H, Li HP, Wang H, Zhang XH, Wu SK, Lee ST. Org Lett. 2004;6:1071. doi: 10.1021/ol035818e. [DOI] [PubMed] [Google Scholar]
- 533.Davis AP, Perry JJ, Wareham RS. Tetrahedron Lett. 1998;39:4569. [Google Scholar]
- 534.Kondo Si, Suzuki T, Yano Y. Tetrahedron Lett. 2002;43:7059. [Google Scholar]
- 535.Sowadski JM, Handschumacher MD, Murthy HMK, Foster BA, Wyckoff HW. J Mol Biol. 1985;186:417. doi: 10.1016/0022-2836(85)90115-9. [DOI] [PubMed] [Google Scholar]
- 536.Kim EE, Wyckoff HW. Clin Chim Acta. 1989;186:175. doi: 10.1016/0009-8981(90)90035-q. [DOI] [PubMed] [Google Scholar]
- 537.Hough E, Hansen LK, Birknes B, Jynge K, Hansen S, Hordvik A, Little C, Dodson E, Derewenda Z. Nature. 1989;338:357. doi: 10.1038/338357a0. [DOI] [PubMed] [Google Scholar]
- 538.Kim EE, Wyckoff HW. J Mol Biol. 1991;218:449. doi: 10.1016/0022-2836(91)90724-k. [DOI] [PubMed] [Google Scholar]
- 539.Coleman JE. Annu Rev Biophys Biomol Struct. 1992;21:441. doi: 10.1146/annurev.bb.21.060192.002301. [DOI] [PubMed] [Google Scholar]
- 540.Coleman JE. Annu Rev Biochem. 1992;61:897. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 541.Hansen S, Hansen LK, Hough E. J Mol Biol. 1992;225:543. doi: 10.1016/0022-2836(92)90938-g. [DOI] [PubMed] [Google Scholar]
- 542.Fenton DE, Okawa H. J Chem Soc, Dalton Trans. 1993:1349. [Google Scholar]
- 543.Hansen S, Hough E, Svensson LA, Wong YL, Martin SF. J Mol Biol. 1993;234:179. doi: 10.1006/jmbi.1993.1572. [DOI] [PubMed] [Google Scholar]
- 544.Karlin KD. Science. 1993;261:701. doi: 10.1126/science.7688141. [DOI] [PubMed] [Google Scholar]
- 545.Lipscomb WN, Straeter N. Chem Rev. 1996;96:2375. doi: 10.1021/cr950042j. [DOI] [PubMed] [Google Scholar]
- 546.Hawthorne MF, Yang X, Zheng Z. Pure Appl Chem. 1994;66:245. [Google Scholar]
- 547.Ganjali MR, Mizani F, Emami M, Niasari MS, Shamsipur M, Yousefi M, Javanbakht M. Electroanalysis. 2003;15:139. [Google Scholar]
- 548.Ganjali MR, Mizani F, Salavati-Niasari M. Anal Chim Acta. 2003;481:85. [Google Scholar]
- 549.Swamy KMK, Kwon SK, Lee HN, Kumar SMS, Kim JS, Yoon J. Tetrahedron Lett. 2007;48:8683. [Google Scholar]
- 550.Chin J, Chung S, Kim DH. J Am Chem Soc. 2002;124:10948. doi: 10.1021/ja026920u. [DOI] [PubMed] [Google Scholar]
- 551.Receptors 498, 501, 504, and 507 in this section are identical to compounds 18, 65, 70, and 66, respectively, in section 4.1.1. They are shown again to facilitate direct comparison between the free base and metalated receptors.
- 552.Motekaitis RJ, Martell AE. Inorg Chem. 1992;31:5534. [Google Scholar]
- 553.Jurek PE, Martell AE, Motekaitis RJ, Hancock RD. Inorg Chem. 1995;34:1823. [Google Scholar]
- 554.Motekaitis RJ, Martell AE. Inorg Chem. 1994;33:1032. [Google Scholar]
- 555.Lu Q, Reibenspies JJ, Martell AE, Motekaitis RJ. Inorg Chem. 1996;35:2630. doi: 10.1021/ic951384a. [DOI] [PubMed] [Google Scholar]
- 556.Lu Q, Reibenspies JH, Martell AE, Motekaitis RJ. Inorg Chem. 1996;35:7462. doi: 10.1021/ic960914f. [DOI] [PubMed] [Google Scholar]
- 557.Lu Q, Reibenspies JH, Carroll RI, Martell AE, Clearfiled A. Inorg Chim Acta. 1998;270:207. [Google Scholar]
- 558.English JB, Martell AE, Motekaitis RJ, Murase I. Inorg Chim Acta. 1997;258:183. [Google Scholar]
- 559.Nation DA, Martell AE, Carroll RI, Clearfield A. Inorg Chem. 1996;35:7246. doi: 10.1021/ic960661q. [DOI] [PubMed] [Google Scholar]
- 560.Pauwels TF, Lippens W, Herman GG, Goeminne AM. Polyhedron. 1998;17:1715. [Google Scholar]
- 561.Fabbrizzi L, Marcotte N, Stomeo F, Taglietti A. Angew Chem, Int Ed. 2002;41:3811. doi: 10.1002/1521-3773(20021018)41:20<3811::AID-ANIE3811>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 562.Marcotte N, Taglietti A. Supramol Chem. 2003;15:617. [Google Scholar]
- 563.Barker JE, Liu Y, Martin ND, Ren T. J Am Chem Soc. 2003;125:13332. doi: 10.1021/ja036407w. [DOI] [PubMed] [Google Scholar]
- 564.Tobey SL, Jones BD, Anslyn EV. J Am Chem Soc. 2003;125:4026. doi: 10.1021/ja021390n. [DOI] [PubMed] [Google Scholar]
- 565.Tobey SL, Anslyn EV. J Am Chem Soc. 2003;125:14807. doi: 10.1021/ja030507k. [DOI] [PubMed] [Google Scholar]
- 566.Tobey SL, Anslyn EV. Org Lett. 2003;5:2029. doi: 10.1021/ol034427x. [DOI] [PubMed] [Google Scholar]
- 567.Zhong Z, Anslyn EV. Angew Chem, Int Ed. 2003;42:3005. doi: 10.1002/anie.200351165. [DOI] [PubMed] [Google Scholar]
- 568.Jang YJ, Jun EJ, Lee YJ, Kim YS, Kim JS, Yoon J. J Org Chem. 2005;70:9603. doi: 10.1021/jo0509657. [DOI] [PubMed] [Google Scholar]
- 569.Kim SY, Hong JI. Tetrahedron Lett. 2009;50:1951. [Google Scholar]
- 570.Shang XF, Lin H, Cai ZS, Lin HK. Talanta. 2007;73:296. doi: 10.1016/j.talanta.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 571.Oh DJ, Han MS, Ahn KH. Supramol Chem. 2007;19:315. [Google Scholar]
- 572.Oh DJ, Ahn KH. Org Lett. 2008;10:3539. doi: 10.1021/ol801290g. [DOI] [PubMed] [Google Scholar]
- 573.Koike T, Kimura E. J Am Chem Soc. 1991;113:8935. [Google Scholar]
- 574.Koike T, Kajitani S, Nakamura I, Kimura E, Shiro M. J Am Chem Soc. 1995;117:1210. [Google Scholar]
- 575.Aoki S, Kagata D, Shiro M, Takeda K, Kimura E. J Am Chem Soc. 2004;126:13377. doi: 10.1021/ja040095v. [DOI] [PubMed] [Google Scholar]
- 576.Aoki S, Jikiba A, Takeda K, Kimura E. J Phys Org Chem. 2004;17:489. [Google Scholar]
- 577.Fujioka H, Koike T, Yamada N, Kimura E. Heterocycles. 1996;42:775. [Google Scholar]
- 578.Aoki S, Kimura E. J Am Chem Soc. 2000;122:4542. [Google Scholar]
- 579.Kimura E, Aoki S, Koike T, Shiro M. J Am Chem Soc. 1997;119:3068. [Google Scholar]
- 580.Aoki S, Zulkefeli M, Shiro M, Kohsako M, Takeda K, Kimura E. J Am Chem Soc. 2005;127:9129. doi: 10.1021/ja050876b. [DOI] [PubMed] [Google Scholar]
- 581.Gunning P, Benniston AC, Peacock RD. Chem Commun. 2004:2226. doi: 10.1039/b409132a. [DOI] [PubMed] [Google Scholar]
- 582.Benniston AC, Gunning P, Peacock RD. J Org Chem. 2005;70:115. doi: 10.1021/jo048621o. [DOI] [PubMed] [Google Scholar]
- 583.Turygin DS, Subat M, Raitman OA, Selector SL, Arslanov VV, König B, Kalinina MA. Langmuir. 2007;23:2517. doi: 10.1021/la0624079. [DOI] [PubMed] [Google Scholar]
- 584.Jose DA, Stadlbauer S, König B. Chem--Eur J. 2009;15:7404. doi: 10.1002/chem.200900339. [DOI] [PubMed] [Google Scholar]
- 585.Ge R, Lin H, Xu X, Sun X, Lin H, Zhu S, Ji B, Li F, Wu H. J Inorg Biochem. 2004;98:917. doi: 10.1016/j.jinorgbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 586.Ng PL, Lee CS, Kwong HL, Chan ASC. Inorg Chem Commun. 2005;8:769. [Google Scholar]
- 587.Bazzicalupi C, Bencini A, Bianchi A, Danesi A, Giorgi C, Lodeiro C, Pina F, Santarelli S, Valtancoli B. Chem Commun. 2005:2630. doi: 10.1039/b502229k. [DOI] [PubMed] [Google Scholar]
- 588.Gunning PT. Org Biomol Chem. 2005;3:3877. doi: 10.1039/b510262f. [DOI] [PubMed] [Google Scholar]
- 589.Bazzicalupi C, Bencini A, Bianchi A, Borsari L, Danesi A, Giorgi C, Lodeiro C, Mariani P, Pina F, Santarelli S, Tamayo A, Valtancoli B. Dalton Trans. 2006:4000. doi: 10.1039/b603505a. [DOI] [PubMed] [Google Scholar]
- 590.Seo JS, Sung ND, Hynes RC, Chin J. Inorg Chem. 1996;35:7472. [Google Scholar]
- 591.Han MS, Kim DH. Angew Chem, Int Ed. 2002;41:3809. doi: 10.1002/1521-3773(20021018)41:20<3809::AID-ANIE3809>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 592.Han MS, Kim DH. Bioorg Med Chem Lett. 2003;13:1079. doi: 10.1016/s0960-894x(03)00055-6. [DOI] [PubMed] [Google Scholar]
- 593.Morgan BP, He S, Smith RC. Inorg Chem. 2007;46:9262. doi: 10.1021/ic701374h. [DOI] [PubMed] [Google Scholar]
- 594.Ojida A, Park Sk, Mito-oka Y, Hamachi I. Tetrahedron Lett. 2002;43:6193. [Google Scholar]
- 595.Wongkongkatep J, Miyahara Y, Ojida A, Hamachi I. Angew Chem, Int Ed. 2006;45:665. doi: 10.1002/anie.200503107. [DOI] [PubMed] [Google Scholar]
- 596.Ojida A, Mito-oka Y, Sada K, Hamachi I. J Am Chem Soc. 2004;126:2454. doi: 10.1021/ja038277x. [DOI] [PubMed] [Google Scholar]
- 597.Ojida A, Mitooka Y, Inoue M, Hamachi I. J Am Chem Soc. 2002;124:6256. doi: 10.1021/ja025761b. [DOI] [PubMed] [Google Scholar]
- 598.Ojida A, Kohira T, Hamachi I. Chem Lett. 2004;33:1024. [Google Scholar]
- 599.Ojida A, Inoue Ma, Mito-oka Y, Tsutsumi H, Sada K, Hamachi I. J Am Chem Soc. 2006;128:2052. doi: 10.1021/ja056585k. [DOI] [PubMed] [Google Scholar]
- 600.Ojida A, Miyahara Y, Kohira T, Hamachi I. Biopolymers. 2004;76:177. doi: 10.1002/bip.10574. [DOI] [PubMed] [Google Scholar]
- 601.Ojida A, Hamachi I. Bull Chem Soc Jpn. 2006;79:35. [Google Scholar]
- 602.Ojida A, Inoue M, Mitooka Y, Hamachi I. J Am Chem Soc. 2003;125:10184. doi: 10.1021/ja036317r. [DOI] [PubMed] [Google Scholar]
- 603.Ojida A, Miyahara Y, Wongkongkatep J, Tamaru Si, Sada K, Hamachi I. Chem--Asian J. 2006;1:555. doi: 10.1002/asia.200600137. [DOI] [PubMed] [Google Scholar]
- 604.Ojida A, Nanaka H, Miyahara Y, Tamaru Si, Sada K, Hamachi I. Angew Chem, Int Ed. 2006;45:5518. doi: 10.1002/anie.200601315. [DOI] [PubMed] [Google Scholar]
- 605.Ojida A, Takashima I, Kohira T, Nonaka H, Hamachi I. J Am Chem Soc. 2008;130:12095. doi: 10.1021/ja803262w. [DOI] [PubMed] [Google Scholar]
- 606.Yoshimura I, Miyahara Y, Kasagi N, Yamane H, Ojida A, Hamachi I. J Am Chem Soc. 2004;126:12204. doi: 10.1021/ja045962a. [DOI] [PubMed] [Google Scholar]
- 607.Yamaguchi S, Yoshimura I, Kohira T, Tamaru S, Hamachi I. J Am Chem Soc. 2005;127:11835. doi: 10.1021/ja052838y. [DOI] [PubMed] [Google Scholar]
- 608.Lee DH, Im JH, Son SU, Chung YK, Hong JI. J Am Chem Soc. 2003;125:7752. doi: 10.1021/ja034689u. [DOI] [PubMed] [Google Scholar]
- 609.Lee DH, Kim SY, Hong JI. Angew Chem, Int Ed. 2004;43:4777. doi: 10.1002/anie.200453914. [DOI] [PubMed] [Google Scholar]
- 610.Lee JH, Park J, Lah MS, Chin J, Hong JI. Org Lett. 2007;9:3729. doi: 10.1021/ol071306e. [DOI] [PubMed] [Google Scholar]
- 611.Lee DH, Kim SY, Hong JI. Tetrahedron Lett. 2007;48:4477. [Google Scholar]
- 612.Cho HK, Lee DH, Hong JI. Chem Commun. 2005:1690. doi: 10.1039/b417845a. [DOI] [PubMed] [Google Scholar]
- 613.Rhee HW, Choi HY, Han K, Hong JI. J Am Chem Soc. 2007;129:4524. doi: 10.1021/ja070026r. [DOI] [PubMed] [Google Scholar]
- 614.Koulov AV, Stucker KA, Lakshmi C, Robinson JP, Smith BD. Cell Death Differ. 2003;10:1357. doi: 10.1038/sj.cdd.4401315. [DOI] [PubMed] [Google Scholar]
- 615.Lakshmi C, Hanshaw RG, Smith BD. Tetrahedron. 2004;60:11307. [Google Scholar]
- 616.Hanshaw RG, Hilkert SM, Jiang H, Smith BD. Tetrahedron Lett. 2004;45:8721. [Google Scholar]
- 617.Hanshaw RG, O'Neil EJ, Foley M, Carpenter RT, Smith BD. J Mater Chem. 2005;15:2707. [Google Scholar]
- 618.McDonough MJ, Reynolds AJ, Lee WYG, Jolliffe KA. Chem Commun. 2006:2971. doi: 10.1039/b606917g. [DOI] [PubMed] [Google Scholar]
- 619.Lee HN, Swamy KMK, Kim SK, Kwon JY, Kim Y, Kim SJ, Yoon YJ, Yoon J. Org Lett. 2007;9:243. doi: 10.1021/ol062685z. [DOI] [PubMed] [Google Scholar]
- 620.Lee HN, Xu Z, Kim SK, Swamy KMK, Kim Y, Kim SJ, Yoon J. J Am Chem Soc. 2007;129:3828. doi: 10.1021/ja0700294. [DOI] [PubMed] [Google Scholar]
- 621.Jose DA, Mishra S, Ghosh A, Shrivastav A, Mishra SK, Das A. Org Lett. 2007;9:1979. doi: 10.1021/ol0705797. [DOI] [PubMed] [Google Scholar]
- 622.Ghosh A, Shrivastav A, Jose DA, Mishra SK, Chandrakanth CK, Mishra S, Das A. Anal Chem. 2008;80:5312. doi: 10.1021/ac8005022. [DOI] [PubMed] [Google Scholar]
- 623.Su GY, Liu ZP, Xie ZJ, Qian F, He WJ, Guo ZJ. Dalton Trans. 2009:7888. doi: 10.1039/b914802g. [DOI] [PubMed] [Google Scholar]
- 624.Tang LJ, Li Y, Zhang H, Guo ZL, Qian JH. Tetrahedron Lett. 2009;50:6844. [Google Scholar]
- 625.Rodriguez L, Lima JC, Parola AJ, Pina F, Meitz R, Aucejo R, García-España E, Llinares JM, Soriano C, Alarcon J. Inorg Chem. 2008;47:6173. doi: 10.1021/ic7023956. [DOI] [PubMed] [Google Scholar]
- 626.Chen Z, Wang X, Chen J, Yang X, Li Y, Guo Z. New J Chem. 2007;31:357. [Google Scholar]
- 627.Ambrosi G, Formica M, Fusi V, Giorgi L, Guerri A, Macedi E, Micheloni M, Paoli P, Pontellini R, Rossi P. Inorg Chem. 2009;48:5901. doi: 10.1021/ic900231h. [DOI] [PubMed] [Google Scholar]
- 628.Khatua S, Choi SH, Lee J, Kim K, Do Y, Churchill DG. Inorg Chem. 2009;48:2993. doi: 10.1021/ic8022387. [DOI] [PubMed] [Google Scholar]
- 629.Wu JS, Wang F, Liu WM, Wang PF, Wu SK, Wu XH, Zhang XH. Sens Actuators, B. 2007;125:447. [Google Scholar]
- 630.Ion L, Morales D, Nieto S, Perez J, Riera L, Riera V, Miguel D, Kowenicki RA, McPartlin M. Inorg Chem. 2007;46:2846. doi: 10.1021/ic0621650. [DOI] [PubMed] [Google Scholar]
- 631.Wallace KJ, Daari R, Belcher WJ, Abouderbala LO, Boutelle MG, Steed JW. J Organomet Chem. 2003;666:63. [Google Scholar]
- 632.Tovilla JA, Vilar R, White AJP. Chem Commun. 2005:4839. doi: 10.1039/b509565d. [DOI] [PubMed] [Google Scholar]
- 633.Tovilla JA, Carlqvist P, Benet-Buchholz J, Maseras F, Vilar R. Supramol Chem. 2007;19:599. [Google Scholar]
- 634.Mendoza C, Benet-Buchholz J, Pericas MA, Vilar R. Dalton Trans. 2009:2974. doi: 10.1039/b819612e. [DOI] [PubMed] [Google Scholar]
- 635.Bondy CR, Gale PA, Loeb SJ. J Am Chem Soc. 2004;126:5030. doi: 10.1021/ja039712q. [DOI] [PubMed] [Google Scholar]
- 636.Mizukami S, Nagano T, Urano Y, Odani A, Kikuchi K. J Am Chem Soc. 2002;124:3920. doi: 10.1021/ja0175643. [DOI] [PubMed] [Google Scholar]
- 637.Zhang T, Anslyn EV. Org Lett. 2006;8:1649. doi: 10.1021/ol060279+. [DOI] [PubMed] [Google Scholar]
- 638.Glazier SA, Arnold MA. Anal Chem. 1988;60:2540. doi: 10.1021/ac00173a024. [DOI] [PubMed] [Google Scholar]
- 639.Glazier SA, Arnold MA. Anal Chem. 1991;63:754. doi: 10.1021/ac00008a003. [DOI] [PubMed] [Google Scholar]
- 640.DeMeulenaere RL, Onsrud P, Arnold MA. Electroanalysis. 1993;5:833. [Google Scholar]
- 641.Fluri K, Koudelka J, Simon W. Helv Chim Acta. 1992;75:1012. [Google Scholar]
- 642.Chaniotakis NA, Jurkschat K, Ruehlemann A. Anal Chim Acta. 1993;282:345. [Google Scholar]
- 643.Tsagatakis JK, Chaniotakis NA, Jurkschat K. Helv Chim Acta. 1994;77:2191. [Google Scholar]
- 644.Liu D, Chen WC, Yang RH, Shen GL, Yu RQ. Anal Chim Acta. 1997;338:209. [Google Scholar]
- 645.Liu D, Chen WC, Yang RH, Yu RQ. Chin Chem Lett. 1997;8:251. [Google Scholar]
- 646.Sasaki S, Ozawa S, Citterio D, Yamada K, Suzuki K. Talanta. 2004;63:131. doi: 10.1016/j.talanta.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 647.Bruce JI, Dickins RS, Govenlock LJ, Gunnlaugsson T, Lopinski S, Lowe MP, Parker D, Peacock RD, Perry JJB, Aime S, Botta M. J Am Chem Soc. 2000;122:9674. [Google Scholar]
- 648.Dickins RS, Aime S, Batsanov AS, Beeby A, Botta M, Bruce JI, Howard JAK, Love CS, Parker D, Peacock RD, Puschmann H. J Am Chem Soc. 2002;124:12697. doi: 10.1021/ja020836x. [DOI] [PubMed] [Google Scholar]
- 649.Massue J, Quinn SJ, Gunnlaugsson T. J Am Chem Soc. 2008;130:6900. doi: 10.1021/ja800361e. [DOI] [PubMed] [Google Scholar]
- 650.Best MD, Anslyn EV. Chem--Eur J. 2003;9:51. doi: 10.1002/chem.200390003. [DOI] [PubMed] [Google Scholar]
- 651.Mameri S, Charbonniere LJ, Ziessel RF. Inorg Chem. 2004;43:1819. doi: 10.1021/ic030295s. [DOI] [PubMed] [Google Scholar]
- 652.Charbonniere LJ, Schurhammer R, Mameri S, Wipff G, Ziessel RF. Inorg Chem. 2005;44:7151. doi: 10.1021/ic051033o. [DOI] [PubMed] [Google Scholar]
- 653.Li SH, Yuan WT, Zhu CQ, Xu JG. Anal Biochem. 2004;331:235. doi: 10.1016/j.ab.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 654.Hou F, Wang X, Jiang C. Anal Sci. 2005;21:231. doi: 10.2116/analsci.21.231. [DOI] [PubMed] [Google Scholar]
- 655.Hou F, Miao Y, Jiang C. Spectrochim Acta, Part A. 2005;61A:2891. doi: 10.1016/j.saa.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 656.Peng Q, Hou F, Ge X, Jiang C, Gong S. Anal Chim Acta. 2005;549:26. [Google Scholar]
- 657.Duerkop A, Turel M, Lobnik A, Wolfbeis OS. Anal Chim Acta. 2006;555:292. [Google Scholar]
- 658.Schaeferling M, Wolfbeis OS. Chem--Eur J. 2007;13:4342. doi: 10.1002/chem.200601509. [DOI] [PubMed] [Google Scholar]
- 659.Miao Y, Liu J, Hou F, Jiang C. J Lumin. 2005;116:67. [Google Scholar]
- 660.Yin C, Gao F, Huo F, Yang P. Chem Commun. 2004:934. doi: 10.1039/b315965e. [DOI] [PubMed] [Google Scholar]
- 661.Yin C, Huo F, Yang P. Sens Actuators, B. 2005;B109:291. [Google Scholar]
- 662.Rudkevich DM, Stauthamer WPRV, Verboom W, Engbersen JFJ, Harkema S, Reinhoudt DN. J Am Chem Soc. 1992;114:9671. [Google Scholar]
- 663.Rudkevich DM, Verboom W, Brzozka Z, Palys MJ, Stauthamer WPRV, van Hummel GJ, Franken SM, Harkema S, Engbersen JFJ, Reinhoudt DN. J Am Chem Soc. 1994;116:4341. [Google Scholar]
- 664.Antonisse MMG, Snellink-Rueel BHM, Yigit I, Engbersen JFJ, Reinhoudt DN. J Org Chem. 1997;62:9034. [Google Scholar]
- 665.Antonisse MMG, Snellink-Ruel BHM, Engbersen JFJ, Reinhoudt DN. Sens Actuators, B. 1998;B47:9. [Google Scholar]
- 666.Wroblewski W, Wojciechowski K, Dybko A, Brzozka Z, Egberink RJM, Snellink-Ruel BHM, Reinhoudt DN. Sens Actuators, B. 2000;B68:313. [Google Scholar]
- 667.Wroblewski W, Wojciechowski K, Dybko A, Brzozka Z, Egberink RJM, Snellink-Ruel BHM, Reinhoudt DN. Anal Chim Acta. 2001;432:79. [Google Scholar]
- 668.Wroblewski W, Wojciechowski K, Dybko A, Brzozka Z, Egberink RJM, Snellink-Ruel BHM, Reinhoudt DN. Sens Actuators, B. 2001;B78:315. [Google Scholar]
- 669.Cametti M, Dalla Cort A, Mandolini L, Nissinen M, Rissanen K. New J Chem. 2008;32:1113. [Google Scholar]
- 670.Rudkevich DM, Brzozka Z, Palys M, Visser HC, Verboom W, Reinhoudt DN. Angew Chem. 1994;106:467. [Google Scholar]
- 671.Rudkevich DM, Verboom W, Reinhoudt DN. J Org Chem. 1994;59:3683. [Google Scholar]
- 672.Visser HC, Rudkevich DM, Verboom W, de Jong F, Reinhoudt DN. J Am Chem Soc. 1994;116:11554. [Google Scholar]
- 673.Chrisstoffels LAJ, de Jong F, Reinhoudt DN. Chem--Eur J. 2000;6:1376. doi: 10.1002/(sici)1521-3765(20000417)6:8<1376::aid-chem1376>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 674.Lacy SM, Rudkevich DM, Verboom W, Reinhoudt DN. Tetrahedron Lett. 1994;35:5953. [Google Scholar]
- 675.Molina P, Tarraga A, Caballero A. Eur J Inorg Chem. 2008:3401. [Google Scholar]
- 676.Beer PD, Chen Z, Goulden AJ, Graydon A, Stokes SE, Wear T. J Chem Soc, Chem Commun. 1993:1834. [Google Scholar]
- 677.Chen Z, Graydon AR, Beer PD. J Chem Soc, Faraday Trans. 1996;92:97. [Google Scholar]
- 678.Beer PD, Drew MGB, Graydon AR, Smith DK, Stokes SE. J Chem Soc, Dalton Trans. 1995:403. [Google Scholar]
- 679.Beer PD, Graydon AR, Johnson AOM, Smith DK. Inorg Chem. 1997;36:2112. doi: 10.1021/ic961401b. [DOI] [PubMed] [Google Scholar]
- 680.Pratt MD, Beer PD. Polyhedron. 2003;22:649. [Google Scholar]
- 681.Beer PD, Chen Z, Drew MGB, Kingston J, Ogden M, Spencer P. J Chem Soc, Chem Commun. 1993:1046. [Google Scholar]
- 682.Beer PD, Chen Z, Drew MGB, Johnson AOM, Smith DK, Spencer P. Inorg Chim Acta. 1996;246:143. [Google Scholar]
- 683.Beer PD, Drew Michael GB, Smith DK. J Organomet Chem. 1997;543:259. [Google Scholar]
- 684.Lloris JM, Martínez-Máñez R, Padilla-Tosta M, Pardo T, Soto JS, Tendero MJL. J Chem Soc, Dalton Trans. 1998:3657. [Google Scholar]
- 685.Lloris JM, Martínez-Máñez R, Padilla-Tosta ME, Pardo T, Soto J. Helv Chim Acta. 1999;82:1445. [Google Scholar]
- 686.Padilla-Tosta ME, Martínez-Máñez R, Pardo T, Soto J, Tendero MJL. Chem Commun. 1997:887. [Google Scholar]
- 687.Beer PD, Cadman J, Lloris JM, Martínez-Máñez R, Padilla ME, Pardo T, Smith DK, Soto J. J Chem Soc, Dalton Trans. 1999:127. [Google Scholar]
- 688.Beer PD, Cadman J, Lloris JM, Martínez-Máñez R, Soto J, Pardo T, Marcos MD. Dalton Trans. 2000:1805. [Google Scholar]
- 689.Lloris JM, Martínez-Máñez R, Soto J, Pardo T. J Organomet Chem. 2001;637-639:151. [Google Scholar]
- 690.Sancenón F, Benito A, Hernandez FJ, Lloris JM, Martínez-Máñez R, Pardo T, Soto J. Eur J Inorg Chem. 2002:866. [Google Scholar]
- 691.Scherer M, Sessler JL, Gebauer A, Lynch V. Chem Commun. 1998:85. [Google Scholar]
- 692.Sessler JL, Zimmerman RS, Kirkovits GJ, Gebauer A, Scherer M. J Organomet Chem. 2001;637-639:343. [Google Scholar]
- 693.Denuault G, Gale PA, Hursthouse MB, Light ME, Warriner CN. New J Chem. 2002;26:811. [Google Scholar]
- 694.Coles SJ, Denuault G, Gale PA, Horton PN, Hursthouse MB, Light ME, Warriner CN. Polyhedron. 2003;22:699. [Google Scholar]
- 695.Arroyo M, Birkin PR, Gale PA, García-Garrido SE, Light ME. New J Chem. 2008;32:1221. [Google Scholar]
- 696.Buda M, Ion A, Moutet JC, Saint-Aman E, Ziessel R. J Electroanal Chem. 1999;469:132. [Google Scholar]
- 697.Reynes O, Maillard F, Moutet JC, Royal G, Saint-Aman E, Stanciu G, Dutasta JP, Gosse I, Mulatier JC. J Organomet Chem. 2001;637-639:356. [Google Scholar]
- 698.Reynes O, Moutet JC, Pecaut J, Royal G, Saint-Aman E. New J Chem. 2002;26:9. [Google Scholar]
- 699.Reynes O, Bucher C, Moutet JC, Royal G, Saint-Aman E. Chem Commun. 2004:428. doi: 10.1039/b314236a. [DOI] [PubMed] [Google Scholar]
- 700.Oton F, Tarraga A, Velasco MD, Molina P. Dalton Trans. 2005:1159. doi: 10.1039/b419082c. [DOI] [PubMed] [Google Scholar]
- 701.Oton F, Tarraga A, Espinosa A, Velasco MD, Bautista D, Molina P. J Org Chem. 2005;70:6603. doi: 10.1021/jo050458p. [DOI] [PubMed] [Google Scholar]
- 702.Oton F, Tarraga A, Espinosa A, Velasco MD, Molina P. J Org Chem. 2006;71:4590. doi: 10.1021/jo0604540. [DOI] [PubMed] [Google Scholar]
- 703.Zapata F, Caballero A, Espinosa A, Tarraga A, Molina P. J Org Chem. 2008;73:4034. doi: 10.1021/jo800296c. [DOI] [PubMed] [Google Scholar]
- 704.Oton F, Espinosa A, Tarraga A, Ramirez de Arellano C, Molina P. Chem--Eur J. 2007;13:5742. doi: 10.1002/chem.200601757. [DOI] [PubMed] [Google Scholar]
- 705.Basurto S, Riant O, Moreno D, Rojo J, Torroba T. J Org Chem. 2007;72:4673. doi: 10.1021/jo0702589. [DOI] [PubMed] [Google Scholar]
- 706.Delavaux-Nicot B, Guari Y, Douziech B, Mathieu R. J Chem Soc, Chem Commun. 1995:585. [Google Scholar]
- 707.Li C, Wang L, Deng L, Yu H, Huo J, Ma L, Wang J. J Phys Chem B. 2009;113:15141. doi: 10.1021/jp9078376. [DOI] [PubMed] [Google Scholar]
- 708.Beer PD, Hesek D, Kingston JE, Smith DK, Stokes SE, Drew MGB. Organometallics. 1995;14:3288. [Google Scholar]
- 709.Beer PD, Drew MGB, Hesek D, Shade M, Szemes F. Chem Commun. 1996:2161. [Google Scholar]
- 710.Beer PD, Drew MG, Hesek D, Nam KC. Chem Commun. 1997:107. [Google Scholar]
- 711.Beer PD, Hesek D, Nam KC, Drew MGB. Organometallics. 1999;18:3933. [Google Scholar]
- 712.Gale PA, Chen Z, Drew MGB, Heath JA, Beer PD. Polyhedron. 1998;17:405. [Google Scholar]
- 713.Evans AJ, Matthews SE, Cowley AR, Beer PD. Dalton Trans. 2003:4644. [Google Scholar]
- 714.Metay E, Duclos MC, Pellet-Rostaing S, Lemaire M, Schulz J, Kannappan R, Bucher C, Saint-Aman E, Chaix C. Eur J Org Chem. 2008:4304. [Google Scholar]
- 715.Metay E, Duclos MC, Pellet-Rostaing S, Lemaire M, Schulz J, Kannappan R, Bucher C, Saint-Aman E, Chaix C. Supramol Chem. 2009;21:68. [Google Scholar]
- 716.Guo DS, Liu ZP, Ma JP, Huang RQ. Tetrahedron Lett. 2007;48:1221. [Google Scholar]
- 717.Beer PD, Davis JJ, Drillsma-Milgrom DA, Szemes F. Chem Commun. 2002:1716. doi: 10.1039/b205340n. [DOI] [PubMed] [Google Scholar]
- 718.Valerio C, Fillaut JL, Ruiz J, Guittard J, Blais JC, Astruc D. J Am Chem Soc. 1997;119:2588. [Google Scholar]
- 719.Ruiz J, Medel MJR, Daniel MC, Blais JC, Astruc D. Chem Commun. 2003:464. doi: 10.1039/b211772j. [DOI] [PubMed] [Google Scholar]
- 720.Daniel MC, Ruiz J, Astruc D. J Am Chem Soc. 2003;125:1150. doi: 10.1021/ja020833k. [DOI] [PubMed] [Google Scholar]
- 721.Daniel MC, Ba F, Aranzaes JR, Astruc D. Inorg Chem. 2004;43:8649. doi: 10.1021/ic0493999. [DOI] [PubMed] [Google Scholar]
- 722.Labande A, Astruc D. Chem Commun. 2000:1007. [Google Scholar]
- 723.Labande A, Ruiz J, Astruc D. J Am Chem Soc. 2002;124:1782. doi: 10.1021/ja017015x. [DOI] [PubMed] [Google Scholar]
- 724.Daniel MC, Ruiz J, Nlate S, Blais JC, Astruc D. J Am Chem Soc. 2003;125:2617. doi: 10.1021/ja021325d. [DOI] [PubMed] [Google Scholar]
- 725.Astruc D, Daniel MC, Ruiz J. Chem Commun. 2004:2637. doi: 10.1039/b410399h. [DOI] [PubMed] [Google Scholar]
- 726.Aranzaes JR, Belin C, Astruc D. Angew Chem, Int Ed. 2006;45:132. doi: 10.1002/anie.200502291. [DOI] [PubMed] [Google Scholar]
- 727.Casado CM, Cuadrado I, Alonso B, Moran M, Losada J. J Electroanal Chem. 1999;463:87. [Google Scholar]
- 728.Alonso B, Casado CM, Cuadrado I, Moran M, Kaifer AE. Chem Commun. 2002:1778. doi: 10.1039/b205619d. [DOI] [PubMed] [Google Scholar]
- 729.Beer PD, Chen Z, Goulden AJ, Grieve A, Hesek D, Szemes F, Wear T. J Chem Soc, Chem Commun. 1994:1269. [Google Scholar]
- 730.Szemes F, Hesek D, Chen Z, Dent SW, Drew MGB, Goulden AJ, Graydon AR, Grieve A, Mortimer RJ, Wear TJ, Weightman JS, Beer PD. Inorg Chem. 1996;35:5868. [Google Scholar]
- 731.Beer PD, Dent SW, Hobbs GS, Wear TJ. Chem Commun. 1997:99. [Google Scholar]
- 732.Beer PD, Szemes F, Balzani V, Sala CM, Drew MGB, Dent SW, Maestri M. J Am Chem Soc. 1997;119:11864. [Google Scholar]
- 733.Beer PD, Cadman J. New J Chem. 1999;23:347. [Google Scholar]
- 734.Vickers MS, Martindale KS, Beer PD. J Mater Chem. 2005;15:2784. [Google Scholar]
- 735.Beer PD, Timoshenko V, Maestri M, Passaniti P, Balzani V. Chem Commun. 1999:1755. [Google Scholar]
- 736.Beer PD, Dent SW. Chem Commun. 1998:825. [Google Scholar]
- 737.Watanabe S, Onogawa O, Komatsu Y, Yoshida K. J Am Chem Soc. 1998;120:229. [Google Scholar]
- 738.Deetz MJ, Smith BD. Tetrahedron Lett. 1998;39:6841. [Google Scholar]
- 739.Duff T, Grussing A, Thomas JL, Duati M, Vos JG. Polyhedron. 2003;22:775. [Google Scholar]
- 740.Yu X, Lin H, Cai Z, Lin H. Tetrahedron Lett. 2007;48:8615. [Google Scholar]
- 741.Cookson J, Vickers MS, Paul RL, Cowley AR, Beer PD. Inorg Chim Acta. 2008;361:1689. [Google Scholar]
- 742.Lin TP, Chen CY, Wen YS, Sun SS. Inorg Chem. 2007;46:9201. doi: 10.1021/ic701300a. [DOI] [PubMed] [Google Scholar]
- 743.Mizuno T, Wei WH, Eller LR, Sessler JL. J Am Chem Soc. 2002;124:1134. doi: 10.1021/ja017298t. [DOI] [PubMed] [Google Scholar]
- 744.Plitt P, Gross DE, Lynch VM, Sessler JL. Chem--Eur J. 2007;13:1374. doi: 10.1002/chem.200601514. [DOI] [PubMed] [Google Scholar]
- 745.Shang XF, Li J, Lin H, Jiang P, Cai ZS, Lin HK. Dalton Trans. 2009:2096. doi: 10.1039/b804445g. [DOI] [PubMed] [Google Scholar]
- 746.Padilla-Tosta ME, Lloris JM, Martínez-Máñez R, Pardo T, Soto J, Benito A, Marcos MD. Inorg Chem Commun. 2000;3:45. [Google Scholar]
- 747.Padilla-Tosta ME, Lloris JM, Martínez-Máñez R, Pardo T, Sancenón F, Soto J, Marcos MD. Eur J Inorg Chem. 2001:1221. [Google Scholar]
- 748.Morzherin Y, Rudkevich DM, Verboom W, Reinhoudt DN. J Org Chem. 1993;58:7602. [Google Scholar]
- 749.Cameron BR, Loeb SJ. Chem Commun. 1997:573. [Google Scholar]
- 750.Shi Y, Schneider HJ. J Chem Soc, Perkin Trans 2. 1999:1797. [Google Scholar]
- 751.Budka J, Lhotak P, Michlova V, Stibor I. Tetrahedron Lett. 2001;42:1583. [Google Scholar]
- 752.Stibor I, Budka J, Michlova V, Tkadlecova M, Pojarova M, Curinova P, Lhotak P. New J Chem. 2008;32:1597. [Google Scholar]
- 753.Miao R, Zheng QY, Chen CF, Huang ZT. Tetrahedron Lett. 2005;46:2155. [Google Scholar]
- 754.Chawla HM, Singh SP. Tetrahedron. 2007;64:741. [Google Scholar]
- 755.Nam KC, Kim DS, Yang YS. Bull Korean Chem Soc. 1998;19:1133. [Google Scholar]
- 756.Nam KC, Kang SO, Ko SW. Bull Korean Chem Soc. 1999;20:953. [Google Scholar]
- 757.Nam KC, Kang SO, Jeong HS, Jeon S. Tetrahedron Lett. 1999;40:7343. [Google Scholar]
- 758.Beer PD, Drew MGB, Gradwell K. J Chem Soc, Perkin Trans 2. 2000:511. [Google Scholar]
- 759.Hamdi A, Nam KC, Ryu BJ, Kim JS, Vicens J. Tetrahedron Lett. 2004;45:4689. [Google Scholar]
- 760.Chen QY, Chen CF. Eur J Org Chem. 2005:2468. [Google Scholar]
- 761.Chen CF, Chen QY. New J Chem. 2006;30:143. [Google Scholar]
- 762.Quinlan E, Matthews SE, Gunnlaugsson T. Tetrahedron Lett. 2006;47:9333. [Google Scholar]
- 763.Quinlan E, Matthews SE, Gunnlaugsson T. J Org Chem. 2007;72:7497. doi: 10.1021/jo070439a. [DOI] [PubMed] [Google Scholar]
- 764.Beer PD, Drew MGB, Knubley RJ, Ogden MI. J Chem Soc, Dalton Trans. 1995:3117. [Google Scholar]
- 765.Cooper JB, Drew MGB, Beer PD. J Chem Soc, Dalton Trans. 2000:2721. [Google Scholar]
- 766.Wolf NJ, Georgiev EM, Yordanov AT, Whittlesey BR, Koch HF, Roundhill DM. Polyhedron. 1999;18:885. [Google Scholar]
- 767.Gupta VK, Ludwig R, Agarwal S. Anal Chim Acta. 2005;538:213. [Google Scholar]
- 768.Kivlehan F, Mace WJ, Moynihan HA, Arrigan DWM. Anal Chim Acta. 2007;585:154. doi: 10.1016/j.aca.2006.11.078. [DOI] [PubMed] [Google Scholar]
- 769.Niikura K, Anslyn EV. J Chem Soc, Perkin Trans 2. 1999:2769. [Google Scholar]
- 770.Atilgan S, Akkaya EU. Tetrahedron Lett. 2004;45:9269. [Google Scholar]
- 771.Sebo L, Diederich F, Gramlich V. Helv Chim Acta. 2000;83:93. [Google Scholar]
- 772.Kim SK, Moon BS, Park JH, Seo YI, Koh HS, Yoon YJ, Lee KD, Yoon J. Tetrahedron Lett. 2005;46:6617. [Google Scholar]
- 773.Lhotak P, Svoboda J, Stibor I. Tetrahedron. 2006;62:1253. [Google Scholar]
- 774.Hoffman JL, Bock RM. Biochemistry. 1970;9:3542. doi: 10.1021/bi00820a007. [DOI] [PubMed] [Google Scholar]
- 775.Formoso C. Biochem Biophys Res Commun. 1973;50:999. doi: 10.1016/0006-291x(73)91505-2. [DOI] [PubMed] [Google Scholar]
- 776.Formoso C. Biopolymers. 1974;13:909. doi: 10.1002/bip.1974.360130507. [DOI] [PubMed] [Google Scholar]
- 777.Boger J, Knowles JR. J Am Chem Soc. 1979;101:7631. [Google Scholar]
- 778.Eliseev AV, Schneider HJ. Angew Chem. 1993;105:1423. [Google Scholar]
- 779.Eliseev AV, Schneider HJ. Angew Chem, Int Ed. 1993;105:1331. [Google Scholar]
- 780.Eliseev AV, Schneider HJ. J Am Chem Soc. 1994;116:6081. [Google Scholar]
- 781.Vizitiu D, Thatcher GRJ. J Org Chem. 1999;64:6235. [Google Scholar]
- 782.Schwinte P, Darcy R, O'Keeffe F. J Chem Soc, Perkin Trans 2. 1998:805. [Google Scholar]
- 783.Ghosh M, Sanders TC, Zhang R, Seto CT. Org Lett. 1999;1:1945. doi: 10.1021/ol990301h. [DOI] [PubMed] [Google Scholar]
- 784.Ghosh M, Zhang R, Lawler RG, Seto CT. J Org Chem. 2000;65:735. [Google Scholar]
- 785.Cotner ES, Smith PJ. J Org Chem. 1998;63:1737. [Google Scholar]
- 786.Hauser SL, Johanson EW, Green HP, Smith PJ. Org Lett. 2000;2:3575. doi: 10.1021/ol006503r. [DOI] [PubMed] [Google Scholar]
- 787.Yuan DQ, Izuka A, Fukudome M, Rekharsky MV, Inoue Y, Fujita K. Tetrahedron Lett. 2007;48:3479. [Google Scholar]
- 788.Gale PA, Sessler JL, Král V, Lynch V. J Am Chem Soc. 1996;118:5140. [Google Scholar]
- 789.Gale PA, Sessler JL, Allen WE, Tvermoes NA, Lynch V. Chem Commun. 1997:665. [Google Scholar]
- 790.Sessler JL, Cho WS, Gross DE, Shriver JA, Lynch VM, Marquez M. J Org Chem. 2005;70:5982. doi: 10.1021/jo050662c. [DOI] [PubMed] [Google Scholar]
- 791.Anzenbacher P, Jr, Jursikova K, Lynch VM, Gale PA, Sessler JL. J Am Chem Soc. 1999;121:11020. [Google Scholar]
- 792.Sessler JL, Anzenbacher P, Jr, Miyaji H, Jursikova K, Bleasdale ER, Gale PA. Ind Eng Chem Res. 2000;39:3471. [Google Scholar]
- 793.Schmidtchen FP. Org Lett. 2002;4:431. doi: 10.1021/ol017137u. [DOI] [PubMed] [Google Scholar]
- 794.Custelcean R, Delmau LH, Moyer BA, Sessler JL, Cho WS, Gross D, Bates GW, Brooks SJ, Light ME, Gale PA. Angew Chem, Int Ed. 2005;44:2537. doi: 10.1002/anie.200462945. [DOI] [PubMed] [Google Scholar]
- 795.Gross DE, Schmidtchen FP, Antonius W, Gale PA, Lynch VM, Sessler JL. Chem--Eur J. 2008;14:7822. doi: 10.1002/chem.200800899. [DOI] [PubMed] [Google Scholar]
- 796.Gale PA, Twyman LJ, Handlin CI, Sessler JL. Chem Commun. 1999:1851. [Google Scholar]
- 797.Linn MM, Poncio DC, Machado VG. Tetrahedron Lett. 2007;48:4547. [Google Scholar]
- 798.Miyaji H, Sato W, Sessler JL. Angew Chem, Int Ed. 2000;39:1777. doi: 10.1002/(sici)1521-3773(20000515)39:10<1777::aid-anie1777>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 799.Miyaji H, Anzenbacher P, Jr, Sessler JL, Bleasdale ER, Gale PA. Chem Commun. 1999:1723. [Google Scholar]
- 800.Anzenbacher P, Jr, Jursikova K, Sessler JL. J Am Chem Soc. 2000;122:9350. [Google Scholar]
- 801.Shao S, Guo Y, He L, Jiang S, Yu X. Tetrahedron Lett. 2003;44:2175. [Google Scholar]
- 802.Yoo J, Kim MS, Hong SJ, Sessler JL, Lee CH. J Org Chem. 2009;74:1065. doi: 10.1021/jo802059c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 803.Sessler JL, Gale PA, Genge JW. Chem--Eur J. 1998;4:1095. [Google Scholar]
- 804.Král V, Sessler JL, Shishkanova TV, Gale PA, Volf R. J Am Chem Soc. 1999;121:8771. [Google Scholar]
- 805.Shishkanova TV, Sapurina I, Stejkal J, Král V, Volf R. Anal Chim Acta. 2005;553:160. [Google Scholar]
- 806.Sessler JL, Král V, Shishkanova TV, Gale PA. Proc Natl Acad Sci U S A. 2002;99:4848. doi: 10.1073/pnas.062633799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.Gale PA, Hursthouse MB, Light ME, Sessler JL, Warriner CN, Zimmerman RS. Tetrahedron Lett. 2001;42:6759. [Google Scholar]
- 808.Szymanska I, Radecka H, Radecki J, Gale PA, Warriner CN. J Elect Chem. 2006;591:223. [Google Scholar]
- 809.Yang W, Yin Z, Wang CH, Huang C, He J, Zhu X, Cheng JP. Tetrahedron. 2008;64:9244. [Google Scholar]
- 810.Warriner CN, Gale PA, Light ME, Hursthouse MB. Chem Commun. 2003:1810. [PubMed] [Google Scholar]
- 811.Nishiyabu R, Anzenbacher P., Jr J Am Chem Soc. 2005;127:8270. doi: 10.1021/ja051421p. [DOI] [PubMed] [Google Scholar]
- 812.Nishiyabu R, Anzenbacher P., Jr Org Lett. 2006;8:359. doi: 10.1021/ol0521782. [DOI] [PubMed] [Google Scholar]
- 813.Danil de Namor AF, Shehab M. J Phys Chem A. 2004;108:7324. [Google Scholar]
- 814.Danil de Namor AF, Shehab M, Abbas I, Withams MV, Zvietcovich-Guerra J. J Phys Chem B. 2006;110:12653. doi: 10.1021/jp060859o. [DOI] [PubMed] [Google Scholar]
- 815.Danil de Namor AF, Shehab M, Khalife R, Abbas I. J Phys Chem B. 2007;111:12177. doi: 10.1021/jp073863o. [DOI] [PubMed] [Google Scholar]
- 816.Danil de Namor AF, Abbas I, Hammud HH. J Phys Chem B. 2006;110:2142. doi: 10.1021/jp056492p. [DOI] [PubMed] [Google Scholar]
- 817.Valik M, Král V, Herdtweck E, Schmidtchen FP. New J Chem. 2007;31:703. [Google Scholar]
- 818.Piatek P, Lynch VM, Sessler JL. J Am Chem Soc. 2004;126:16073. doi: 10.1021/ja045218q. [DOI] [PubMed] [Google Scholar]
- 819.Sessler JL, An D, Cho WS, Lynch V, Yoon DW, Hong SJ, Lee CH. J Org Chem. 2005;70:1511. doi: 10.1021/jo048480q. [DOI] [PubMed] [Google Scholar]
- 820.Sessler JL, An D, Cho WS, Lynch V, Marquez M. Chem--Eur J. 2005;11:2001. doi: 10.1002/chem.200400894. [DOI] [PubMed] [Google Scholar]
- 821.Anzenbacher P, Jr, Nishiyabu R, Palacios MA. Coor Chem Rev. 2006;250:2929. [Google Scholar]
- 822.Nishiyabu R, Palacios MA, Dehaen W, Anzenbacher P., Jr J Am Chem Soc. 2006;128:11496. doi: 10.1021/ja0622150. [DOI] [PubMed] [Google Scholar]
- 823.Gu R, Depraetere S, Kotek J, Budka J, Wagner-Wysiecka E, Biernat JF, Dehaen W. Org Biomol Chem. 2005;3:2921. doi: 10.1039/b507508d. [DOI] [PubMed] [Google Scholar]
- 824.Dehaen W, Gale PA, García-Garrido SE, Kostermans M, Light ME. New J Chem. 2007;31:691. [Google Scholar]
- 825.Sessler JL, Camiolo S, Gale PA. Coord Chem Rev. 2003;240:17. [Google Scholar]
- 826.Aoyama Y, Nonaka S, Motomura T, Toi H, Ogoshi H. Chem Lett. 1991:1241. [Google Scholar]
- 827.Král V, Schmidtchen FP, Lang K, Berger M. Org Lett. 2002;4:51. doi: 10.1021/ol0168667. [DOI] [PubMed] [Google Scholar]
- 828.Kejik Z, Zaruba K, Michalik D, Sebek J, Dian J, Pataridis S, Volka K, Král V. Chem Commun. 2006:1533. doi: 10.1039/b518239e. [DOI] [PubMed] [Google Scholar]
- 829.Starnes SD, Arungundram S, Saunders CH. Tetrahedron Lett. 2002;43:7785. [Google Scholar]
- 830.Pasternack RF, Gibbs EJ, Antebi A, Bassner S, De Poy L, Turner DH, Williams A, Laplace F, Lansard MH, Merienne C, Perree-Fauvet M. J Am Chem Soc. 1985;107:8179. doi: 10.1021/ja00312a061. [DOI] [PubMed] [Google Scholar]
- 831.Hodinar A, Jyo A. Chem Lett. 1988:993. [Google Scholar]
- 832.Kuroda Y, Hatakeyama H, Inakoshi N, Ogoshi H. Tetrahedron Lett. 1993;34:8285. [Google Scholar]
- 833.Kuroda Y, Hatakeyama H, Seshimo H, Ogoshi H. Supramol Chem. 1994;3:267. [Google Scholar]
- 834.Beer PD, Drew MGB, Hesek D, Jagessar R. J Chem Soc, Chem Commun. 1995:1187. [Google Scholar]
- 835.Cormode DP, Murray SS, Cowley AR, Beer PD. Dalton Trans. 2006:5135. doi: 10.1039/b609817g. [DOI] [PubMed] [Google Scholar]
- 836.Cormode DP, Drew MGB, Jagessar R, Beer PD. Dalton Trans. 2008:6732. doi: 10.1039/b807153e. [DOI] [PubMed] [Google Scholar]
- 837.Jagessar RC, Shang M, Scheidt WR, Burns DH. J Am Chem Soc. 1998;120:11684. [Google Scholar]
- 838.Burns DH, Calderon-Kawasaki K, Kularatne S. J Org Chem. 2005;70:2803. doi: 10.1021/jo047756r. [DOI] [PubMed] [Google Scholar]
- 839.Calderon-Kawasaki K, Kularatne S, Li YH, Noll BC, Scheidt WR, Burns DH. J Org Chem. 2007;72:9081. doi: 10.1021/jo701443c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 840.Lee C, Lee DH, Hong JI. Tetrahedron Lett. 2001;42:8665. [Google Scholar]
- 841.Bao X, Zhang H, Zhang Z, Wu L, Li Z. Inorg Chem Commun. 2007;10:728. [Google Scholar]
- 842.Imada T, Kijima H, Takeuchi M, Shinkai S. Tetrahedron Lett. 1995;36:2093. [Google Scholar]
- 843.Imada T, Kijima H, Takeuchi M, Shinkai S. Tetrahedron. 1996;52:2817. [Google Scholar]
- 844.Sessler JL, Davis JM. Acc Chem Res. 2001;34:989. doi: 10.1021/ar980117g. [DOI] [PubMed] [Google Scholar]
- 845.Kumar MR, Chandrashekar TKJ. J Inclusion Phenom Macrocycl Chem. 1999;35:553. [Google Scholar]
- 846.Král V, Furuta H, Shreder K, Lynch V, Sessler JL. J Am Chem Soc. 1996;118:1595. [Google Scholar]
- 847.Furuta H, Cyr MJ, Sessler JL. J Am Chem Soc. 1991;113:6677. [Google Scholar]
- 848.Král V, Sessler JL, Furuta H. J Am Chem Soc. 1992;114:8704. [Google Scholar]
- 849.Král V, Sessler JL. Tetrahedron. 1995;51:539. [Google Scholar]
- 850.Sessler JL, Furuta H, Král V. Supramol Chem. 1993;1:209. [Google Scholar]
- 851.Iverson BL, Thomas RE, Král V, Sessler JL. J Am Chem Soc. 1994;116:2663. [Google Scholar]
- 852.Sessler JL, Genge JW, Král V, Iverson BL. Supramol Chem. 1996;8:45. [Google Scholar]
- 853.Sessler JL, Král V, Genge JW, Thomas RE, Iverson BL. Anal Chem. 1998;70:2516. doi: 10.1021/ac971214a. [DOI] [PubMed] [Google Scholar]
- 854.Král V, Andrievsky A, Sessler JL. J Chem Soc, Chem Commun. 1995:2349. [Google Scholar]
- 855.Sessler JL, Cyr M, Furuta H, Král V, Mody T, Morishima T, Shionoya M, Weghorn S. Pure Appl Chem. 1993;65:393. [Google Scholar]
- 856.Sessler JL, Davis JM, Král V, Kimbrough T, Lynch V. Org Biomol Chem. 2003;1:4113. doi: 10.1039/b306964h. [DOI] [PubMed] [Google Scholar]
- 857.Král V, Lang K, Kralova J, Dvorak M, Martasek P, Chin AO, Andrievsky A, Lynch V, Sessler JL. J Am Chem Soc. 2006;128:432. doi: 10.1021/ja0518474. [DOI] [PubMed] [Google Scholar]
- 858.Charvatova J, Matejka P, Král V, Deyl Z. J Chromatogr A. 2001;921:99. doi: 10.1016/s0021-9673(01)00747-6. [DOI] [PubMed] [Google Scholar]
- 859.Charvatova J, Král V, Deyl Z. Electrophoresis. 2002;23:237. doi: 10.1002/1522-2683(200202)23:2<237::AID-ELPS237>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 860.Furuta H, Morishima T, Král V, Sessler JL. Supramol Chem. 1993;3:5. [Google Scholar]
- 861.Sridevi B, Narayanan SJ, Rao R, Chandrashekar TK. Inorg Chem. 2000;39:3669. doi: 10.1021/ic000031v. [DOI] [PubMed] [Google Scholar]
- 862.Sessler JL, Katayev E, Pantos GD, Ustynyuk YA. Chem Commun. 2004:1276. doi: 10.1039/b403665d. [DOI] [PubMed] [Google Scholar]
- 863.Sessler JL, Katayev E, Pantos GD, Scherbakov P, Reshetova MD, Khrustalev VN, Lynch VM, Ustynyuk YA. J Am Chem Soc. 2005;127:11442. doi: 10.1021/ja0522938. [DOI] [PubMed] [Google Scholar]
- 864.Katayev EA, Pantos GD, Reshetova MD, Khrustalev VN, Lynch VM, Ustynyuk YA, Sessler JL. Angew Chem, Int Ed. 2005;44:7386. doi: 10.1002/anie.200502393. [DOI] [PubMed] [Google Scholar]
- 865.Katayev EA, Boev NV, Khrustalev VN, Ustynyuk YA, Tananaev IG, Sessler JL. J Org Chem. 2007;72:2886. doi: 10.1021/jo0624849. [DOI] [PubMed] [Google Scholar]
- 866.Katayev EA, Sessler JL, Khrustalev VN, Ustynyuk YA. J Org Chem. 2007;72:7244. doi: 10.1021/jo071106g. [DOI] [PubMed] [Google Scholar]
- 867.Katayev EA, Boev NV, Myshkovskaya E, Khrustalev VN, Ustynyuk YA. Chem--Eur J. 2008;14:9065. doi: 10.1002/chem.200800860. [DOI] [PubMed] [Google Scholar]
- 868.Sessler JL, Maeda H, Mizuno T, Lynch VM, Furuta H. J Am Chem Soc. 2002;124:13474. doi: 10.1021/ja0273750. [DOI] [PubMed] [Google Scholar]
- 869.Andrievsky A, Ahuis F, Sessler JL, Vögtle F, Gudat D, Moini M. J Am Chem Soc. 1998;120:9712. [Google Scholar]