

Abstract
Hallmarks of inflammation in various cardiovascular diseases, notably atherosclerosis, have been observed for a long time. However, evidence for an (auto)antigen-driven process at these sites of inflammation has come forward only recently. Heat shock proteins (HSPs) have been identified as playing either immunologically mediated disease promoting or protective roles. HSP60 has been shown to trigger innate and adaptive immune responses that initiate the earliest still reversible inflammatory stage of atherosclerosis. HSP60 is structurally highly conserved and abundantly expressed by prokaryotic and eukaryotic cells under stressful conditions. Beneficial protective immunity to microbial HSP60 acquired by infection or vaccination and bona fide autoimmunity to biochemically altered autologous HSP60 is present in all humans. In vitro and in vivo experiments have demonstrated that classical atherosclerosis risk factors can act as endothelial stressors that provoke the simultaneous expression of adhesion molecules and of HSP60 in mitochondria, in cytoplasm, and on the cell surface, where it acts as a “danger signal” for cellular and humoral immune reactions. Hence, protective, preexisting anti-HSP60 immunity may have to be “paid for” by harmful (auto)immune cross-reactive attack on arterial endothelial cells maltreated by atherosclerosis risk factors. These experimentally and clinically proven findings are the basis for the autoimmune concept of atherosclerosis.
Keywords: atherosclerosis, endothelium, immune system, risk factors, stress, heat shock protein
Typical cellular hallmarks of chronic inflammation and infections, notably infiltration by mononuclear cells, are also present in the cardiovascular system, as has been known for more than 150 years. However, until quite recently, it was not clear whether these inflammatory immunologic processes are primary or secondary in nature.1 One of the reasons for this uncertainty may have been the fact that most investigations in humans were conducted on surgical or autopsy specimens representing very advanced stages of cardiovascular disease (CVD) and thus did not provide information on the initial mechanisms triggering these processes. In complex situations, such as in atherosclerosis, it was also difficult to appreciate that different, clinically well-proven risk factors may provoke a similar, or even identical, pathophysiological outcome. The main thrust to resolve this dilemma was to delineate the array of nonspecific and specific humoral and cellular inflammatory reactions taking place within the afflicted vascular territories. The availability of animal models that, at least partly, mimic human CVD was of utmost importance for this progress. In recent decades, the aim has been to identify exogenous or autologous antigens that may induce the local cardiovascular immune reactions. Among the candidates for such antigens are infectious agents, such as Chlamydia pneumoniae, as well as autoantigens, such as biochemically altered ones (eg, oxidized low-density lipoprotein [oxLDL], phospholipids, and heat shock proteins [HSPs]). In this review, we will focus on the role of HSPs in atherosclerosis.
HSPs
Under physiological conditions, HSPs fulfill important intracellular tasks with regard to protein folding and transport. Under stress, HSPs may act as chaperones preventing protein denaturation and loss of function. As the name implies, the expression of HSPs was first demonstrated as a response to increased temperature.2 Recently, a first attempt for a consistent and clear nomenclature for the HSPs and related chaperone genes in the human database has been achieved.3 HSPs are classified according to their molecular mass into families ranging from less than 5 kDa to more than 100 kDa.4 HSPs are structurally highly conserved from prokaryotic to eukaryotic cells. HSP60 of different bacterial species display greater than 95% sequence homology at both DNA and protein levels. An overall 55% homology exists between human and bacterial HSP60 that can even reach 72% at certain domains of the 573-amino-acid-long molecule. All animals and humans show protective innate and adaptive immunity against HSP60. Because of the antigenic similarity between prokaryotic and eukaryotic HSPs, the body is confronted with the dilemma that HSP60 should be recognized and reacted against to fight infection, but autologous HSP60 should still be tolerated to avoid autoimmunity.5
Under physiological conditions, animals and humans seem to be tolerant against autologous HSP60. How this state of tolerance is activated and maintained and under what circumstances it may be broken (eg, by an accumulating life-long infectious load or by immunization together with adjuvants) is still a matter of debate. With respect to central tolerance, it has been shown that HSPs are among the molecules that are promiscuously expressed by thymic epithelial cells.6 In addition, extracellular sampling by circulating dendritic cells migrating into the thymus can be considered.7 Also, postthymic peripheral failsafe mechanisms must be operative to prevent the onset of overt autoimmunity against HSP60.8
Among the CVDs, the role of HSP60 has mainly been studied for atherosclerosis. HSP60 is a mitochondrially expressed stress protein that can be translocated to the cytosol and, later, transported to the cell surface and shed to the environment.9 Hence, the plasma concentration of soluble HSP60 (sHSP60) shows a wide range related to genetic, biological, and psychological factors in CVD, and patients with borderline hypertension10 and patients with early CVD show elevated levels of sHSP60.11 Translocation of HSP60 to the cell surface is a significant stress response that correlates with apoptosis and exacerbation of the disease state.12,13 HSP60 can activate both the innate immune system (via toll-like receptor 4) and the adaptive immune system.14
HSP in Atherosclerosis
Early Human Atherosclerotic Lesions
The predominantly inflammatory nature of initial atherosclerotic lesions is supported by both clinical and experimental facts that are, however, still often neglected. Although hypercholesterolemia is a proven atherosclerosis risk factor, more than 60% of patients are normocholesterolemic. Furthermore, activated T cells (mainly CD4+) are the first invaders of the arterial intima in early human atherosclerotic lesions,15-17 only later followed by macrophages and smooth muscle cells (SMCs), the latter 2 often transformed into foam cells prevailing in late, complicated plaques and in early xanthoma. When healthy babies, children, and young adults (aged 8 months to 16 years) were studied, mononuclear cell infiltration at predilection sites again started with T cells, followed by macrophages, SMCs, and a few scattered mast cells. In a cohort of very young children (8 months to 8 years), macrophages did not show the characteristics of foam cells, and extracellular lipid deposits were not yet detectible in the intima.18 In another study on young subjects (15 to 34 years old) collected from the American Pathobiological Determinants of Atherosclerosis in Youth study,19 the same phenotypic observations summarized above could again be confirmed with the additional feature of an intricate network of dendritic cells present in the arterial intima providing the prerequisites for a local immune reaction.16
The Autoimmune Concept of Atherosclerosis
Experimental and clinical facts summarized below have provided the basis for the development of the autoimmune concept of atherosclerosis20 that is schematically depicted in Figures 1 and 2.
Figure 1.
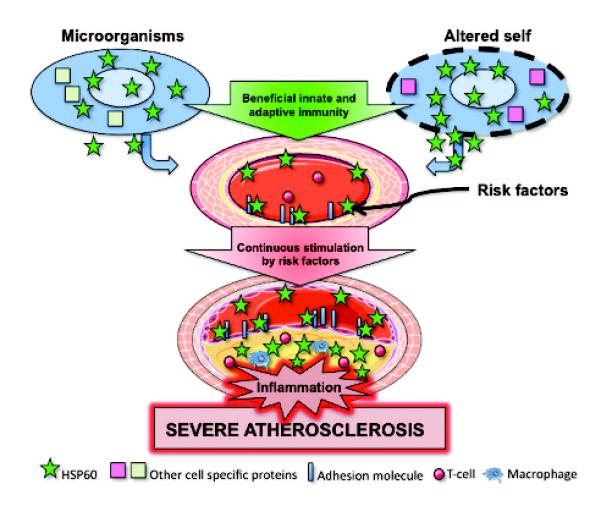
Schematic overview of the autoimmune concept of atherosclerosis. Under physiological conditions, human vascular ECs do not express HSP60 (green stars) on their surface. However, when ECs are stressed by classical atherosclerosis risk factors, this leads to the simultaneous expression of HSP60 and adhesion molecules. HSP60 expression on the EC surface may serve as a target for preexisting cross-reactive antimicrobial human (cross)reactive HSP60 immunity or bona fide autoimmune reactions induced by biochemically altered autologous HSP60. Continuous stress by persisting risk factors leads to severe atherosclerosis. Partly adapted from Wick et al20 and Servier Medical Art.
Figure 2.

Schematic overview of HSP60 expression/function in early and late atherosclerosis. Stress-induced EC surface expression of adhesion molecules and HSP60 enables T cells, monocytes, and dendritic cells to adhere to the ECs and transmigrate into the intima. The T cells (mostly CD4+) are the first cells entering the intima during early atherosclerosis. Antigen recognition can be performed both by professional antigen-presenting cells (dendritic cells and macrophages) and by ECs and SMCs expressing MHC class I and class II (induced by interferon-γ). In addition, circulating anti-HSP60 antibodies are present. During progression from an early lesion to a severe plaque, EC damage via anti-HSP60 antibodies, an increased number of macrophages and SMCs often loaded with lipids (foam cells), and neovascularization can be demonstrated. The foam cells may rupture and release their contents into the lesional area, leading to the characteristic formation of cholesterol crystals. Also, the concentration of sHSP60 and anti-HSP60 antibodies is elevated in the serum of subjects with severe atherosclerosis. Anti-HSP60 antibodies can lyse the ECs in a complement-mediated fashion or via antibody-dependent cellular cytotoxicity (ADCC). In humans, the pathogenetic role of HSP60-specific T cells and antibodies, respectively, has still not been completely elucidated. However, studies in experimental animals have provided evidence for both mechanisms to contribute to atherogenesis. IL indicates interleukin; IFN, interferon; ADCC, antibody-dependent cellular cytotoxicity. Partly adapted from Servier Medical Art.
All humans develop protective, beneficial adaptive immunity against the phylogenetically highly conserved microbial HSP60 antigen via infection or vaccination in addition to the immunity against organism-specific epitopes. Under physiological conditions, vascular endothelial cells (ECs) do not express HSP60. However, when stressed by classical atherosclerosis risk factors, the simultaneous expression of adhesion molecules and HSP60 by ECs leads to a (cross)reaction against and destruction of these target cells by preexisting cellular and humoral immunity against HSP60, entailing intimal infiltration by mononuclear cells. If atherosclerosis risk factors persist, these early, still reversible inflammatory stage of atherosclerosis proceeds to plaque formation with deleterious consequences. Supplemental Table I (available online at http://atvb.ahajournals.org) lists the most important contributions from various laboratories on the association of CVD with anti-HSP antibodies, sHSPs, and HSP-specific T cells that appeared since our last extensive review on this topic in 2004.20
Self-HSP-Reactive T Cells and B Cells
Because upregulation of HSP60 is part of an inflammatory response, self-HSP60-reactive T cells with a regulatory phenotype should be part of the physiological termination of the inflammatory response. T cells reacting to self-HSP epitopes have been found in both human and animal studies. An important prerequisite for stimulation of HSP-reactive T cells is the presentation of the corresponding peptides by major histocompatibility complex (MHC) molecules. Self-HSP peptides have access to both MHC class I and class II molecules.
As already mentioned, we have shown that CD4+ T cells are the first T-cell population in early human lesions. Many of the CD4+ cells are HLA-DR and CD25 positive, and a majority of intima infiltrating T cells carry the T-cell receptor α/β, but an unexpectedly high proportion of these T cells are T-cell receptor γ/δ positive. Both SMCs and ECs are HLA-DR positive when activated, interferon-γ-producing T cells are present in the vicinity,15-17 suggesting that SMCs and ECs may participate as antigen presenting cells, in the perpetuation of the autoimmune reaction. Interestingly, the T-cell reaction against human HSP60 (hHSP60) is significantly increased in intralesional T cells (mainly displaying an oligoclonally restricted T-cell receptor α/β repertoire) compared with peripheral T cells (polyclonal repertoire) of the same individual.21 Moreover, carotid plaques of atherosclerosis patients harbor in vivo–activated CD4+ T cells that react specifically to self-HSP60 peptides.22 In addition to cross-reactive T cells, cross-reactive B cells epitopes have also been shown to serve as autoimmune targets in incipient atherosclerosis.23
MHC class II antigen expression on ECs overlying atherosclerotic lesions has been found in low-density lipoprotein-receptor (LDLr)/interferon-γ double knockout mice.24 In mice immunized with heat-killed Mycobacterium tuberculosis, around 20% of all mycobacterium-reactive CD4+ α/β T cells were specific for HSP60,25 and immunization with mycobacterial HSP60 (mHSP65 as a paradigmatic example of bacterial HSP60) induced cross-reactive CD8+ α/β T cells.26 Both α/β- and γ/δ-positive HSP60-reactive T cells may play a role in the pathogenesis of human autoimmune diseases (ie, rheumatoid arthritis, multiple sclerosis, Behcet disease, and atherosclerosis).27 For example, CD8+ α/β T cells recognize HSP60 peptides on MHC class I in vitro, and after transfer to α/β T-cell-deficient mice, these T cells can induce autoimmune disease with severe damage of the gut epithelium.27 After stimulation with mHSP65 peptides in vitro, HSP60-specific CD8+ T cells recognize and lyse host cells exposed to external stress factors. However, recognition and lysis are prevented when HSP60 expression is blocked in target cells with antisense nucleotides.28
Much less is known about the pathogenic role of B cells and anti-HSP60 antibodies in atherogenesis than about the role of T cells. Immunohistological analyses of early and more advanced human atherosclerotic lesions revealed only low numbers of B cells compared with T cells. Furthermore, B cells can mostly only be found in the media and in the adventitia of atherosclerotic plaques but not within early lesion.16,17,29 However, studies on atherosclerotic aneurysms have demonstrated the presence of plasma cells and occasional lymphoid follicles.30 Also, aged apolipoprotein E–null (ApoE−/−) mice showed adventitial formation of inflammatory follicle-like structures that contain proliferating B cells and plasma cells,31 indicating that the adventitia may be a site of local adaptive immune reactions during atherogenesis. Nevertheless, recent studies of the effect of B-cell depletion on the development of atherosclerosis have provided contradictory results. In most instances, B-cell depletion entailed an aggravation of the disease,32-34 whereas several other reports described an amelioration of atherosclerosis.33,35 This discrepancy may be explained by an increased formation of atheroprotective (auto)antibodies (eg, anti-oxLDL)32 in the former or of atherogenic (auto)antibodies (eg, anti-HSP60) in the latter case.
hHSP60 can induce naíve mouse B cells to proliferate and secrete interleukin-10 and interleukin-6. hHSP60-treated B cells can upregulate the expression of MHC class II and accessory molecules CD69, CD40, and B7-2. In addition, sHSP60 can activate B cells via toll-like receptor 4–MyD88 signaling.36 Furthermore, hHSP60 can inhibit mouse B-cell apoptosis, spontaneous or induced, via MyD88 signaling (toll-like receptor 4 is not required in this instance). Inhibition of apoptosis by hHSP60 is associated with upregulation of the antiapoptotic molecules Bcl-2, Bcl-xL, and survivin. Importantly, B cells incubated with hHSP60 manifested prolonged survival following transfer into recipient mice.37 In conclusion, these findings show that both T and B cells can regulate HSP60 immune responses and thus induce autoreactive T and B cells recognizing self-HSP60 peptides, but with respect to primary atherogenic mechanisms, the balance seems to be tilted toward T cells.
Experimental Observations
Up to now, a pathogenic role in atherogenesis is proven only for HSP60, and the induction of atherosclerosis in experimental animals has so far been achieved only by immunization with prokaryotic and eukaryotic HSP60. In the present context, mHSP65 is always used as a paradigmatic and potent representative of bacterial HSP60. An induction of atherosclerosis was successfully demonstrated in normocholesterolemic rabbits after immunizations with mHSP65.38 T-cell depletion in mHSP65 immunized rabbits leads to a significant decrease of atherosclerotic lesions.39 This is also the case for nonimmunized hypercholesterolemic atherosclerosis-prone (LDLr−/−) mice crossed with lymphocyte-deficient (RAG-1−/−) mice. In the progeny of these double knockout mice, early lesion development was reduced by 54%, indicating that lymphocytes play an important role in early atherosclerosis.40 Immunization of rabbits with mHSP65 had an even more pronounced effect when these were simultaneously fed a cholesterol-rich (Western) diet.41 Although the early inflammatory stage of atherosclerosis in normocholesterolemic mHSP65-immunized rabbits was still reversible during a 32-week observation period, the lesions persisted in a similarly immunized hypercholesterolemic group.41 This was later corroborated in atherosclerosis-resistant wild-type C57BL/6J mice fed a Western diet combined with mHSP65 immunization and in LDLr−/− mice fed normal chow diet together with mHSP65 immunizations (Figure 3).42,43
Figure 3.
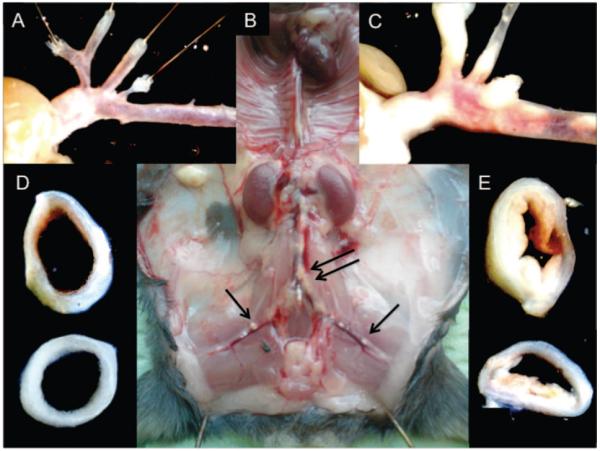
Plaque formation in an atherosclerosis-prone animal model, the ApoE−/− mouse. A, The aortic arch of a 22-week-old female ApoE−/− mouse fed normal chow diet. B, A 22-week-old female ApoE−/− mouse fed a cholesterol-rich Western diet for 14 weeks and in addition having been injected 4 times with mHSP65 (50 μg/100 mL). Black arrows indicate visible plaque formation. C, A higher magnification of the aortic arch and branching arteries (of B). D, A cross-section of the aortic arch (of A). E, A cross-section of the aortic arch (of B).
Lesion-derived T cells from normocholesterolemic mHSP65-immunized rabbits show a significantly increased reactivity against mHSP65 as compared with autologous peripheral blood T cells.44 Passive transfer of T cells from mHSP65 immunized atherosclerotic LDLr−/− mice into non-immunized synergeneic LDLr−/− recipients leads to the development of disease in the latter.45 Interestingly, passive transfer of a mouse monoclonal antibody (II-13) recognizing an epitope consisting of amino acid residues 288 to 366 of HSP60, or of affinity polyclonal anti-hHSP60 antibodies purified from the serum of atherosclerotic patients can also induce atherosclerosis in ApoE−/− recipient mice.46 Importantly, immunization of hypercholesterolemic ApoE−/− mice with homologous malondialdehyde–low-density lipoprotein had the opposite effect, ie, a disease ameliorating effect.47
Exerting endothelial stress in rats or rabbits, eg, by intravenous injection of bacterial lipopolysaccharide, leads to the simultaneous expression of adhesion molecules and HSP60 at known predilection sites for the later development of atherosclerotic lesions in ECs at arterial branching points subjected to turbulent blood flow conditions.48 This process can be visualized in vitro and in vivo by molecular imaging techniques (Supplemental Figure I).49
In Vitro Observations
HSP60 is encoded in the nucleus but is expressed in mitochondria. Under stressful conditions, HSP60 is transported into the cytosol and then appears on the cell surface, where it acts as a “danger signal” for innate and adaptive immunity. The latter has been proven with immunofluorescence,50 atomic force microscopy,9 and metabolic labeling.51 In our hands, all classical atherosclerosis risk factors studied so far first lead to the simultaneous endothelial expression of adhesion molecules and HSP60, allowing for an interaction of HSP60-specific T cells and (auto)antibodies with the target cells, in this case the stressed ECs. Interestingly, the threshold for adhesion molecule and HSP60 expression on confrontation with such stress factors is lower in arterial as compared with venous ECs because the former have been subjected to lifelong “prestress” by the higher arterial blood pressure. This has been demonstrated in an arterio-venous carotid bypass model. These venous conduits subjected to the new arterial blood flow and pressure conditions developed neointimal lesions within 1 to 2 weeks after surgery, finally leading to complete restenosis.52 The first event in these pathological processes is the ECs expression of HSP60 and adhesion molecules and T cells infiltrating the intima.20 Stressed ECs, but not unstressed ones, are lysed by anti-HSP60 monoclonal or affinity purified polyclonal human anti-HSP60 antibodies in a complement-mediated fashion or via antibody-dependent cellular cytotoxicity.53 Moreover, HSP60 may also function as an inducer of anti-EC antibodies able to trigger cytotoxic and apoptotic responses when recognized by the related autoantibodies. Depending on the HSP60 epitope specificity, it appears that anti-EC antibodies with HSP60 reactivity may differ in their functional effects.54
EC Stressors
The role of classical atherosclerosis risk factors has been proven in abundant numbers of clinical, experimental, and in vitro studies. Because the autoimmune concept of atherosclerosis in essence relies on 2 assumptions—(1) preexistent immunity to HSP60 and (2) pathological stress-induced transformation of ECs to HSP60-expressing targets for this immunity—it was deemed of interest to scrutinize the role of different risk factors with respect to the latter phenomenon. This stressor effect was first observed for high (blood) pressure in vivo and in vitro in rats,55 as well as for lipopolysaccharide in vivo in rabbits48 and in vitro on human ECs.56 These latter data are also in agreement with the correlation of the lifelong infectious load with anti-HSP60 immunity and the appearance of atherosclerosis.20
Recently, we have obtained in vitro evidence that oxLDL, ie, an inducer of foam cell formation, and advanced glycation end products as a surrogate for diabetic metabolic conditions, also act as endothelial stressors. oxLDL showed a more pronounced HSP60-inducing capacity compared with advanced glycation end products (Figure 4). For a considerable period of time, cigarette smoke extract appeared to be our most potent endothelial stressor. Using cigarette smoke extract in vitro and calibrating its concentration via the nicotine content mimicking blood levels in mild, intermediate, or heavy smokers, we were able to achieve abundant expression of HSP60, adhesion molecules, proinflammatory cytokines, and at higher concentrations autophagy and necrosis.57 As a final step in this series of experiments on the mode of action of endothelial stressors, we have just recently finished a study on the potential stressor effect of an infection with C. pneumoniae and found this to be the most potent EC stressor that we have observed so far, even exceeding the effect of cigarette smoke extract. C. pneumoniae also induces excessive expression of adhesion molecules and proinflammatory cytokines. Furthermore, ongoing experiments in our laboratory have also revealed a downregulation of the expression of antioxidant molecules and, most importantly, of autophagy-associated molecules, notably ATG 2, ATG 5, and ATG 12. Thus, for a certain period of time, this intercellular endothelial stressing organism seems to be able to create a subtle equilibrium of the host cell metabolism, providing appropriate conditions for its own survival.
Figure 4.
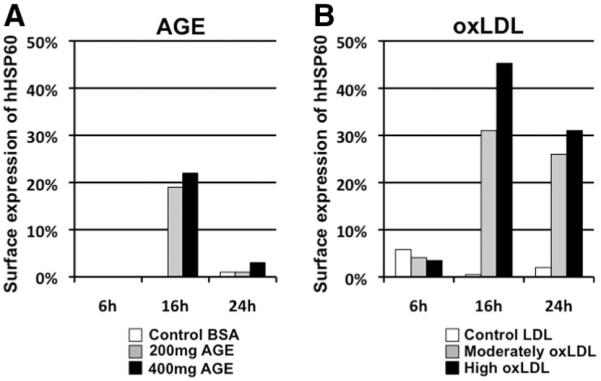
hHSP60 surface expression of human umbilical vein endothelial cells (HUVECs) treated with metabolic stressors. (A) Comparison of the effect of 2 different concentrations of advanced glycation end product (AGE) (200 mg/mL and 400 mg/mL) to native bovine serum albumin (BSA) (400 mg/mL). (B) oxLDL (moderate or high) was compared with native low-density lipoprotein (LDL) (60 μg/mL). Measurements were performed after 6, 16, and 24 hours of stimulation.
Damage of ECs and their sloughing off into the blood-stream has been demonstrated for several atherosclerotic risk factors (eg, cigarette smoking and hyperlipidemia), especially in areas of turbulent blood flow conditions. This results in increased endothelial turnover with increased EC apoptosis or necrosis. Interestingly, this damage seems to be continuously repaired. This reendothelialization can occur both by out-growing neighboring ECs and by circulating endothelial progenitor cells.58,59 We hypothesize—but have no experimental proof—that ECs at atherosclerosis predilection sites are especially prone to this sequence of events.
Clinical Observations
All healthy humans display innate and adaptive anti-HSP60 immunity, the latter induced by infection, by vaccination, or as bona fide autoimmunity against biochemically altered autologous HSP60, probably derived from damaged or necrotic ECs.20,60 For example, it has been speculated that in genetically predisposed individuals the same antigen(s), especially mycobacterial antigen(s), eg, HSP60 (Mtb-HSP60), may induce different immune responses, leading to the development of sarcoidosis and tuberculosis, respectively.61
The use of circulating sHSP60 concentrations or anti-hHSP60 antibody titers as prognostic biomarkers for the risk of developing CVD has been discussed during the last decade. In a prospective follow-up study of 195 healthy subjects, significantly higher anti-hHSP60 antibody titers were found in individuals with future CVD compared with individuals without cardiovascular events.62 Similarly, a large population-based study with 826 subjects showed elevated sHSP60 levels in individuals with prevalent/incident carotid atherosclerosis, with serum levels that correlated with common carotid artery intima-media thickness (IMT).63 These data were later confirmed in a prospective follow-up study.11 Furthermore, patients with borderline hypertension and coronary heart disease present with elevated levels of sHSP6064,65 and increased titers of anti-hHSP65 and anti-hHSP70 antibodies are associated with established hypertension.10 Increased serum levels of sHSP60, sHSP72, and inflammatory markers can be correlated with the extent of cardiac and microvascular dysfunction in patients with angiographically normal coronary arteries.66
A significant correlation between the titers of anti-hHSP65 antibodies and sHSP60 with carotid atherosclerosis lesion size has been found in a large prospective longitudinal atherosclerosis prevention study originally comprising 1000 volunteers of both sexes, aged 40 to 79 years (the BRUNECK study).67,68 In follow-up studies on this cohort, it was then shown that anti-HSP60 reactivity of peripheral blood T cells correlates with increased IMT in clinically healthy male youngsters but not in men aged 50 to 69 from this cohort,69 indicating a more prominent role of specific cellular immunity to HSP60 in the early stages of atherosclerosis. Furthermore, the anti-hHSP60 antibody titer was not only identified as a new early biomarker for morbidity but also for mortality from atherosclerosis.70 In Western blots, these anti-hHSP60 antibodies not only react with recombinant bacterial HSP60 but also cross-react with recombinant hHSP60. Interestingly, anti-HSP60 antibody titers drop after myocardial infarction, a phenomenon that is probably due to immune complex formation with hHSP60 released from the damaged myocardial tissue.71 This possible sequence of events has been suggested and proven later in experiments in rats on induction of cardiac hypoxia, as mentioned above.72
A cross-sectional study involving 17 to 18 year-old clinically healthy men (the Atherosclerosis Risk Factors in Male Youngsters study),73 revealed an unexpected and alarmingly high incidence of increased arterial IMT as a hallmark of the first inflammatory stage of the disease already afflicting 28% of these young men. This parameter showed a significant correlation with cigarette smoking, followed by a significant correlation with HSP60-specific reactivity of peripheral T cells, surprisingly only then followed by a correlation with the diastolic blood pressure and finally, least significantly, with anti-hHSP60 antibody titers.73 Furthermore, T-cell reactivity against hHSP60 correlates with increased IMT in these youngsters.69 This indicates a prominent role of specific cellular immunity to HSP60 in very early stages of atherosclerosis. In a study investigating 19- to 21-year-old young healthy female volunteers (the Atherosclerosis Risk Factors in Female Youngsters study),74 19% had an increased arterial IMT that correlated significantly with passive smoking and, again similar to the Atherosclerosis Risk Factors in Male Youngsters study, anti-HSP60 reactivity of peripheral T cells. In this group, no correlation between increased IMT and anti-HSP60 antibodies emerged.74
Lifelong infectious load has been discussed as correlated with antimicrobal HSP60 antibody titers and with atherosclerosis. More detailed studies on anti-HSP60 antibody reactivity have revealed a linear correlation of the antibody titer to non-HSP60 antigens of C. pneumoniae and Helicobacter pylori with the antibacterial HSP60 titer and atherosclerosis.75,76 Cross-reactivity of plasma anti-GroEL and anti-Porphyromonas gingivalis antibodies with hHSP60 have also been demonstrated in atherosclerosis patients.77 It therefore seems that anti-HSP60 immunity is a common denominator for the association of infectious load with atherosclerosis. In contrast, no association with ECs dysfunction and the presence and severity of coronary artery disease and antibody response to C. pneumoniae IgG or human or Chlamydia HSP60 has been documented, arguing against the suggestion that infection contributes to disease progression.78 In our hands, anti-cytomegalovirus antibody titers do not correlate with anti-HSP60 antibody titers.76 However, this is not surprising, taking into account the fact that viruses do not encode their own HSP60 but that the HSP60 found in viral envelopes originate from the surface of the host cells.
In summary, the results point to a possible primary pathogenetic role of HSP60-specific T cells in the earliest stages of the disease, whereas HSP60 antibodies seem to lead to later aggravation and perpetuation. Our laboratory has recently confirmed this assumption in mice showing that T cells from draining lymph nodes and spleens in ApoE−/− animals immunized with mHSP65 harbor mHSP65 peptide–specific T cells. In humans, we have isolated intralesional T cells both from late and, more importantly, early, clinically still inapparent lesions (Figure 5). The reactivity of intralesional T cells against HSP60 significantly exceeded that obtained with T cells from the peripheral blood from the same donors.
Figure 5.

Immunohistochemical staining of early, clinically inapparent human atherosclerotic lesion. A, Under stress conditions, ECs (green: von Willebrand factor) express hHSP60 (red). Yellow represents colocalization between vWF and HSP60. Blue indicates DNA. B, Adhesion molecules (green: vascular cell adhesion molecule-1) and hHSP60 (red) simultaneously expressed on the EC surface. Blue indicates DNA. The intima of the early lesion contain a higher number of T cells (C) (red: CD3) compared with macrophages (D) (brown: CD68). Original magnification: ×250 (A), ×600 (B), and ×400 (C and D). *Lumen.
HSP Vaccination
In the last few years, several studies have provoked an increased interest in mucosal tolerization against HSP60 aiming to suppress atherogenesis.79,80 Both of these studies could successfully demonstrate a reduction in aortic plaques by this approach. However, only full-length mHSP65 (not defined proatherogenic peptides) was applied, therefore not taking into account the importance of bacterial-human cross-reactive epitopes. Full-length HSP60 and a HSP60 peptide (253 to 268) have also been orally administrated to LDLr−/− mice before the induction of atherosclerosis, which resulted in a 80% reduction in plaque size in the carotid arteries and in a 27% reduction in plaque size at the aortic root.81 The reduction in plaque size correlated with an increase in the number of T-regulatory cells,81 which are known to be protective in atherosclerosis.82,83 Nasal treatment with mHSP65 can effectively attenuate atherosclerosis in rabbits fed a Western diet.84 Moreover, a promising cross-reactive B-cell epitope on both mHSP60 and hHSP60 involved in early atherogenesis has recently been demonstrated.23 However, whether immune reactions against this epitope are involved in the pathogenesis of atherosclerosis remains to be elucidated. oxLDL is another example in which modulation of autoimmunity against atherosclerotic-associated autoantigens represents a novel and promising target for prevention and treatment of CVDs. Similar to HSP60, oxLDL is targeted by both antibody mediated and cellular immune responses. The immune activation is primarily of the proinflammatory Th1-type and inhibition of Th1 immunity reduces atherosclerosis in experimental animals. Atherosclerosis vaccines based on different antigens derived from oxLDL have been developed to modulate these processes.85
In conclusion, these studies suggest that vaccination with HSP60, or more preferably with atheroprotective HSP60 peptides, is a promising idea for the prevention and treatment of atherosclerosis. We are now in the process of delineating atherogenic HSP60 peptides in the murine system to use these for the development of an antiatherosclerosis vaccine via the induction of mucosal tolerance. Although the tolerizing approach in mice may form the basis for the subsequent development of such a vaccine in humans, it is rather improbable that the same HSP60 peptide candidates will emerge as atherogenic in both species. Here, our own data obtained with T cells from early human atherosclerotic lesions will be of crucial importance.
Open Issues
There are still many other open questions to be answered with respect to the role of adaptive and innate immunity to HSP60 in atherogenesis: (1) Why are healthy people tolerant to autologous HSP60? (2) Does sensitization of HSP60-specific T cells take place in situ or in regional lymph nodes? (3) How can bona fide beneficial anti-HSP60 immunity, ie, for elimination of damaged and dead cells, be distinguished from pathogenetically relevant immune reactions? (4) Which atherogenic HSP60 epitopes are recognized by T cells isolated from early, clinically still inapparent human atherosclerotic lesions? (5) What is the exact sequence of gene expression in early versus late atherosclerotic lesion as assessed by microarray techniques? (6) With respect to the endothelial stressor-effect of classical atherosclerotic risk factors, what is the role of infection with C. pneumoniae? (7) Although binding of HSPs to toll-like receptors and trigging of the MyD88 signal-transduction pathway has now been unequivocally demonstrated, what is the possible pathogenic significance of this fact? (8) Finally, because data from clinical studies showed an increasing statistical significance of the reactivity of HSP60-specific T cells in the peripheral blood with decreasing age of the cohorts, we deem it important to perform similar studies in children, ie, revealing the very earliest stages of atherosclerotic disease.
Supplementary Material
Acknowledgments
We are grateful to Drs Ziad Mallat, Willem van Eden, and Andrew C. Newby for their critical reading of and valuable comments on the manuscript. We are also grateful to the group of Dr. Günther Jürgens from the Institute of Physiological Medicine at the Medical University of Graz, Austria, for supplying us with oxLDL and for great technical assistance.
Sources of Funding
The work of our group has been supported by the Austrian Research Fund (FWF; P19881-B05); the European Union Framework Program 6 (MOLSTROKE, LSHM-CT-2004-005206, EVGN; LSHM-CT-2003-S03254); the European Union Framework Program 7, Large Scale Integrated Project: Novel approaches to reconstitute normal immune function at old age (TOLERAGE Health research grant; HEALTH-F4-2008 to 202156); the ERANET PathoGenoMics Program (European Initiative to Fight Chlamydial Infections by Unibased Genomics); the Propter Homines Foundation, FL, and the Medizinische Forschungsförderung Innsbruck (Project 9443, to M.C.W.).
Footnotes
Disclosures
None.
References
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Milkman R. Temperature effects on day old Drosophila pupae. J Gen Physiol. 1962;45:777–799. doi: 10.1085/jgp.45.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig EA, Gambill BD, Nelson RJ. Heat shock proteins: molecular chaperones of protein biogenesis. Microbiol Rev. 1993;57:402–414. doi: 10.1128/mr.57.2.402-414.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young RA, Elliott TJ. Stress proteins, infection, and immune surveillance. Cell. 1989;59:5–8. doi: 10.1016/0092-8674(89)90861-1. [DOI] [PubMed] [Google Scholar]
- 6.Derbinski J, Gabler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M, Peltonen L, Walter J, Kyewski B. Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J Exp Med. 2005;202:33–45. doi: 10.1084/jem.20050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derbinski J, Kyewski B. How thymic antigen presenting cells sample the body’s self-antigens. Curr Opin Immunol. 22:592–600. doi: 10.1016/j.coi.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Teh CE, Daley SR, Enders A, Goodnow CC. T-cell regulation by casitas B-lineage lymphoma (Cblb) is a critical failsafe against autoimmune disease due to autoimmune regulator (Aire) deficiency. Proc Natl Acad Sci U S A. 107:14709–14714. doi: 10.1073/pnas.1009209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfister G, Stroh CM, Perschinka H, Kind M, Knoflach M, Hinterdorfer P, Wick G. Detection of HSP60 on the membrane surface of stressed human endothelial cells by atomic force and confocal microscopy. J Cell Sci. 2005;118:1587–1594. doi: 10.1242/jcs.02292. [DOI] [PubMed] [Google Scholar]
- 10.Pockley AG, De Faire U, Kiessling R, Lemne C, Thulin T, Frostegard J. Circulating heat shock protein and heat shock protein antibody levels in established hypertension. J Hypertens. 2002;20:1815–1820. doi: 10.1097/00004872-200209000-00027. [DOI] [PubMed] [Google Scholar]
- 11.Xiao Q, Mandal K, Schett G, Mayr M, Wick G, Oberhollenzer F, Willeit J, Kiechl S, Xu Q. Association of serum-soluble heat shock protein 60 with carotid atherosclerosis: clinical significance determined in a follow-up study. Stroke. 2005;36:2571–2576. doi: 10.1161/01.STR.0000189632.98944.ab. [DOI] [PubMed] [Google Scholar]
- 12.Gupta S, Knowlton AA. Cytosolic heat shock protein 60, hypoxia, and apoptosis. Circulation. 2002;106:2727–2733. doi: 10.1161/01.cir.0000038112.64503.6e. [DOI] [PubMed] [Google Scholar]
- 13.Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, Torre-Amione G, Knowlton AA. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293:H2238–H2247. doi: 10.1152/ajpheart.00740.2007. [DOI] [PubMed] [Google Scholar]
- 14.Schoneveld AH, Hoefer I, Sluijter JP, Laman JD, de Kleijn DP, Pasterkamp G. Atherosclerotic lesion development and Toll like receptor 2 and 4 responsiveness. Atherosclerosis. 2008;197:95–104. doi: 10.1016/j.atherosclerosis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Kleindienst R, Xu Q, Willeit J, Waldenberger FR, Weimann S, Wick G. Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing α/β or γ/δ receptor in human atherosclerotic lesions. Am J Pathol. 1993;142:1927–1937. [PMC free article] [PubMed] [Google Scholar]
- 16.Millonig G, Malcom GT, Wick G. Early inflammatory-immunological lesions in juvenile atherosclerosis from the Pathobiological Determinants of Atherosclerosis in Youth (PDAY)-study. Atherosclerosis. 2002;160:441–448. doi: 10.1016/s0021-9150(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 17.Xu QB, Oberhuber G, Gruschwitz M, Wick G. Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin Immunol Immunopathol. 1990;56:344–359. doi: 10.1016/0090-1229(90)90155-j. [DOI] [PubMed] [Google Scholar]
- 18.Millonig G, Niederegger H, Rabl W, Hochleitner BW, Hoefer D, Romani N, Wick G. Network of vascular-associated dendritic cells in intima of healthy young individuals. Arterioscler Thromb Vasc Biol. 2001;21:503–508. doi: 10.1161/01.atv.21.4.503. [DOI] [PubMed] [Google Scholar]
- 19.Wissler RW, Strong JP, Pathological Determinants of Atherosclerosis in Youth Research Group Risk factors and progression of atherosclerosis in youth. Am J Pathol. 1998;153:1023–1033. doi: 10.1016/s0002-9440(10)65647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wick G, Knoflach M, Xu Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu Rev Immunol. 2004;22:361–403. doi: 10.1146/annurev.immunol.22.012703.104644. [DOI] [PubMed] [Google Scholar]
- 21.Rossmann A, Henderson B, Heidecker B, Seiler R, Fraedrich G, Singh M, Parson W, Keller M, Grubeck-Loebenstein B, Wick G. T-cells from advanced atherosclerotic lesions recognize hHSP60 and have a restricted T-cell receptor repertoire. Exp Gerontol. 2008;43:229–237. doi: 10.1016/j.exger.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Benagiano M, D’Elios MM, Amedei A, Azzurri A, van der Zee R, Ciervo A, Rombola G, Romagnani S, Cassone A, Del Prete G. Human 60-kDa heat shock protein is a target autoantigen of T cells derived from atherosclerotic plaques. J Immunol. 2005;174:6509–6517. doi: 10.4049/jimmunol.174.10.6509. [DOI] [PubMed] [Google Scholar]
- 23.Perschinka H, Wellenzohn B, Parson W, van der Zee R, Willeit J, Kiechl S, Wick G. Identification of atherosclerosis-associated conformational heat shock protein 60 epitopes by phage display and structural alignment. Atherosclerosis. 2007;194:79–87. doi: 10.1016/j.atherosclerosis.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 24.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-γ on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–460. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann SH, Vath U, Thole JE, Van Embden JD, Emmrich F. Enumeration of T cells reactive with Mycobacterium tuberculosis organisms and specific for the recombinant mycobacterial 64-kDa protein. Eur J Immunol. 1987;17:351–357. doi: 10.1002/eji.1830170308. [DOI] [PubMed] [Google Scholar]
- 26.Koga T, Wand-Wurttenberger A, DeBruyn J, Munk ME, Schoel B, Kaufmann SH. T cells against a bacterial heat shock protein recognize stressed macrophages. Science. 1989;245:1112–1115. doi: 10.1126/science.2788923. [DOI] [PubMed] [Google Scholar]
- 27.Zugel U, Kaufmann SH. Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin Microbiol Rev. 1999;12:19–39. doi: 10.1128/cmr.12.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinhoff U, Zugel U, Wand-Wurttenberger A, Hengel H, Rosch R, Munk ME, Kaufmann SH. Prevention of autoimmune lysis by T cells with specificity for a heat shock protein by antisense oligonucleotide treatment. Proc Natl Acad Sci U S A. 1994;91:5085–5088. doi: 10.1073/pnas.91.11.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramshaw AL, Parums DV. Immunohistochemical characterization of inflammatory cells associated with advanced atherosclerosis. Histopathology. 1990;17:543–552. doi: 10.1111/j.1365-2559.1990.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 30.Walton LJ, Powell JT, Parums DV. Unrestricted usage of immunoglobulin heavy chain genes in B cells infiltrating the wall of atherosclerotic abdominal aortic aneurysms. Atherosclerosis. 1997;135:65–71. doi: 10.1016/s0021-9150(97)00152-4. [DOI] [PubMed] [Google Scholar]
- 31.Moos MP, John N, Grabner R, Nossmann S, Gunther B, Vollandt R, Funk CD, Kaiser B, Habenicht AJ. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2386–2391. doi: 10.1161/01.ATV.0000187470.31662.fe. [DOI] [PubMed] [Google Scholar]
- 32.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–753. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, Kehry M, Dunn R, Agrotis A, Tipping P, Bobik A, Toh BH. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. 2010;185:4410–4419. doi: 10.4049/jimmunol.1000033. [DOI] [PubMed] [Google Scholar]
- 34.Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2002;22:1892–1898. doi: 10.1161/01.atv.0000039169.47943.ee. [DOI] [PubMed] [Google Scholar]
- 35.Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van Vre E, Esposito B, Vilar J, Sirvent J, Van Snick J, Tedgui A, Tedder TF, Mallat Z. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207:1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen-Sfady M, Nussbaum G, Pevsner-Fischer M, Mor F, Carmi P, Zanin-Zhorov A, Lider O, Cohen IR. Heat shock protein 60 activates B cells via the TLR4-MyD88 pathway. J Immunol. 2005;175:3594–3602. doi: 10.4049/jimmunol.175.6.3594. [DOI] [PubMed] [Google Scholar]
- 37.Cohen-Sfady M, Pevsner-Fischer M, Margalit R, Cohen IR. Heat shock protein 60, via MyD88 innate signaling, protects B cells from apoptosis, spontaneous and induced. J Immunol. 2009;183:890–896. doi: 10.4049/jimmunol.0804238. [DOI] [PubMed] [Google Scholar]
- 38.Xu Q, Dietrich H, Steiner HJ, Gown AM, Schoel B, Mikuz G, Kaufmann SH, Wick G. Induction of arteriosclerosis in normocholesterolemic rabbits by immunization with heat shock protein 65. Arterioscler Thromb. 1992;12:789–799. doi: 10.1161/01.atv.12.7.789. [DOI] [PubMed] [Google Scholar]
- 39.Metzler B, Mayr M, Dietrich H, Singh M, Wiebe E, Xu Q, Wick G. Inhibition of arteriosclerosis by T-cell depletion in normocholesterolemic rabbits immunized with heat shock protein 65. Arterioscler Thromb Vasc Biol. 1999;19:1905–1911. doi: 10.1161/01.atv.19.8.1905. [DOI] [PubMed] [Google Scholar]
- 40.Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. 2001;108:251–259. doi: 10.1172/JCI11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu Q, Kleindienst R, Schett G, Waitz W, Jindal S, Gupta RS, Dietrich H, Wick G. Regression of arteriosclerotic lesions induced by immunization with heat shock protein 65-containing material in normocholesterolemic, but not hypercholesterolemic, rabbits. Atherosclerosis. 1996;123:145–155. doi: 10.1016/0021-9150(96)05800-5. [DOI] [PubMed] [Google Scholar]
- 42.Afek A, George J, Gilburd B, Rauova L, Goldberg I, Kopolovic J, Harats D, Shoenfeld Y. Immunization of low-density lipoprotein receptor deficient (LDL-RD) mice with heat shock protein 65 (HSP-65) promotes early atherosclerosis. J Autoimmun. 2000;14:115–121. doi: 10.1006/jaut.1999.0351. [DOI] [PubMed] [Google Scholar]
- 43.George J, Shoenfeld Y, Afek A, Gilburd B, Keren P, Shaish A, Kopolovic J, Wick G, Harats D. Enhanced fatty streak formation in C57BL/6J mice by immunization with heat shock protein-65. Arterioscler Thromb Vasc Biol. 1999;19:505–510. doi: 10.1161/01.atv.19.3.505. [DOI] [PubMed] [Google Scholar]
- 44.Xu Q, Kleindienst R, Waitz W, Dietrich H, Wick G. Increased expression of heat shock protein 65 coincides with a population of infiltrating T lymphocytes in atherosclerotic lesions of rabbits specifically responding to heat shock protein 65. J Clin Invest. 1993;91:2693–2702. doi: 10.1172/JCI116508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.George J, Afek A, Gilburd B, Shoenfeld Y, Harats D. Cellular and humoral immune responses to heat shock protein 65 are both involved in promoting fatty-streak formation in LDL-receptor deficient mice. J Am Coll Cardiol. 2001;38:900–905. doi: 10.1016/s0735-1097(01)01440-1. [DOI] [PubMed] [Google Scholar]
- 46.Foteinos G, Afzal AR, Mandal K, Jahangiri M, Xu Q. Anti-heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E-deficient mice via endothelial damage. Circulation. 2005;112:1206–1213. doi: 10.1161/CIRCULATIONAHA.105.547414. [DOI] [PubMed] [Google Scholar]
- 47.George J, Afek A, Gilburd B, Levkovitz H, Shaish A, Goldberg I, Kopolovic Y, Wick G, Shoenfeld Y, Harats D. Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis. 1998;138:147–152. doi: 10.1016/s0021-9150(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 48.Seitz CS, Kleindienst R, Xu Q, Wick G. Coexpression of heat-shock protein 60 and intercellular-adhesion molecule-1 is related to increased adhesion of monocytes and T cells to aortic endothelium of rats in response to endotoxin. Lab Invest. 1996;74:241–252. [PubMed] [Google Scholar]
- 49.Wick MC, Mayerl C, Backovic A, van der Zee R, Jaschke W, Dietrich H, Wick G. In vivo imaging of the effect of LPS on arterial endothelial cells: molecular imaging of heat shock protein 60 expression. Cell Stress Chaperones. 2008;13:275–285. doi: 10.1007/s12192-008-0044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Q, Schett G, Seitz CS, Hu Y, Gupta RS, Wick G. Surface staining and cytotoxic activity of heat-shock protein 60 antibody in stressed aortic endothelial cells. Circ Res. 1994;75:1078–1085. doi: 10.1161/01.res.75.6.1078. [DOI] [PubMed] [Google Scholar]
- 51.Soltys BJ, Gupta RS. Immunoelectron microscopic localization of the 60-kDa heat shock chaperonin protein (Hsp60) in mammalian cells. Exp Cell Res. 1996;222:16–27. doi: 10.1006/excr.1996.0003. [DOI] [PubMed] [Google Scholar]
- 52.Zou Y, Dietrich H, Hu Y, Metzler B, Wick G, Xu Q. Mouse model of venous bypass graft arteriosclerosis. Am J Pathol. 1998;153:1301–1310. doi: 10.1016/S0002-9440(10)65675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schett G, Xu Q, Amberger A, Van der Zee R, Recheis H, Willeit J, Wick G. Autoantibodies against heat shock protein 60 mediate endothelial cytotoxicity. J Clin Invest. 1995;96:2569–2577. doi: 10.1172/JCI118320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alard JE, Dueymes M, Youinou P, Jamin C. Modulation of endothelial cell damages by anti-Hsp60 autoantibodies in systemic autoimmune diseases. Autoimmun Rev. 2007;6:438–443. doi: 10.1016/j.autrev.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 55.Hochleitner BW, Hochleitner EO, Obrist P, Eberl T, Amberger A, Xu Q, Margreiter R, Wick G. Fluid shear stress induces heat shock protein 60 expression in endothelial cells in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2000;20:617–623. doi: 10.1161/01.atv.20.3.617. [DOI] [PubMed] [Google Scholar]
- 56.Mayr M, Metzler B, Kiechl S, Willeit J, Schett G, Xu Q, Wick G. Endothelial cytotoxicity mediated by serum antibodies to heat shock proteins of Escherichia coli and Chlamydia pneumoniae: immune reactions to heat shock proteins as a possible link between infection and atherosclerosis. Circulation. 1999;99:1560–1566. doi: 10.1161/01.cir.99.12.1560. [DOI] [PubMed] [Google Scholar]
- 57.Henderson B, Csordas A, Backovic A, Kind M, Bernhard D, Wick G. Cigarette smoke is an endothelial stressor and leads to cell cycle arrest. Atherosclerosis. 2008;201:298–305. doi: 10.1016/j.atherosclerosis.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 58.Bai X, Wang X, Xu Q. Endothelial damage and stem cell repair in atherosclerosis. Vascul Pharmacol. 2010;52:224–229. doi: 10.1016/j.vph.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 59.Kirton JP, Xu Q. Endothelial precursors in vascular repair. Microvasc Res. 2010;79:193–199. doi: 10.1016/j.mvr.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 60.Rajaiah R, Moudgil KD. Heat-shock proteins can promote as well as regulate autoimmunity. Autoimmun Rev. 2009;8:388–393. doi: 10.1016/j.autrev.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dubaniewicz A. Mycobacterium tuberculosis heat shock proteins and autoimmunity in sarcoidosis. Autoimmun Rev. 2010;9:419–424. doi: 10.1016/j.autrev.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 62.Hoppichler F, Koch T, Dzien A, Gschwandtner G, Lechleitner M. Prognostic value of antibody titre to heat-shock protein 65 on cardiovascular events. Cardiology. 2000;94:220–223. doi: 10.1159/000047320. [DOI] [PubMed] [Google Scholar]
- 63.Xu Q, Schett G, Perschinka H, Mayr M, Egger G, Oberhollenzer F, Willeit J, Kiechl S, Wick G. Serum soluble heat shock protein 60 is elevated in subjects with atherosclerosis in a general population. Circulation. 2000;102:14–20. doi: 10.1161/01.cir.102.1.14. [DOI] [PubMed] [Google Scholar]
- 64.Pockley AG, Wu R, Lemne C, Kiessling R, de Faire U, Frostegard J. Circulating heat shock protein 60 is associated with early cardiovascular disease. Hypertension. 2000;36:303–307. doi: 10.1161/01.hyp.36.2.303. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, He M, Cheng L, Chen Y, Zhou L, Zeng H, Pockley AG, Hu FB, Wu T. Elevated heat shock protein 60 levels are associated with higher risk of coronary heart disease in Chinese. Circulation. 2008;118:2687–2693. doi: 10.1161/CIRCULATIONAHA.108.781856. [DOI] [PubMed] [Google Scholar]
- 66.Giannessi D, Colotti C, Maltinti M, Del Ry S, Prontera C, Turchi S, Labbate A, Neglia D. Circulating heat shock proteins and inflammatory markers in patients with idiopathic left ventricular dysfunction: their relationships with myocardial and microvascular impairment. Cell Stress Chaperones. 2007;12:265–274. doi: 10.1379/CSC-272.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Willeit J, Kiechl S. Prevalence and risk factors of asymptomatic extracranial carotid artery atherosclerosis: a population-based study. Arterioscler Thromb. 1993;13:661–668. doi: 10.1161/01.atv.13.5.661. [DOI] [PubMed] [Google Scholar]
- 68.Xu Q, Willeit J, Marosi M, Kleindienst R, Oberhollenzer F, Kiechl S, Stulnig T, Luef G, Wick G. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis. Lancet. 1993;341:255–259. doi: 10.1016/0140-6736(93)92613-x. [DOI] [PubMed] [Google Scholar]
- 69.Knoflach M, Kiechl S, Mayrl B, Kind M, Gaston JS, van der Zee R, Faggionato A, Mayr A, Willeit J, Wick G. T-cell reactivity against HSP60 relates to early but not advanced atherosclerosis. Atherosclerosis. 2007;195:333–338. doi: 10.1016/j.atherosclerosis.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 70.Xu Q, Kiechl S, Mayr M, Metzler B, Egger G, Oberhollenzer F, Willeit J, Wick G. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis: clinical significance determined in a follow-up study. Circulation. 1999;100:1169–1174. doi: 10.1161/01.cir.100.11.1169. [DOI] [PubMed] [Google Scholar]
- 71.Hoppichler F, Lechleitner M, Traweger C, Schett G, Dzien A, Sturm W, Xu Q. Changes of serum antibodies to heat-shock protein 65 in coronary heart disease and acute myocardial infarction. Atherosclerosis. 1996;126:333–338. doi: 10.1016/0021-9150(96)05931-x. [DOI] [PubMed] [Google Scholar]
- 72.Schett G, Metzler B, Kleindienst R, Amberger A, Recheis H, Xu Q, Wick G. Myocardial injury leads to a release of heat shock protein (hsp) 60 and a suppression of the anti-hsp65 immune response. Cardiovasc Res. 1999;42:685–695. doi: 10.1016/s0008-6363(99)00012-7. [DOI] [PubMed] [Google Scholar]
- 73.Knoflach M, Kiechl S, Kind M, Said M, Sief R, Gisinger M, van der Zee R, Gaston H, Jarosch E, Willeit J, Wick G. Cardiovascular risk factors and atherosclerosis in young males: ARMY study (Atherosclerosis Risk-Factors in Male Youngsters) Circulation. 2003;108:1064–1069. doi: 10.1161/01.CIR.0000085996.95532.FF. [DOI] [PubMed] [Google Scholar]
- 74.Knoflach M, Kiechl S, Penz D, Zangerle A, Schmidauer C, Rossmann A, Shingh M, Spallek R, Griesmacher A, Bernhard D, Robatscher P, Buchberger W, Draxl W, Willeit J, Wick G. Cardiovascular risk factors and atherosclerosis in young women: atherosclerosis risk factors in female youngsters (ARFY study) Stroke. 2009;40:1063–1069. doi: 10.1161/STROKEAHA.108.525675. [DOI] [PubMed] [Google Scholar]
- 75.Burian K, Kis Z, Virok D, Endresz V, Prohaszka Z, Duba J, Berencsi K, Boda K, Horvath L, Romics L, Fust G, Gonczol E. Independent and joint effects of antibodies to human heat-shock protein 60 and Chlamydia pneumoniae infection in the development of coronary atherosclerosis. Circulation. 2001;103:1503–1508. doi: 10.1161/01.cir.103.11.1503. [DOI] [PubMed] [Google Scholar]
- 76.Mayr M, Kiechl S, Willeit J, Wick G, Xu Q. Infections, immunity, and atherosclerosis: associations of antibodies to Chlamydia pneumoniae, Helicobacter pylori, and cytomegalovirus with immune reactions to heat-shock protein 60 and carotid or femoral atherosclerosis. Circulation. 2000;102:833–839. doi: 10.1161/01.cir.102.8.833. [DOI] [PubMed] [Google Scholar]
- 77.Ford PJ, Gemmell E, Hamlet SM, Hasan A, Walker PJ, West MJ, Cullinan MP, Seymour GJ. Cross-reactivity of GroEL antibodies with human heat shock protein 60 and quantification of pathogens in atherosclerosis. Oral Microbiol Immunol. 2005;20:296–302. doi: 10.1111/j.1399-302X.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- 78.Hoymans VY, Bosmans JM, Van Herck PL, Ieven MM, Vrints CJ. Implications of antibodies to heat-shock proteins in ischemic heart disease. Int J Cardiol. 2008;123:277–282. doi: 10.1016/j.ijcard.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 79.Harats D, Yacov N, Gilburd B, Shoenfeld Y, George J. Oral tolerance with heat shock protein 65 attenuates Mycobacterium tuberculosis-induced and high-fat-diet-driven atherosclerotic lesions. J Am Coll Cardiol. 2002;40:1333–1338. doi: 10.1016/s0735-1097(02)02135-6. [DOI] [PubMed] [Google Scholar]
- 80.Maron R, Sukhova G, Faria AM, Hoffmann E, Mach F, Libby P, Weiner HL. Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation. 2002;106:1708–1715. doi: 10.1161/01.cir.0000029750.99462.30. [DOI] [PubMed] [Google Scholar]
- 81.van Puijvelde GH, van Es T, van Wanrooij EJ, Habets KL, de Vos P, van der Zee R, van Eden W, van Berkel TJ, Kuiper J. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2677–2683. doi: 10.1161/ATVBAHA.107.151274. [DOI] [PubMed] [Google Scholar]
- 82.Mallat Z, Gojova A, Brun V, Esposito B, Fournier N, Cottrez F, Tedgui A, Groux H. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2003;108:1232–1237. doi: 10.1161/01.CIR.0000089083.61317.A1. [DOI] [PubMed] [Google Scholar]
- 83.Taleb S, Tedgui A, Mallat Z. Regulatory T-cell immunity and its relevance to atherosclerosis. J Intern Med. 2008;263:489–499. doi: 10.1111/j.1365-2796.2008.01944.x. [DOI] [PubMed] [Google Scholar]
- 84.Xiong Q, Li J, Jin L, Liu J, Li T. Nasal immunization with heat shock protein 65 attenuates atherosclerosis and reduces serum lipids in cholesterol-fed wild-type rabbits probably through different mechanisms. Immunol Lett. 2009;125:40–45. doi: 10.1016/j.imlet.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 85.Nilsson J, Fredrikson GN, Bjorkbacka H, Chyu KY, Shah PK. Vaccines modulating lipoprotein autoimmunity as a possible future therapy for cardiovascular disease. J Intern Med. 2009;266:221–231. doi: 10.1111/j.1365-2796.2009.02150.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.