

Abstract
Apratoxins are cytotoxic marine natural products that prevent cotranslational translocation early in the secretory pathway. We showed that apratoxins downregulate receptors and growth factor ligands, giving a one–two punch to cancer cells, particularly those that rely on autocrine loops. Through total synthesis, we tested the effects of amino acid substitutions, including alanine scanning, on the downregulation of receptor tyrosine kinases and vascular endothelial growth factor A (VEGF-A) and probed the stereospecificity of target engagement by epimerization of selected chiral centers. Differential effects on two types of secretory molecules suggest that the apratoxins' substrate selectivity with respect to inhibition of secretion may be tuned through structural modifications to provide tailored therapy. Our structure–activity relationship studies and medicinal chemistry efforts led to a potent inhibitor with in vivo efficacy in a colorectal tumor xenograft model without irreversible toxicity exerted by apratoxin A, demonstrating that this novel mechanism of action has therapeutic potential.
Keywords: Antitumor agents, natural products, receptor tyrosine kinases, growth factors, secretory pathway, total synthesis
Cancer cells are characterized by aberrant pro-growth signaling, usually induced upon ligand binding to their corresponding receptor tyrosine kinases (RTKs), which are overexpressed in many cancers.1 Consequently, RTKs have become prime therapeutic targets resulting in the development of specific small molecules (e.g., erlotinib) and antibodies (e.g., cetuximab and panitumumab) targeting epidermal growth factor receptors.2 However, drug resistance is a clinical problem, and combination therapy or the use of broader-spectrum RTK inhibitors (e.g., sorafenib and sunitinib), targeting vascular endothelial growth factor (VEGF) receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR), may be more advantageous.3−5 Alternatively, the ligands themselves may be targeted, which has led to the development and FDA approval of an anti-VEGF-A antibody (bevacizumab) for colorectal cancer treatment.6,7 While studying the mechanism of action of the marine natural product apratoxin A (Figure 1),8 belonging to a class of potent cytotoxins produced by marine cyanobacteria,9−12 we have raised the possibility that inhibition of the secretory pathway at the level of cotranslational translocation by apratoxins and, in general, may be exploited for anticancer therapy.13 Inhibition of this process prevents export of receptors and secretory molecules from the cytoplasm, leading to receptor (including RTK) depletion and additionally should prevent secretion of the corresponding ligands, growth factors, and cytokines.14 This one–two punch is expected to have unusually potent antiproliferative activity and may be an effective alternative to combination therapy with multiple (or broad-spectrum) RTK and growth factor inhibitors and may be particularly useful to treat cancers where autocrine loops, that is, uncontrolled proliferation stimulated by secreted growth factors, play a major role (e.g., colorectal cancer).15 Here, we test this hypothesis through a combination of medicinal chemistry based on the apratoxin scaffold, molecular in vitro and in vivo pharmacology, and colorectal tumor xenograft efficacy studies.
Figure 1.
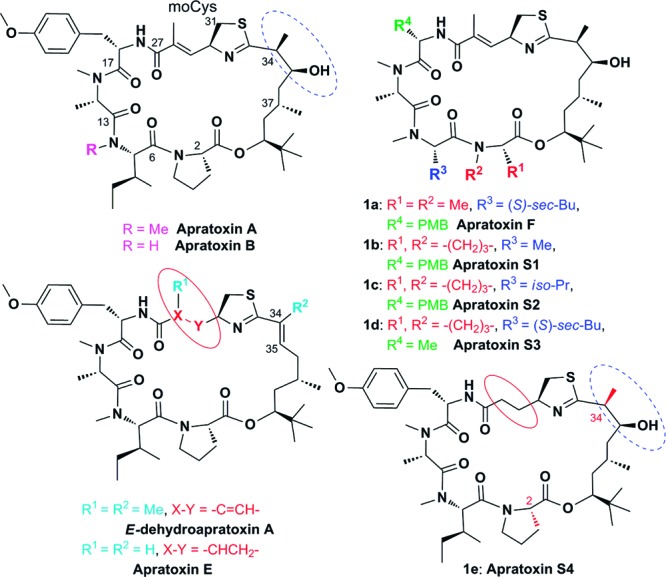
Selected known apratoxins and synthetic targets 1a–e produced through alanine or valine incorporations within apratoxin A at various positions (apratoxins F and S1–S3) or through “hybridization” of apratoxins A and E (apratoxin S4). Me, methyl; Bu, butyl; Pr, propyl; and PMB, p-methoxybenzyl. Two epimers of 1e (2-epi-1e, apratoxin S5; and 34-epi-1e, apratoxin S6), minor reaction products during 1e synthesis, were included in our SAR studies.
Since we found that apratoxin A activity was reversible,13 it became evident that apratoxins have to be administered on a chronic schedule at a low dose rather than given as a bolus injection; the latter was initially reported to cause toxicity without significant antitumor activity.8 Our preliminary data for chronic administration of apratoxin A (Figure S3 in the Supporting Information), recently confirmed by others,12 indicate that this compound shows some antitumor efficacy in vivo but is not well tolerated. The therapeutic window is small; we found a 50% death rate (day 16) at the “therapeutic” concentration of 0.25 mg/kg (daily ip) with irreversible toxicity occurring after 2 weeks of treatment (Figure S3 in the Supporting Information). The main question remained whether this toxicity is mechanism-related or an off-target effect. Here, we describe the synthesis and biological evaluation of synthetic analogues of apratoxin A, one of which shows greater potency and efficacy than the parent compound and is much better tolerated in vivo, providing the groundwork for second-generation apratoxins with more promising anticancer potential and validating this novel mechanism of action for cancer therapy.
Our goal was 3-fold: We aimed (1) to define critical and tunable elements for inhibition of cotranslational translocation, (2) to extend our previous receptor studies and prove that transport of receptors and their corresponding ligands are inhibited by apratoxins, and (3) to design a synthetic analogue with improved potency and less toxic side effects in vivo. After the report of the first total synthesis,16 structure–activity relationship (SAR) studies by other groups have focused on the polyketide (C33–C43) chain.17,18 Apratoxin cytotoxicity is highly sensitive to certain structural modifications to this unit. Reversal of the configuration at C37 from 37S to 37R or removal of the methyl group at C37 abolished the antiproliferative activity of apratoxins, shown for the oxazoline analogue;17 however, the C-34 epimer of apratoxin A showed equipotent activity.18 Depending on the cell line, the C34–C35 dehydration product of apratoxin A possessed 12–29-fold reduced cytotoxicity.11 However, the potency of apratoxin E (Figure 1), while also unsaturated at C34–C35, is only 6–15-fold less cytotoxic than apratoxin A,11 suggesting that the modification in the moCys unit (C27–C31), viz. removal of the methyl group and/or α,β-unsaturation, may increase activity if the original, hydrated configuration of C34–C35 as in apratoxin A can be restored. We also hypothesized that the α,β-unsaturated system in the moCys unit may be responsible for nonspecific (off-target) toxicity due to conjugate addition of cellular nucleophiles. Thus, we aimed to synthesize such an apratoxin A/E hybrid (1e) with hopefully improved anticancer activity and also tumor selectivity. Furthermore, we interrogated modifications in the peptide portion, where it is known that lack of N-methylation of isoleucine reduces activity (apratoxin B)9 and replacement of proline by N-methyl alanine largely retains activity (1a, apratoxin F)12 (Figure 1). We carried out an alanine scan (without changing the N-alkylation pattern) and synthesized a valine analogue to more broadly probe the hydrophobicity requirements of the R3 position (Ile → Val → Ala; Figure 1), which led to the first synthesis of the natural product apratoxin F (1a) and positional congeners (1b–d), providing new insight into the SAR of apratoxins.
Scheme 1 illustrates the main synthetic procedures to obtain the desired apratoxin analogues, closely paralleling reported synthesis strategies and retrosynthetic analysis for apratoxin A with minor modifications.19,20 Aldehyde 2(19−21) was treated with Roush's (E)-crotylboronate 3(22) to give 4. Protection of the hydroxyl group with Troc gave 5, and removal of PMB group with DDQ, followed by esterification with Fmoc-Pro-OH/Fmoc-N-Me-Ala-OH using the Yamaguchi method,23 provided ester 6. Acid 7 was obtained when 6 was oxidized with OsO4/oxone/NaIO4 oxidation system.24 Coupling of 6 with modified cysteine amine 8 gave the key intermediates 9. Using the Kelly method (PPh3(O)/Tf2O in CH2Cl2),17,19,20,25 the thiazoline ring was formed, and Troc was removed immediately thereafter. Subsequent removal of allyl from 10 with Pd(PPh3)4/N-methylaniline provided acid 11, which was coupled with tripeptide 12 (see the Supporting Information) using HATU/DIEA to give protected linear compounds 13. Cleavage of O-allyl ester by Pd(PPh3)4/N-methylaniline and removal of Fmoc by Et2NH/MeCN afforded cyclization precursors, whose final macrolactamization with HATU/DIEA under high dilution concentration provided apratoxins 1a–e in 18–52% yield. We used the same strategy to synthesize apratoxin A for direct comparison in our biological assays.
Scheme 1. Preparation of the Analogues of Apratoxin A.

Reagents and conditions: (a) MS 4 Å, toluene, −78 °C. (b) TrocCl, DMAP, pyridine, CH2Cl2. (c) DDQ, CH2Cl2–H2O. (d) Cl3C6H2COCl, DIEA, THF, DMAP, toluene. (e) OsO4, oxone, NaIO4. (f) HATU, DIEA, CH2Cl2. (g) Ph3P(O), Tf2O, CH2Cl2, 0 °C. (h) Zn, NH4OAc, THF. (i) Pd(PPh3)4, N-methylaniline, THF. (j) HATU, DIEA, CH2Cl2. (k) Pd(PPh3)4, N-methylaniline, THF. (l) Et2NH, MeCN. (m) HATU, DIEA, CH2Cl2. Troc, 2,2,2-trichloroethoxycarbonyl; DMAP, 4-(dimethylamino)pyridine; DDQ, 2,3-dichloro-4,5-dicyanobenzoquinone; Fmoc, 9-fluorenylmethoxycarbonyl; Trt, triphenylmethyl; HATU, O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate; DIEA, diisopropylethylamine; and Tf, trifluoromethylsulfonyl.
We found that apratoxin F (1a) was the most potent apratoxin A analogue that arose from an alanine scan; both compounds showed comparable effects on HCT116 colorectal carcinoma cell viability (Table 1) and met proto-oncogene (MET) receptor levels (Figure 2a). In contrast, the other two alanine scan “mutants” 1b and 1d exhibited strongly reduced activity in both assays. Replacement of Ile with Val (1c) did not affect activity. However, apratoxin S4 (1e)—the apratoxin A/E hybrid (Figure 1)—was superior as compared with all other apratoxins (Figure 2a,b and Table 1), consistent with our hypothesis. As a result of synthesis scale-up, we also identified two minor reaction products that we characterized as 2-epi-1e and 34-epi-1e (Figure 1 and see the Supporting Information), which we included in our SAR studies. While 2-epi-1e lost activity by ca. 200-fold, the two 34-epimers of 1e were almost equally active. In all cases, cell viability (Table 1) correlated well with the reduction of receptor levels based on immunoblot analysis (Figure 2a), suggesting that the configuration at C-2 is crucial for potent cytotoxicity, while the C-34 configuration is irrelevant as recently found for apratoxin A.18
Table 1. Activities of Apratoxins on HCT116 Cell Viability and VEGF-A Secretion.
| apratoxin | IC50 (nM)a cell viability | IC50 (nM)b VEGF-A secretion |
|---|---|---|
| A | 5.97 | 1.49 |
| E | 184 | 9.10 |
| F (1a) | 4.92 | 0.461 |
| S1 (1b) | 373 | 20.7 |
| S2 (1c) | 4.13 | 0.322 |
| S3 (1d) | 1700 | 340 |
| S4 (1e) | 1.14 | 0.308 |
| S5 (2-epi-1e) | 258 | 112 |
| S6 (34-epi-1e) | 1.58 | 0.391 |
Determined after 48 h (n = 4).
Determined after 12 h (n = 3).
Figure 2.
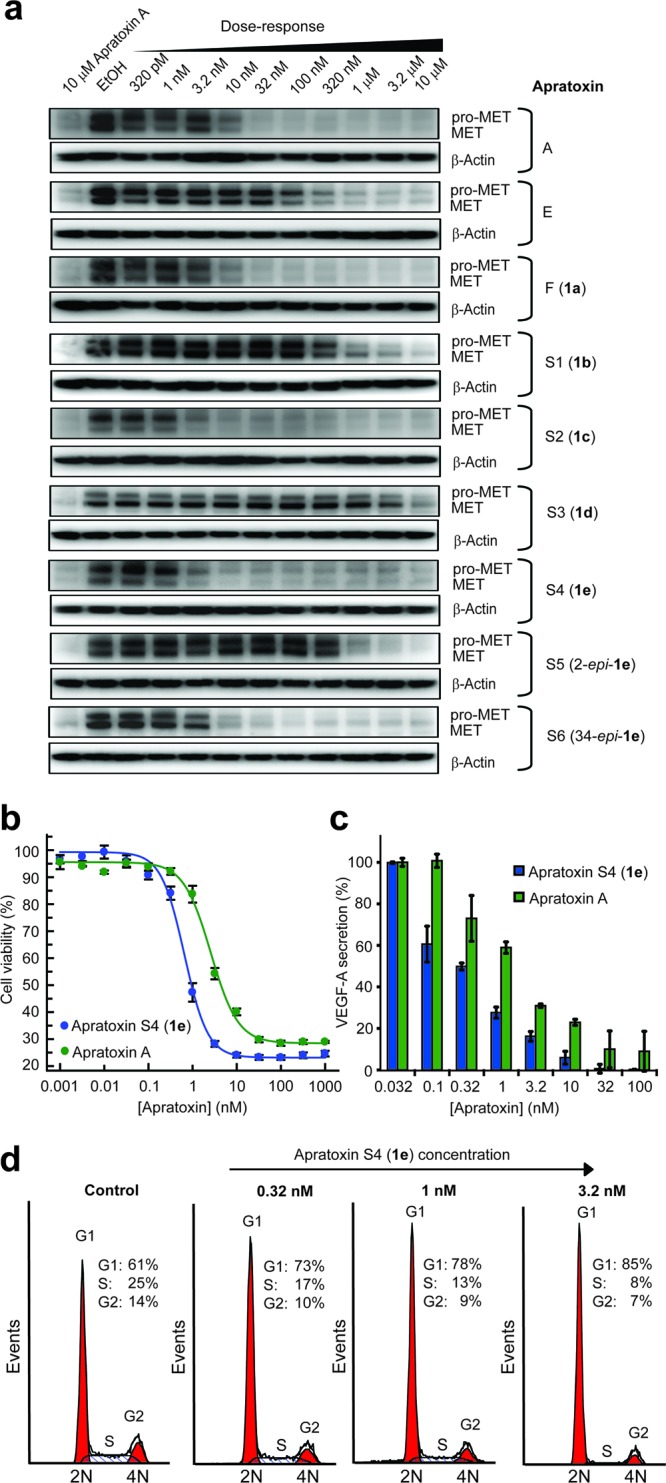
Cellular activity of apratoxins (HCT116 cells). (a) SAR by immunoblot analysis for RTK levels (MET) after 24 h. Comparison of apratoxin A and S4 (1e) effects on (b) cell viability (48 h, n = 4) and (c) VEGF-A secretion (12 h, n = 3). Error bars indicate SD. (d) Dose-dependent cell cycle effects of 1e determined by DNA content analysis, demonstrating induction of G1 arrest (24 h).
Next, we extended our SAR studies to our hypothesized effects of apratoxins on growth factor secretion and focused on the angiogenic drug target VEGF-A.26−28 Indeed, all apratoxins inhibited the secretion of VEGF-A (Table 1 and Figure 2c), confirming our hypothesis that apratoxins prevent the export of receptors and the secretion of ligands, which is also consistent with our data for the antiangiogenic properties of apratoxins.29 Furthermore, depending on the apratoxin structure, the IC50 values were 2–20-fold lower than the IC50 values for cell viability, suggesting that VEGF-A is more susceptible to inhibition of secretion as compared with receptor proteins tested. This indicates that reducing levels of VEGF-A (and likely other ligands) alone is not sufficient to inhibit proliferation and that receptor depletion is a necessity. This result also raises the possibility that the apratoxins' substrate selectivity with respect to inhibition of secretion may be tuned through structural modifications, which is a subject of ongoing research in our laboratory.
We then tested our prioritized compound (1e) on the cell cycle to determine if its effects parallel those of apratoxin A. Consistent with data for apratoxin A,29 dose-dependent DNA content analysis of HCT116 cells upon 24 h of treatment revealed that 1e increases the population of cells in G1 phase concomitant with decrease of cells in S phase, starting at subnanomolar concentrations (0.32 nM) with strongest effect at 3.2 nM (Figure 2d), which is the concentration range for the inhibition of VEGF-A and receptor levels and also near the IC50 for cell viability (Table 1). Next, we confirmed that the inhibition of the secretory pathway by apratoxin S4 (1e) occurs at the level of cotranslational translocation from the cytoplasm to the endoplasmic reticlum.13 We used in vitro translation experiments in the presence of microsomal membranes to demonstrate the inhibition by 1e of cotranslational processing events characteristic for secretory molecules, specifically glycosylation (using α-factor mRNA substrate) and signal peptide cleavage (using β-lactamase mRNA substrate). Inhibition of cotranslational processing in a dose-dependent manner is consistent with the mode of action that we had reported for apratoxin A (Figure 3).13 We also used a human receptor cDNA (PDGFR-β) and proved by coupled in vitro transcription/translation that 1e inhibits processing of this cancer-associated RTK (Figure 3).
Figure 3.

Apratoxin S4 (1e) inhibits cotranslational processing in vitro. Products of in vitro translation reactions (rabbit reticulocyte, amino acids, [35S]methionine, canine pancreatic microsomal membranes, mRNA template, and apratoxin 1e) were separated by SDS-PAGE to autoradiographically detect effects on translation and glycosylation of α-factor and signal peptide cleavage of β-lactamase. When PDGFR-β cDNA was used as a template, in vitro transcription/translation was carried out using T7 TNT Quick Master Mix and glycosylation determined by SDS-PAGE followed by autoradiography.
Initial in vivo studies aimed at determining the maximum tolerated dose (MTD) already suggested that 1e is much better tolerated in vivo than apratoxin A, which is consistent with our hypothesis that the conjugated system may be a liability in vivo. While high doses (≥0.375 mg/kg) of apratoxin A led to irreversible toxicity, that is, continued weight loss and eventual death even when ip injection was discontinued, mice treated with apratoxin S4 (1e) quickly recovered when high-dose injections were stopped. Using the same low dose of 0.25 mg/kg of 1e (daily ip for over 2 weeks) used for apratoxin A to treat HCT116 tumor-bearing nu/nu mice (Figure S3 in the Supporting Information), the antitumor effect was much stronger (Figure 4a) without any evident toxicity based on lack of weight loss and histological assessment of kidney and liver tissue. To determine if the mechanism of action in vivo correlates with our in vitro data, we measured receptor levels in the tiny residual tumor tissue and found that VEGFR2 and PDGFR-β levels were depleted (Figure 4b). In contrast, the liver appeared less affected, as we did not observe a significant reduction of VEGFR2 in this tissue of the same mice (Figure 4c), which may indicate preferential uptake of 1e by the tumor and/or enhanced dependency of tumor on the secretory pathway due to more rapid turnover and resynthesis of receptor molecules.
Figure 4.
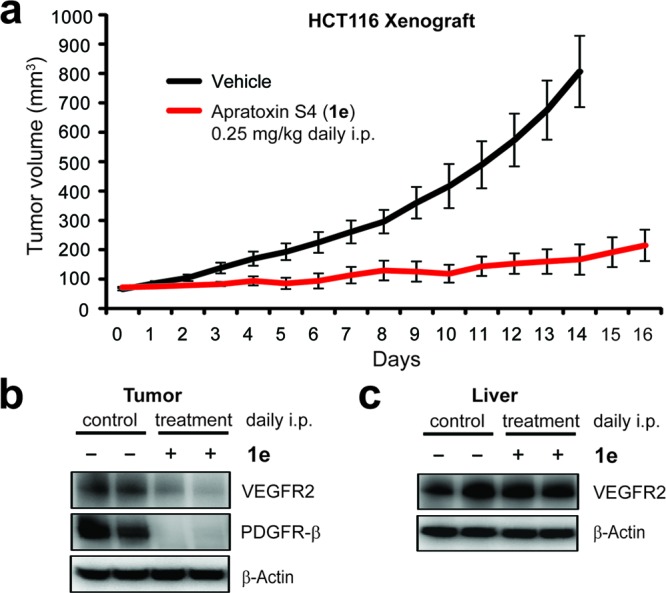
In vivo activity of apratoxin S4 (1e) using a HCT116 xenograft mouse model. (a) Efficacy studies (daily ip). Subcutaneous tumor-bearing mice were injected with 1e (n = 8) or DMSO vehicle (n = 10), and tumor volumes were monitored over time. Error bars indicate SEM. (b) At the end of the efficacy studies, tumors were analyzed by immunoblot analysis for receptor levels. (c) Levels of VEGFR2 were analogously measured in liver tissue of vehicle- vs 1e-treated mice.
In conclusion, apratoxin S4 (1e) is the first viable candidate of the apratoxin family that shows the requisite tumor selectivity and increased antitumor activity and potency. Thus, we have largely separated anticancer activity from nonselective toxicity, indicating for the first time that inhibition of cotranslational translocation is a promising novel mechanism for cancer therapy, particularly to treat cancers that are characterized by aberrant autocrine loops such as colorectal cancer. Furthermore, our SAR studies suggest that through structural modification we may be able to tune apratoxin selectivity toward certain substrates destined for the secretory pathway, which may be exploited to provide tailored therapy and also more selective chemical tools to study the secretory pathway.
Acknowledgments
We thank Prof. B. Law for initial help with tumor implantation.
Glossary
Abbreviations
- MET
met proto-oncogene (hepatocyte growth factor receptor)
- PDGFR
platelet-derived growth factor receptor
- RTK
receptor tyrosine kinase
- SAR
structure–activity relationship
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
- Me
methyl
- Bu
butyl
- Pr
propyl
- PMB
p-methoxybenzyl
- Troc
2,2,2-trichloroethoxycarbonyl
- DMAP
4-(dimethylamino)pyridine
- DDQ
2,3-dichloro-4,5-dicyanobenzoquinone
- Fmoc
9-fluorenylmethoxycarbonyl
- Trt
triphenylmethyl
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- DIEA
diisopropylethylamine
- Tf
trifluoromethylsulfonyl
Supporting Information Available
Synthetic procedures, NMR data and spectra, descriptions of biochemical assays, cell cycle analysis, and in vivo experiments as well as additional figures, schemes, and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally to this work.
This work was supported by the University of Florida Research Opportunity Fund and the Bankhead-Coley Cancer Research Program, Grant 1BG07.
Supplementary Material
References
- Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science 2006, 312, 1175–1178. [DOI] [PubMed] [Google Scholar]
- Lurje G.; Lenz H.-J. EGFR signaling and drug discovery. Oncology 2009, 77, 400–410. [DOI] [PubMed] [Google Scholar]
- Chow L. Q.; Eckhardt S. G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–96. [DOI] [PubMed] [Google Scholar]
- Kling J. Bundling next-generation cancer therapies for synergy. Nat. Biotechnol. 2006, 24, 871–872. [DOI] [PubMed] [Google Scholar]
- Martinelli E.; Troiani T.; Morgillo F.; Rodolico G.; Vitagliano D.; Morelli M. P.; Tuccillo C.; Vecchione L.; Capasso A.; Orditura M.; De Vita F.; Eckhardt S. G.; Santoro M.; Berrino L.; Cardiello F. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin. Cancer Res. 2010, 16, 4990–5001. [DOI] [PubMed] [Google Scholar]
- Hurwitz H.; Fehrenbacher L.; Novotny W.; Cartwright T.; Hainsworth J.; Heim W.; Berlin J.; Baron A.; Griffing S.; Holmgren E.; Ferrara N.; Fyfe G.; Rogers B.; Ross R.; Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [DOI] [PubMed] [Google Scholar]
- Koutras A. K.; Starakis I.; Kyriakopoulou U.; Katsaounis P.; Nikolakopoulos A.; Kalofonos H. P. Targeted therapy in colorectal cancer: Current status and future challenges. Curr. Med. Chem. 2011, 18, 1599–1612. [DOI] [PubMed] [Google Scholar]
- Luesch H.; Yoshida W. Y.; Moore R. E.; Paul V. J.; Corbett T. H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [DOI] [PubMed] [Google Scholar]
- Luesch H.; Yoshida W. Y.; Moore R. E.; Paul V. J.; Corbett T. H. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [DOI] [PubMed] [Google Scholar]
- Gutierrez M.; Suyama T. L.; Engene N.; Wingerd J. S.; Matainaho T.; Gerwick W. H. Apratoxin D, a potent cytotoxic cyclodepsipeptide from Papua New Guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [DOI] [PubMed] [Google Scholar]
- Matthew S.; Schupp P. J.; Luesch H. Apratoxin E, a cytotoxic peptolide from a Guamanian collection of the marine cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2008, 71, 1113–1116. [DOI] [PubMed] [Google Scholar]
- Tidgewell K.; Engene N.; Byrum T.; Media J.; Doi T.; Valeriote F. A.; Gerwick W. H. Evolved diversification of a modular natural product pathway: Apratoxins F and G, two cytotoxic cyclic depsipeptides from a Palmyra collection of Lyngbya bouillonii. ChemBioChem 2010, 11, 1458–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Law B. K.; Luesch H. Apratoxin A reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharmacol. 2009, 76, 91–104. [DOI] [PubMed] [Google Scholar]
- Pleiotropic mechanisms on HSP90 client proteins have been proposed for the apratoxins, although many of these observations may be rationalized by inhibition of the secretory pathway:Shen S.; Zhang P.; Lovchik M. A.; Li Y.; Tang L.; Chen Z.; Zeng R.; Ma D.; Yuan J.; Yu Q. Cyclodepsipeptide toxin promotes the degradation of Hsp90 client proteins through chaperone-mediated autophagy. J. Cell. Biol. 2009, 185, 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan W.-J.; Lai M.-D. Autocrine stimulation in colorectal carcinoma (CRC): Positive autocrine loops in human colorectal carcinoma and applicable significance of blocking the loops. Med. Oncol. 2004, 21, 1–7. [DOI] [PubMed] [Google Scholar]
- Chen J.; Forsyth C. J. Total synthesis of apratoxin A. J. Am. Chem. Soc. 2003, 125, 8734–8735. [DOI] [PubMed] [Google Scholar]
- Ma D.; Zou B.; Cai G.; Hu X.; Liu J. O. Total synthesis of the cyclodepsipeptide apratoxin A and its analogues and assessment of their biological activities. Chem.—Eur. J. 2006, 12, 7615–7626. [DOI] [PubMed] [Google Scholar]
- Doi T.; Numajiri Y.; Takahashi T.; Takagi M.; Shin-ya K. Solid-phase total synthesis of (−)-apratoxin A and its analogues and their biological evaluation. Chem. Asian J. 2011, 6, 180–188. [DOI] [PubMed] [Google Scholar]
- Doi T.; Numajiri Y.; Munakata A.; Takahashi T. Total synthesis of apratoxin A. Org. Lett. 2006, 8, 531–534. [DOI] [PubMed] [Google Scholar]
- Numajiri Y.; Takahashi T.; Doi T. Total synthesis of (−)-apratoxin A, 34-epimer, and its oxazoline analogue. Chem. Asian J. 2009, 4, 111–125. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Chen Z.; Ye T. Synthesis of polyketide segment of apratoxin A. Tetrahedron: Asymmetry 2004, 15, 355–363. [Google Scholar]
- Roush W. R.; Ando K.; Powers D. B.; Palkowitz A. D.; Halterman R. L. Asymmetric synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Preparation of the chiral crotylboronates and reactions with achiral aldehydes. J. Am. Chem. Soc. 1990, 112, 6339–6348. [Google Scholar]
- Inanaga J.; Hirata K.; Saeki H.; Katsuki T.; Yamaguchi M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- Travis B. R.; Narayan R. S.; Borhan B. Osmium tetroxide-promoted catalytic oxidative cleavage of olefins: an organometallic ozonolysis. J. Am. Chem. Soc. 2002, 124, 3824–3825. [DOI] [PubMed] [Google Scholar]
- You S.-L.; Razavi H.; Kelly J. W. A biomimetic synthesis of thiazolines using hexaphenyloxodiphosphonium trifluoromethanesulfonate. Angew. Chem., Int. Ed. 2003, 42, 83–85. [DOI] [PubMed] [Google Scholar]
- Carmeliet P.; Jain R. K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [DOI] [PubMed] [Google Scholar]
- Kerbel R. S. Tumor angiogenesis: Past, present and the near future. Carcinogenesis 2000, 21, 505–515. [DOI] [PubMed] [Google Scholar]
- Luesch H.; Chanda S. K.; Raya R. M.; DeJesus P. D.; Orth A. P.; Walker J. R.; Izpisúa Belmonte J. C.; Schultz P. G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.