

Abstract
Utilization of cyanide as a nitrogen source by Pseudomonas fluorescens NCIMB 11764 occurs via oxidative conversion to carbon dioxide and ammonia, with the latter compound satisfying the nitrogen requirement. Substrate attack is initiated by cyanide oxygenase (CNO), which has been shown previously to have properties of a pterin-dependent hydroxylase. CNO was purified 71-fold and catalyzed the quantitative conversion of cyanide supplied at micromolar concentrations (10 to 50 μM) to formate and ammonia. The specific activity of the partially purified enzyme was approximately 500 mU/mg of protein. The pterin requirement for activity could be satisfied by supplying either the fully (tetrahydro) or partially (dihydro) reduced forms of various pterin compounds at catalytic concentrations (0.5 μM). These compounds included, for example, biopterin, monapterin, and neopterin, all of which were also identified in cell extracts. Substrate conversion was accompanied by the consumption of 1 and 2 molar equivalents of molecular oxygen and NADH, respectively. When coupled with formate dehydrogenase, the complete enzymatic system for cyanide oxidation to carbon dioxide and ammonia was reconstituted and displayed an overall reaction stoichiometry of 1:1:1 for cyanide, O2, and NADH consumed. Cyanide was also attacked by CNO at a higher concentration (1 mM), but in this case formamide accumulated as the major reaction product (formamide/formate ratio, 0.6:0.3) and was not further degraded. A complex reaction mechanism involving the production of isocyanate as a potential CNO monooxygenation product is proposed. Subsequent reduction of isocyanate to formamide, whose hydrolysis occurs as a CNO-bound intermediate, is further envisioned. To our knowledge, this is the first report of enzymatic conversion of cyanide to formate and ammonia by a pterin-dependent oxygenative mechanism.
Cyanide is a potent poison that arises in the environment by anthropogenic and natural means. Large quantities are generated in the electroplating, mining, and coal gasification industries, and the presence of cyanide in cigarette and industrial fire smoke, as well as in automobile exhaust, has also been documented (2, 17). Over 1,000 different plant species generate cyanide as a natural product, as do some fungi and bacteria (38, 41). Despite the occurrence of cyanide in the biosphere, our understanding of the role that microbes play in cyanide recycling is incomplete. This is somewhat surprising since bacteria able to use cyanide as a sole nitrogen source can readily be isolated from nature (1, 8, 15, 39, 40, 45). As far as is known, growth on cyanide requires that it be enzymatically converted to ammonia, which is then assimilated by well-established pathways. One enzyme that does this is cyanide nitrilase, also described as cyanide dihydratase or cyanidase (CNN). CNN catalyzes the single-step hydrolysis of cyanide to ammonia and formic acid (equation 1).
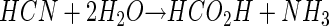 |
(1) |
CNN has been found in several bacteria (20, 26, 32, 46, 47), but whether it confers the ability to grow on cyanide is not clear since a correlation between growth and enzyme production has not been established. This may be because at the high concentrations of substrate required by the enzyme (Km values of 1.5 to 10 mM have been reported [for a review see reference 37]) growth is inhibited. Thus far, mutants defective in CNN production have not been described, which has precluded genetic investigation of the effects on cyanide utilization. At present, the role of CNN is thought to be mainly one of cyanide detoxification.
A second route of cyanide conversion is a route involving oxygenolytic conversion to carbon dioxide and ammonia (equation 2).
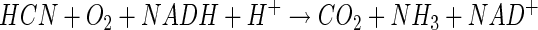 |
(2) |
In this case, substrate decomposition is initiated oxygenatively by an enzyme described as a cyanide oxygenase (CNO) (16, 22, 26). CNO has thus far been found only in Pseudomonas fluorescens strain NCIMB 11764. In contrast to CNN, CNO is able to attack cyanide at much lower concentrations (apparent Km, 3.5 μM) (24). This helps explain the ability of P. fluorescens NCIMB 11764 to grow on cyanide at low nontoxic concentrations, such as those provided under fed-batch conditions (15, 25). It also helps explain how various compounds containing dissociable cyanide as a ligand (e.g., metal-cyano complexes, cyanohydrins) can support growth because free cyanide (predominantly HCN given that the pKa for HCN/CN− is 9.2 [10]) can be enzymatically scavenged. The hypothesis that CNO is absolutely required for growth on cyanide is supported by findings demonstrating that mutant strains that are unable to grow on cyanide have no detectable CNO enzyme activity (26; R. Fernandez, H. Wessler, R. Benjamin, and D. Kunz, Abstr. 101st Gen. Meet. Am. Soc. Microbiol., abstr. K106, 2001).
Isotopic labeling studies which showed that a single atom of molecular oxygen was incorporated during substrate conversion by crude cell extracts indicated that CNO functions as a monooxygenase (44). A second atom of oxygen derived from water was also shown to be incorporated, which allowed the following reaction mechanism to be proposed, in which an unidentified monooxygenated intermediate (X-OH) formed by CNO eventually gives rise to NH3 and CO2 following hydrolysis (equations 3 and 4) (44).
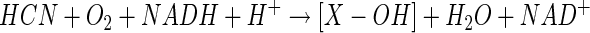 |
(3) |
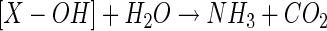 |
(4) |
However, further details regarding the enzymes and possible intermediates involved have not been presented. Results showing that cyanide-grown cells contain elevated levels of both CNO and NAD+-linked formate dehydrogenase (FDH) suggest that formate is a pathway intermediate, but further evidence of this has not been obtained (26).
It was recently reported that CNO displays properties of a pterin-dependent hydroxylase since a soluble cofactor in cell extracts required for enzyme activity could be replaced by reduced pterins (tetrahydrobiopterin and 6-methyltetrahydropterin) (Fig. 1) (24). Here we describe the partial purification of CNO and show that the products of pterin-dependent conversion of cyanide are ammonia and formate. In addition, several reduced pterin species capable of serving as natural cofactors for the enzyme were identified in P. fluorescens NCIMB 11764 cell extracts. When these species were added to separate protein fractions containing partially purified CNO and FDH, the complete enzymatic system for reduced pterin-dependent cyanide oxidation to CO2 and ammonia could be reconstituted.
FIG. 1.

General structures of fully reduced pterins. H4biopterin, 6-l-erythro-(1,2-dihydroxypropyl)-5,6,7,8-tetrahydropterin; H4monapterin, 6-l-threo-(1,2,3-trihydroxypropyl)-5,6,7,8-tetrahydropterin. The partially reduced dihydropterins have double bonds at positions 5 and 6.
(A preliminary account of some of this work has appeared previously [R. Fernandez, B. Venables, and D. Kunz, Abstr. 102nd Gen. Meet. Am. Soc. Microbiol., abstr. K41, 2002].)
MATERIALS AND METHODS
Coenzymes and other chemicals.
l-Biopterin, 7,8-dihydro-l-biopterin (H2biopterin), (6R,S)-5,6,7,8-tetrahydro-l-biopterin (H4biopterin), l-monapterin, 7,8-dihydro-l-monapterin (H2monapterin), (6R,S)-5,6,7,8-tetrahydro-l-monapterin (H4monapterin), d-neopterin, and 7,8-dihydro-d-neopterin (H2neopterin) were all purchased from B. Schirck's Laboratory (Jona, Switzerland). For some experiments H4biopterin and biopterin were obtained from Sigma/Aldrich. 6-Methyl-5,6,7,8-tetrahydropterin (H46-methylpterin) was obtained from Fluka. The pterin concentrations of stock solutions were determined spectrophotometrically by using previously published molar extinction values (35).
KCN (97%) and K13C15N (99 atom% for each isotope) were obtained from Aldrich. K14CN (1.9 GBq/mmol) and sodium [14C]formate (2.1 GBq/mmol) were purchased from Sigma. All cyanide solutions were prepared immediately prior to use and were handled according to Environmental Protection Agency and Texas Commission on Environmental Quality standards.
Growth of bacteria and preparation of cell extracts.
P. fluorescens NCIMB 11764 was grown and induced for enzymatic activity by adding KCN (0.1 mM) to stationary-phase cultures grown in 10 mM glucose-ammonia (1 mM NH4Cl) minimal medium as described previously (25, 39). Cells were harvested (approximately 0.8 g [wet weight]/liter), pelleted by centrifugation at 7,500 × g, and washed in 50 mM Na2HPO4-KH2PO4 phosphate buffer (pH 7.0) (Na-K buffer) before they were stored at −80°C. Large-scale cultivation of cells was performed in a 400-liter APV Crepaco fermentor at 30°C (Department of Biochemistry, University of Wisconsin, Madison). After 24 h of growth, KCN (approximately 0.1 mM) was added to induce enzyme activity, and cells were harvested within 8 h by collection with a Sharples Super centrifuge. The cell paste was stored at −80°C until it was used.
Cell extracts were prepared as described previously (24, 26), and enzyme activities were recovered in 150,000-×-g high-speed supernatants (HSS). Each HSS was concentrated by passage through 15-ml (Centriprep-10) or 2-ml (Centricon-10) ultrafiltration concentrators (nominal molecular mass cutoff, 10 kDa; Amicon). The method of Lowry et al. with bovine serum albumin as a standard was used to quantify protein (29).
Enzyme assays.
Fixed-time measurements for CNO and FDH were determined by measuring the extents of K14CN and [14C]formate conversion to 14CO2, respectively. Reactions were carried out in crimp-sealed vial or bottles as described previously (24, 44). For CNO assays, reaction mixtures (0.25 ml) contained enzyme (0.125 to 0.5 mg protein), NADH (1 mM), H2biopterin (0.5 μΜ), and radiolabeled substrate (1 μCi [48 KBq]; approximately 100 μM) in Na-K buffer. For FDH assays, the conditions were the same except that the reaction mixtures contained [14C]formate, enzyme, and NAD+ (1 mM). The reaction mixtures were incubated with shaking at 30°C, and 14CO2 was quantified after acidification with H2SO4 (0.02 N) by liquid scintillation counting with a Beckman LS 7000 counter (24, 44).
The time-dependent CNO assay was performed at ambient temperature by measuring the rate of KCN-dependent NADH oxidation (ɛ340 = 6.2 mM−1 cm−1) with a Perkin-Elmer Lambda 6 UV/VIS spectrophotometer. The reaction mixtures (0.5 ml) contained protein (12.5 μg to 2 mg), NADH (0.2 mM), H2biopterin (0.5 μM), and KCN (10 to 40 μM) in Na-K buffer. CNO was also assayed by measuring oxygen consumption at 30°C with an O2 electrode (Clark type; Rank Bros., Cambridge, United Kingdom) in reaction mixtures that contained the same components in air-saturated buffer (0.224 mM O2). The oxygen electrode was calibrated by using catechol-2,3-dioxygenase (3). In each case, reactions were initiated by addition of KCN, and the initial rates of NADH and O2 consumption were corrected for background consumption occurring in the absence of KCN. FDH activity was measured spectrophotometrically by monitoring NAD+ reduction at 340 nm. The reaction mixtures (0.4 ml) contained protein (0.2 to 2 mg), 0.4 mM NAD+, and 0.2 mM formate in 50 mM KH2PO4 buffer (pH 7.5). One unit of enzyme activity was defined as 1 nmol/min (1 mU) per mg of protein.
Substrate depletion and product formation assays.
Assays for cyanide degradation and product formation by CNO were performed in sealed vials with shaking at 30°C. For assays conducted with low cyanide concentrations (10 to 50 μM), the reaction mixtures (0.25 to 1 ml) contained H2biopterin (0.5 μM), NADH (0.2 mM), and protein (10 to 50 μg/ml) in Na-K buffer. For assays in which cyanide was supplied at a high concentration (1 mM), the reaction conditions were the same except that NADH was included in the mixtures at a concentration of 2 mM. Cyanide was determined colorimetrically as previously described (25, 28). Ammonia was determined by the indophenol method of Fawcett and Scott (6). To do this, 0.25 ml of sodium phenate, 0.375 ml of 0.01% sodium nitroprusside, and 0.375 ml of 0.2 N NaOCl were added successively to 0.125 ml of a sample, and the absorbance at 630 nm was determined after 30 min (limit of detection, 0.6 nmol). Formate was quantified enzymatically with commercial FDH (Candida lipolytica; 1.4 U/mg; Sigma) following deproteinization of samples with ZnSO4 as described previously (26). The presence of formate in reaction mixtures was independently confirmed by gas chromatography (GC)-mass spectrometry (MS) with a Hewlett-Packard 5992 GC-MS by injecting 1 to 5 μl of a deproteinized sample onto an Alltech Associates EC-1000 capillary column (length, 30 m; inside diameter, 0.25 mm; injector temperature, 150°C; inlet pressure, 5 lb/in2; carrier gas, He; oven temperature, 40°C for 5 min and then ramped to 175°C at a rate of 10°C min−1 and held constant for 24 min). Formamide was determined either colorimetrically or enzymatically (by formate determination following hydrolysis) as described previously (25, 26). Colorimetric determinations were performed with an LKB Ultraspec II UV/VIS spectrophotometer.
Resolution of CNO and FDH.
CNO and FDH were resolved to separate protein fractions by anion-exchange chromatography at 4°C using an Amersham-Pharmacia fast protein liquid chromatograph. HSS (180 ml containing 10.1 mg of protein per ml) was concentrated with a Centriprep-10, and the retentate (designated fraction H; 56 ml containing 29.8 mg of protein per ml) was applied to a Source 30Q anion-exchange column (10 by 24 cm; Amersham-Pharmacia) equilibrated with 50 mM piperazine buffer (pH 10). A linear gradient in which the Na2SO4 concentration increased from 0 to 0.5 M was applied, and fractions (6 ml) were assayed for CNO and FDH activities by measuring the extents of conversion of K14CN and [14C]formate to 14CO2. Fractions showing activity were pooled, desalted in Na-K buffer, and concentrated with 10-kDa-cutoff ultraconcentrators.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) with a 16% polyacrylamide total gel and a 4% polyacrylamide cross-linking gel was carried out with a Mini-Protean (Bio-Rad) system by the procedure of Laemmli (27). The protein-solubilizing buffer contained 150 mM dithiothreitol, 150 mM Trizma (pH 6.8), 21% glycerol, 6% lithium dodecyl sulfate, and 0.003% bromphenol blue. Electrophoresis was conducted in SDS denaturing buffer (25 mM Trizma, 192 mM glycine, 0.1% SDS; pH 8.3) at 200 V for 55 min at 4°C. The low-molecular-mass protein standards (97 to 14 kDa) used for calibrating gels were purchased from Bio-Rad.
Analysis of pterins in cell extracts.
The filtrate (<10 kDa) obtained after HSS was passed through a Centriprep-10 ultraconcentrator was analyzed for pterins by high-performance liquid chromatography (HPLC). Compounds were analyzed in either their reduced or fully oxidized states. Oxidation of compounds was achieved by the procedure of Milstein and Kaufman (33). Filtered samples (pore size, 0.45 μm) were injected onto a Microsorb C18 column (4.6 by 250 mm; Varian), and adsorbed compounds were eluted isocratically at a flow rate of 0.8 ml/min with 5% aqueous methanol. Compounds were detected by fluorescence (excitation at 350 nm and emission at 450 nm) and UV absorption (254 nm) by using a Rainin/Varian liquid chromatograph (LC) equipped with fluorescence (Shimadzu) and UV (Knauer) detectors. Compounds were also analyzed by ion trap MS by using an Agilent Technologies model G2445 LC-MS.
RESULTS
Resolution of CNO and FDH in cell extracts.
Cell extracts (HSS) from cyanide-induced P. fluorescens NCIMB 11764 cells converted both K14CN and [14C]formate to 14CO2 with a yield of more than 70% (Table 1). This coincided with the detection of CNO and FDH in extracts, and each enzyme exhibited a specific activity of approximately 7 mU/mg (data not shown). Following passage of HSS through a 10-kDa-cutoff ultraconcentrator, conversion of K14CN to 14CO2 was no longer observed. However, addition of H4biopterin to recovered proteins in the retentate (fraction H) restored the cyanide-oxidizing activity (Table 1). In addition to H4biopterin, H4monapterin, H2biopterin, and H2monapterin were also found to support enzyme activity (see below). H2biopterin rather than H4biopterin was routinely added to enzymatic reaction mixtures because of its greater stability to air oxidation (35). In addition to the pterin-dependent conversion of cyanide to CO2, fraction H also oxidized [14C]formate in the presence of NAD+. When resolved protein fractions were screened for [14C]cyanide conversion to 14CO2 after anion-exchange chromatography (Fig. 2), no conversion was detected. In contrast, the ability to oxidize [14C]formate to 14CO2 was detected in a single elution peak, which therefore was deduced to contain FDH (Fig. 2, peak A). This peak was collected and designated fraction A, and it contained no CNO activity, as shown by its inability to consume cyanide. SDS-PAGE of fraction A resulted in a predominant protein band at 42 kDa.
TABLE 1.
Resolution of enzyme activities in P. fluorescens NCIMB 11764 cell extracts by anion-exchange chromatography
| Fraction(s)a | % of 14C substrate converted to 14CO2 byb:
|
||
|---|---|---|---|
| CNO
|
FDH ([14C]formate) | ||
| H2biopterin added | K14CN | ||
| HSS | − | 73.5 | 84.6 |
| H | + | 79.8 | 86.4 |
| H | − | 10.5 | 84.8 |
| A | + | 12.1 | 88.2 |
| B | + | 12.3 | 10.2 |
| A + B | + | 83.5 | 90.5 |
| A + B | − | 10.1 | ND |
HSS, supernatant from high-speed centrifugation (150,000 × g); H, retentate after passing HSS through a Centricon-10 filter (>10 kDa molecular size cutoff); A and B, pooled fractions from peaks A and B shown in Fig. 2.
Reaction mixtures were incubated for 15 min and contained the following amounts of protein: HSS, 1 mg/ml; fraction H, 1 mg/ml; fraction A, 0.5 mg/ml; fraction B, 0.5 mg/ml; fractions A + B, 0.25 mg of A and 0.25 mg of B per ml. H2biopterin was provided at a concentration of 0.5 μM. ND, not determined.
FIG. 2.

Separation of CNO- and FDH-containing P. fluorescens NCIMB 11764 protein fractions. Proteins in cell extracts were eluted from a Source Q anion-exchange column at pH 10.0 with increasing concentrations of Na2SO4. FDH and CNO activities were recovered in peaks A and B, respectively. (Inset) SDS-PAGE of active protein fractions. Lane 1, protein molecular weight standards (5 μg); lane 2, induced fraction H (10 μg); lane 3, uninduced fraction H (10 μg); lane 4, peak A (5 μg); lane 5, peak B (10 μg).
Recovery of partially purified CNO.
When the ability to oxidize cyanide was tested by combining separate protein fractions after Source Q chromatography, it was found that combining peak A with a peak that eluted slightly later (Fig. 2, peak B) restored activity (Table 1). Conversion was not observed when H2biopterin was omitted from reaction mixtures, which provided strong evidence that peak B contained one or more proteins responsible for CNO activity. When peak B (designated fraction B) was collected and was tested for the ability to oxidize formate, no activity was detected. However, when this peak was incubated with 50 μM cyanide (representing a 14-fold-greater concentration than the estimated half-saturation constant for CNO [3.5 μM] [24]), substrate disappeared at a high rate. In the absence of H2biopterin or NADH or under anaerobic conditions, substrate consumption by fraction B did not occur. In addition, KCN stimulated oxygen consumption by fraction B, but it did not do this when H2biopterin was omitted from reaction mixtures (Fig. 3A). Also, no oxygen consumption was observed when KCN was added to 0.5 μM H2biopterin in the absence of protein (data not shown). Parallel experiments showed that addition of cyanide to CNO-containing fraction B stimulated NADH consumption (Fig. 3B). The specific activity of the enzyme calculated from rates of NADH consumption was 500 mU/mg of protein (which was approximately the value obtained from initial rates of cyanide disappearance). Since unfractionated cell extracts (fraction H) contained an average CNO activity of 7 mU/mg, the increase in CNO activity in fraction B reflected a 71-fold enrichment of the enzyme. Approximately 13.2% of the original activity (2.2 mg of protein from the original 1,670 mg of protein fractionated) was recovered in fraction B, with about 20% of the activity being lost after 1 month of storage at −80°C. SDS-PAGE analysis of the partially purified enzyme revealed a number of protein bands at sizes ranging from 92 to 17 kDa (Fig. 2, inset). The appearance of a 19-kDa protein coincided with detection of a protein having a similar molecular mass in unfractionated cell extracts from induced cells but not from uninduced cells, suggesting that this species is involved with inducible CNO activity. After continued freezing and thawing the 19-kDa protein gave way to an increase in the amount of a 17-kDa species that presumably represented a smaller polypeptide derivative.
FIG. 3.

Stimulation of oxygen (A) and NADH (B) consumption by cyanide when it was added to partially purified CNO. The reaction mixture contained 37.5 μg of protein and 0.1 mM NADH (A) or 12.5 μg of protein and 0.2 mM NADH (B). Substrates were added at the times indicated by arrows.
CNO reaction products and stoichiometry.
Oxygen consumption measurements showed that 1 molar equivalent of oxygen was consumed per molar equivalent of KCN provided to CNO enriched in fraction B (Fig. 3A). Analyses of reaction mixtures after oxygen consumption had ceased showed that there was no remaining cyanide. Parallel observations were made for NADH consumption; however, in this case 2 molar equivalents of NADH was consumed (Fig. 3B). A search for possible reaction products consistently revealed the presence of formate in the reaction mixtures, and the identity of this compound was confirmed both enzymatically and by GC-MS (H12CO2H [m/z = 46] and H13CO2H [m/z = 47] [retention time, 13.1 min] from K12CN and K13CN, respectively). When cyanide consumption and formate accumulation were quantified, a 1:1 stoichiometry at substrate concentrations ranging from 10 to 50 μM was observed (Table 2). Separate analyses showed that ammonia was also formed and that the amount was equal to the amount of formate. The relationship between substrate depletion and product formation by CNO was further investigated at limiting NADH concentrations. Table 2 shows that the amount of cyanide consumed by CNO-enriched fraction B was equivalent to one-half the amount of NADH provided when equivalent concentrations of the compounds were supplied. In comparison, when there was a twofold excess of NADH over cyanide, 90% of the substrate was converted to formate.
TABLE 2.
Reaction stoichiometry for partially purified CNOa
| KCN provided (μM) | NADH provided (μM) | KCN consumed (μM) | NADH provided/KCN consumed ratio | Formate produced (μM) | Formate produced/KCN consumed ratio |
|---|---|---|---|---|---|
| 10 | 10 | 5.1 | 1.96 | 4.8 | 0.91 |
| 10 | 20 | 10.0 | 2.0 | 9.8 | 0.98 |
| 20 | 20 | 10.1 | 1.98 | 10.6 | 1.04 |
| 20 | 40 | 20.0 | 2.0 | 18.7 | 0.94 |
| 40 | 40 | 20.9 | 1.91 | 18.8 | 0.90 |
| 40 | 80 | 40.0 | 2.0 | 37.6 | 0.94 |
Reaction mixtures containing 50 μg of CNO-enriched fraction B protein per ml were initiated by injecting KCN. After 20 min, samples (50 μl) were removed for determination of the amount of cyanide and 10 μl of saturated ZnSO4 was added simultaneously to stop the reactions before the amount of formate was determined.
The time course of cyanide conversion by partially purified CNO showed that there was a significant lag in the accumulation of formate (Fig. 4). This suggested that potential intermediates could be formed. Careful analysis of reaction mixtures following the consumption of 50 μM cyanide by CNO-enriched fraction B revealed trace quantities of formamide (approximately 7 μM) in addition to formate (approximately 45 μM). Moreover, as the amount of cyanide provided to fraction B was increased, a greater quantity of formamide was detected, suggesting that the amount of substrate provided to CNO had an effect on the amount of formamide that accumulated. This was confirmed by experiments in which CNO was incubated with 1 mM KCN. In this case formamide was detected as the major reaction product (ratio of formamide to formate, 0.6:0.3 [n = 4]). Because oxygen, NADH, and H2biopterin were also required for formamide production in the presence of 1 mM cyanide, we deduced that CNO and not a separate enzyme was responsible for formamide accumulation at the higher cyanide concentration. Separate experiments showed that formamide (50 μM to 1 mM) was not degraded by either CNO-enriched fraction B or unfractionated cell extract provided at an excess protein concentration (4 mg/ml). Hence, the difference between the amount of formamide that accumulated at low substrate concentrations and the amount of formamide that accumulated at high substrate concentrations was interpreted as indicating that formamide is formed as an enzyme-bound intermediate and is susceptible either to hydrolysis or release from the enzyme depending upon the amount of substrate supplied.
FIG. 4.

Time course of cyanide conversion to formate by partially purified P. fluorescens NCIMB 11764 CNO. The reaction mixtures (1 ml) contained 10 μg of protein per ml. Symbols: □, KCN; ♦, formate; ○, KCN with H2B omitted. The values are means for two separate determinations.
Detection of pterins in P. fluorescens NCIMB 11764 cell extracts.
The analysis of filtrates obtained after HSS were passed through 10-kDa-cutoff ultraconcentrators revealed several fluorescent peaks when HPLC was performed (Fig. 5). The two major peaks, peaks b and c, were identified as H2monapterin and H2biopterin, respectively, by comparing their elution times with those of authentic standards and by ion trap LC-MS (H + 1 molecular ions at 256 and 240, respectively). Two additional compounds were also detected (Fig. 5, peaks a and d). The compounds in peaks a and d were identified as H2neopterin (threo isomer of H2monapterin) and threo-H2biopterin, respectively, by their relative mobilities (9). The presence of H2neopterin in peak a was also confirmed by LC-MS (H + 1 molecular ion at 256).
FIG. 5.

HPLC analysis of the filtrate (10 μl) obtained after passage of P. fluorescens NCIMB 11764 HSS through a 10-kDa-cutoff ultraconcentrator. The identities of the peaks, as determined by comparison with authentic standards, are as follows: peak a, H2neopterin; peak b, H2monapterin; peak c, H2biopterin; peak d, threo-H2biopterin.
When tested for the ability to serve as cofactors, both authentic H2biopterin and H2monapterin supported CNO activity. As shown in Table 3, when either compound was provided to the enzyme at a concentration that was 100-fold lower than the concentration of cyanide (0.5 versus 50 μM), the specific activities (calculated from rates of ammonia production) were comparable to those obtained for activity determinations when NADH consumption was measured. Similar activities for ammonia production were observed when H2neopterin, H4monapterin, H4biopterin, and H46-methylpterin were supplied. The corresponding oxidized pterins were not effective in supporting enzyme activity. Also, no activity was observed in the absence of reduced pterin or in controls containing reduced pterin but no protein.
TABLE 3.
CNO-enhancing activities of pterin compounds
| Pterin provided | Activity (mU/mg of protein)a
|
|
|---|---|---|
| Cyanide degraded | Ammonia formed | |
| H4biopterin | 494 ± 92 | 421 ± 67 |
| H2biopterin | 501 ± 38 | 508 ± 88 |
| Biopterin | <10 | <10 |
| H4monapterin | 407 ± 38 | 411 ± 11 |
| H2monapterin | 444 ± 32 | 379 ± 85 |
| Monapterin | <10 | <10 |
| H2neopterin | 412 ± 36 | 410 ± 61 |
| Neopterin | <10 | <10 |
| H46-methylpterin | 425 ± 21 | 385 ± 17 |
| None | <10 | <10 |
Reaction mixtures contained 50 μM KCN, 10 μg of CNO-enriched fraction B protein per ml, and 0.5 μM pterin. After 10 min the amount of remaining cyanide and the amount of ammonia accumulated were determined. The values are means ± average deviations of the means for three separate determinations.
DISCUSSION
The complete oxidation of cyanide to CO2 and NH3 by reconstituted CNO- and FDH-containing protein fractions provides strong evidence that these two enzymes are required for assimilation of cyanide as a nitrogen source by P. fluorescens NCIMB 11764. The requirement for reduced pterin, oxygen, and NADH during conversion of cyanide to formate and ammonia by partially purified CNO provides the first evidence that these products can be generated by an oxygen-dependent mechanism. Thus, the following equations summarize the overall enzymatic process.
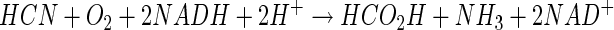 |
(5) |
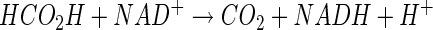 |
(6) |
 |
(7) |
The finding that 2 molar equivalents of NADH was required for cyanide conversion by CNO is consistent with the reaction stoichiometry shown in equation 5. One molar equivalent of NADH can then be regenerated by the action of FDH (equation 6), which accounts for the overall reaction stoichiometry in which 1 molar equivalent of NADH and 1 molar equivalent of O2 are consumed per molar equivalent of cyanide degraded (equation 7). This parallels previously reported values for the reaction stoichiometry and the results of studies showing that CNO and FDH could be coupled (16, 22, 26).
The lag period observed between cyanide consumption and the production of formate (and ammonia) by CNO (Fig. 4) is consistent with a reaction mechanism involving more than a single step. This finding coincides with previous isotopic labeling studies in which both monooxygenative and hydrolytic reactions were shown to occur (equations 3 and 4) (44). Although the identity of the initial monooxygenation product remains unknown, a reductive step in its conversion to formic acid (and ammonia) is envisioned given that a putative monooxygenated species is expected to have a higher oxidation state (presumably +4) than formate (+2) and cyanide (+2). The detection of formamide, whose oxidation state is also +2 and which is known to give rise to formate and ammonia upon hydrolysis, is consistent with this hypothesis. Thus, we propose that 1 molar equivalent of NADH is utilized in the monooxygenation of cyanide and that 1 molar equivalent of NADH is utilized in the apparent reduction of a monooxygenated reaction product to formamide. Support for such a mechanism is based on analogies from studies of the chemical oxidation of cyanide. For example, cyanide attack by singlet oxygen [O(1D2)] reportedly generates isocyanic acid (HNCO) and cyanic acid (HOCN) at a ratio of approximately 10:1 following rearrangement of a proposed oxazarine intermediate (equation 8) (5).
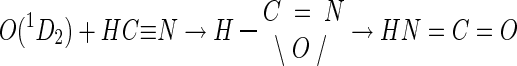 |
(8) |
We hypothesize, therefore, that a related mechanism may be involved in the monoxygenation of cyanide by CNO (equation 9), and because HNCO exists at a higher oxidation state (+4) than formamide, the need for a reductive step becomes self-evident (equation 10). Support for the feasibility of a two-electron reduction of isocyanate to formamide also comes from theoretical considerations (12) as well as documented reports of the catalytic reduction of isocyanates to amides (19). A final step involving hydrolysis of formamide thus completes the process (equation 11).
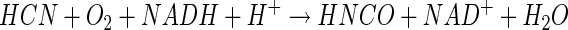 |
(9) |
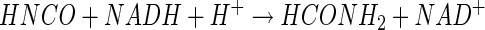 |
(10) |
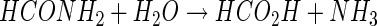 |
(11) |
Although there were no rigorous attempts to identify isocyanate (or cyanate) as a reaction product, results demonstrating that >90% of cyanide carbon could be accounted for as formate and formamide suggest that no significant accumulation takes place. The detection of formate and formamide as CNO-generated reaction products provides a rational basis for explaining the previously reported detection of these metabolites in both intact cell and cell-free incubation mixtures (25, 26). However, repeated attempts to show that formamide is degraded by P. fluorescens NCIMB 11764 have failed (4, 25, 43). We interpret these findings as indicating that P. fluorescens NCIMB 11764 is unable to synthesize an enzyme for formamide decomposition (e.g., formamidase [EC 3.5.1.49]). This implies that formamide hydrolysis must occur on CNO, which would help explain why only trace quantities of formamide were detected when the enzyme was supplied with cyanide at a low substrate concentration. In contrast, the increasingly larger amounts of formamide that accumulated when the enzyme was provided with a larger amount of cyanide (1 mM) are thought to occur because formamide dissociation from the enzyme is favored for unknown reasons under these conditions.
Reasonable parallels between our findings and the properties of classical nitrilases (EC 3.5.5.1) (for reviews see references 23 and 34), which catalyze the single-step conversion of nitriles to carboxylic acids and ammonia, can be drawn (equation 12).
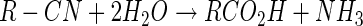 |
(12) |
For example, variable amounts of amide have been reported to be produced along with the acid by several nitrilases (14, 18, 30). In addition, the mercaptoethanol-catalyzed hydrolysis of nitriles, a model for the nitrilase reaction, yields similar results (48). CNN appears to represent a special kind of nitrilase whose substrate specificity is limited to the simplest nitrile, cyanide. Despite its almost certain involvement, formamide cannot serve as a substrate for CNN, and no significant accumulation occurs. These properties are curiously similar to those of CNO, whose substrate range, as far as is known, is also limited to cyanide. The case could thus be made that for both CNO and CNN, formamide represents the first discernible product of substrate attack, in each instance undergoing hydrolysis as an enzyme-bound intermediate.
CNO and FDH were successfully resolved to separate protein fractions (fractions A and B, respectively) by anion-exchange chromatography at high pH (10.0). The enrichment of FDH coincided with detection of a 42-kDa protein (Fig. 2, lane 4), which parallels the subunit size reported for cytosolic FDHs from a number of organisms (7). As far as we are aware, this is the only time that induction of FDH by cyanide has been reported in any organism. The simultaneous induction of FDH with CNO suggests that synthesis of the two enzymes is regulated coordinately. CNO, purified 71-fold, contained a number of protein species (Fig. 2, lane 5), the biochemical functions of which remain to be determined. The recovery of a 19-kDa protein, which was also detected in unfractionated extracts from induced cells (Fig. 2, lanes 2 and 4), parallels the detection by Harris and Knowles (16) of a protein having approximately the same molecular mass (15 to 17 kDa) which was thought to be involved in P. fluorescens NCIMB 11764 cyanide oxidation. The hypothesis that this protein represents one component of a multicomponent enzyme complex is not inconsistent with the typical molecular structure of oxygenase enzymes (13).
Recent findings (24) showing that H4biopterin and H46-methylpterin were each capable of replacing a low-molecular-weight cofactor required for cyanide oxidation by crude cell extracts were interpreted as indicating that CNO functions as a pterin-dependent hydroxylase. We have now confirmed that the partially purified enzyme requires reduced pterins, and we have shown that formate and ammonia are produced as the products of oxygenolytic conversion. The detection of H2monapterin and H2biopterin in cell extracts of P. fluorescens NCIMB 11764 (Fig. 5), each of which is capable of complementing proteins in fraction B (Table 3), suggests that these compounds (and/or the corresponding compounds H4monapterin and H4biopterin, which can spontaneously oxidize to the more stable dihydro forms) can serve as natural cofactors for the enzyme. The occurrence of these compounds in P. fluorescens NCIMB 11764 is consistent with previous reports of the occurrence of monapterin in related Pseudomonas species (11, 42). In addition to reduced monapterin and biopterin, H2neopterin was found also to support enzyme activity, implying that CNO has a rather broad specificity for pterin cofactors (Table 3). These properties parallel those of classical pterin hydroxylases, notably the amino acid hydroxylases (AAH) (e.g., phenylalanine hydroxylase [EC 1.14.16.1]) and nitric oxide synthase (NOS) (EC 1.14.13.39) (for reviews see references 21 and 31). However, there are distinct differences among CNO, AAH, and NOS with regard to the oxidation state of pterin capable of supporting enzyme activity. The latter enzymes, for example, have a rather strict requirement for the fully reduced tetrahydropterins (in most cases H4biopterin serves as the natural cofactor) (31, 36), whereas both the tetrahydro and dihydro forms were equally capable of enhancing the activity of CNO. Nonetheless, since the fully oxidized pterins were unable to support enzyme activity, the oxidation state of the cofactor is apparently important. The small amount of reduced pterin required could also be interpreted as indicating that there is a mechanism for cofactor regeneration, thereby paralleling the properties of NOS and AAH, both of which are capable of regenerating cofactors, albeit by disparate mechanisms. On the other hand, it is possible that both the tetra- and dihydropterins support enzyme activity because their fundamental role in the CNO mechanism is different from their role in the more classical pterin monooxygenases. In the absence of convincing evidence to the contrary, characterization of CNO as a pterin-dependent hydroxylase seems appropriate. The absolute dependence of this enzyme on oxygen, NADH, and reduced pterin for activity is consistent with this class of enzymes. Further studies on the characterization of CNO are expected to reveal important new information concerning the mechanism of oxygen-activated cyanide conversion and the strategies that microorganisms may have evolved for cyanide detoxification in the environment.
Acknowledgments
This work was supported by the National Science Foundation (grant MCB 0136280).
We are indebted to B. J. Venables for assistance with GC-MS and LC-MS analyses, to W. R. Kenealy for large-scale cultivation of cells, and to R. Dickstein for critical review of the manuscript. Invaluable discussions with P. J. Weimer are also gratefully acknowledged.
REFERENCES
- 1.Adje, M. D., and Y. Ohta. 1999. Isolation and characterization of a cyanide-utilizing Burkholderia cepacia strain. World J. Microbiol. Biotechnol. 15:699-704. [Google Scholar]
- 2.Agency for Toxic Substances and Disease Registry. 1993. Cyanide toxicity. Am. Fam. Physic. 48:107-114. [Google Scholar]
- 3.Beechey, R. B., and D. W. Ribbons. 1972. Oxygen electrode measurements. Methods Microbiol. 6B:25-53. [Google Scholar]
- 4.Chen, J.-L. 1998. Ph.D. thesis. University of North Texas, Denton, Tex.
- 5.Crowley, J. N., and J. R. Sodeau. 1989. Reaction between hydrocyanic acid and O(1D2) or O(3P) oxygen atoms in low-temperature matrices. J. Phys. Chem. 93:3100-3103. [Google Scholar]
- 6.Fawcett, J. K., and J. E. Scott. 1960. A rapid and precise method for the determination of urea. J. Clin. Pathol. 13:156-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferry, J. G. 1990. Formate dehydrogenase: microbiology, biochemistry and genetics, p. 117-141. In G. A. Codd, L. Dijkhuizen, and F. R. Tabita (ed.), Autotrophic microbiology and one-carbon metabolism. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 8.Finnegan, I., S. Toerien, L. Abbot, F. Smit, and H. G. Raubenheimer. 1991. Identification and characterization of an Acinetobacter sp. capable of assimilation of a range of cyano-metal complexes, free cyanide ions and similar organic nitriles. Appl. Microbiol. Biotechnol. 36:142-144. [Google Scholar]
- 9.Fukushima, T., and J. C. Nixon. 1980. Analysis of reduced forms of biopterin in biological tissues and fluids. Anal. Biochem. 102:176-188. [DOI] [PubMed] [Google Scholar]
- 10.Fuller, W. H. 1984. Cyanide in the environment with particular attention to the soil, p. 19-44. In D. Van Zyl (ed.), Cyanide and the environment, vol. 1. Geotechnical Engineering Program, Colorado State University, Fort Collins, Colo.
- 11.Guroff, G., and C. A. Rhoads. 1969. Phenylalanine hydroxylation by Pseudomonas species (ATCC 11299a). J. Biol. Chem. 244:142-146. [PubMed] [Google Scholar]
- 12.Hammerich, O., and V. D. Parker. 1977. The electrochemistry of cyanates and related compounds, p. 321-342. In S. Patai (ed.), The chemistry of cyanates and their thio derivatives. John Wiley & Sons, Chichester, United Kingdom.
- 13.Harayama, S., M. Kok, and E. L. Neidle. 1992. Functional and evolutionary relationships among diverse oxygenases. Annu. Rev. Microbiol. 46:565-601. [DOI] [PubMed] [Google Scholar]
- 14.Harper, D. B. 1977. Microbial metabolism of aromatic nitriles. Enzymology of C-N cleavage by Nocardia sp. (Rhodochrous group) N C I B 11216. Biochem. J. 165:309-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris, R., and C. J. Knowles. 1983. Isolation and growth of a Pseudomonas species that utilizes cyanide as a source of nitrogen. J. Gen. Microbiol. 129:1005-1011. [DOI] [PubMed] [Google Scholar]
- 16.Harris, R. E., and C. Knowles. 1983. The conversion of cyanide to ammonia by extracts of a strain of Pseudomonas fluorescens that utilizes cyanide as a source of nitrogen for growth. FEMS Microbiol. Lett. 20:337-341. [Google Scholar]
- 17.Homan, E. R. 1987. Reactions, processes and materials with potential for cyanide exposure, p. 1-21. In B. Ballantyne and T. C. Marrs (ed.), Clinical and experimental toxicology of cyanides. IOP Publishing Ltd. (Wright), Bristol, United Kingdom.
- 18.Hook, R. H., and W. G. Robinson. 1964. Ricinine nitrilase. II. Purification and properties. J. Biol. Chem. 239:4263-4267. [PubMed] [Google Scholar]
- 19.Howell, H. G. 1983. The hydrogenation of isocyanates. Synth. Commun. 13:635-637. [Google Scholar]
- 20.Ingvorsen, K., B. Hojer-Pedersen, and S. E. Godtfredsen. 1991. Novel cyanide-hydrolyzing enzyme from Alcaligenes xylosoxidans subsp. denitrificans. Appl. Environ. Microbiol. 57:1783-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kappock, T. J., and J. P. Caradonna. 1996. Pterin-dependent amino acid hydroxylases. Chem. Rev. 96:2659-2756. [DOI] [PubMed] [Google Scholar]
- 22.Knowles, C. J. 1988. Cyanide utilization and degradation by microorganisms. CIBA Found. Symp. 140:3-15. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi, M., and S. Shimizu. 1994. Versatile nitrilases: nitrile-hydrolyzing enzymes. FEMS Microbiol. Lett. 120:217-224. [Google Scholar]
- 24.Kunz, D. A., R. Fernandez, and P. Parab. 2001. Evidence that bacterial cyanide oxygenase is a pterin-dependent hydroxylase. Biochem. Biophys. Res. Commun. 287:514-518. [DOI] [PubMed] [Google Scholar]
- 25.Kunz, D. A., O. Nagappan, J. Silva-Avalos, and G. T. Delong. 1992. Utilization of cyanide as a nitrogenous substrate by Pseudomonas fluorescens NCIMB 11764: evidence for multiple pathways of metabolic conversion. Appl. Environ. Microbiol. 58:2022-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunz, D. A., C.-S Wang, and J.-L. Chen. 1994. Alternative routes of enzymic cyanide metabolism in Pseudomonas fluorescens NCIMB 11764. Microbiology 140:1705-1712. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 28.Lambert, J. L., J. Ramasamy, and J. V. Paukstells. 1975. Stable reagents for the colorimetric determination of cyanide by modified König reactions. Anal. Chem. 47:916-918. [Google Scholar]
- 29.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 30.Mahadevan, S., and K. Thimann. 1964. Nitrilase. II. Substrate specificity and possible mode of action. Arch. Biochem. Biophys. 107:62-68. [DOI] [PubMed] [Google Scholar]
- 31.Marletta, M. A., A. R. Hurshman, and K. M. Rusche. 1998. Catalysis by nitric oxide synthase. Curr. Opin. Chem. Biol. 2:656-663. [DOI] [PubMed] [Google Scholar]
- 32.Meyers, P. R., D. E. Rawlings, D. R. Woods, and G. G. Lindsey. 1993. Isolation and characterization of a cyanide dihydratase from Bacillus pumilus C1. J. Bacteriol. 175:6105-6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milstien, S., and S. Kaufman. 1989. The biosynthesis of tetrahydrobiopterin in rat brain. J. Biol. Chem. 264:8066-8073. [PubMed] [Google Scholar]
- 34.Pace, H. C., and C. Brenner. 2001. The nitrilase superfamily: classification, structure and function. Genome Biol. 2:0001.1-0001.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfleiderer, W. 1985. Chemistry of naturally occurring pterins, p. 43-113. In R. L. Blakely and S. J. Benkovic (ed.), Folates and pterins, vol. 2. Chemistry and biochemistry of pterins. John Wiley & Sons, New York, N.Y.
- 36.Presta, A., U. Siddhanta, C. Wu, N. Sennequier, L. Huang, H. M. Abu-Soud, S. Erzurum, and D. J. Stuehr. 1998. Comparative functioning of dihydro- and tetrahydropterins in supporting electron transfer, catalysis, and subunit dimerization in inducible nitric oxide synthase. Biochemistry 37:298-310. [DOI] [PubMed] [Google Scholar]
- 37.Raybuck, S. 1992. Microbes and microbial enzymes for cyanide degradation. Biodegradation 3:3-18. [DOI] [PubMed] [Google Scholar]
- 38.Reed, R. E. 1988. Cyanide compounds in plants and their effects on animals, p. 47-50. In D. Van Zyl (ed.), Cyanide and the environment, vol. 1. Geotechnical Engineering Program, Colorado State University, Fort Collins, Colo.
- 39.Silva-Avalos, J., M. G. Richmond, 0. Nagappan, and D. A. Kunz. 1990. Degradation of the metal-cyano complex tetracyanonickelate(II) by cyanide-utilizing bacterial isolates. Appl. Environ. Microbiol. 56:3664-3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skrowronski, B., and G. A. Strobel. 1969. Cyanide resistance and cyanide utilization by a strain of Bacillus pumilus. Can. J. Microbiol. 15:93-98. [DOI] [PubMed] [Google Scholar]
- 41.Taylor, C., A., and M. H. Ralphs. 1988. The importance of poisonous plants as forages in the prairies and Southwest, p. 363-375. In L. F. James, M. H. Ralphs, and D. B. Nielsen (ed.), The ecology and economic impact of poisonous plants on livestock production. Westview Press, Boulder, Colo.
- 42.Viscontini, M., and R. Bühler-Moor. 1968. Isolierung von l-monapterin aus Pseudomonas roseus fluorescens. Helv. Chim. Acta 51:1548-1554. [DOI] [PubMed] [Google Scholar]
- 43.Wang, C.-S. 1995. Ph.D. thesis. University of North Texas, Denton, Tex.
- 44.Wang, C.-S., D. A. Kunz, and B. J. Venables. 1996. Incorporation of molecular oxygen and water during enzymatic oxidation of cyanide by Pseudomonas fluorescens NCIMB 11764. Appl. Environ. Microbiol. 62:2195-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ware, G. C., and H. A. Painter. 1955. Bacterial utilization of cyanide. Nature 175:900. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe, A., K. Yano, K. Ikebukuro, and I. Karube. 1998. Cyanide hydrolysis in a cyanide-degrading bacterium, Pseudomonas stutzeri AK61, by cyanidase. Microbiology 144:1677-1682. [DOI] [PubMed] [Google Scholar]
- 47.White, J. M., D. D. Jones, D. Huang, and J. J. Gauthier. 1988. Conversion of cyanide to formate and ammonia by a pseudomonad obtained from industrial wastewater. J. Ind. Microbiol. 3:263-272. [Google Scholar]
- 48.Zervos, C., and E. H. Cordes. 1971. Mercaptoethanol catalysis for hydrolysis of N-benzyl-3-cyanopyridinium bromide. A model for the nitrilase reaction. J. Org. Chem. 36:1661-1667. [Google Scholar]