

Abstract
Objective
Nucleotide-binding oligomerization domain protein 2 (NOD2) stimulates diverse inflammatory responses resulting in differential cellular phenotypes. To identify the role of NOD2 in vascular arterial obstructive diseases, we investigated the expression and pathophysiological role of NOD2 in a vascular injury model of neointimal hyperplasia.
Methods and Results
We first analyzed for neointimal hyperplasia following femoral artery injury in NOD2+/+ and NOD2−/− mice. NOD2−/− mice showed a 2.86-fold increase in neointimal formation that was mainly composed of SM α-actin positive cells. NOD2 was expressed in vascular smooth muscle cells (VSMCs) and NOD2−/− VSMCs showed increased cell proliferation in response to mitogenic stimuli, PDGF-BB or fetal bovine serum (FBS), compared with NOD2+/+ VSMCs. Furthermore, NOD2 deficiency markedly promoted VSMCs migration in response to PDGF-BB and this increased cell migration was attenuated by a PI3 kinase inhibitor. However, PKC and JNK inhibitors exerted negligible effects. Moreover, muramyl dipeptide-stimulated NOD2 prevented PDGF-BB-induced VSMCs migration.
Conclusions
Functional NOD2 is expressed in VSMCs, and NOD2 deficiency promoted VSMCs proliferation, migration, and neointimal formation after vascular injury. These results provide evidence for the involvement of NOD2 in vascular homeostasis and tissue injury, serving as a potential molecular target in the modulation of arteriosclerotic vascular disease.
Introduction
NOD2, initially described as a susceptibility gene for Crohn’s disease and inflammatory bowel diseases,1, 2 is an intracellular protein containing leucine-rich repeats similar to those found in Toll-like receptors (TLRs). NOD2 is the intracellular pathogen recognition receptor (PRR) responsible for recognition of bacterial peptidoglycans from gram-positive and gram-negative bacteria, through its interaction with muramyl dipeptide (MDP).3 The expression of NOD2 has been reported in myeloid cells, particularly macrophages, neutrophils, and dendritic cells, as well as in Paneth cells in the small intestine.4–5 Moreover, NOD2 expression can be induced by the proinflammatory cytokines TNF-α and IFN-γ in cultured intestinal epithelial cells.4 However, NOD2 expression and novel functions have been suggested in other cell types, including adipocytes, gingival and pulp fibroblasts, and vascular endothelial cells.6–10
The proliferation and migration of VSMCs from the media into the intima contribute to pathological conditions, including restenosis after angioplasty, post-transplantation coronary artery disease, and hypertensive vasculopathy.11 These events can be induced by cytokines and growth factors, such as PDGF after vascular injury.12 PDGF stimulates intracellular signal molecules with SH2 domains, including Src, PI3 kinase, and ras/raf-1.13 Src and PI3 kinase perform crucial functions related to actin reorganization, growth, and migration in response to PDGF in the VSMCs, and these activities are mediated by the activation of a family of serine/threonine-specific protein kinases and the mitogen-activated protein kinases (MAPK).14 Interestingly, agonists of TLRs, including TLR3 and TLR9, have been suggested to regulate the production of PDGF.15 In addition, enhanced expression of TLRs (TLR1, TLR2, and TLR4) has been shown in human atherosclerotic plaques.16 Animal experiments have also demonstrated an absence of TLR2 or TLR4, in atherosclerotic-susceptible mice (low-density lipoprotein receptor-deficient or apoplipoprotein E deficient mice, respectively)17, 18 resulted in a reduction in atherosclerotic lesion formation, and that femoral artery injury in the presence of a TLR4 agonist (lipopolysaccharide) augmented neointimal formation19. Taken together, these studies suggest a role for PRRs in the pathobiology of vascular disease.
In this study, we find that NOD2 is expressed in VSMCs, and that NOD2 regulates the proliferation and migration of cells exposed to factors such as PDGF-BB. We also show that NOD2 contributes to vascular homeostasis, and that the absence of NOD2 enhances neointimal formation after vascular injury.
Materials and Methods
For any of the Materials and Methods not described in detail, please see the supplementary information provided online.
Animals
NOD2−/− mice were purchased from the Jackson laboratory (Bar Harbor, ME) on a C57BL/6 (N7) genetic background, and the mice were further bred with C57BL/6 mice, and then maintained by crossing either heterozygous or homozygous mutant mice within our animal facility at Harvard Medical School. The Standing Committee on Animal Care at Harvard Medical School approved all animal experimentation protocols of this study under the guidelines of our approved IACUC protocol.
Femoral Artery Injury
Endoluminal injury to the mouse left common femoral artery was performed as described.20 The contra-lateral right femoral arteries of the same mice undergoing endoluminal injury were used as non-injured control vessels. Non-injured and injured femoral arteries were harvested 28 days after femoral artery injury and fixed with 10% formalin. Paraffin-embedded tissues were sectioned as described.21
Histological Analysis and Immunohistochemistry
Femoral arteries were harvested for histological and morphometric analyses. Vessel sections were stained for elastin (Sigma-Aldrich, St Louis, MO) and the intimal and medial areas were measured using NIH Image software. Vessel sections were stained for VSMCs and leukocytes, using antibodies against SM α-actin (Sigma-Aldrich, St Louis, MO) and CD45 (BD Biosciences, Franklin Lakes, NJ), respectively.
Reagents and Antibodies
Protein kinase inhibitors (Enzo Life Science, Plymouth Meeting, PA), mouse and human PDGF-BB (PeproTech Inc., Rocky Hill, NJ), and MDP (Sigma-Aldrich, St Louis, MO) were used in the studies. All signaling antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). All siRNA reagents were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Culture of VSMCs
Primary NOD2+/+ and NOD2−/− VSMCs from mice (mVSMCs) were obtained with the use of collagenase and elastase digestion of aortas as described previously.22 Human primary aortic SMCs (hVSMCs) were purchased from Clonetics Corp. (San Diego, CA).
Western Immunoblotting
Western imunoblotting was performed as previously described.23
Transient Transfection
The wild type mouse and human VSMCs were transfected with mouse and human NOD2 siRNA and control siRNA as described by the manufacturer (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The cells were used for migration and proliferation assays 48 hours after transfection.
Cell Migration and Proliferation Assays
Cell migration was assessed using gelatin-coated 24-transwell chambers and polycarbonate membranes, 8 µm pores (Corning, Lowell, MA). Cell proliferation was determined using Ez-Cytox Cell Viability Assay Kit (Daeillab Service Co. Ltd., Seoul, Korea).
Quantitative Real-time RT-PCR
Total RNA was isolated from mouse and human VSMCs and tissues (spleen and aorta) using Trizol reagent (Invitrogen, Carlsbad, CA), and reverse transcription was performed using SuperScript™ III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Quantitative real-time RT-PCR was performed using TaqMan kit in StepOnePlus™ Real-time PCR System (Applied Biosystems, Foster City, CA). Premade primers and probes were purchased from Applied Biosystems and assay IDs were as follows: mouse NOD2 (Mm00467543_m1), human NOD2 (Hs00223394_m1), mouse NOD1 (Mm00805062_m1), mouse TLR2 (Mm00442346_m1), and mouse TLR4 (Mm00445274_m1). Expression of mouse and human target genes was normalized to mouse peptidyl-prolyl isomerase A (mPPIA) or human GAPDH expression levels, respectively.
Statistical Analysis
Data are represented as mean ± SD. For comparisons between two groups, we used Student’s two-tailed unpaired t test. For comparisons of timed series experiments, we performed Student’s paired t tests. For comparisons between more than two sets of experimental conditions, we used a one-way analysis of variance (ANOVA). If significant, the ANOVA was followed by a protected Fisher’s Least Significant Difference test and a Scheffe’s F-test. Statistically significant differences were accepted at p<0.05.
Results
To investigate the role of NOD2 in the vasculature, we examined the formation of neointimal lesions after femoral artery injury in NOD2+/+ and NOD2−/− mice. There were no differences in the structure of non-injured femoral arteries between NOD2+/+ and NOD2−/− mice (Figure 1A). The vessel size (inside area of external elastic lamina [EEL]) of injured femoral arteries was not different between NOD2+/+ (43.21±0.82 mm2, n=10) and NOD2−/− mice (41.86±0.56 mm2, n=16) 28 days after injury. The medial areas were also similar between NOD2+/+ (12.33±0.23 mm2, n=10) and NOD2−/− mice (11.57±0.19 mm2, n=16). In the injured vessels of NOD2+/+ mice, neointimal thickening (area inside of internal elastic lamina [IEL] excluding luminal area) was evident, although small (5.01±0.86 mm2, n=10). In contrast, we observed a robust increase in intimal thickening in NOD2−/− mice after injury (16.85±0.75 mm2, n=16) (Figure 1A and 1B). An absence of NOD2 increased the intimal/media ratio 2.86 fold, 1.54±0.22 in NOD2−/− compared with 0.37±0.21 in NOD2+/+ mice (Figure 1C). NOD2−/− neointima were composed mainly of SM α-actin positive cells. Both groups of mice showed sparse CD45 positive inflammatory cells after vascular injury in the neointima (Figure 1D). The arrows (red) highlight CD45 positive cells. These results suggest that NOD2 has a broad influence on the vascular response to injury, and an important physiological role in VSMCs.
Figure 1. NOD2 deficiency accelerates neointima formation after vascular injury in mice.

A, Femoral arteries were harvested 28 days after injury. Verhoeff’s staining for elastin was performed on sections from NOD2+/+ and NOD2−/− mice. Arrows indicate IEL and EEL of the vessels. Lu represents the vessel lumen. B, Quantitative morphometric analysis of intimal area in NOD2+/+ (n=10) and NOD2−/− (n=16) vessels after injury (*p<0.05, NOD2+/+ vs NOD2−/−). C, Intima/media area ratio in NOD2−/− (n=16) compared with NOD2+/+ (n=10) mice (*p<0.05). Values are mean ± SD in B and C. D, Immunostaining was performed using antibodies against SM α-actin for VSMCs or CD45 for inflammatory cells after injury. Red colored cells represent SM α-actin positive cells. Red arrows point to CD45 positive cells.
In VSMCs, expression of NOD2 was not previously reported. Hence, we investigated whether NOD2 is expressed in VSMC, and whether NOD2 is induced by stimuli known to increase its expression in other cell types. Total RNA was isolated from the aortas of NOD2+/+ and NOD2−/− mice. As shown in Figure 2A, mRNA expression of NOD2 was detected in NOD2+/+ aortas, but not in NOD2−/− aortas. However, mRNA expression of other PRRs (NOD1, TLR2, and TLR4) was not different in the aortas of NOD2+/+ and NOD2−/− mice (Figure 2A). Spleen was used as a positive control for mRNA expression of NOD2 and other PRRs. To investigate NOD2 expression in VSMCs, we harvested the cells from aortas of littermate mice. NOD2 mRNA was detected in NOD2+/+ VSMCs, but not in NOD2−/− VSMCs (Figure 2B). In NOD2−/− VSMCs, expression levels of NOD1 and TLR2 mRNA were enhanced compared with NOD2+/+ VSMCs. However, TLR4 expression was slightly decreased in NOD2−/− VSMCs (Figure 2B). Some of these changes in PRR mRNA levels were modest, thus we will need to confirm whether these changes translate into an alteration in protein levels. Selective VSMC genes (calponins, caldesmon, SM Myh11) were expressed similarly in both NOD2+/+ and NOD2−/− VSMCs (Online Figure I).
Figure 2. NOD2 mRNA is expressed in VSMCs.
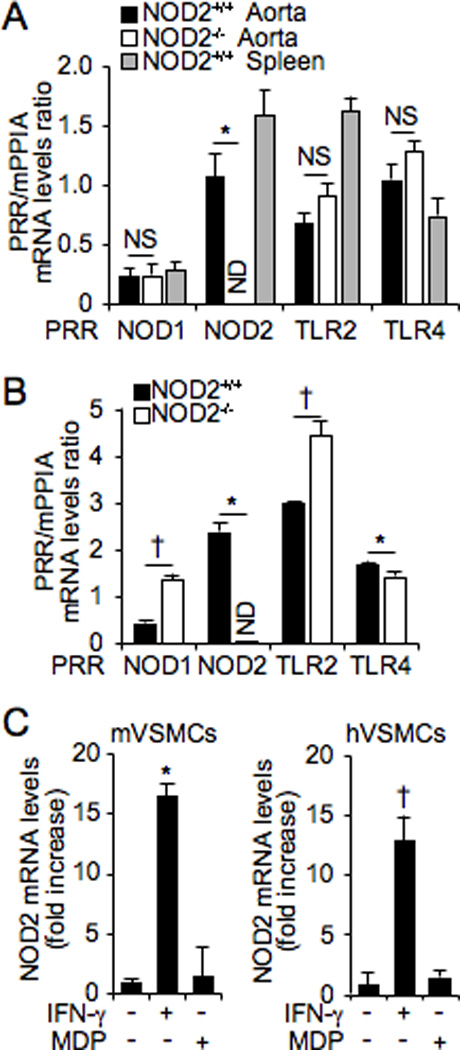
Quantitative real-time PCR was performed to assess mRNA levels of PRRs in NOD2+/+ and NOD2−/− aortas (A) and VSMCs (B). Expression levels of PRRs mRNA are divided by expression of the control gene, mPPIA, and shown as a ratio of PRR/mPPIA. *p<0.05, down-regulation of gene expression in NOD2−/− vs NOD2+/+ aorta and VSMCs. †p<0.05, up-regulation of gene expression in NOD2−/− vs NOD2+/+ aorta and VSMCs. NOD2+/+ spleen was used as a positive control in (A). NS, not significant; ND, not detectable. C, Expression of NOD2 mRNA in VSMCs, after exposure to IFN-γ (10 ng/mL) or MDP (1 µg/mL), was performed by quantitative real-time PCR in mouse primary (m) and human primary (h) VSMCs. For both mouse and human NOD2 mRNA, expression was normalized by an internal control gene, either mPPIA (for mouse NOD2) or hGAPDH (for human NOD2), and shown as a fold increase. *p<0.05 vs no IFN-γ in mVSMCs. †p<0.05 vs no IFN-γ in hVSMCs. For all of the real-time PCR experiments, values are presented as mean ± SD, n=3.
Proinflammatory cytokines, TNF-α and IFN-γ, are known to regulate NOD2 expression in other cell types, including intestinal epithelial cells.4 To investigate induction of NOD2 mRNA, we treated mVSMCs and hVSMCs with IFN-γ and MDP. Interestingly, expression of NOD2 mRNA was increased by IFN-γ in VSMCs, but not by the NOD2 ligand, MDP (Figure 2C). These data suggest that NOD2 mRNA is expressed in VSMCs, and may have a role in VSMCs physiologic and pathophysiologic conditions.
To investigate potential mechanisms by which an absence of NOD2 leads to increased neointimal formation after vascular injury, we assessed VSMC proliferation, migration, and cell death in vitro. NOD2−/− VSMCs exhibited increased cell proliferation compared to NOD2+/+ VSMCs in response to mitogenic FBS stimulation (Online Figure II). We further analyzed whether NOD2 deficiency could increase cell proliferation in response to PDGF-BB, which is a well-known potent growth factor and chemoattractant for VSMCs, and released at sites of vessel injury.12 The proliferation of VSMCs was enhanced in NOD2−/− (27.00±4.64%) compared with NOD2+/+ cells 4 days after PDGF-BB treatment (Figure 3A). In addition, treatment with siRNA-mediated NOD2 knockdown increased VSMC proliferation (Figure 3B). This increased proliferation of NOD2 deficient cells was not due to a difference in cell adhesion after plating, as adherence was 77% in NOD2−/− cells and 75% in NOD2+/+ cells (p>0.05, NS). Real-time RT-PCR was performed to verify down-regulation of NOD2 expression by mouse NOD2 siRNA (Figure 3C).
Figure 3. NOD2 deficiency promotes proliferation of mouse VSMCs.
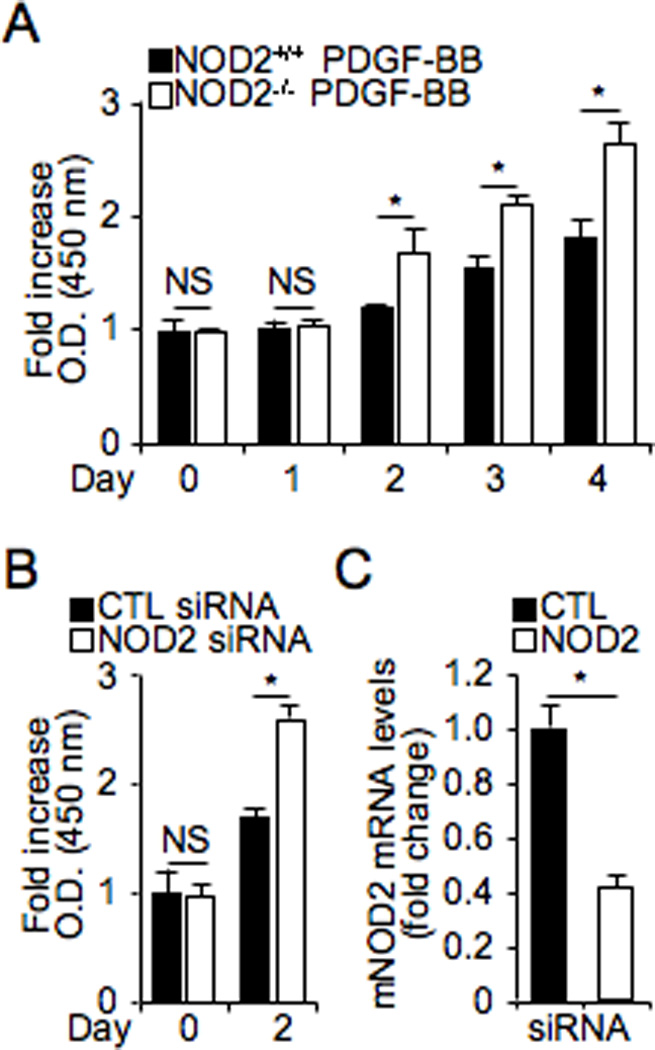
A, Mouse NOD2+/+ and NOD2−/− VSMCs were plated, FBS-starved for 2 days, and stimulated with PDGF-BB (10 ng/mL). Proliferation was measured at each time point, and each group was normalized to its own control at day 0. *p<0.05, NOD2+/+ vs NOD2−/− VSMCs. B, Control (CTL) and NOD2 siRNA were transfected into NOD2+/+ VSMCs, FBS-starved for 2 days and incubated for 2 days with 10% FBS. Proliferation of VSMCs was counted at 0 and 2 days using Ez-Cytox Cell Viability Assay Kit. Values are mean ± SD, n=9. *p<0.05, control siRNA vs NOD2 siRNA transfected NOD2+/+ VSMCs. C, Real-time PCR was performed for NOD2 mRNA levels in control (CTL) and NOD2 siRNA transfected cells. Expression levels of NOD2 mRNA were normalized by expression of mPPIA, and graphed as a fold change compared with control transfected cells.
Next, the involvement of NOD2 deficiency in cell migration was measured using gelatin-coated 24-transwell chambers. NOD2−/− VSMCs showed a 4.05±0.53 fold increased migration in response to PDGF-BB compared with NOD2+/+ VSMCs, 24 hours after PDGF-BB treatment, and the number of migrated cells were counted and represented as a graph (Figure 4A and 4B). Similarly, we observed increased hVSMCs migration in human NOD2 siRNA-treated cells in response to PDGF-BB, compared with control siRNA-treated hVSMCs (Figure 4C and 4D). Real-time RT-PCR was performed to verify down-regulation of human NOD2 expression by human NOD2 siRNA (Figure 4E). These results indicate that in the absence of NOD2, VSMCs proliferate and migrate more rapidly in response to PDGF-BB. However, PDGF-BB did not alter mRNA expression of NOD2 (Online Figure III). Interestingly, cells treated with lysophosphatidylcholine (LPC) and H2O2, which are well known to induce cell death in VSMCs, showed only modest differences in viability between NOD2+/+ and NOD2−/− VSMCs (Online Figure IV). These effects on viability were not nearly as dramatic as the enhanced proliferation and migration in NOD2−/− compared with NOD2+/+ VSMCs. Cell viability of NOD2−/− VSMCs was slightly increased by low concentrations of LPC, but decreased by H2O2, compared with NOD2+/+ cells (Online Figure IV). These results suggest that NOD2 plays an important role in the prevention of VSMC proliferation, and more dramatically migration, without markedly affecting cell death.
Figure 4. NOD2 deficiency increases migration of VSMCs in response to PDGF-BB.
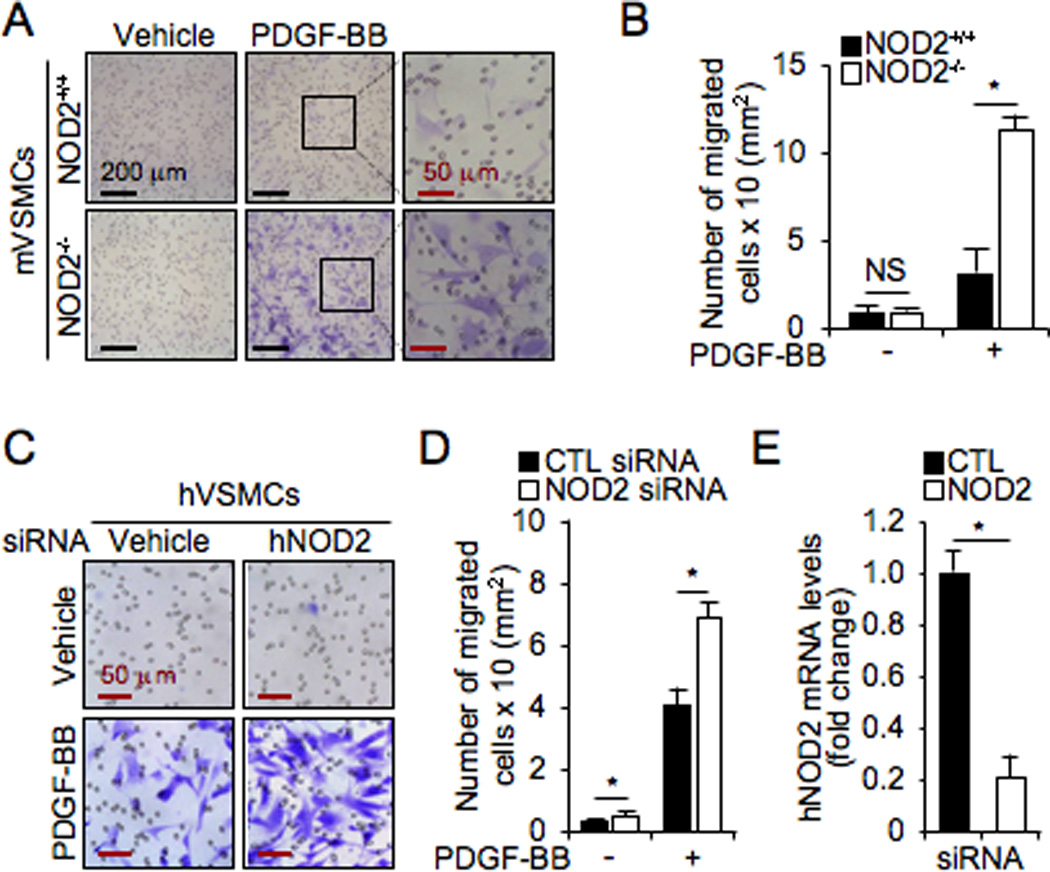
A, The migration assay for VSMCs was analyzed using 24 trans-well chamber plates in response to PDGF-BB (10 ng/mL). Migrated mVSMCs were stained by Crystal Violet staining kit. B, The number of migrated cells was measured and represented as a graph. Values are mean ± SD, n=6. *p<0.05 NOD2+/+ vs NOD2−/− VSMCs in the presence of PDGF-BB. NS, not significant. C, Cells were transfected with control (CTL) or human siRNA in human primary VSMCs. Cell migration was measured in the absence or the presence of PDGF-BB. D, The number of migrated cells was measured and represented as a graph. Values are mean ± SD, n=6. *p<0.05 NOD2 siRNA vs control siRNA in hVSMCs. E, Real-time PCR was performed for human NOD2 to assess its levels in control (CTL) and human NOD2 siRNA transfected cells. Expression levels of NOD2 mRNA were normalized by expression of hGAPDH and graphed as a fold change compared with control transfected cells.
PDGF-BB stimulates many intracellular signaling molecules including PI3K.13–14 To identify which signaling pathways mediate VSMC migration in the absence of NOD2, chemical inhibitors of PKC, PI3K, and JNK were administrated to NOD2+/+ and NOD2−/− VSMCs before PDGF-BB treatment, and migration was measured (Figure 5A). Remarkably, enhanced migration of NOD2−/− VSMCs was abolished when exposed to a PI3K inhibitor, LY294002, compared with NOD2+/+ VSMCs, in response to PDGF-BB stimulation (Figure 5A). The PKC inhibitor, GF10203X, did not alter the enhanced migration of NOD2−/− VSMCs. A JNK inhibitor, SP600125, blunted the migration of VSMCs to PDGF-BB in both NOD2+/+ and NOD2−/− cells, however NOD2−/− cells maintained their increased migration response in comparison with NOD2+/+ cells. Other signaling inhibitors such as p38 and ERK inhibitors did not alter the migration of NOD2−/− VSMCs (Online Figure V). Thus, we focused on the PI3K pathway and its downstream target Akt. Levels of p-Akt (Ser473 and Thr308) were acutely induced by PDGF-BB in NOD2+/+ VSMCs, however the levels returned to baseline by 15 minutes after PDGF-BB stimulation (Figure 5B and 5C). In contrast, p-Akt levels (Ser473 and Thr308) were much more dramatically induced by PDGF-BB in NOD2−/− VSMCs, and these p-Akt levels remained elevated throughout the time course (Figure 5B and 5C). Moreover, basal levels of p-Akt were not different between NOD2+/+ and NOD2−/− cells. Taken together, these data suggest that the PI3K signaling pathway is involved in the enhanced migration of NOD2−/− compared with NOD2+/+ VSMCs.
Figure 5. Inhibition of the PI3 kinase signaling pathway prevents increased migration in NOD2−/− VSMCs.
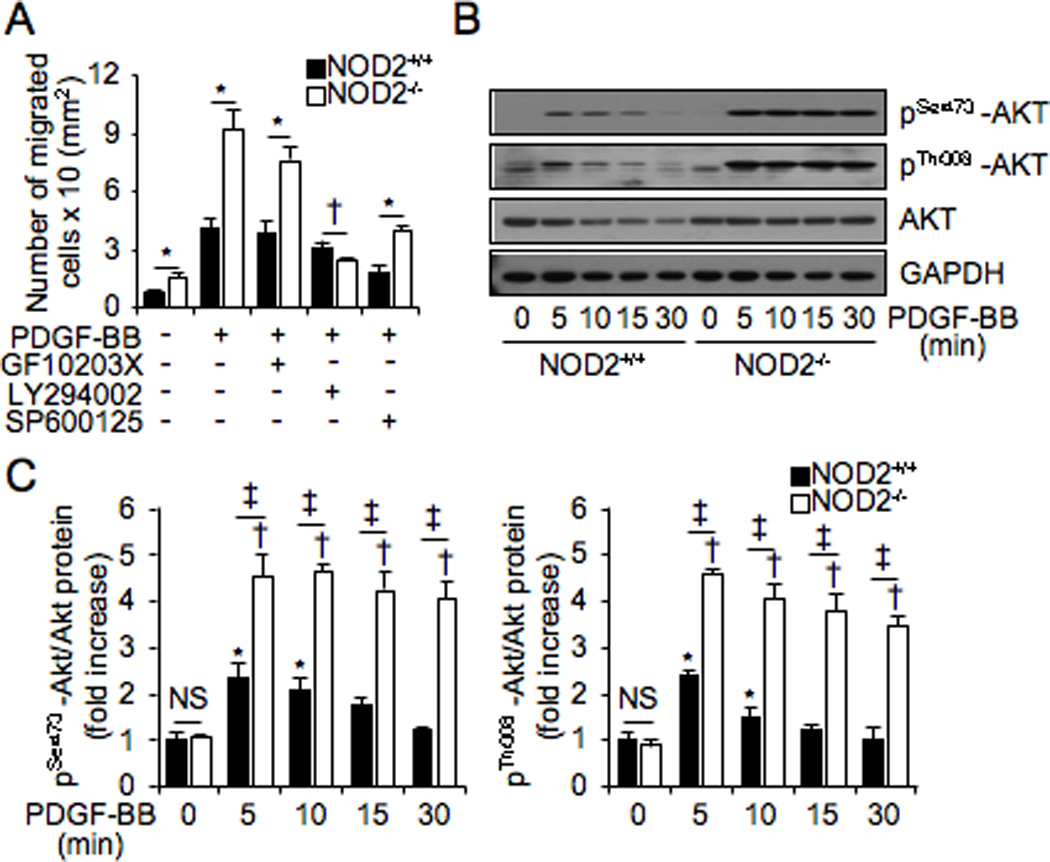
A, VSMCs migration assay was performed to measured migration of NOD2+/+ and NOD2−/− VSMCs in response to PDGF-BB or PDGF-BB plus PKC inhibitor (GF10203X, 1 µM), PI3K inhibitor (LY294002, 10 µM), or JNK inhibitor (SP600125, 10 µM). *p<0.05, increased migration of NOD2−/− vs NOD2+/+ VSMCs. †p<0.05, decreased migration of NOD2−/− vs NOD2+/+ VSMCs. Values are mean ± SD, n=6. B, Phosphorylated Akt (p-Akt Ser473 and p-Akt Thr308), total Akt, and GAPDH were determined at the indicated time points after PDGF-BB treatment by Western blotting. This panel denotes representative blots of three independent experiments, which are quantitated in (C). C, Normalized phosphorylation of Akt levels by total Akt expression are represented as a graph. *p<0.05 vs PDGF-BB (0 minute) in NOD2+/+ VSMCs, †p<0.05 vs PDGF-BB (0 minute) in NOD2−/− VSMCs, and ‡p<0.05, increased phosphorylation of Akt (Ser473 or Thr308) in NOD2−/− vs NOD2+/+ VSMCs. NS, not significant. Values are mean ± SD, n=3.
To confirm the role of endogenous NOD2 on VSMC migration, we treated mouse and human primary VSMCs with a NOD2 ligand, MDP. Interestingly, MDP abrogated the migration of mouse (65.70±8.96%) and human (51.01±8.60%) primary VSMCs in response to PDGF-BB (Figure 6A and 6B). Thus, intracellular NOD2 may be a key regulator of VSMC migration, and activated NOD2 could be a target molecule to prevent VSMCs from migrating under pathophysiologic conditions.
Figure 6. Activation of NOD2 inhibits migration of VSMCs in response to PDGF-BB.

A, Mouse and human VSMCs were pretreated with MDP (2 µg/mL) or vehicle (− MDP) for 30 minutes, followed by administration of PDGF-BB (20 ng/mL) or vehicle (− PDGF-BB). After 12 hours +/− PDGF-BB, migrated cells were stained by Crystal Violet staining kit. B, The number of migrated cells were measured and represented as a graph. Values are mean ± SD, n=4. *p<0.05 PDGF-BB vs PDGF-BB plus MDP in mouse or human VSMCs.
Discussion
PRRs are a class of innate immune response-expressed proteins that respond to pathogen-associated molecular patterns, yet they also respond to endogenous stress signals termed damage-associated molecular patterns.24–26 These stress signals may involve non-infectious danger signals.27–28 TLRs have been proposed to be a link between inflammation and atherosclerosis, and while infectious agents may accelerate atherosclerosis, they are not required for lesion formation29. Taking into account these findings, along with the previously mentioned studies demonstrating an absence of TLR2 or TLR4 in atherosclerotic mice reduce lesion formation17, 18, and that expose to a TLR4 agonist during femoral artery injury increases lesion formation19, we hypothesized that NOD2 may have an important role in vascular biology, particularly after injury. In contrast to a deficiency of TLR2 or TLR4, NOD2−/− mice developed much larger neointimal lesions after vascular injury compared with NOD2+/+ mice (Figure 1A–1C), and these lesions were predominantly composed of VSMCs (Figure 1D). In this study, we demonstrate for the first time that NOD2 mRNA is expressed in aortas and primary VSMCs of mice and humans (Figure 2B and 2C), suggesting NOD2 is important in vascular homeostasis and prevents neointimal formation. Interestingly, NOD2 expression has been previously shown in endothelial cells, another important cell type in vascular pathogenesis.9–10
Next, we wanted to determine whether the expression of NOD2 in VSMCs is functionally important, thus contributing to increased neointimal formation in NOD2−/− mice. NOD2 deficiency promoted increased proliferation and migration of VSMCs (Figures 3 and 4), supporting the concept that NOD2 may have an important protective role in vascular physiology. The effects of NOD2 deficiency on proliferation and migration of VSMCs were much more impressive than the effects of apoptotic agonists (Online Figure IV). We also confirmed that cell proliferation and migration were enhanced by an acute downregulation of NOD2, as siRNA for NOD2 had similar effects as genetically deficient NOD2 VSMCs (Figures 3 and 4).
In NOD2−/− VSMCs, there is evidence of increased expression of NOD1 and TLR2, and a slight reduction in TLR4 (Figure 2B). We questioned whether the increased expression of NOD1 and TLR2 may be a compensatory response for the absence of NOD2. Cartwright and colleagues reported that selective NOD1 agonists cause a systemic inflammatory response, organ injury, and dysfunction of VSMCs, and Yang and colleagues showed that TLR2 expression in VSMCs promoted inflammation within the arterial wall.30–31 However, in the present study, inflammatory cells were sparse in NOD2 deficient mice after vascular injury (Figure 1D), suggesting a different pathophysiology from prior studies assessing injury due to NOD1 agonists and TLR2. However, we cannot totally exclude the fact that a compensatory increase in NOD1 and TLR2 may contribute to vascular lesion formation.
Interestingly, Cole and colleagues established that the other PRR with a protective role in the arterial wall is TLR3.32 They demonstrated that TLR3 activated with Poly(I:C), a TLR3 ligand, decreased neointimal formation in response to carotid injury, using a perivascular collar model. At the same time, mice deficient in both TLR3 and ApoE (ApoE−/−/TLR3−/−) developed accelerated early atherosclerotic lesion formation in the aortic root, compared with control mice (ApoE−/−/TLR3+/+). TLR3 is known to recognize double-stranded RNA, carried by some viruses, and recently single-stranded RNA has been shown to be an agonist of NOD2, along with NOD2 being protective during viral infections.33 These data, in conjunction with the vascular injury models, suggest that PRRs, such as NOD2 and TLR3, are more complex than just innate immune receptors. Moreover, while PRRs are important for the innate immune response, in the presence of non-infectious danger signals they can be quite different in their functions. For instance, PRRs can be either protective (NOD2 and TLR3) or injurious (NOD1, TLR2, and TLR4) in the vascular wall.
Neointimal formation is due, in part, to VSMC proliferation and migration mediated by cytokines and growth factors, such as PDGF, in the development of atherosclerosis and restenosis.1 In addition to increasing proliferation, migration is markedly increased in NOD2−/− VSMCs in response to PDGF-BB (Figure 4A and 4B). In particular, the PI3K signaling pathway performs crucial functions related to actin reorganization, growth, and migration in response to PDGF-BB in VSMCs, and these activities are mediated by the activation of a family of serine/threonine-specific protein kinases.12 In Figure 5A, increasing migration in of NOD2−/− VSMCs was blocked in the presence of a PI3K inhibitor. Interestingly, NOD2−/− VSMCs showed sustained Akt phosphorylation which is the downstream target molecule of PI3K in response to PDGF-BB (Figure 5B–C). These data suggest that NOD2 may negatively regulate the PDGF-BB signaling pathway, even though PDGF-BB did not increase mRNA expression of NOD2 (Online Figure III). However, more detailed mechanism(s) need to be elucidated. Another very important finding was that MDP prevented PDGF-BB-induced migration of mouse and human VSMCs (Figure 6). These findings indicate that modulation of NOD2 signaling in VSMCs may be beneficial in treating vascular diseases involving excessive VSMCs proliferation and migration, such as occurs with restenosis after vascular injury.
Courivaud and colleagues analyzed the role of NOD2 in the pathogenesis of atherosclerosis in humans, and reported no correlation between NOD2 gene polymorphisms and atherosclerosis after renal transplantation.34 They assessed the frequency of NOD2 gene polymorphisms from leukocytes of renal transplant recipients. Thus, it would be interesting to further assess the level of expression and the function of NOD2 polymorphisms in VSMCs from patients that develop other forms of vascular diseases, as the role of NOD2 may vary depending on disease models or patient populations.
In summary, NOD2 is expressed in mouse and human VSMCs. NOD2−/− mice develop more pronounced neointimal formation in response to vascular injury, and the proliferation and migration of NOD2−/− VSMCs to PDGF-BB stimulation is greater than in NOD2+/+ VSMCs. Moreover, activation of NOD2 in mouse and human VSMCs decreases their migratory response to PDGF-BB, suggesting that NOD2 plays a critical role in the function of VSMCs. These findings advance our understanding of PRRs in vascular injury, and provide evidence that NOD2 may be an important therapeutic target in vascular diseases involving excessive VSMCs proliferation and migration.
Supplementary Material
Acknowledgements
We thank B. Ith for his technical assistance with histology, and the helpful suggestions of Dr. GM Hunninghake regarding the statistical analyses.
Sources of Funding
This work was supported by grants NRF-331-2008-1-C00216 from National Research Foundation of Korea (to S.W. Chung) and RO1 HL060788 from NIH (to M.A. Perrella).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP. Association of NOD2 LRR variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 2.Ogura Y, Bonen DK, Inohara N, Nicolae L, Chen FF. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 3.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A. NOD2 is a general sensor of peptidoglycan through MDP detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 4.Rosenstiel P, Kuhbacher T, Seegert D, Schreiber S. TNF-α and IFN-γ regulate the expression of the NOD2 in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–1009. doi: 10.1053/gast.2003.50157. [DOI] [PubMed] [Google Scholar]
- 5.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nunez G, Keshav S. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 6.Stroh T, Batra A, Glauben R, Bereswill S, Siegmund B. NOD1 and 2: regulation of expression and function in preadipocytes. J Immunol. 2008;181:3620–3627. doi: 10.4049/jimmunol.181.5.3620. [DOI] [PubMed] [Google Scholar]
- 7.Hirao K, Yumoto H, Nakanishi T, Matsuo T. Roles of TLR2, TLR4, and NOD2 in pulp fibroblasts. J Dent Res. 2009;88:762–767. doi: 10.1177/0022034509341779. [DOI] [PubMed] [Google Scholar]
- 8.Hosokawa I, Hosokawa Y, Ozaki K, Yumoto H, Nakae H, Matsuo T. Proinflammatory effects of MDP on human gingival fibroblasts. J Periodontal Res. 2010;45:193–199. doi: 10.1111/j.1600-0765.2009.01217.x. [DOI] [PubMed] [Google Scholar]
- 9.Davey MP, Martin TM, Planck SR, Lee J, Zamora D, Rosenbaum JT. Human endothelial cells express NOD2/CARD15 and increase IL-6 secretion in response to muramyl dipeptide. Microvasc Res. 2006;71:103–107. doi: 10.1016/j.mvr.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Oh HM, Lee HJ, Seo GS, Choi EY, Kweon SH, Chun CH, Han WC, Lee KM, Lee MS, Choi SC, Jun CD. Induction and localization of NOD2 protein in human endothelial cells. Cell Immunol. 2005;237:37–44. doi: 10.1016/j.cellimm.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 12.Miyazawa K, Kikuchi S, Fukuyama J, Hamano S, Ujiie A. Inhibition of PDGF- and TGF-β1-induced collagen synthesis, migration and proliferation by tranilast in VSMCs from spontaneously hypertensive rats. Atherosclerosis. 1995;118:213–221. doi: 10.1016/0021-9150(95)05607-6. [DOI] [PubMed] [Google Scholar]
- 13.Ronnstrand L, Heldin CH. Mechanisms of PDGF-induced chemotaxis. Int J Cancer. 2001;91:757–762. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1136>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.Mahabeleshwar GH, Kundu GC. Syk suppresses the cell motility and NF-κB-mediated secretion of PA by inhibiting the PI3K activity in breast cancer cells. J Biol Chem. 2003;278:6209–6221. doi: 10.1074/jbc.M208905200. [DOI] [PubMed] [Google Scholar]
- 15.Chow EK, O'connell RM, Schilling S, Wang XF, Fu XY, Cheng G. TLR agonists regulate PDGF-BB production and cell proliferation through TGF-β/type I IFN crosstalk. EMBO J. 2005;24:4071–4081. doi: 10.1038/sj.emboj.7600867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of TLRs in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- 17.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of TLR4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by TLR2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vink A, Schoneveld AH, van der Meer JJ, van Middelaar BJ, Sluijter JPS, Smeets MB, Quax PHA, Lim SK, Borst C, Pasterkamp G, de Kleijn DPV. In vivo evidence for a role of TLR4 in the development of intimal lesions. Circulation. 2002;106:1985–1990. doi: 10.1161/01.cir.0000032146.75113.ee. [DOI] [PubMed] [Google Scholar]
- 20.Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- 21.Wei J, Gorman TE, Liu X, Ith B, Tseng X, Chen Z, Simon DI, Layne MD, Yet SF. Increased neointima formation in CRP2–deficient mice in response to vascular injury. Circ. Res. 2005;97:1323–13331. doi: 10.1161/01.RES.0000194331.76925.5c. [DOI] [PubMed] [Google Scholar]
- 22.Hu Y, Zou Y, Dietrich H, Wick G, Xu Q. Inhibition of neointima hyperplasia of mouse vein grafts by locally applied suramin. Circulation. 1999;100:861–868. doi: 10.1161/01.cir.100.8.861. [DOI] [PubMed] [Google Scholar]
- 23.Chung SW, Chen YH, Perrella MA. Role of Ets-2 in the regulation of HO-1 by endotoxin. J Biol Chem. 2005;280:4578–4588. doi: 10.1074/jbc.M409125200. [DOI] [PubMed] [Google Scholar]
- 24.Janeway CA., Jr The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13:11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 26.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 27.hang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: TLRs. Free Radic Biol Med. 2010;48:1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright SD, Burton C, Hernandez M, Hassing H, Montenegro J, Mundt S, Patel S, Card DJ, Hermanowski-Vosatka A, Bergstrom JD, Sparrow CP, Detmers PA, Chao YS. Infectious agents are not necessary for murine atherogenesis. J Exp Med. 2000;191:1437–1442. doi: 10.1084/jem.191.8.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cartwright N, Murch O, McMaster SK, Paul-Clark MJ, van Heel DA, Ryffel B, Quesniaux VF, Evans TW, Thiemermann C, Mitchell JA. Selective NOD1 agonists cause shock and organ injury/dysfunction in vivo. Am J Respir Crit Care Med. 2007;175:595–603. doi: 10.1164/rccm.200608-1103OC. [DOI] [PubMed] [Google Scholar]
- 31.Yang X, Coriolan D, Schultz K, Golenbock DT, Beasley D. TLR2 mediates persistent chemokine release by Chlamydia pneumoniae-infected VSMCs. Arterioscler Thromb Vasc Biol. 2005;25:2308–2314. doi: 10.1161/01.ATV.0000187468.00675.a3. [DOI] [PubMed] [Google Scholar]
- 32.Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, Davies AH, Flavell RA, Feldmann M, Monaco C. Unexpected protective role for TLR3 in the arterial wall. Proc Natl Acad Sci U S A. 2011;108:2372–2377. doi: 10.1073/pnas.1018515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. Activation of innate immune antiviral responses by NOD2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Courivaud C, Ferrand C, Ducloux D. No evidence of association between NOD2 gene polymorphism and atherosclerotic events after renal transplantation. Transplantation. 2006;81:1212–1215. doi: 10.1097/01.tp.0000202846.17619.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.