

Abstract
Background
A myriad of new chemicals has been introduced into our environment and exposure to these agents can damage cells and induce cytotoxicity through different mechanisms, including damaging DNA directly. Analysis of global transcriptional and phenotypic responses in the yeast S. cerevisiae provides means to identify pathways of damage recovery upon toxic exposure.
Results
Here we present a phenotypic screen of S. cerevisiae in liquid culture in a microtiter format. Detailed growth measurements were analyzed to reveal effects on ~5,500 different haploid strains that have either non-essential genes deleted or essential genes modified to generate unstable transcripts. The pattern of yeast mutants that are growth-inhibited (compared to WT cells) reveals the mechanisms ordinarily used to recover after damage. In addition to identifying previously-described DNA repair and cell cycle checkpoint deficient strains, we also identified new functional groups that profoundly affect MMS sensitivity, including RNA processing and telomere maintenance.
Conclusions
We present here a data-driven method to reveal modes of toxicity of different agents that impair cellular growth. The results from this study complement previous genomic phenotyping studies as we have expanded the data to include essential genes and to provide detailed mutant growth analysis for each individual strain. This eukaryotic testing system could potentially be used to screen compounds for toxicity, to identify mechanisms of toxicity, and to reduce the need for animal testing.
Introduction
The DNA damage response in budding yeast S. cerevisiae is well characterized, especially regarding its response to the alkylating agent methyl methanesulfonate (MMS) [1-8]. In addition to the ~150 yeast proteins directly involved in DNA repair [9], a plethora of proteins with other biological functions are necessary for recovery after damage [1,2]. The mechanistic relevance of many of these proteins in cellular recovery is still not fully understood. Yeast, as a eukaryotic model system, serves as an eminent tool to develop new methods to unravel pathways for modulating the toxicity of agents, especially those agents with unknown modes of action. Several tests, such as the Ames test or the RAD54-GFP Greenscreen [10], exist to determine the genotoxicity of compounds. However, these tests do not always reveal the agents' modes of genotoxicity or the consequential cellular responses elicited by the interactions between the agent and cellular components other than DNA. In addition, these tests are notorious for false positives in predicting the toxicity of an agent for mammalian cells, as revealed later by animal testing. To decipher the mode of toxicity by different toxicants, powerful tools such as genomic phenotyping have been developed [1,2,11-16]. Such methodology is used to determine growth under various conditions for an entire panel of 4,852 yeast strains with single non-essential genes deleted. Of the estimated 6,000 genes in S. cerevisiae, 80% are non-essential for growth in rich media; the remaining are essential genes that cannot be deleted and are thus more difficult to study. The subset of essential genes is more highly conserved between species [17] and may therefore be of more relevance in understanding how humans react to toxicants. Essential genes can be studied in hemizygous diploid strains [18] and in haploid strains with either conditional expression of genes or with decreased levels of transcripts [19,20]. We have queried the essential genes in the Decreased Abundance by mRNA Perturbation (DAmP) library of haploid strains [19,21]; transcript levels in the DAmP library were reduced by tagging the 3' UTR of the transcripts with a sequence that elicits nonsense-mediated decay [22].
By using arrayed assays of growing liquid cultures in a microtiter format, sensitive detection of toxicity is achieved. Previous studies using liquid assays in microtiter plates were not high throughput enough to allow screening of the entire yeast genome [23], and although high throughput analysis has been achieved by others, that was only by pooling strains tagged with a specific DNA sequence 'bar-code'. That method detects differences in fast-growing strains, but slow-growing strains are depleted from the pool and are thus quantified with less precision. However, this obstacle may be overcome by deep sequencing of the 'bar-codes' instead of the more common detection by microarrays [24,25].
Here we present a sensitive yet robust and highly automated liquid culture method that we have used as a screen to reveal modes of damage recovery in a eukaryotic system. By combining our data with protein-protein interaction maps, and using databases of functional categories, we have discovered novel biological pathways important for the recovery of cells in response to toxicants. Importantly, the screen has the potential to increase our understanding of toxicity modulating pathways for many different agents. The eukaryotic testing system we present here could be used to screen novel compounds for toxicity and thus reduce the need for animal testing.
Results
Experimental system to query genotoxic agents
To systematically characterize biological responses to toxic agents, we set up a system where yeast strains were exposed to increasing doses of the alkylating agent MMS. Mutations in 5,528 S. cerevisiae genes, representing ~ 92% of the S. cerevisiae genome [26], are now available as individual haploid yeast strains; each strain carries either a complete gene deletion or an insertion in the 3' UTR of a gene to destabilize its transcript, thus reducing its steady state level [19,21]. The yeast strains were grown in 96 well plates to stationary phase and then diluted into MMS-containing media (0, 0.004, 0.008, 0.012 and 0.016% MMS). Growth kinetics were recorded in the presence of the DNA damaging agent by optical density measurements at room temperature every 4 hours between 12 and 48 h after transfer to MMS-containing medium (Figure 1), and every strain was queried in at least three independent experiments. Three control strains with known MMS sensitivity were present in every plate: rad14Δ, rev1Δ and mag1Δ were present at three known locations, and WT was present at three other locations (Figure 1, 2A). The dose response for each strain was calculated by determination of the area under the growth curve at each dose (see Methods section for details). In this study, the dose response was required to behave in a close-to linear manner to be considered valid, such that only when the dose response of a strain could be fitted to a straight line (R2>0.7) were the data included (Figure 2B). For 89.3% of the deletion strains, and 76.9% of the DAmP strains, two or more experiments fulfilled this criterion, resulting in 87.3% coverage of the tested 5,528 yeast strains.
Figure 1.
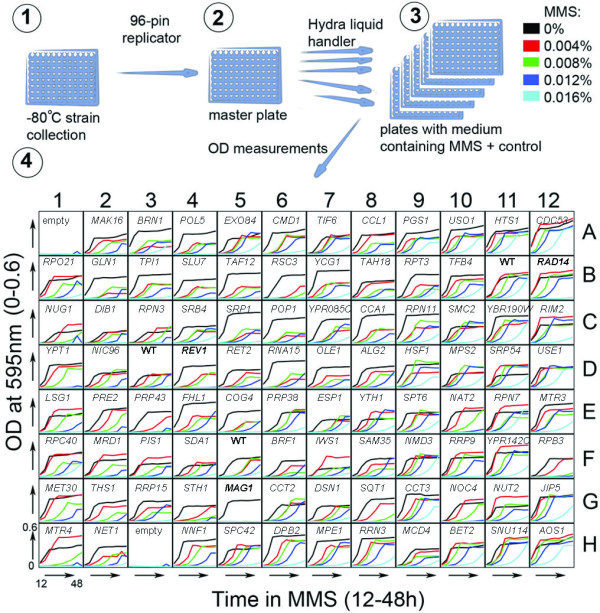
Schematic of the experimental procedure. (1) Cells are kept at -80°C from where they are pin-replicated and (2) grown to stationary phase in a master plate. (3) Once in stationary phase, the cultures are robotically diluted in YPD media (using a Hydra liquid handler) containing increasing doses of MMS (0-0.016% final concentration). (4) After incubation at 25°C for 12 hours, optical densities of all cultures are measured every 4 h until 48 h post-treatment. Growth curves are plotted and data is analyzed. As an example, plate 1 from the DAmP library is shown with the name of gene deleted in each strain given above the growth curves. Every plate contains control strains (in bold), WT (B11, D3, F5), rad14Δ (B12), rev1Δ (D4) and mag1Δ (G5), as well as at least two empty wells, containing media only.
Figure 2.

Determination of sensitivity phenotype of the tested strains. A) Growth curves of the controls - WT and the sensitive strains rad14Δ, rev1Δ, and mag1Δ - during 12-48 h after insult with increasing doses of alkylating agent MMS. A few data points at 32 h were undersampled and showed large variation. These points were omitted from the graphs. B) The area under the growth curves were plotted against the dose to calculate the dose that result in 50% growth inhibition (GI50). The data for the four control strains are shown. Error bars represents the s.e.m. C) Histograms of showing the distribution of GI50 from the entire tested panel (both deletion and DAmP strains), with the GI50 of the four control strains forming the limits between severe, intermediate and slight sensitivity to MMS. The WT strain has been transfected with a plasmid conferring G418 resistance that alters the sensitivity of the strain slightly. D) Pie charts of sensitivities of the strains lacking (left) non-essential genes (deletion strains) and (right) essential genes (DAmP strains).
For each strain, the dose that gave 50% growth inhibition (GI50) was calculated based on a total of a median of 150 measurements (10 time points × 5 doses × 2-4 replicates). Only replicates that passed the quality criterion above of R2>0.7 were included in the calculations. The GI50 for WT haploid yeast was determined as 0.01 ± 0.002% MMS (average ± s.e.m.). The GI50 for the three control strains were 0.008 ± 0.001%, 0.006 ± 0.0005%, and 0.003 ± 0.0007% for rad14Δ, rev1Δ, and mag1Δ respectively (Figure 2B). The reproducibility between experiments was high (the average R2 for the GI50 between replicates was 0.88 ± 0.03). To make a quantitative comparison of the tested strains to previous studies, all strains were categorized as showing severe, intermediate, and slight or no sensitivity to MMS. This categorization was based on the comparison between GI50 values of the tested strains, WT and the three control strains used as standards to indicate the thresholds for slight (rad14Δ), intermediate (rev1Δ) and severe (mag1Δ) sensitivity (Figure 2C).
In total, 258 (6%) of the 4,331 deletion strains that passed the quality criterion were determined to be more sensitive to MMS than WT; among these strains 18 (7.0%) showed severe, 87 (33.7%) showed intermediate, and 153 (59.3%) showed slight MMS sensitivity (Figure 2D, Table S1, Additional file 1). A much higher fraction of the DAmP strains with hypomorphic mutations in essential genes demonstrated sensitivity to MMS compared to the deletion strains. Among the 675 DAmP strains that passed the quality criterion, 222 (33%) were MMS sensitive; among these strains 13 (5.9%) showed severe, 69 (31.1%) showed intermediate, and 140 (63.1%) showed slight MMS sensitivity (Figure 2D, Table S1, Additional file 1). The environmental stress response (ESR) genes [27] only modestly overlap with the genes deleted in the MMS sensitive strains; the ESR genes make up 19% of the genes deleted in sensitive strains, whereas the ESR comprises 16% of the entire genome (p = 0.002). Further, the WT used here was a modified version of BY4741 (with a plasmid conferring G418 resistance). This strain was confirmed as being slightly more sensitive than the original BY4741 (p-value < 0.01, t-test). The assay has its most sensitive range in detecting strains with GI50 between 0.002 and 0.008, where the data points to calculate the GI50 cover the entire range from control growth (100%) to no growth (0%). The confidence in calculating the GI50 of resistant strains decreases, as the growth is not as inhibited by the tested doses. However, we also identified 152 strains from both libraries (145 deletion strains, 7 DAmP strains) that showed some resistance to MMS compared to WT (Figure S1, Additional file 2). The criterion for resistance is described in the Methods section. No GO term was significantly enriched (FDR<0.05) among the genes that conferred resistance when deleted. As in previous studies, this method has been unable to reproducibly identify resistant strains [1,2].
The data was also used to calculate the time required for the cultures to demonstrate visible growth (lag time) and the MMS dependency of the lag time (Figure S2, Additional file 2). Most strains, including the four control strains had a lag time 10-0 h. "Slow-growers" were defined as having a lag time exceeding 20 h. A large fraction (40%) of the "slow-growers" came from the relatively small DAmP library. Among the sensitive strains, a significant proportion (26.0%) were "slow-growers", which is significantly higher (p < 10-47) than in the entire collection (6.7%). This observation was further confirmed by a comparison with other "slow-growers" identified elsewhere [28]. In this set 18.6% of the sensitive strains were identified as "slow-growers", again significantly higher (p < 10-9) than in the entire collection (8.6%). The MMS dependency of the lag time represents an alternative measure of MMS sensitivity (Figure S2C, Additional file 2).
The MMS sensitivities and the lag times of the individual strains in this liquid assay are listed in Table S1 and S2 (Additional file 1).
Functional enrichment
To determine the biological functions involved in the recovery after being exposed to the DNA damaging agents MMS, we sought enrichment among gene ontology (GO) functional categories (see Methods) in the selection of genes deleted in the MMS sensitive strains compared to the entire genome. Among the functions that are highly enriched in the MMS sensitive strains are, as expected, DNA repair, cell cycle and transcription, but in addition the unexpected categories of telomere maintenance and RNA processing are also very highly enriched in the MMS sensitive strains (abstracted data in Table 1 complete data in Table S3, Additional file 1). Previous genomic phenotyping on solid agar did not find 'telomere maintenance' enriched among MMS sensitive strains [2]. However, re-analysis of the previous data with updated gene annotations revealed that this function and related GO categories are in fact among the most significantly enriched terms in that data set (Table S4, Additional file 1). Moreover, the RNA processing functions revealed in the current study derive primarily from screening the essential genes, which was not possible at the time of the previous study [2].
Table 1.
Enriched GO terms among the toxicity-modulating genes
| GO IDDescription | P-value | Adjusted P-value | x/X* (%) | n/N** (%) | |
|---|---|---|---|---|---|
| 6974 | response to DNA damage stimulus | 6.8E-19 | 2.1E-16 | 15.3 | 4.8 |
| 6259 | DNA metabolic process | 3.0E-16 | 4.7E-14 | 19.0 | 7.6 |
| 6281 | DNA repair | 2.9E-16 | 4.7E-14 | 12.8 | 3.9 |
| 22402 | cell cycle process | 6.6E-12 | 5.4E-10 | 17.4 | 7.9 |
| 6394 | RNA processing | 2.5E-09 | 1.5E-07 | 16.0 | 7.9 |
| 7001 | chromosome organization and biogenesis | 4.4E-09 | 2.4E-07 | 14.4 | 6.8 |
| 6365 | rRNA processing | 1.3E-08 | 6.4E-07 | 9.2 | 3.5 |
| 6260 | DNA replication | 1.4E-08 | 6.5E-07 | 7.8 | 2.7 |
| 278 | mitotic cell cycle | 2.2E-08 | 9.3E-07 | 11.4 | 5.1 |
| 6351 | transcription, DNA-dependent | 2.4E-08 | 9.8E-07 | 11.2 | 4.9 |
| 6302 | double-strand break repair | 2.5E-08 | 9.8E-07 | 4.3 | 1.0 |
| 34470 | ncRNA processing | 3.4E-08 | 1.3E-06 | 11.2 | 5.0 |
| 65004 | protein-DNA complex assembly | 1.1E-07 | 3.7E-06 | 5.9 | 1.9 |
| 723 | telomere maintenance | 2.0E-05 | 3.7E-04 | 3.4 | 1.0 |
| 6395 | RNA splicing | 2.4E-05 | 4.4E-04 | 5.7 | 2.4 |
| 7059 | chromosome segregation | 4.0E-04 | 5.0E-03 | 5.3 | 2.5 |
| 51656 | establishment of organelle localization | 3.9E-03 | 3.4E-02 | 2.5 | 1.0 |
| 7127 | meiosis I | 4.4E-03 | 3.8E-02 | 3.0 | 1.3 |
* number of toxicity-modulating genes with GO term/number of toxicity-modulating genes × 100
** number of genes with GO term/number of genes × 100
Table only shows non-redundant GO terms containing between 50 and 500 genes, with FDR adjusted p-value <0.05 and an enrichment of at least 2. For complete table, see supplemental table S3 (Additional file 1).
Protein-protein interaction networks
To interpret the results in a wider biological context, the protein products of all toxicity-modulating genes were mapped onto the S. cerevisiae protein-protein interaction network [29]. One large interconnected network with several well-defined sub-networks were identified (Figure 3A). As previously assumed [2], non-essential and essential toxicity-modulating gene products were present in the same biological networks. The networks were highly connected as each protein had an average number of 2.2 protein-protein interactions (p < 0.001, permutation test). In a random sample of equal size, each protein had on average 1.2 protein-protein interactions. The identified sub-networks are discussed below.
Figure 3.

Yeast protein-protein interaction networks. A) Network of proteins whose absence or reduced levels render cells sensitive to MMS B) Sub-network of these proteins involved in RNA processing. Non-essential proteins are in red, essential proteins in green. The size (inverse to the GI50-value) reflects the sensitivity to MMS of the corresponding yeast strains. Classification of severe, intermediate and slight sensitivity was established according to GI50 values (GI50<0.003%, GI50<0.006% and GI50<0.008%, respectively).
DNA repair and replication
As expected, many strains deficient in DNA repair proteins are identified as MMS sensitive. In concordance with previous results [1,2], this set of toxicity-modulating genes include members of the RAD52 epistasis group, such as RAD50, RAD51, RAD52, RAD59, RAD54 and RAD57. Together with XRS2, these gene products are required for homologous recombination repair of DNA double-strand breaks (reviewed in [30]). Other DNA repair pathways that were important for cellular recovery after MMS include both the base excision repair pathway (MAG1, APN1) and the nucleotide excision repair pathway (RAD14, SSL1, TFB1, RAD26), including associated factors (RAD9, RAD24, DEF1). Rad14p is the yeast homolog of damage binding protein XPA [31], and Ssl1p and Tfb1p are subunits of the TFIIH complex essential for NER [32,33]. Rad9p and Rad24p are checkpoint proteins required for NER [34]. A branch of NER, transcription coupled repair (TCR), is effective on the transcribed strand of DNA. This pathway is represented by RAD26 and DEF1. Rad26p is the yeast homolog of CSB, a DNA dependent ATPase [35] and Def1p is required for the ubiquitination and subsequent proteolytic degradation of RNA pol II [36,37]. The involvement of transcription coupled repair for DNA methylation damage is surprising in light of previous reports stating that TCR does not act on methylated DNA bases in mammalian cells [38]. The post-replication repair error-free prone pathway was also represented in our dataset, albeit by mutants that showed only slight MMS sensitivity (POL32, MMS2, RAD6, UBC13, RAD52, RAD5, RAD18).
We also identified three essential genes that encode three subunits of the Replication Factor C (RFC), namely RFC5, RFC1 and RFC3, as being important for MMS-induced damage recovery. The RFC complex is involved in both DNA repair and DNA replication, acting as a "clamp loader" to load Pol30p (the yeast homolog of PCNA) onto DNA; RFC is also thought to contribute to the maintenance of a DNA replication checkpoint during S phase [39].
Telomere maintenance
DNA damaging agents can induce gross chromosomal rearrangements and MMS is known to induce chromosomal aberrations in the form of telomere additions and translocations [40]. In the last few years, various genome-wide screens have shown that more than 350 genes affect the regulation of telomere length [41-43]. The RAD52 epistasis group provides a telomerase-independent mechanism of telomere maintenance, and is heavily represented among the toxicity-modulating proteins, as mentioned above. Besides the RAD52 epistasis group, deletion of other non-essential genes involved in telomere maintenance also results in MMS sensitivity. For instance, severe MMS sensitivity results upon deletion of SGS1 encoding a DNA helicase of the RecQ family that is required for recombination-mediated telomere lengthening [44,45]. The Sgs1p N-terminal physically interacts with Top3p [46] and Rmi1p [47], two other proteins that when lacking cause severe cellular sensitivity to MMS. Intermediate sensitivity results from deletion of EST1, encoding a protein associated with the telomere template RNA sequence (TLC1 RNA) used to add TG-repeats to form telomeric DNA that is part of the telomerase complex, and is essential for effective telomerase function [43,48]. Deletion of YKU80 also results in intermediate MMS sensitivity; this gene encodes a subunit of the Ku heterodimer, a DNA repair complex that also binds TLC1 RNA [40].
DAmP mutations in three essential genes related with 'telomere maintenance' were also found to result in a MMS sensitive phenotype. These essential genes were as follows: RAP1, encoding a protein that caps chromosome ends to prevent telomere fusion [49,50]; TEL2, encoding a protein that binds specifically to single-stranded telomeric DNA repeats and is required for telomere length regulation and telomere position effect [51]; DDC2, encoding a protein that interacts directly with Mec1p and Mec3p that are part of the essential component of the telomere checkpoint pathway, activated in the presence of DNA damage to induce a delay in cell cycle progression [52].
RNA processing
One of the major categories of cellular functions for essential genes is RNA processing. Approximately 10% of the entire S. cerevisiae genome is involved in one of various RNA-related processes [53], including mRNA splicing and export, tRNA modification, translation, rRNA processing, and RNA degradation. By screening the essential genes in the DAmP library, we found the GO term 'RNA processing' highly enriched among the toxicity-modulating proteins. A total of 61 strains sensitive to MMS had defects in proteins associated with this biological process; these are integrated within 2 subnetworks (Figure 3B). One network comprised of 20 essential proteins is primarily involved in rRNA processing and ribosome biogenesis (Figure 3B). Among this set there are two proteins that convey severe sensitivity to MMS when levels are reduced, namely Rnt1p and Prp43p. Rnt1p is an RNA endonuclease and Prp43p is an RNA helicase; both are involved in cleavage of the 3'-end of pre-rRNAs [54-56]. Prp43p is also involved in the release of lariat-introns from the spliceosome processing of pre-mRNAs [57]. As described below, many more proteins involved in mRNA splicing were shown to affect the recovery of cells from MMS-induced damage.
mRNA splicing
Seventeen proteins in the sub-networks are involved in nuclear mRNA splicing via the spliceosome (Figure 3B). mRNA splicing is a complex reaction involving dozens of proteins, and consisting of two consecutive catalytic reactions divided into three coordinated stages [58]. Toxicity-modulating genes were found to be involved in each of the three stages as follows: in the assembly and activation of the spliceosome (CDC40, BRR2, CLF1, LSM4, LSM8, PRP40, SMX3, PRP39); in the catalysis stage (PRP4, MSL5, PRP2, PRP24, DIB, SNU56, YHC1); and in the release, disassembly and snRNP recycling stage (PRP43 and PRP22).
In addition to mRNA splicing, MMS sensitivity was produced upon reduced expression of genes involved in other kinds of RNA splicing, such as snoRNA splicing (CWC24) and tRNA splicing (PTA1, NUP116, NUP49, SEN15, SEN2, POP1, POP6, POP4, RPR2, SPB1). The surprising finding that RNA splicing of all kinds is required for cellular survival after damage with MMS could be a reflection of the need for spliced gene products to help cells recover. In S. cerevisiae only ~ 280 genes contain introns (5% of all genes). Twenty of the 506 toxicity-modulating genes have introns (table 2), corresponding to ~ 4%, indicating no significant enrichment for intron-containing genes among the toxicity-modulating genes. While most intron-containing genes are implicated in the ribosomal machinery, and some are involved in meiosis [59], there was no significant enrichment of any biological process among the 20 toxicity-modulating intron-containing genes. However, it should be noted that three of these 20 gene products have well-described roles in DNA repair (Mms2p, Ubc13p and Rad14p), suggesting a highly specific role of these particular spliced gene products after DNA damage. Yeast strains with any one of these three genes deleted are sensitive to a number of genotoxic agents, including MMS, 4-NQO and UV [2]. Exactly why so many different kinds of RNA processing are important for the recovery of cell growth after exposure to MMS is not yet clear, but the extent to which the RNA-processing deficient mutants are sensitive and the fact that so many kinds of RNA are involved points to a fundamentally important biological mechanism. We are currently investigating why deficient RNA processing of various types renders cells so sensitive to DNA damaging agents.
Table 2.
Toxicity-modulating genes containing introns.
| ORF | Common name | GI50 (%MMS) |
|---|---|---|
| YGL087C* | MMS2 | 0.004 |
| YDR367W | 0.005 | |
| YDR092W* | UBC13 | 0.005 |
| YNL096C** | RPS7B | 0.005 |
| YNL038W | GPI15 | 0.006 |
| YNL112W | DBP2 | 0.006 |
| YLL050C | COF1 | 0.006 |
| YDL075W* | RPL31A | 0.007 |
| YFR045W* | 0.007 | |
| YBL018C | POP8 | 0.007 |
| YJL191W** | RPS14B | 0.007 |
| YMR033W | ARP9 | 0.008 |
| YKR094C** | RPL40B | 0.008 |
| YMR079W | SEC14 | 0.008 |
| YMR116C* | ASC1 | 0.008 |
| YNL162W** | RPL42A | 0.008 |
| YLR078C | BOS1 | 0.008 |
| YHR041C* | SRB2 | 0.008 |
| YML094W* | GIM5 | 0.008 |
| YMR201C* | RAD14 | 0.008 |
* Sensitive in Begley et al 2004.
** Not sensitive in Begley et al 2004.
Reproducing previous data
The results obtained in this liquid culture-based screen were compared to previous results using an agar-based colony growth screen [2]; we found reasonable concordance between the datasets for the sensitive strains, especially for the strains with a sensitivity score higher than 7, the score of rad14Δ (R2 = 0.4). The strains with a sensitivity score less than 7 are not reliably detected in the liquid assay (line fitted to the data with R2 = 0.0) (Figure 4A). In the previous data set, only the non-essential genes were tested by growing individual yeast strains on solid agar; each strain was assigned a sensitivity score ranging from 0 (no sensitivity) to 30 (highest sensitivity) based on the extent of growth on MMS-containing agar relative to the WT strain. Compared to the liquid culture screen, the solid agar screen, which was far more labor-intensive, found many more strains sensitive to MMS. In the current study we defined the thresholds of severe, intermediate and slight sensitivity based on the scores of the three control strains mag1Δ, rev1Δ and rad14Δ, respectively. After the application of these thresholds, the previous study identified a total 588 out of 4852 deletion strains tested (12%) as being sensitive to MMS: 30 strains showed severe sensitivity, 43 strains showed intermediate sensitivity, and 515 strains showed slight sensitivity. The highest correspondence between the assays is found for strains that show severe or intermediate sensitivity in at least one of the assays. Of the 105 strains with severe/intermediate sensitivity in the liquid assay, 73% (77/105) were also associated with severe/intermediate sensitivity in the agar assay. Among the 73 strains with severe/intermediate sensitivity in the agar assay, 51% (37/73) were determined to have severe/intermediate sensitivity in the liquid assay. Strains with slight MMS sensitivity in the agar screen were within the variation of the WT in the liquid assay presented here. Among the 153 slightly sensitive strains in the liquid assay, 97 (63%) were sensitive in the agar assay, whereof 61 strains showed intermediate sensitivity and 10 strains showed severe sensitivity. Despite the fact that the liquid assay identified fewer sensitive strains than the agar assay (among the non-essential deletion strains) we found 84 strains that had not shown MMS-sensitivity in the agar assay. Among these genes, four were classified as resulting in severe sensitivity when deleted; these are as follows: TAT1 (YBR069C), an amino acid transporter; EMP24 (YGL200C), involved in ER to Golgi transport; YOR331C encoding a protein of unknown function, localizing to endosomes [60]; and also the as yet uncharacterized YNL086W.
Figure 4.
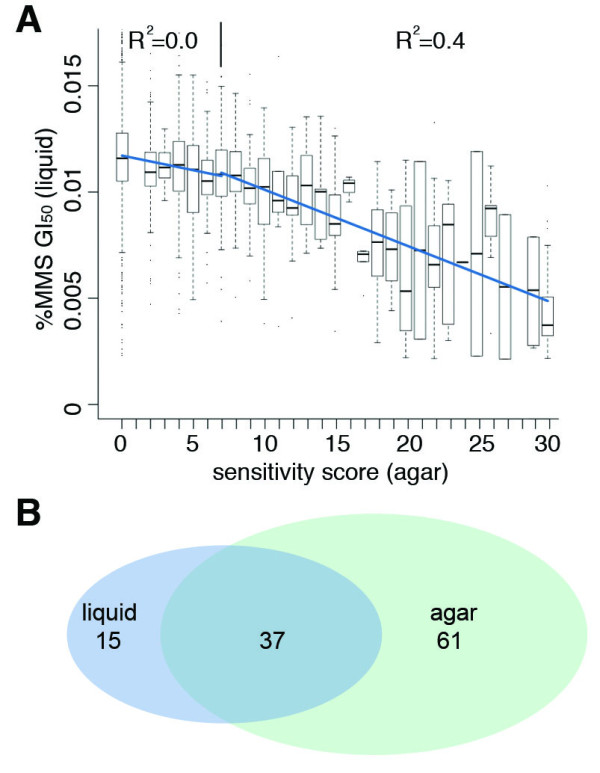
Comparison between results of liquid genomic phenotyping (this study) and previous results using a solid agar assay (Begley et al, 2004). A) A boxplot of the previous dataset (x-axis) where the data ranges from 0 (no sensitivity to MMS) and 30 (high sensitivity to MMS) and the dataset presented here. The bold line represents the median, the box contains 50% of the data, the whiskers extend to 1.5 times the inter-quartile range, and outliers are represented by dots. Two lines (blue) are fitted with linear regression to the data, one in the range of sensitivity scores 0-7 and one in the range of sensitivity scores 7-20. B) Venn diagram showing the overlap in GO terms enriched in sensitive strains from the liquid assay and the agar assay.
Finally, it is very important to note that despite fewer strains being identified in the liquid assay, most (71%) of the enriched functional categories (Bonferroni adjusted p-value<0.0001) in the list of toxicity-modulating genes resulting from the liquid assay (Table S3, Additional file 1) were also found in the list resulting from the reanalysis of previous dataset (Table S4, Additional file 1) (Figure 4B). The main categories uniquely present in the liquid assay can be summarized as processing of different species of RNA, whereas the liquid assay results are lacking a significant enrichment for vesicle transport genes. It should also be noted that since the mutant libraries were screened under different growth conditions (liquid versus agar) we expected to see differences in the pathways detected.
Different modes of toxicity found through growth patterns
The detailed growth curves obtained in this study allow further categorization of the sensitive yeast strains. The sensitivity phenotype was associated with several distinct growth patterns. The shape of growth curves of each strain in various MMS doses provides a wealth of information (Figure 5). After 0.008% MMS exposure, the WT strain shows a prolonged lag phase, but then starts growing exponentially and reaches the plateau at the same level as the untreated cells (Figure 2A, 5). The growth data of the sensitive strains after 0.008% MMS exposure was subjected to self-organizing map (SOM) analysis to split the data in three classes (Figure 5A). Other doses were also examined but at higher doses, the dynamic range is lost, as many strains do not grow at all; at the lower dose (0.004% MMS), less effect is seen, but the strains still group into similar categories. The three classes evident at the 0.008% MMS dose show distinct growth patterns characterized by: (i) lacking a MMS-induced lag-phase at this dose (n = 184); (ii) showing slower growth compared to WT (n = 110); and (iii) showing a very prolonged lag-phase and a slower recovery rate (n = 186). The average GI50 value calculated based on all five doses (0-0.016% MMS) were calculated to be 0.0067 ± 0.0016, 0.0071 ± 0.0010 and 0.0053 ± 0.0016% MMS for clusters (i), (ii) and (iii) respectively (Tables S5-7, Additional file 1). In other words, all three categories are in fact MMS sensitive compared to WT despite the fact that at the 0.008% MMS dose only category (iii) appears to be sensitive, and category (i) appears to be resistant. This underscores the importance of monitoring the effects of a range of doses, and a range of exposure times.
Figure 5.

The strains with intermediate sensitivity are divided into classes based on their growth phenotype after exposure to 0.008% MMS. A) Through self-organizing maps, three classes were identified based on the growth curves after MMS exposure. WT is shown as a reference in light blue. The black lines show the average values within the class for each time point. B) Hierarchical clustering and heatmaps of the three clusters in A. The growth of the WT strain is shown at the top.
Notably, most of the genes (75/110, 68%) mutated in the class (ii) strains are essential. GO enrichment analysis of the different classes reveals that several functional categories are enriched (Table S8-10, Additional file 1). In particular, class (i) is overrepresented by response to DNA stimulus, DNA repair and DNA replication. The most prominent groups of enrichment in class (ii) are RNA processing and cell cycle. Class (iii) has the most widely distributed functional diversity. The cluster is enriched (FDR<0.05) for 125 GO categories, representing most enriched categories found in the entire dataset of sensitive strains.
The strains in class (iii) are registered by most assays and these results correlate well with previous datasets (78% recognized in our previous study, 51/65 non-essential gene deletion strains). The strains represented in both class (i) and (ii) are expected to be more difficult to detect in assays employing just one late time point to measure sensitivity. However, although class (i) and (ii) show a smaller overlap with previous data than does class (iii), 63% of the non-essential genes in class (i) and 66% of the non-essential genes in class (ii) were in fact detected in the previous dataset that used one late time-point to assess toxicity [2]. Thus, methods relying on a single time point have a slightly lower resolution in detecting the growth patterns of class (i) and (ii). The dynamics of the growth curves make these clusters easy to identify using the method described here.
The complete dataset is available as a database with a web-interface available at http://genomicphenotyping.mit.edu/svensson/2011 (Figure S3, Additional file 2).
Discussion
In this study, we have measured growth curves after exposure to the DNA damaging agent MMS for a collection of yeast mutant strains deficient in 5,528 essential and non-essential genes. Compared to previous studies using similar genomic phenotyping [1,2], we have expanded the data to include essential genes, and to include detailed growth analysis of each strain; growth was measured at 10 time-points after treatment with a toxicant, in biological triplicates. By testing compounds in a eukaryotic system, an estimate of the toxicity in eukaryotic cells is given, as well as details regarding the way the cell responds to the toxicant, in this case MMS. We show here that genomic phenotyping is a valuable tool to decipher the modes of toxicity conferred by a DNA damaging agent. This was demonstrated by our identification of several novel toxicity-modulating genes, including those involved in RNA processing and telomere maintenance.
The fact that the toxicity-modulating proteins are found within protein-protein interaction networks of significantly higher connectivity than expected (p > 0.001) raises our confidence that the novel candidates are truly needed for cells to recover after MMS-induced damage. This includes the proteins involved in different kinds of RNA processing. It was recently shown that certain tRNA-modifications can influence cell survival after exposure to DNA damaging agents, in both yeast and human cells [61,62]. Here we also identify mRNA, snoRNA and tRNA splicing as being required for survival after DNA damage, even though relatively few yeast transcripts are spliced [59,63]. From this study, it remains inconclusive whether RNA splicing in general is important for helping the cell better handle MMS-induced damage or whether the processing of a few specialized transcripts may provide MMS resistance; such specialized targets include mRNA transcripts from the MMS2, UBC13 and RAD14 genes, three DNA repair genes all of which are spliced in yeast [59]. However this does not explain why snoRNA and tRNA splicing is required for MMS-resistance.
In addition to genes encoding mRNA, snoRNA and tRNA processing proteins, one of the prominent groups of genes resulting in MMS sensitive strains when deleted, is involved in the rRNA metabolic process, consisting of 'rRNA catabolic process' and 'rRNA processing'. Forty-one out of the 262 (16%) genes of this GO category are toxicity-modulating. The majority of the toxicity-modulating rRNA-related genes are essential in yeast (34/41), which is presumably the reason why these pathways were not identified in previous screens.
Another cellular function highlighted in this study is telomere maintenance. In yeast, many of the telomere maintenance proteins also have functions in DNA damage responses, such as Tel1p and Mec1p, which are homologs of the human ATM and ATR kinases that are activated in response to DNA damage. Yeast telomeres are maintained differently than their metozoan counterparts. The components of the mammalian shelterin complex that protects the telomere ends have no direct homologs in budding yeast, although yeast shelterin-like proteins have been described [64].
The fact that a substantial proportion of the MMS sensitive strains have a slow growing phenotype under normal conditions, could reflect that this subset of the "sensitive" strains are identified as a consequence of the accumulated stress exceeding a viability threshold with the additional DNA damage. However, for the majority of the sensitive strains, this is not the case.
We have further shown that the DAmP strains are very well suited to studying essential genes in this type of damage-sensitivity screening. Given the essential role of these genes, it is not surprising that reduced levels of the transcripts lead to a reduction in growth rate for several of the DAmP strains. Compared to the diploid hemizygous strains [65], the DAmP strains show a higher proportion of toxicity-modulating genes (data not shown). This observation is consistent with previous results using the drug methotrexate [20]. Compared to previous studies of genomic phenotyping, the information provided by this study is richer in data sampling, thus resulting in the possibility to further dissect the modes of toxicity and differentiate between patterns of sensitivity. New modes of sensitivity can be detected through understanding of the dynamics of the growth. Types of sensitivities that could go undetected in other systems can be scored here, as demonstrated by our self-organizing map analysis. Interestingly, the majority of the genes (68%) that were present in class (ii), were essential and primarily members of the relatively small DAmP library. This pattern of MMS sensitivity that is only apparent at higher MMS doses may be explained by the fact that lower levels of transcript expressed in the DAmP may be able to maintain sufficient protein levels to handle low levels of cellular damage but then fail at higher levels of damage.
Conclusions
To conclude, we present here a data-driven method to reveal modes of toxicity of different agents that impair cellular growth. This eukaryotic testing system could potentially be used to screen compounds for toxicity, to identify mechanisms of toxicity, and to reduce the need for animal testing.
Methods
Strains
S. cerevisiae strain haploid BY4741, diploid BY4743 were purchased from Research Genetics. As previously described [1], strain BY4741 was transformed with plasmind pYE13g (American Type Culture Collection) which confers G418 resistance. Deletion, DAmP and hemizygous library were purchased from Open Biosystems. The deletion library consists of a collection of 4,852 haploid strains where each strain has a single ORF replaced with the KanMX4 module, which confers G-418 resistance. These strains are in the BY4741 background (MATa his3Δ leu2Δ met15Δ ura3Δ ).
Cell culture
96-well master plates containing individual deletion strains were grown to stationary phase in 150ul YPD (10 g yeast extract, 20 g peptone, 20 g dextrose/liter), containing G-418 (Sigma) at 200ug/ml. Three wells of WT yeast and three control strains with known sensitivity were added into the plates. Settled cells were resuspended and a 1600X dilution of the cell suspension was inoculated with five doses (0, 0.004, 0.008, 0.012 and 0.016%) of MMS (Sigma) using a 96-pin Hydra (Robins Scientific). Cells were incubated for 48 h at 30°C. After 12 h, the OD600 was measured every 4 h using a Victor3 (Perkin Elmer). Comparison to cultures grown in bulk revealed small differences in growth patterns (data not shown).
Data analysis
Files with raw data were analyzed with in-house developed scripts in R (http://www.r-project.org). The OD measurements of empty wells were subtracted from all wells. Growth curves for the 48 hours after addition of MMS were drawn for the 5 doses for the individual yeast strains. The area under the curve (AUC) was calculated for each dose (including the mock-treated sample). For each strain, the dose-specific AUC was plotted against the dose. A line was fitted by linear regression and the goodness-of-fit (R2) was used to estimate linearity of the response. The slope revealed by the regression was used to determine the dose leading to 50% growth inhibition, GI50, by GI50= -0.5/slope (Figure 1B). R-scripts to regenerate the analysis are available in supplementary material together with instructions to access the raw data (Additional file 3). The visualization of the heat maps was done in R. Self-organizing maps were implemented through the SOM package. Functional enrichment was performed in Bingo 2.0.
Sensitivity thresholds were calculated based on the average GI50 of the three control strains (mag1Δ, rev1Δ, rad14Δ ). The resistance threshold was determined as GI50_average + (GI50_average - GI50_rad14Δ).
The data is available at a searchable database http://genomicphenotyping.mit.edu/svensson/2011 (Figure S3, Additional file 2).
Reducing the number of time-points
To assess how essential it was to measure cell density every 4 hours between hour 12 and 48 of the 48 h time course, we determined the loss of information resulting from removal of the data for several time-points (Figure S4, Additional file 2). For practical reasons it is important to note that removal of several measurements at intermediate times had only a limited effect on the reproducibility of the data. The goodness-of fit was 0.97 between the full dataset (with 10 time-points) and a reduced dataset (with six time-points). The coverage was determined by the percentage of tested strains that passed the linearity criterion as R2>0.7 using the selected time-points only. Using the more practical six point time-course, the coverage was still 84% versus 89.3% with the full non-essential dataset. On the other hand, only considering single observations (at 24 or 48 h) had drastic negative effects on the reproducibility of the data.
Network analysis
Yeast interaction networks were retrieved from [29] and loaded into Cytoscape v2.6.1 [66]. Functional enrichment was determined by the plug-in Bingo2.0 [67].
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LDS, RCF and JPS designed the experiments, LQP, JPS conducted the experiments, LQP, JPS, RCF, YAA and PC analyzed the data, LQP, JPS and LDS wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Supplementary tables. This file includes 10 additional tables to supplement the text.
Supplementary tables. This file includes additional figures to supplement the text.
R script. Text file containing the R code to regenerate the analysis.
Contributor Information
J Peter Svensson, Email: peter.svensson@ki.se.
Laia Quirós Pesudo, Email: pesudol@mit.edu.
Rebecca C Fry, Email: rfry@unc.edu.
Yeyejide A Adeleye, Email: yeyejide.adeleye@unilever.com.
Paul Carmichael, Email: paul.carmichael@unilever.com.
Leona D Samson, Email: lsamson@mit.edu.
Acknowledgements
This research was supported by Unilever and by NIH grant CA055042 and ES002109. JPS was supported by a Swedish Research Council Fellowship. LQP was supported by a Spanish Ministry of Science and Innovation Fellowship. LDS is an American Cancer Society Research Professor. The authors would like to thank Emma Wang, Peter Kemble and Siobhan McRee for technical assistance.
References
- Begley TJ, Rosenbach AS, Ideker T, Samson LD. Damage recovery pathways in Saccharomyces cerevisiae revealed by genomic phenotyping and interactome mapping. Molecular Cancer Research. 2002;1(2):103–112. [PubMed] [Google Scholar]
- Begley TJ, Rosenbach AS, Ideker T, Samson LD. Hot spots for modulating toxicity identified by genomic phenotyping and localization mapping. Molecular Cell. 2004;16(1):117–125. doi: 10.1016/j.molcel.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Jelinsky SA, Estep P, Church GM, Samson LD. Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes. Molecular and Cellular Biology. 2000;20(21):8157–8167. doi: 10.1128/MCB.20.21.8157-8167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinsky SA, Samson LD. Global response of Saccharomyces cerevisiae to an alkylating agent. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(4):1486–1491. doi: 10.1073/pnas.96.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(26):16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanway D, Chin JK, Xia G, Oshiro G, Winzeler EA, Romesberg FE. Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10605–10610. doi: 10.1073/pnas.152264899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash L, Prakash S. Isolation and Characterization of Mms-Sensitive Mutants of Saccharomyces-Cerevisiae. Genetics. 1977;86(1):33–55. doi: 10.1093/genetics/86.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman CT, Mak HC, McCuine S, Tagne JB, Agarwal M, Ozier O, Begley TJ, Samson LD, Ideker T. A systems approach to mapping DNA damage response pathways. Science. 2006;312(5776):1054–1059. doi: 10.1126/science.1122088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, D.C.: ASM Press; 1995. [Google Scholar]
- Cahill PA, Knight AW, Billinton N, Barker MG, Walsh L, Keenan PO, Williams CV, Tweats DJ, Walmsley RM. The GreenScreen((R)) genotoxicity assay: a screening validation programme. Mutagenesis. 2004;19(2):105–119. doi: 10.1093/mutage/geh015. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z. et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418(6896):387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. Genes required for ionizing radiation resistance in yeast. Nature Genetics. 2001;29(4):426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G. The chemical genomic portrait of yeast: Uncovering a phenotype for all genes. Science. 2008;320(5874):362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, Cheung KH, Sheehan A, Symoniatis D, Umansky L, Heidtman M, Nelson FK, Iwasaki H, Hager K, Gerstein M, Miller P, Roeder GS, Snyder M. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402(6760):413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ, Boone C. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126(3):611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Dudley AM, Janse DM, Tanay A, Shamir R, Church GM. A global view of pleiotropy and phenotypically derived gene function in yeast. Molecular Systems Biology. 2005. [DOI] [PMC free article] [PubMed]
- Mnaimneh S, Davierwala AP, Haynes J, Moffat J, Peng WT, Zhang W, Yang X, Pootoolal J, Chua G, Lopez A, Trochesset M, Morse D, Krogan NJ, Hiley SL, Li Z, Morris Q, Grigull J, Mitsakakis N, Roberts CJ, Greenblatt JF, Boone C, Kaiser CA, Andrews BJ, Hughes TR. Exploration of essential gene functions via titratable promoter alleles. Cell. 2004;118(1):31–44. doi: 10.1016/j.cell.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169(4):1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nature Methods. 2008;5(8):711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Costanzo M, Heisler LE, Paw J, Kaper F, Andrews BJ, Boone C, Giaever G, Nislow C. Yeast Barcoders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nature Methods. 2008;5(8):719–725. doi: 10.1038/nmeth.1231. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, Weissman JS, Krogan NJ. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123(3):507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Parker R. Aberrant mRNAs with extended 3 ' UTRs are substrates for rapid degradation by mRNA surveillance. Rna-a Publication of the Rna Society. 1999;5(10):1299–1307. doi: 10.1017/s1355838299990829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint M, Conconi A. High-throughput and sensitive assay to measure yeast cell growth: a bench protocol for testing genotoxic agents. Nature Protocols. 2006;1(4):1922–1928. doi: 10.1038/nprot.2006.304. [DOI] [PubMed] [Google Scholar]
- Smith AM, Ammar R, Nislow C, Giaever G. A survey of yeast genomic assays for drug and target discovery. Pharmacology & Therapeutics. 2010;127(2):156–164. doi: 10.1016/j.pharmthera.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, Roth FP, Giaever G, Nislow C. Quantitative phenotyping via deep barcode sequencing. Genome Research. 2009;19(10):1836–1842. doi: 10.1101/gr.093955.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423(6937):241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Molecular Biology of the Cell. 2000;11(12):4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone Lab - SGA Technology. http://www.utoronto.ca/boonelab/sga_technology/index.shtml
- Lee I, Li Z, Marcotte EM. An improved, bias-reduced probabilistic functional gene network of baker's yeast, Saccharomyces cerevisiae. Plos One. 2007;2(10):e988. doi: 10.1371/journal.pone.0000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. Role of RAD52 Epistasis Group Genes in Homologous Recombination and Double-Strand Break Repair. Microbiol Mol Biol Rev. 2002;66(4):630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankmann M, Prakash L, Prakash S. Yeast Rad14 and Human Xeroderma-Pigmentosum Group-a DNA-Repair Genes Encode Homologous Proteins. Nature. 1992;355(6360):555–558. doi: 10.1038/355555a0. [DOI] [PubMed] [Google Scholar]
- Feaver WJ, Svejstrup JQ, Bardwell L, Bardwell AJ, Buratowski S, Gulyas KD, Donahue TF, Friedberg EC, Kornberg RD. Dual Roles of a Multiprotein Complex from Saccharomyces-Cerevisiae in Transcription and DNA-Repair. Cell. 1993;75(7):1379–1387. doi: 10.1016/0092-8674(93)90624-Y. [DOI] [PubMed] [Google Scholar]
- Habraken Y, Sung P, Prakash S, Prakash L. Transcription factor TFIIH and DNA endonuclease Rad2 constitute yeast nucleotide excision repair factor 3: Implications for nucleotide excision repair and Cockayne syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):10718–10722. doi: 10.1073/pnas.93.20.10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SR, Teng YM, Lowndes NF, Waters R. RAD9, RAD24, RAD16 and RAD26 are required for the inducible nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers from the transcribed and non-transcribed regions of the Saccharomyces cerevisiae MFA2 gene. Mutation Research-DNA Repair. 2001;485(3):229–236. doi: 10.1016/S0921-8777(01)00061-1. [DOI] [PubMed] [Google Scholar]
- Vangool AJ, Verhage R, Swagemakers SMA, Vandeputte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JHJ. Rad26, the Functional Saccharomyces-Cerevisiae Homolog of the Cockayne-Syndrome-B Gene Ercc6. Embo Journal. 1994;13(22):5361–5369. doi: 10.1002/j.1460-2075.1994.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard H, Wellinger RE, Aguilera A. A new connection of mRNP biogenesis and export with transcription-coupled repair. Nucleic Acids Research. 2007;35(12):3893–3906. doi: 10.1093/nar/gkm373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid J, Svejstrup JQ. DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro. Journal of Biological Chemistry. 2004;279(29):29875–29878. doi: 10.1074/jbc.C400185200. [DOI] [PubMed] [Google Scholar]
- Plosky B, Samson L, Engelward BP, Gold B, Schlaen B, Millas T, Magnotti M, Schor J, Scicchitano DA. Base excision repair and nucleotide excision repair contribute to the removal of N-methylpurines from active genes. DNA Repair. 2002;1(8):683–696. doi: 10.1016/S1568-7864(02)00075-7. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Ando S, Shimomura T, Matsumoto K. Rfc5, a replication factor C component, is required for regulation of Rad53 protein kinase in the yeast checkpoint pathway. Molecular and Cellular Biology. 1997;17(10):5905–5914. doi: 10.1128/mcb.17.10.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE. Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes & Development. 2003;17(19):2384–2395. doi: 10.1101/gad.1125903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askree SH, Yehuda T, Smolikov S, Gurevich R, Hawk J, Coker C, Krauskopf A, Kupiec M, McEachern MJ. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(23):8658–8663. doi: 10.1073/pnas.0401263101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatbonton T, Imbesi M, Nelson M, Akey JM, Ruderfer DM, Kruglyak L, Simon JA, Bedalov A. Telomere Length as a Quantitative Trait: Genome-Wide Survey and Genetic Mapping of Telomere Length-Control Genes in Yeast. PLoS Genet. 2006;2(3):e35. doi: 10.1371/journal.pgen.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar L, Yosef N, Sela Y, Sharan R, Ruppin E, Kupiec M. A genome-wide screen for essential yeast genes that affect telomere length maintenance. Nucl Acids Res. 2009;37(12):3840–3849. doi: 10.1093/nar/gkp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kozak M, Martin JD, Pennock E, Johnson FB. Evidence that a RecQ helicase slows senescence by resolving recombining telomeres. Plos Biology. 2007;5(6):1334–1344. doi: 10.1371/journal.pbio.0050160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillard-Wetherell K, Combs KA, Groden J. BLM helicase complements disrupted type II telomere lengthening in telomerase-negative sgs1 yeast. Cancer Research. 2005;65(13):5520–5522. doi: 10.1158/0008-5472.CAN-05-0632. [DOI] [PubMed] [Google Scholar]
- Gangloff S, Mcdonald JP, Bendixen C, Arthur L, Rothstein R. The Yeast Type-I Topoisomerase Top3 Interacts with Sgs1, a DNA Helicase Homolog - a Potential Eukaryotic Reverse Gyrase. Molecular and Cellular Biology. 1994;14(12):8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Zhang CY, Desai R, Morozov P, Delgado-Cruzata L, Rothstein R, Freyer GA, Boone C, Brown GW. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. Embo Journal. 2005;24(11):2024–2033. doi: 10.1038/sj.emboj.7600684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53–56. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- Negrini S, Ribaud V, Bianchi A, Shore D. DNA breaks are masked by multiple Rap1 binding in yeast: implications for telomere capping and telomerase regulation. Genes & Development. 2007;21(3):292–302. doi: 10.1101/gad.400907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, Marcand P. Rap1 prevents telomere fusions by nonhomologous end joining. Embo Journal. 2005;24(17):3117–3127. doi: 10.1038/sj.emboj.7600778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge KW, Zakian VA. TEL2, an essential gene required for telomere length regulation and telomere position effect in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16(6):3094–3105. doi: 10.1128/mcb.16.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto S, Glowczewski L, Berman J. MEC3, MEC1, and DDC2 Are Essential Components of a Telomere Checkpoint Pathway Required for Cell Cycle Arrest during Senescence in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13(8):2626–2638. doi: 10.1091/mbc.02-02-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, Ideker T, Guthrie C, Krogan NJ. A Genetic Interaction Map of RNA-Processing Factors Reveals Links between Sem1/Dss1-Containing Complexes and mRNA Export and Splicing. Molecular Cell. 2008;32(5):735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braglia P, Kawauchi J, Proudfoot NJ. Co-transcriptional RNA cleavage provides a failsafe termination mechanism for yeast RNA polymerase I. Nucl Acids Res. 2011;39(4):1439–1448. doi: 10.1093/nar/gkq894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elela SA, Ares M. Depletion of yeast RNase III blocks correct U2 3' end formation and results in polyadenylated but functional U2 snRNA. Embo Journal. 1998;17(13):3738–3746. doi: 10.1093/emboj/17.13.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertschy B, Schneider C, Gnadig M, Schafer T, Tollervey D, Hurt E. RNA Helicase Prp43 and Its Co-factor Pfa1 Promote 20 to 18 S rRNA Processing Catalyzed by the Endonuclease Nob1. Journal of Biological Chemistry. 2009;284(50):35079–35091. doi: 10.1074/jbc.M109.040774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Semlow DR, Staley JP. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(22):10020–10025. doi: 10.1073/pnas.0906022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Query CC, Konarska MM. Nought May Endure but Mutability : Spliceosome Dynamics and the Regulation of Splicing. 2008. pp. 657–666. [DOI] [PMC free article] [PubMed]
- Juneau K, Palm C, Miranda M, Davis RW. High-density yeast-tiling array reveals previously undiscovered introns and extensive regulation of rneiotic splicing. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(5):1522–1527. doi: 10.1073/pnas.0610354104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425(6959):686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Molecular Cell. 2007;28(5):860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, Brophy JAN, Chan CTY, Atmore KA, Begley U, Paules RS, Dedon PC, Begley TJ, Samson LD. Human AlkB Homolog ABH8 Is a tRNA Methyltransferase Required for Wobble Uridine Modification and DNA Damage Survival. Molecular and Cellular Biology. 2010;30(10):2449–2459. doi: 10.1128/MCB.01604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura F, Kawaguchi N, Sese J, Toyoda A, Hattori M, Morishita S, Ito T. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17846–17851. doi: 10.1073/pnas.0605645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti D, Clerici M, Anbalagan S, Martina M, Lucchini G, Longhese MP. Shelterin-Like Proteins and Yku Inhibit Nucleolytic Processing of Saccharomyces cerevisiae Telomeres. Plos Genetics. 2010;6(5) doi: 10.1371/journal.pgen.1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(3):793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables. This file includes 10 additional tables to supplement the text.
Supplementary tables. This file includes additional figures to supplement the text.
R script. Text file containing the R code to regenerate the analysis.