

Abstract
Hypoxia inducible factor-1 (HIF-1) is a transcriptional factor responsible for cellular and tissue adaption to low oxygen tension. HIF-1, a heterodimer consisting of a constitutively expressed β subunit and an oxygen-regulated α subunit, regulates a series of genes that participate in angiogenesis, iron metabolism, glucose metabolism, and cell proliferation/survival. The activity of HIF-1 is controlled by post-translational modifications on different amino acid residues of its subunits, mainly the alpha subunit. Besides in ischemic stroke (see review [1]), emerging evidence has revealed that HIF-1 activity and expression of its down-stream genes, such as vascular endothelial growth factor and erythropoietin, are altered in a range of neurodegenerative diseases. At the same time, experimental and clinical evidence has demonstrated that regulating HIF-1 might ameliorate the cellular and tissue damage in the neurodegenerative diseases. These new findings suggest HIF-1 as a potential medicinal target for the neurodegenerative diseases. This review focuses on HIF-1α protein modifications and HIF-1’s potential neuroprotective roles in Alzheimer’s (AD), Parkinson’s (PD), Huntington’s diseases (HD), and amyotrophic lateral sclerosis (ALS).
Keywords: AD, ALS, EPO, HD, HIF-1, PD, VEGF, protein modification, prolyl hydroxylase inhbitor, iron chelator, 2-oxoglutarate analogues, cobalt, neurodegenerative diseases
1. Introduction
The oxygen tension-dependent transcriptional factor, hypoxia inducible factor-1 (HIF-1), is responsible for the induction of genes that facilitate the adaption and survival of cells exposed to hypoxia [2]. HIF-1 activation induces a diverse range of target genes, encompassing a wide variety of cellular processes, including angiogenesis, erythropoiesis, energy metabolism, cell proliferation, and cell cycle control [3]. HIF-1 can improve the redox environment [4], increase blood oxygen and glucose supply, and affect iron metabolism by regulating its target genes.
The brain consumes a large quantity of oxygen and demonstrates a high vulnerability at conditions with impaired oxygen supply. It has been suggested that reduced oxygen supply plays a key role in neurodegeneration during the aging process [5]. Pathological processes such as oxidative stress, impaired oxygen or glucose supply, and disruption of iron homeostasis are common in neurodegenerative diseases [6–8]. This raises the possibility that HIF-1 is a potential therapeutic target for these neurodegenerative diseases. In this review, we focus on the mechanisms of HIF-1 regulation and roles of HIF-1 in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Alzheimer’s (AD), Parkinson’s (PD), and Huntington’s diseases (HD). We also summarized recent drug development based on HIF-1 pathway in neurodegenerative diseases.
2. HIF-1 modifications, expression, and activation
HIF-1 is a heterodimetric complex consisting of a constitutively expressed β subunit (also known as the aryl hydrocarbon receptor nuclear translocation, ARNT) and a hypoxia inducible subunit, α subunit [2]. Both subunits belong to the basic helix-loop-helix (bHLH)-PER-ARNT-SIM (PAS) protein family. The bHLH and PAS motifs are required for dimerization between HIF-1α and HIF-1β. HIF-1α possesses a unique oxygen-dependent degradation domain (ODDD), which mediates oxygen-regulated stability, and two transactivation domains, N-terminal (N-TAD) and C-terminal (C-TAD) [9–10]. Under hypoxia, HIF-1α is stabilized and translocated to the nucleus where it dimerizes with HIF-1β [3]. The activated HIF-1 complex subsequently binds to hypoxic response elements (HREs) in the regulatory regions of genes and recruits co-transactivation factors such as CBP/p300 [CBP: CREB (cAMP-response element-binding protein)-binding protein/E1A-binding protein]. It induces transcription of more than a hundred genes with various functions. The activity of HIF-1 is mainly controlled through regulating the protein level of HIF-1α [3]. Although the transcription and the synthesis of HIF-1α are constitutive, its protein degradation and transcriptional activity are regulated by post-translational modifications on different amino acid residues in different domains as discussed in the following and summarized in Scheme 1 and Table 1.
Scheme 1.
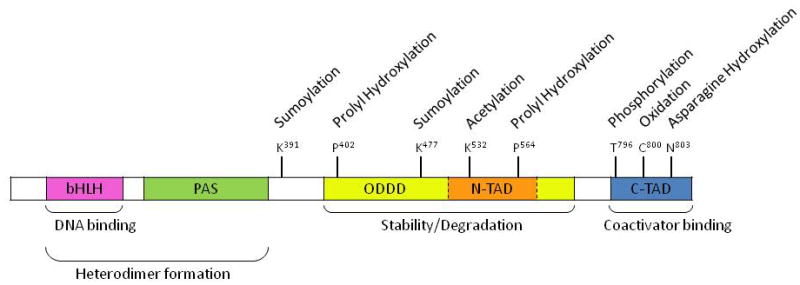
Domain structure and major modification sites of HIF-1α.
Table 1.
Summary of HIF-1α protein modifications.
| Modifications | Enzymes or Oxidants Involved | Target Amino Acids | Outcomes | Reference |
|---|---|---|---|---|
| Prolyl hydroxylation | Prolyl hydroxylase domains (PHDs) | P402 and 564 | Recognition signal for binding of pVHL E3 ligase | [109] |
|
| ||||
| Polyubiquitination | pVHL E3 ligase | K | Signal for Degradation | [18] [110] |
|
| ||||
| Acetylation | Arrest-defective-1 (ARD1) | K532 | ↑Interaction of HIF-1α with pVHL E3 ligase | [21] |
|
| ||||
| Asparagine hydroxylation | Factor inhibiting HIF-1 (FIH-1) | N803 | ↓Interaction of HIF-1α with CBP/p300 | [111] |
|
| ||||
| Phosphorylation | MAPK | T796 | ↑Transcriptional activity | [23] |
| S641 and 643 | ↑Transcriptional activity and nuclear localization | [26] | ||
| S551, T555 and S589 | Target for degradation | [26] | ||
|
| ||||
| Oxidation | Hydroxyl radical | C800 | HIF-1α degradation | [28] |
| H2O2 | HIF-1α degradation | [27] | ||
|
| ||||
| Sumoylation | E3 SUMO ligase | Within ODD | ↓Transcriptional activity | [34] |
| E3 SUMO ligase | K391 and 477 | ↑Stability and transcriptional activity | [112] | |
| RWD-containing sumoylation enhancer (RSUME) and SUMO conjugase Ubc9 | HIF-1α stabilization | [32] | ||
HIF-1α protein can be hydroxylated. Prolyl hydroxylation determines HIF-1α degradation in normoxia. HIF-1α is hydroxylated on the prolyl residues 402 and 564 within the ODDD [11]. Both sites of modification contain a conserved LXXLAP motif and the hydroxylation is taken place at their 4-positions [11]. The hydroxylation is mediated by a family of prolyl hydroxylases, namely PHD1, PHD2, and PHD3 [12], among which PHD2 has the highest specificity for hydroxylation of HIF-1α [11]. These PHD enzymes are 2-oxoglutarate-dependent dioxygenases and require oxygen as well as Fe2+ as cofactors [13–14]. Hydroxylation promotes interaction of HIF-1α with the von Hippel-Lindau (pVHL) ubiquitin E3 ligase complex [15–16]. HIF-1α is thereby poly-ubiquitinated and degraded by the 26S proteasome [17–18]. Since the PHDs employ non-heme iron in the catalytic moiety, it can be predicted that shortage of intracellular iron will result in inhibition of activity of PHDs and HIF-1 upregulation. As the result, iron chelators have been used to upregulate HIF-1α expression [19].
At the transcriptional level, oxygen availability enables hydroxylation of the asparagine residue 803 in the C-TAD of HIF-1α by factor inhibiting HIF (FIH-1), which blocks the association of HIF-1α with transcriptional co-activators CBP/p300 and thus inhibits its transcriptional activity [20].
HIF-1α can be acetylated on lysine residue 532 in the ODDD domain. Acetylation may affect HIF-1α hydroxylation and ubiquitination [21]. K532 acetylation by the ARrest-Defective-1 (ARD-1) enhances the interaction between HIF-1α and pVHL and, thus, leads to increased ubiquitination and concomitant proteasomal degradation [21]. Mutation of lysine 532 to arginine has been reported to result in increased stability of overexpressed HIF-1α [22]. Artificial maintenance of the acetylated state of HIF-1α by deacetylase inhibitor can lead to increased degradation of HIF-1α [22]. All evidence indicates that acetylation of HIF-1α is a signal for destabilization.
Direct phosphorylation on threonine 796 by mitogen-activated protein kinase (MAPK) has recently been identified as a novel modification on HIF-1α and is able to increase HIF-1 activity [23]. One mechanism to explain the increased transcriptional activity is that HIF-1β binds preferentially to the phosphorylated form of HIF-1α [24]. Moreover, it has been reported that phosphorylation at T796 in HIF-1α increases the affinity of the interaction between HIF-1α and the transcriptional co-activator CBP/p300 [25]. Consistent with this, it is found that phosphorylation of T796 prevents the hydroxylation of N803 by FIH [25]. More recently, it is suggested that direct phosphorylation of HIF-1α by MAPK is not correlated with its transcriptional activity, but that phosphorylation of the HIF-1α co-activator p300 by MAPK increases the interaction between the HIF-1α C-TAD and p300 [22]. Furthermore, HIF-1α S641 and S643 have been identified by mass spectroscopy with in vitro phosphorylated recombinant HIF-1α to be MAPK targets [26]. Inhibition of these phosphorylation sites has been shown to impair both transcriptional activity and the nuclear localization of HIF-1α [26]. In addition, phosphorylation of HIF-1α by glycogen synthase kinase (GSK)-3β may target HIF-1α for proteasomal degradation [26].
HIF-1α can be oxidized. Hydroxyl radical and hydrogen peroxide can destabilize HIF-1α protein in both normoxic and hypoxic conditions [27–28]. The oxidized HIF-1α protein might be recognized and degraded by the ubiquitin-independent 20S proteasomal pathway, which primarily degrades cellular oxidized proteins under oxidative stress conditions [29]. Therefore, in addition to the 26S proteasomal pathway, 20S proteasomal pathway may play an important role in the degradation of HIF-1α in ischemic conditions [1]. Moreover, studies have revealed that S-nitrosation stabilizes HIF-1α protein and S-nitrosation of cysteine 800 of HIF-1α promotes its interaction with CBP/p300, thus enhancing HIF-1α activation [30]. However, a fluorescein polarization-based binding assay has demonstrated that S-nitrosation of HIF-1α inhibits its p300 binding, which is contrary to a previous report [31].
Sumoylation of HIF-1α has been reported to regulate its protein level and transcriptional activity. Increasing the activity of RWD-containing sumoylation enhancer (RSUME) could promote the sumoylation of HIF-1α by Ubc9 SUMO conjugase and stabilize HIF-1α during hypoxia [32]. However, desumoylation by SUMO-Specific Protease 1 (SENP-1) has been shown to be essential for the stabilization of HIF-1α [33]. Sumoylation has also been found to work with ubiquitination to reduce HIF-1 protein level and transcriptional activity [33–34].
In summary, HIF-1α protein level and transcriptional activity are highly regulated through residue modifications such as hydroxylation, acetylation, phosphorylation, oxidation, and sumoylation.
3. HIF-1 and amyotrophic lateral sclerosis (ALS)
Lou Gehrig’s disease, or ALS, affects approximately 2 in 100,000 people in the world. The characteristics of ALS are damaged upper and lower motor neurons, causing weakness, muscle atrophy, fasciculations, spasticity, hypo- or hyper-reflexia, and extensor plantar responses [35]. Hypoxia, or an impaired oxygen supply, is a possible contributor to motor neuron death. In occupations typically leading to intermittent and topical suppression of blood flow around motor neuron axon at tissue levels, there is a two-fold increase in ALS risk [36]. In addition to occupation-induced hypoxia, chronically-reduced vascular perfusion by aging or other factors can produce either chronic or episodic deficits in glucose (hypoglycemia) and oxygen (hypoxia) [37]. This lack of glucose and oxygen fails to meet the energy requirement of motor neurons, which will in turn induce neuronal death and lead to the occurrence of ALS. Indeed, results from animal experiments have shown that in a ALS animal model, SOD1G93A mutant mice, hypoxia is the major cause of motor neuron death [38].
It is well known that one of the primary cellular responses to hypoxia is activation of the HIF-1-vascular endothelial growth factor (VEGF) pathway. VEGF can induce angiogenesis and increase blood supply to motor neurons during hypoxia through action on endothelial cells [39]. This would be beneficial in ALS. However, it is implied that this pathway is impaired in people with higher risk to ALS and in ALS patients. There is a negative correlation between the VEGF level and the severity of hypoxemia in patients with ALS, which suggests a deregulation of VEGF in ALS [40–41]. It has been shown that motor neurons are prone to death in subjects with genetically impaired response to hypoxia [36]. ALS-like symptoms and neuropathology can be produced in mice by a targeted deletion of the hypoxia response element that eliminates the expression of VEGF in response to hypoxia [42]. This indicates that an impaired HIF-1-VEGF pathway may contribute to the pathogenesis of ALS. Furthermore, overexpression of VEGF in SOD1G93A mutant mice delays the degeneration of motor neurons and neuronal death and prolongs the survival of these ALS mice [43].
The mechanism for the protective effect of VEGF in ALS possibly comprises two pathways: its angiogenetic effect and its direct action on motor neurons. VEGF acts on vascular endothelial cells to induce the proliferation and migration of endothelial cells and to form new micro-vessels [39]. The newly formed micro-vessels can increase the blood supply to motor neurons in brain and spinal cords [39]. By this mechanism, VEGF can prevent the death of motor neurons and postpone the occurrence of ALS. Also, VEGF acts directly on motor neurons as neurotrophic or neuroprotective factors [44]. VEGF supports the survival of primary motor neurons and a motor-like cell line in vitro and could protect these motor neurons from hypoxia-induced cell death by binding with neuropilin-1, a receptor known to be involved in axon guidance during development [42, 45]. Through these direct actions, VEGF can increase motor neurotrophy and activate the growth of motor neuron axon during the pathogenesis of ALS, thereby postponing the progression of ALS.
Besides VEGF, erythropoietin (EPO) may also mediate the protective effect of HIF-1. It has been reported that EPO levels are significantly decreased in cerebrospinal fluids from patients with differing severity and duration of ALS [46], while EPO immunoreacitivity is significantly increased in midbrain, brain stem, and cortex of the SOD1G93A mutant mice [47]. In this SOD1G93A mutant mouse ALS model, administration of EPO suppresses the onset and progression of ALS by preventing motor neuron death and inflammation [48–49]. These EPO neuroprotective effects indicate that EPO administration may be a new therapeutic approach to ALS.
Since increasing VEGF and EPO expression are both beneficial for the survival of motor neurons, their transcriptional regulator, HIF-1, may also serve as a potent therapeutic strategy for ALS. Indeed, the iron chelator M30, which up-regulates HIF-1 expression, protects NSC-34 motor neuron cells from oxidative damage in vitro and significantly delays the onset of ALS in SOD1G93A mutant mice [50] (Table 2).
Table 2.
Summary of HIF-1 inducers and their mechanisms responsible for HIF-1’s neuroprotection in neurodegenerative diseases.
| Diseases | Chemicals | Mechanisms of action | Positive effects | Involvement of HIF-1 target genes |
|---|---|---|---|---|
| ALS | M30 | Novel iron chelator; sequestering of ferrous iron, preventing formation of a catalytically active centre in the HIF hydroxylases thereby inducing HIF-1 activation | ↑Microvessel formation and blood supply ↑Motor neurotrophy |
VEGF and EPO |
| AD | DFO | Iron chelators; sequestering of ferrous iron, preventing formation of a catalytically active centre in the HIF hydroxylases thereby inducing HIF-1 activation | ↑Redox homeostasis ↑Oxygen delivery and supply ↑Glucose uptake |
VEGF, EPO, and GLUT-1,3 |
| M30 | ||||
| PD | Cobalt | Displacing the free ferrous at the active site of hydroxylases thereby inhibiting HIF-1 hydroxylation | ↑ DA Synthesis and secretion ↑Iron homeostasis ↑Mitochondria functions |
VEGF and EPO |
| DFO | Iron chelator; sequestering of ferrous iron, preventing formation of a catalytically active centre in the HIF hydroxylases thereby inducing HIF-1 activation | |||
| FG0041 3,4-DHB | Analogues of 2-oxoglutarate; bind to the active site of HIF hydroxylase, inhibiting HIF hydroxylase | |||
| HD | Cobalt | Displacing the free ferrous at the active site of hydroxylases thereby inhibiting HIF-1 hydroxylation | ↑Production of ATP and NADPH ↑Glia cells activity |
|
| DFO Clioquinol M30 | Iron chelators; sequestering of ferrous iron, preventing formation of a catalytically active centre in the HIF hydroxylases thereby inducing HIF-1 activation |
4. HIF-1 and Alzheimer’s disease (AD)
The incidence of Alzheimer’s disease is 15% among those 65 years and older, and is close to 50% for those aged over 85 years [51]. Patients with AD display loss of synapses and neurons, as well as extra-cellular senile plaques and intracellular neurofibrillary tangles (NFTs) [52]. Senile plaques consist largely of aggregated amyloid-β peptide (Aβ), which is liberated from the holoprotein, amyloid precursor protein (APP), by sequential cleavages mediated by the β-secretase β-site APP cleavage enzyme 1 (BACE1) and the γ-secretase complex [53]. NFTs are made up a highly phosphorylated form of the microtubule-associated protein tau [54]. Both Aβ accumulation and NFTs are pathological hallmarks of Alzheimer’s disease which are responsible for induction of oxidative stress, neuroinflammation, and calcium deregulation [55–56].
Results from both basic research and clinical trials have confirmed that HIF-1 activation may be a potent strategy to postpone the pathogenesis and ameliorate the outcomes of AD. The HIF-1 inducer, M30, has been reported to increase the HIF-1α protein level and elevate expression of the HIF-1 target genes, VEGF and EPO. It has been reported that M30 simultaneously attenuates tau phosphorylation and protects cortical neurons against Aβ (25–35) toxicity [57]. Overexpression of HIF-1α has been shown to protect the rat sympathetic nerves-like cell line PC12, the central nervous system cell line B12, and primary cultured cortical neurons from Aβ-induced neurotoxicity [58], possibly through activating glycolytic and hexose monophosphate shunt-related enzymes. Clinical applications of HIF-1 inducers have also demonstrated the neuroprotective effect of HIF-1 in AD (Table 2). Deferoxamine (DFO), a widely used HIF-1 inducer, has been used in clinical trial in AD patients and slowed cognitive decline [59]. These studies suggest that increasing HIF-1 activity can prevent neuron death and ameliorate these symptoms of AD.
The mechanism responsible for neuroprotective effects of HIF-1 in AD pathogenesis may rely on its regulation of various physiological processes. HIF-1 activation promotes cellular response to low oxygen, reduced glucose supply, and oxidative stress. As a result, it contributes to cell survival [3]. It is known that reduced cerebral blood flow [60], impaired glucose uptake and metabolism [61], and increased oxidative stress [62] are involved in the pathogenesis of AD. HIF-1 may mitigate the deleterious effects by the factors discussed below.
It has been shown that individuals who have suffered severe hypoxia or ischemia are more susceptible to developing AD [63]. Reduced blood oxygenation has been found in people with higher AD risk [64]. Cerebral blood flow reduction is present in the early stages of AD pathogenesis [60]. In vitro results have also shown that primary cortical neurons treated with Aβ and hypoxia exposure exhibit a higher cell death than treatment with Aβ alone [65]. In brains of transgenicAD mice, hypoxia elevates the β-cleavage of amyloid-β precursor protein and increases the expression of γ-secretase, which in turn increases Aβ generation and senile plaque formation [66]. HIF-1 can induce the erythropoiesis and angiogenesis to increase oxygen delivery and supply [3]. Thus, an increase of HIF-1 activity might reduce the risk of AD by suppressing the Aβ generation and AD occurrence induced by hypoxia.
Accumulating data have shown that impaired glucose uptake and metabolism may increase the risk of AD [61]. The reduced glucose uptake is due to decreased expressions of neuronal glucose transporters (GLUT-1 and GLUT-3) [67]. Since GLUT-1 and GLUT-3 are target genes of HIF-1, induction of HIF-1α expression may restore the glucose uptake and postpone the progression of AD.
Both Aβ-accumulation and NFTs induce oxidative stress in neurons, which in turn cause severe neuronal death [62]. HIF-1 may prevent neuronal death in the AD brain through elevating glycolysis and the hexose monophosphate shunt and increasing the reducing ability of neurons [4, 58].
HIF-1 target gene EPO may also mediate its neuroprotective effects. Erythropoietin is found to be secreted by neurons and to act on neurons directly through its receptor [68–69]. It has been shown that EPO is able to block the Aβ-generated neuronal apoptosis and that this protection is completely abolished by co-treatment with an anti-EPO neutralizing antibody [70]. In animal studies, EPO has been found to reduce the cognitive and behavioral symptoms of mechanical brain injury [71]. Recently, EPO has come into consideration as a possible treatment for AD.
Through regulating target genes expression, HIF-1 may prevent AD occurrence, postpone AD progression, and ameliorate AD symptoms. However, many important questions remain unanswered. First, it is still unclear whether the suppression of HIF-1 activity can induce AD, although the impairment of its down-streamed pathways has been widely found to contribute to the occurrence of AD, AD progression, and neuron death in patients. Second, besides causing oxidative stress, Aβ and NFTs also induce caspases and other neuronal apoptosis-related pathways [72]. Whether HIF-1 interacts with these pathways during AD pathogenesis is unclear. Third, most of the present available results are from models with activated HIF-1. Results from HIF-1α knock-out models are needed to identify whether HIF-1 is essential for the protective effects of Aβ preconditioning or iron chelators. Moreover, it is known that inflammatory responses mediated by micro-glia contribute to the progression of AD [73]. Since HIF-1 can modulate the inflammatory response of macrophages in the blood [74], further research on HIF-1’s functions in the inflammatory processes of glial cells in AD pathogenesis will be helpful for understanding the applications of HIF-1 in clinical trials of AD.
5. HIF-1 and Parkinson’s disease (PD)
Parkinson’s disease (PD) affects 1.5–2% in the population over 60 years of age [75]. It mainly results from a progressive loss of dopaminergic neurons in the substantia nigra, causing classical motor symptoms of bradykinesia, rigidity and resting tremors [76]. Thus, we may expect that restoration of dopamine (DA) synthesis and secretion levels may help to ameliorate the PD patients’ symptoms. Furthermore, it is suggested that mitochondrial dysfunction and the associated oxidative stress is the main mechanism responsible for neurodegeneration in PD [77]. DAergic neurons are particularly prone to oxidative damage due to high levels of inherent reactive oxygen species that are produced during DA synthesis or its breakdown by monoamine oxidases [78–79].
Parkinson’s disease can be induced in primates and mice by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is converted into an active toxin 1-methyl-4-phenylpyridinium (MPP+) in astrocytes. Accumulation of MPP+ is able to inhibit oxidative phosphorylation via inhibition of complex I in mitochondrial electron transport chain, resulting in energy failure and nigrostrital neuron death. It is reported that MPTP treatment inhibits HIF-1α accumulation in dopaminergic cell lines PC12 and in mice [80], suggesting that HIF-1 activity is impaired in PD. The most direct linkage between HIF-1 and PD is the tyrosine hydroxylase (TH), which is the rate-limiting enzyme in the synthesis of DA in DAergic neurons [81]. Promoter activity studies suggest that the TH gene is under the control of a hypoxia response element [82–83]. Hypoxia and DFO, both are known to upregulate HIF-1, can increase TH expression in rat brains [84–85]. Meanwhile, a conditional knock-down of HIF-1α in mice results in a 40% decrease of TH expression in substantia nigra [86]. Injection of the PHD inhibitor FG0041 increases HIF-1 activity and the extracellular DA level in the striatum of the rat brain [87]. Furthermore, administration of HIF-1 activator cobalt chloride raises the DA release [87]. Beside the increased DA synthesis, the secretion of DA is also increased through K+-depolarization in the FG0041 and cobalt chloride-treated rats [87]. Therefore, through increasing synthesis and secretion of DA, HIF-1 may prevent the PD pathological process.
Recent studies have also demonstrated that HIF-1 may protect DAergic neurons through alteration in iron homeostasis and defense against oxidative stress and mitochondrial dysfunction. The 2-oxoglutarate analogue 3,4-dihydroxybenzoate (DHB) effectively inhibits PHDs activities. It has been found that 3,4-DHB treatment is able to stabilize HIF-1α protein, leading to the elevation of HIF-1 target genes expression, such as ferroportin and heme oxygenase (HO)-1, in the substantia nigra. Both ferroportin and HO-1 are involved in an attenuation of iron accumulation as a result of MPTP administration [88]. The research group also reported that the iron chelator clioquinol increases HIF-1α within DAergic neurons and protects against MPTP-induced nigral cell death [88]. Moreover, DFO is shown to prevent neurotoxicity in the MPTP-mouse model of PD [89]. In addition, Ben-Shachar et al. have demonstrated that intracerebroventricular-injection of DFO protects the rat brain from DAergic neurodegeneration induced by 6-hydroxydopamine [90].
HIF-1 target genes EPO and VEGF have been shown to contribute to the protection of neurons from PD pathogenesis. EPO is neuroprotective against the DAergic neurotoxin, 6-hydroxydopamine, in both the DAergic cell line MN9D and primary DAergic neurons [91]. In an embryonic rat explant model, DAergic neurons, particularly, exhibit increased survival in response to VEGF application [92], suggesting that VEGF may promote the growth of DAergic neurons. Primary midbrain neural precursor cells from mice with conditional inactivated HIF-1 in substantia nigra show a reduced differentiation to DAergic neurons, whereas VEGF partially recovers DAergic proliferation and differentiation in these cells [86]. In addition, a mouse model of pan-neuronal overexpression of VEGF demonstrates that VEGF provides protection against MPTP toxicity in nigral cells [88]. In conclusion, many reports have revealed that HIF-1 can increase dopamine synthesis and DAergic neuron growth. All the experimental evidence supports the concept that HIF-1 may have a neuroprotective effect in PD brain.
6. HIF-1 and Huntington’s disease (HD)
Huntington’s disease (HD) results from genetically programmed degeneration of neurons, specifically in the basal ganglia at the base of the brain [93]. Mutated huntingtin protein seems responsible for the neuronal death in HD [94]. Mutant huntingtin protein leads to transcriptional repression of many genes, including those that control mitochondrial energy metabolism such as peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) [95]. The impaired mitochondrial biogenesis, induced by suppression of PGC-1α, plays an important role in HD pathogenesis. Indeed, deletion of PGC-1α leads to similar striatal degeneration and behavioral abnormalities in animal models of HD. By contrast, restored mitochondrial proliferation and biogenesis by overexpression PGC-1α prevents the striatal degeneration induced by transgenic- expressed mutant huntingtin protein [96]. In addition to its deleterious effects in neurons, huntingtin protein in glial cells also induces HD pathogenesis through mitochondria dysfunction [97]. One of the most important neuroprotective effects of astroglial cells is the uptake of glutamate, which prevents neurons from excitotoxicity induced by glutamate [98]. Huntingtin protein is able to induce mitochondrial dysfunction and suppress glutamate uptake in primary astrocytes, which reduces the neuroprotective ability of astrocytes [99]. Using the transgenic mouse model of HD brain (R6/2 mice), Lieven et al. have reported that expression of astroglial glutamate transporter-1 is lower and the glutamate uptake ability is suppressed [100]. In this way, huntingtin protein-induced mitochondrial metabolism dysfunction in the brain serves as the primary cause of HD pathogenesis.
The research on huntingtin protein-induced mitochondrial dysfunction raises the hypothesis that an alternate strategy for compensating for mitochondrial energy deficits may prevent mutant huntingtin protein-induced neuronal death. HIF-1 regulates glycolysis and hexose monophosphate shunt, which increases the production of adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide phosphate (NADPH) for cell survival. Although in glycolysis and in the hexose monophosphate shunt, cells need more glucose to produce ATP to meet their energy requirements, these two metabolism pathways can make up for the energy shortage induced by a mitochondrial biogenesis defect. Therefore, increasing HIF-1 activity may prevent neurons from mutant huntingtin protein damage.
Many strategies for HIF-1 activation have been used to prevent HD pathogenesis. The mitochondrial toxin 3-nitropropionic acid is widely used to induce mitochondrial dysfunction and HD in animal models. Three HIF-1 inducers, CoCl2, mimosine, and DFO, have been reported to significantly attenuate cytotoxicity induced by 3-nitropropionic acid in glioma cells. Another metal chelator, clioquinol, increases HIF-1α expression and reduces cell death and mutant protein accumulation in both a tissue culture model and a transgenic Huntington’s animal model [101–102]. Several plausible mechanisms that may mediate HIF-1’s protective effects have been proposed. HIF-1 activation induces the expression of glucose transporters as well as glycolytic enzymes. The enhanced glucose transport coupled with glycolytic pathways may compensate for the reduced ATP supply from compromised mitochondria. Moreover, the end product of glycolytic pathways is pyruvate and it is an antioxidant capable of scavenging radicals [103]. Therefore, HIF-1 induction may provide cytoprotection by increasing energy supply and contributing to cellular redox homeostasis. In addition, it has been shown that the protective effect of HIF-1 is not PGC-1α dependent, suggesting that HIF-1 may ameliorate the damage through its own target genes [104]. This indicates that HIF-1 may prevent neuronal death under conditions of HD and therefore increasing HIF-1 activity may be a strategy for prevention or therapy of HD.
7. Recent drug development based on HIF-1 induction in neurodegenerative diseases
HIF-1 hydroxylases include prolyl hydroxylases (PHDs) and asparaginyl hydroxylase (FIH). PHDs can induce HIF-1α degradation while FIH inhibits HIF-1 activity. Inhibiting the hydroxylases has been shown to provide protective effects in neurodegenerative diseases. The following discusses the recent findings on the hydroxylase inhibitors and related drug development progress.
The ferrous ion bound at the active sites is essential for the activity of PHDs [105]. Cobalt displaces the single free ferrous at the active site and thus deactivates the hydroxylases [11]. Therefore, cobalt is able to stabilize HIF-1α. It has been reported that cobalt treatment can increase DA release in rats [87]. Moreover, the preconditioning of rat C6 astroglia cells with the CoCl2 provides extensive cytoprotective effects against metabolic insults induced by mitochondrial toxin 3-nitropropionic acid [102].
Iron chelators have been traditionally regarded to provide neuroprotection by sequestering redox-active iron and thereby prevent hydroxyl radical formation [106]. More and more evidence demonstrates that iron chelators’ neuroprotection may result, at least partly, from inhibiting the activation of PHDs [106]. The hexadentate siderophore DFO (Fig. 1.1) possesses a high and specific affinity for iron [107]. The sequestering of ferrous ion by DFO prevents formation of a catalytically active center in the PHDs and induces HIF-1 activation [11]. DFO has been demonstrated to confer neuroprotection by inducing HIF-1 expression in a variety of animal ischemia models [19]. Intracereventricular injection of DFO protects against dopaminergic neurodegeneration induced by 6-hydroxydopamine [90] and prevents MPTP induced neurotoxicity in mouse models of PD [89]. Compromised mitochondrial function in neurons and glial cells has been observed in HD and AD. Preconditioning of C6 astroglial cells with DFO can activate HIF-1 and thereby attenuate cytotoxicity induced by mitochondrial inhibitor 3-nitropropionic acid [102].
Fig. 1.
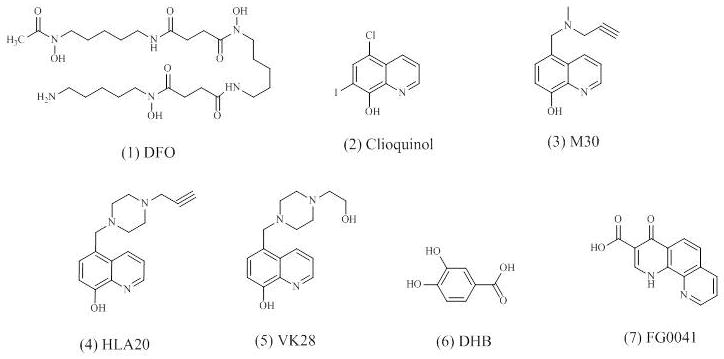
Chemical structures of HIF hydroxylase inhibitors.
Another example of iron chelators is clioquinol (5-chloro-7-iodo-8-hydroxyquinoline) (Fig. 1.2). Clioquinol is a lipophilic bioavailable metal chelator and has been investigated for the treatment of neurodegenerative diseases [106]. Clioquinol-treated transgenic HD mice (R6/2) demonstrate improved behavioral and pathologic phenotypes, including decreased huntingtin aggregate accumulation as well as enhanced motor functions and survival [101]. It is also reported that clioquinol protects against MPTP-induced nigral dopaminergic cell loss and up-regulates HIF-1α within these neurons [88], indicating that it may provide neuroprotection in PD.
Furthermore, novel compounds compromising aromatic heterocyclic related to pyridine derivatives, such as 8-hydroxyquinolines, are developed as selective PHDs inhibitors [19]. Hydroxyquinolines can exert their physiological properties through bidentate chelation of metal ions [19]. Among a series of novel multifunctional iron chelators derived from 8-hydroxyquinolines, the compound M30 (Fig. 1.3) is found to be the most potent, nontoxic, lipophilic, and BBB-permeable selective iron chelator [106]. M30 and another iron chelator HLA20 (Fig. 1.4) are both designed from the brain-permeable iron chelator VK28 (Fig. 1.5) and chemically attached to the neuroprotective N-propargyl moiety of the anti-Parkinson drug rasageline. As a result, both M30 and HLA20 inherit some of the neuroprotective properties from VK28 and rasageline [106]. M30 is able to activate the HIF-1 signaling pathway and increase the transcription of HIF-1-dependent genes, including VEGF, EPO, enolase-1, p21, and TH, in rat primary cortical cells [57]. Both M30 and HLA20 enhance HIF-1α mRNA and protein expression as well as its nuclear translocation in motor-neuron-like cell line, NSC-34 cells [50]. Furthermore, both M30 and HLA20 increase the levels of the endogenous HIF-1-dependent genes, including enolase-1, VEGF, and brain derived neurotrophic factor (BDNF) in NSC-34 cells [50]. In addition, treatment with M30 provides benefits in G93A-SOD1-mutant ALS transgenic mice, increasing their survival and delaying the onset of neurological dysfunction [50].
Since both PHDs and FIH are members of the Fe(II)- and 2-oxoglutarate-dependent oxygenase superfamily, besides cobalt and iron chelators, analogues of 2-oxoglutarate can also inhibit hydroxylation and induce HIF-1α protein [11]. Analogues of 2-oxoglutarate are more selective for PHDs and FIH than simple iron chelators [11]. Two examples of 2-oxoglutarate analogues are 3,4-DHB (Fig. 1.6) and FG0041 (Fig. 1.7). It has been reported that 3,4-DHB is significantly more selective in inhibiting FIH than PHDs [105]. It protects against MPTP-induced neurotoxicity both in vitro and in vivo [19]. In addition, 3,4-DHB also elevates the expression of HIF-dependent genes such as heme oxygenase-1 and manganese superoxide dismutase in the presence of MPTP [88]. FG0041 increases HIF-1 activity and the extracellular DA level in the striatum of the rat brain [87]. Both 3,4-DHB and FG0041 may prevent the PD pathological process by inducing HIF-1 activation.
In summary, both iron chelators and 2-oxoglutarate analogues have demonstrated neuroprotective effects and they may become potential treatments against neurodegenerative diseases. Iron chelators may also exert their neuroprotective effects through other mechanisms besides inhibiting PHDs and inducing HIF-1 signaling pathway. Therefore, understanding the exact mechanisms responsible for iron chelators’ protective effects will help the development of new therapeutic strategies in treating neurodegenerative diseases. Furthermore, since PHD inhibitors display different potencies to the three PHD subtypes [108], it may be possible to develop PHD inhibitors with different subtype selectivity which exhibit different biological properties. Another issue to consider when developing PHD inhibitors for neurodegenerative diseases is the cellular concentration of 2-oxoglutarate in the brain tissue of interest. Study has shown that 2-oxoglutarate may decrease the cellular potency of a competitive PHD inhibitor [108]. Thus, to specifically inhibit the intracellular PHDs, a competitive inhibitor with a low Ki should be developed.
Conclusions
HIF-1’s protein level and transcriptional activity are largely regulated by post-translational modifications on different amino acid residues of the α subunit. Many lines of evidence have demonstrated that HIF-1 is involved in the pathogenesis of AD, PD, ALS, and HD. Extensive experimental studies have revealed that activating HIF-1 by inhibiting the activation of PHDs can provide neuroprotection, ameliorate the outcomes, or prevent the pathogenesis in these pathological conditions. The beneficial effects of HIF-1 arise mainly from the increased expression of HIF-1 target genes, which combat oxidative stress, improve blood oxygen and glucose supply, promote glucose metabolism, regulate iron homeostasis, activate the synthesis of dopamine, and block cell death signal pathways. Increasing HIF-1 activity may be an important potential strategy for preventing the onset or ameliorating the pathogenesis of these diseases. The applications of HIF-1 inducers in neurodegenerative models have shown positive effects in many cases and are worth pursuing further.
Scheme 2.
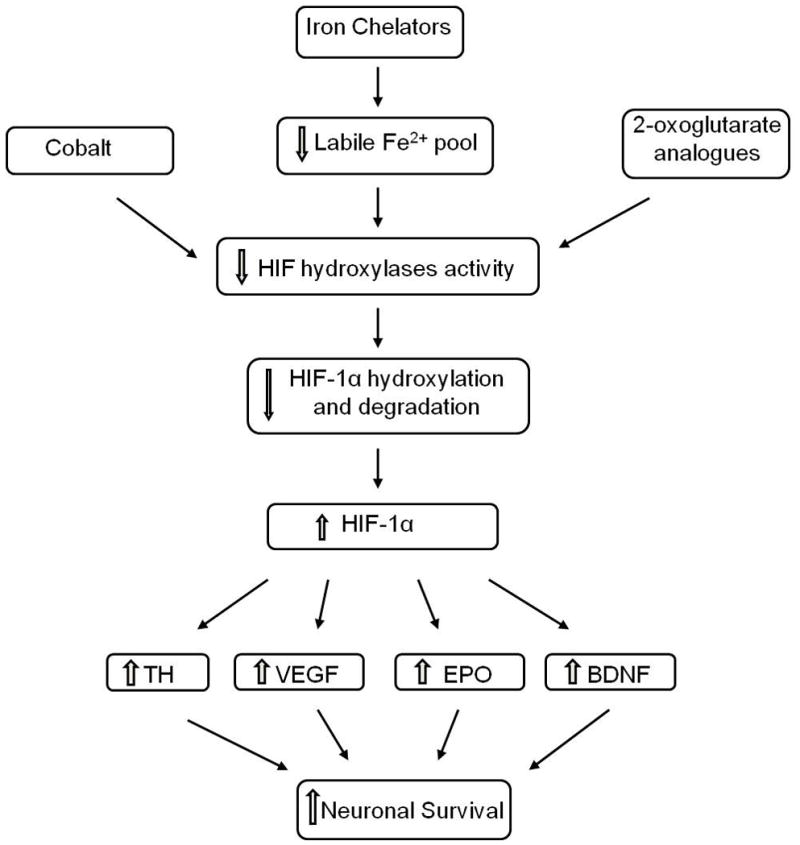
Summary of neuroprotective effects of HIF hydroxylase inhibitors associated with HIF-1 activity.
Acknowledgments
This research was supported in part by grants from NIH (R01NS058807, PO1AG17490, and AG037319) and Alzheimer Association.
Abbreviation
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- EPO
erythropoietin
- HD
Huntington’s disease
- HIF-1
hypoxia inducible factor 1
- PD
Parkinson’s disease
- VEGF
vascular endothelial growth factor
References
- 1.Shi H. Hypoxia inducible factor 1 as a therapeutic target inischemic stroke. Curr Med Chem. 2009;16(34):4593–4600. doi: 10.2174/092986709789760779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci US A. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2006;70(5):1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 4.Guo S, Miyake M, Liu KJ, Shi H. Specific inhibition of hypoxia inducible factor 1 exaggerates cell injury induced by in vitro ischemia through deteriorating cellular redox environment. J Neurochem. 2009;108(5):1309–1321. doi: 10.1111/j.1471-4159.2009.05877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogunshola OO, Antoniou X. Contribution of hypoxia to Alzheimer’s disease: is HIF-1α a mediator of neurodegeneration? Cell Mol Life Sci. 2009;66(22):3555–3563. doi: 10.1007/s00018-009-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Correia SC, Moreira PI. Hypoxia-inducible factor 1: a new hope to counteract neurodegeneration? J Neurochem. 2010;112(1):1–12. doi: 10.1111/j.1471-4159.2009.06443.x. [DOI] [PubMed] [Google Scholar]
- 7.Gironi M, Bianchi A, Russo A, Alberoni M, Ceresa L, Angelini A, Cursano C, Mariani E, Nemni R, Kullmann C, Farina E, Martinelli Boneschi F. Oxidative Imbalance in Different Neurodegenerative Diseases with Memory Impairment. Neurodegener Dis. 2011;8(3):129–37. doi: 10.1159/000319452. [DOI] [PubMed] [Google Scholar]
- 8.Benarroch EE. Brain iron homeostasis and neurodegenerative disease. Neurology. 2009;72(16):1436–1440. doi: 10.1212/WNL.0b013e3181a26b30. [DOI] [PubMed] [Google Scholar]
- 9.Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the α subunit. J Biol Chem. 1997;272(17):11205–11214. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- 10.Ruas JL, Poellinger L, Pereira T. Functional analysis of hypoxia-inducible factor-1α-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J Biol Chem. 2002;277(41):38723–38730. doi: 10.1074/jbc.M205051200. [DOI] [PubMed] [Google Scholar]
- 11.Hewitson KS, McNeill LA, Schofield CJ. Modulating the hypoxia-inducible factor signaling pathway: applications from cardiovascular disease to cancer. Curr Pharm Des. 2004;10(8):821–833. doi: 10.2174/1381612043452884. [DOI] [PubMed] [Google Scholar]
- 12.Srinivas V, Zhang LP, Zhu XH, Caro J. Characterization of an oxygen/redox-dependent degradation domain of hypoxia-inducible factor α (HIF-α) proteins. Biochem Biophys Res Commun. 1999;260(2):557–561. doi: 10.1006/bbrc.1999.0878. [DOI] [PubMed] [Google Scholar]
- 13.Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. HIF α targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292(5516):464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 14.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 15.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 16.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2(7):423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 17.Salceda S, Caro J. Hypoxia-inducible factor-1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272(36):22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 18.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1α-pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296(5574):1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 19.Weinreb O, Amit T, Mandel S, Kupershmidt L, Youdim MB. Neuroprotective multifunctional iron chelators: from redox-sensitive process to novel therapeutic opportunities. Antioxid Redox Signal. 2010;13(6):919–949. doi: 10.1089/ars.2009.2929. [DOI] [PubMed] [Google Scholar]
- 20.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 21.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell. 2002;111(5):709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 22.Brahimi-Horn C, Mazure N, Pouyssegur J. Signalling via the hypoxia-inducible factor-1α requires multiple posttranslational modifications. Cell Signal. 2005;17(1):1–9. doi: 10.1016/j.cellsig.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Gradin K, Takasaki C, Fujii-Kuriyama Y, Sogawa K. The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J Biol Chem. 2002;277(26):23508–23514. doi: 10.1074/jbc.M201307200. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Tomida A, Tsuruo T. Dephosphorylated hypoxia-inducible factor 1α as a mediator of p53-dependent apoptosis during hypoxia. Oncogene. 2001;20(41):5779–5788. doi: 10.1038/sj.onc.1204742. [DOI] [PubMed] [Google Scholar]
- 25.Lancaster DE, McNeill LA, McDonough MA, Aplin RT, Hewitson KS, Pugh CW, Ratcliffe PJ, Schofield CJ. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem J. 2004;383(Pt. 3):429–437. doi: 10.1042/BJ20040735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimova EY, Michiels C, Kietzmann T. Kinases as upstream regulators of the HIF system: their emerging potential as anti-cancer drug targets. Curr Pharm Des. 2009;15(33):3867–3877. doi: 10.2174/138161209789649358. [DOI] [PubMed] [Google Scholar]
- 27.Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J Biol Chem. 1996;271(50):32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 28.Liu Q, Berchner-Pfannschmidt U, Moller U, Brecht M, Wotzlaw C, Acker H, Jungermann K, Kietzmann T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc Natl Acad Sci US A. 2004;101(12):4302–4307. doi: 10.1073/pnas.0400265101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278(1):311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 30.Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1α protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549(1–3):105–109. doi: 10.1016/s0014-5793(03)00807-x. [DOI] [PubMed] [Google Scholar]
- 31.Cho H, Ahn DR, Park H, Yang EG. Modulation of p300 binding by posttranslational modifications of the C-terminal activation domain of hypoxia-inducible factor-1α. FEBS Lett. 2007;581(8):1542–1548. doi: 10.1016/j.febslet.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 32.Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, Holsboer F, Arzt E. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1α during hypoxia. Cell. 2007;131(2):309–323. doi: 10.1016/j.cell.2007.07.044. [DOI] [PubMed] [Google Scholar]
- 33.Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of hypoxia-inducible factor-1αduring hypoxia. Cell. 2007;131(3):584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berta MA, Mazure N, Hattab M, Pouyssegur J, Brahimi-Horn MC. SUMOylation of hypoxia-inducible factor-1α reduces its transcriptional activity. Biochem Biophys Res Commun. 2007;360(3):646–652. doi: 10.1016/j.bbrc.2007.06.103. [DOI] [PubMed] [Google Scholar]
- 35.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 36.Vanacore N, Cocco P, Fadda D, Dosemeci M. Job strain, hypoxia and risk of amyotrophic lateral sclerosis: Results from a death certificate study. Amyotroph Lateral Scler. 2010;11(5):430–434. doi: 10.3109/17482961003605796. [DOI] [PubMed] [Google Scholar]
- 37.Skene JP, Cleveland DW. Hypoxia and Lou Gehrig. Nat Genet. 2001;28(2):107–108. doi: 10.1038/88805. [DOI] [PubMed] [Google Scholar]
- 38.Tankersley CG, Haenggeli C, Rothstein JD. Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. J Appl Physiol. 2007;102(3):926–932. doi: 10.1152/japplphysiol.00193.2006. [DOI] [PubMed] [Google Scholar]
- 39.Bogaert E, Van Damme P, Van Den Bosch L, Robberecht W. Vascular endothelial growth factor in amyotrophic lateral sclerosis and other neurodegenerative diseases. Muscle Nerve. 2006;34(4):391–405. doi: 10.1002/mus.20609. [DOI] [PubMed] [Google Scholar]
- 40.Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, Tonnel AB, Lassalle P, Just N. Paradoxical response of VEGF expression to hypoxia in CSF of patients with ALS. J Neurol Neurosurg Psychiatry. 2006;77(2):255–257. doi: 10.1136/jnnp.2005.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devos D, Moreau C, Lassalle P, Perez T, De Seze J, Brunaud-Danel V, Destee A, Tonnel AB. Just N Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 2004;62(11):2127–2129. doi: 10.1212/01.wnl.0000129913.44351.a3. [DOI] [PubMed] [Google Scholar]
- 42.Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D, Carmeliet P. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28(2):131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007;27(2):304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci US A. 2000;97(18):10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92(6):735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 46.Janik P, Kwiecinski H, Sokolowska B, Niebroj-Dobosz I. Erythropoietin concentration inserum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J Neural Transm. 2010;117(3):343–347. doi: 10.1007/s00702-009-0354-2. [DOI] [PubMed] [Google Scholar]
- 47.Chung YH, Joo KM, Kim YS, Lee KH, Lee WB, Cha CI. Enhanced expression of erythropoietin in thecentral nervous system of SOD1G93A transgenic mice. Brain Res. 2004;1016(2):272–280. doi: 10.1016/j.brainres.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 48.Koh SH, Kim Y, Kim HY, Cho GW, Kim KS, Kim SH. Recombinant human erythropoietin suppresses symptom onset and progression of G93A-SOD1 mouse model of ALS by preventing motor neuron death and inflammation. Eur J Neurosci. 2007;25(7):1923–1930. doi: 10.1111/j.1460-9568.2007.05471.x. [DOI] [PubMed] [Google Scholar]
- 49.Grunfeld JF, Barhum Y, Blondheim N, Rabey JM, Melamed E, Offen D. Erythropoietin delays disease onset in an amyotrophic lateral sclerosis model. Exp Neurol. 2007;204(1):260–263. doi: 10.1016/j.expneurol.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB. Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2009;23(11):3766–3779. doi: 10.1096/fj.09-130047. [DOI] [PubMed] [Google Scholar]
- 51.Bonda DJ, Lee HG, Camins A, Pallas M, Casadesus G, Smith MA, Zhu X. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 2011;10(3):275–279. doi: 10.1016/S1474-4422(11)70013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 54.Palmer AM. Neuroprotective therapeutics for Alzheimer’s disease: progress and prospects. Trends Pharmacol Sci. 2011;32(3):141–147. doi: 10.1016/j.tips.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 55.Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J Alzheimers Dis. 2001;3(1):75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- 56.Moreira PI, Honda K, Zhu X, Nunomura A, Casadesus G, Smith MA, Perry G. Brain and brawn: parallels in oxidative strength. Neurology. 2006;66(2 Suppl 1):S97–101. doi: 10.1212/01.wnl.0000192307.15103.83. [DOI] [PubMed] [Google Scholar]
- 57.Avramovich-Tirosh Y, Bar-Am O, Amit T, Youdim MB, Weinreb O. Up-regulation of hypoxia-inducible factor (HIF) -1α and HIF-target genes in cortical neurons by the novel multifunctional iron chelator anti-Alzheimer drug, M30. Curr Alzheimer Res. 2010;7(4):300–306. doi: 10.2174/156720510791162403. [DOI] [PubMed] [Google Scholar]
- 58.Soucek T, Cumming R, Dargusch R, Maher P, Schubert D. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid β peptide. Neuron. 2003;39(1):43–56. doi: 10.1016/s0896-6273(03)00367-2. [DOI] [PubMed] [Google Scholar]
- 59.Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337(8753):1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 60.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009;118(1):103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoyer S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv Exp Med Biol. 2004;541:135–152. doi: 10.1007/978-1-4419-8969-7_8. [DOI] [PubMed] [Google Scholar]
- 62.Taupin PA. dual activity of ROS and oxidative stress on adult neurogenesis and Alzheimer’s disease. Cent Nerv Syst Agents Med Chem. 2010;10(1):16–21. doi: 10.2174/187152410790780172. [DOI] [PubMed] [Google Scholar]
- 63.Desmond DW, Moroney JT, Sano M, Stern Y. Incidence of dementia after ischemic stroke: results of a longitudinal study. Stroke. 2002;33(9):2254–2260. doi: 10.1161/01.str.0000028235.91778.95. [DOI] [PubMed] [Google Scholar]
- 64.Fleisher AS, Podraza KM, Bangen KJ, Taylor C, Sherzai A, Sidhar K, Liu TT, Dale AM, Buxton RB. Cerebral perfusion and oxygenation differences in Alzheimer’s disease risk. Neurobiol Aging. 2009;30(11):1737–1748. doi: 10.1016/j.neurobiolaging.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Egashira N, Iwasaki K, Ishibashi M, Hatip-Al-Khatib I, Wolozin B, Mishima K, Irie K, Fujiwara M. Hypoxia enhances β-amyloid-induced apoptosis in rat cultured hippocampal neurons. Jpn J Pharmacol. 2002;90(4):321–327. doi: 10.1254/jjp.90.321. [DOI] [PubMed] [Google Scholar]
- 66.Vu K, Weksler B, Romero I, Couraud PO, Gelli A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by Cryptococcus neoformans. Eukaryot Cell. 2009;8(11):1803–1807. doi: 10.1128/EC.00240-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582(2):359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maiese K, Chong ZZ, Li F, Shang YC. Erythropoietin: elucidating new cellular targets that broaden therapeutic strategies. Prog Neurobiol. 2008;85(2):194–213. doi: 10.1016/j.pneurobio.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26(9):943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- 70.Chong ZZ, Li F, Maiese K. Erythropoietin requires NF-κB and its nuclear translocation to prevent early and late apoptotic neuronal injury during β-amyloid toxicity. Curr Neurovasc Res. 2005;2(5):387–399. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mala H, Alsina CG, Madsen KS, Sibbesen EC, Stick H, Mogensen J. Erythropoietin improves place learning in an 8-arm radial maze in fimbria-fornix transected rats. Neural Plast. 2005;12(4):329–340. doi: 10.1155/NP.2005.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 73.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 74.Zhou J, Dehne N, Brune B. Nitric oxide causes macrophage migration via the HIF-1-stimulated small GTPases Cdc42 and Rac1. Free Radic Biol Med. 2009;47(6):741–749. doi: 10.1016/j.freeradbiomed.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 75.Sullivan AM, Toulouse A. Neurotrophic factors for the treatment of Parkinson’s disease. Cytokine Growth Factor Rev. 2011 doi: 10.1016/j.cytogfr.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 76.Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, Obeso JA, Rascol O, Schapira A, Voon V, Weiner DM, Tison F, Bezard E. Priorities in Parkinson’s disease research. Nat Rev Drug Discov. 2011;10(5):377–393. doi: 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- 77.Fujita K, Nakabeppu Y, Noda M. Therapeutic effects of hydrogen in animal models of Parkinson’s disease. Parkinsons Dis. 2011;2011:307875. doi: 10.4061/2011/307875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cohen G, Farooqui R, Kesler N. Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proc Natl Acad Sci US A. 1997;94(10):4890–4894. doi: 10.1073/pnas.94.10.4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haavik J. L-DOPA is a substrate for tyrosine hydroxylase. J Neurochem. 1997;69(4):1720–1728. doi: 10.1046/j.1471-4159.1997.69041720.x. [DOI] [PubMed] [Google Scholar]
- 80.Agani FH, Pichiule P, Chavez JC, LaManna JC. The role of mitochondria in the regulation of hypoxia-inducible factor-1 expression during hypoxia. J Biol Chem. 2000;275(46):35863–35867. doi: 10.1074/jbc.M005643200. [DOI] [PubMed] [Google Scholar]
- 81.Haavik J, Toska K. Tyrosine hydroxylase and Parkinson’s disease. Mol Neurobiol. 1998;16(3):285–309. doi: 10.1007/BF02741387. [DOI] [PubMed] [Google Scholar]
- 82.Millhorn DE, Raymond R, Conforti L, Zhu W, Beitner-Johnson D, Filisko T, Genter MB, Kobayashi S, Peng M. Regulation of gene expression for tyrosine hydroxylase in oxygen sensitive cells by hypoxia. Kidney Int. 1997;51(2):527–535. doi: 10.1038/ki.1997.73. [DOI] [PubMed] [Google Scholar]
- 83.Schnell PO, Ignacak ML, Bauer AL, Striet JB, Paulding WR, Czyzyk-Krzeska MF. Regulation of tyrosine hydroxylase promoter activity by the von Hippel-Lindau tumor suppressor protein and hypoxia-inducible transcription factors. J Neurochem. 2003;85(2):483–491. doi: 10.1046/j.1471-4159.2003.01696.x. [DOI] [PubMed] [Google Scholar]
- 84.Czyzyk-Krzeska MF, Furnari BA, Lawson EE, Millhorn DE. Hypoxia increases rate of transcription and stability of tyrosine hydroxylase mRNA in pheochromocytoma (PC12) cells. J Biol Chem. 1994;269(1):760–764. [PubMed] [Google Scholar]
- 85.Nguyen MV, Pouvreau S, El Hajjaji FZ, Denavit-Saubie M, Pequignot JM. Desferrioxamine enhances hypoxic ventilatory response and induces tyrosine hydroxylase gene expression in the rat brainstem in vivo. J Neurosci Res. 2007;85(5):1119–1125. doi: 10.1002/jnr.21202. [DOI] [PubMed] [Google Scholar]
- 86.Milosevic J, Maisel M, Wegner F, Leuchtenberger J, Wenger RH, Gerlach M, Storch A, Schwarz J. Lack of hypoxia-inducible factor-1αimpairs midbrain neural precursor cells involving vascular endothelial growth factor signaling. J Neurosci. 2007;27(2):412–421. doi: 10.1523/JNEUROSCI.2482-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Witten L, Sager T, Thirstrup K, Johansen JL, Larsen DB, Montezinho LP, Mork A. HIF prolyl hydroxylase inhibition augments dopamine release in the rat brain in vivo. J Neurosci Res. 2009;87(7):1686–1694. doi: 10.1002/jnr.21988. [DOI] [PubMed] [Google Scholar]
- 88.Lee DW, Rajagopalan S, Siddiq A, Gwiazda R, Yang L, Beal MF, Ratan RR, Andersen JK. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. J Biol Chem. 2009;284(42):29065–29076. doi: 10.1074/jbc.M109.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lan J, Jiang DH. Desferrioxamine and vitamin E protect against iron and MPTP-induced neurodegeneration in mice. J Neural Transm. 1997;104(4–5):469–481. doi: 10.1007/BF01277665. [DOI] [PubMed] [Google Scholar]
- 90.Ben-Shachar D, Eshel G, Finberg JP, Youdim MB. The iron chelator desferrioxamine (Desferal) retards 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons. J Neurochem. 1991;56(4):1441–1444. doi: 10.1111/j.1471-4159.1991.tb11444.x. [DOI] [PubMed] [Google Scholar]
- 91.Signore AP, Weng Z, Hastings T, VanLaar AD, Liang Q, Lee YJ, Chen J. Erythropoietin protects against 6-hydroxydopamine-induced dopaminergic cell death. J Neurochem. 2006;96(2):428–443. doi: 10.1111/j.1471-4159.2005.03587.x. [DOI] [PubMed] [Google Scholar]
- 92.Silverman WF, Krum JM, Mani N, Rosenstein JM. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience. 1999;90(4):1529–1541. doi: 10.1016/s0306-4522(98)00540-5. [DOI] [PubMed] [Google Scholar]
- 93.Cummins A, Eggert J, Pruitt R, Collins JS. Huntington disease: Implications for practice. Nurse Pract. 2011;36(2):41–47. doi: 10.1097/01.NPR.0000392796.01760.e2. [DOI] [PubMed] [Google Scholar]
- 94.Crook ZR, Housman D. Huntington’s disease: can mice lead the way to treatment? Neuron. 2011;69(3):423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Quintanilla RA, Jin YN, Fuenzalida K, Bronfman M, Johnson GV. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtin-expressing cells: possible role of peroxisome proliferator-activated receptor-γ(PPARγ) in the pathogenesis of Huntington disease. J Biol Chem. 2008;283(37):25628–25637. doi: 10.1074/jbc.M804291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jin YN, Johnson GV. The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. J Bioenerg Biomembr. 2010;42(3):199–205. doi: 10.1007/s10863-010-9286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsiao HY, Chern Y. Targeting glial cells to elucidate the pathogenesis of Huntington’s disease. Mol Neurobiol. 2010;41(2–3):248–255. doi: 10.1007/s12035-009-8097-5. [DOI] [PubMed] [Google Scholar]
- 98.Estrada Sanchez AM, Mejia-Toiber J, Massieu L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch Med Res. 2008;39(3):265–276. doi: 10.1016/j.arcmed.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 99.Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171(6):1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8(5):807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- 101.Nguyen T, Hamby A, Massa SM. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc Natl Acad Sci US A. 2005;102(33):11840–11845. doi: 10.1073/pnas.0502177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang YT, Ju TC, Yang DI. Induction of hypoxia inducible factor-1 attenuates metabolic insults induced by 3-nitropropionic acid in rat C6 glioma cells. J Neurochem. 2005;93(3):513–525. doi: 10.1111/j.1471-4159.2005.03032.x. [DOI] [PubMed] [Google Scholar]
- 103.Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997;11(5):388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- 104.Niatsetskaya Z, Basso M, Speer RE, McConoughey SJ, Coppola G, Ma TC, Ratan RR. HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington’s disease and Alzheimer’s disease. Antioxid Redox Signal. 2010;12(4):435–443. doi: 10.1089/ars.2009.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hewitson KS, Schofield CJ. The HIF pathway as a therapeutic target. Drug Discov Today. 2004;9(16):704–711. doi: 10.1016/S1359-6446(04)03202-7. [DOI] [PubMed] [Google Scholar]
- 106.Weinreb O, Mandel S, Bar-Am O, Amit T. Iron-chelating backbone coupled with monoamine oxidase inhibitory moiety as novel pluripotential therapeutic agents for Alzheimer’s disease: a tribute to Moussa Youdim. J Neural Transm. 2011;118(3):479–492. doi: 10.1007/s00702-011-0597-6. [DOI] [PubMed] [Google Scholar]
- 107.Whitnall M, Richardson DR. Iron: a new target for pharmacological intervention in neurodegenerative diseases. Semin Pediatr Neurol. 2006;13(3):186–197. doi: 10.1016/j.spen.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 108.Thirstrup K, Christensen S, Moller HA, Ritzen A, Bergstrom AL, Sager TN, Jensen HS. Endogenous 2-oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors. Pharmacol Res. 2011;64(3):268–273. doi: 10.1016/j.phrs.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 109.Masson N, Ratcliffe PJ. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O2 levels. J Cell Sci. 2003;116(Pt 15):3041–3049. doi: 10.1242/jcs.00655. [DOI] [PubMed] [Google Scholar]
- 110.Groulx I, Lee S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol. 2002;22(15):5319–5336. doi: 10.1128/MCB.22.15.5319-5336.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277(29):26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 112.Bae SH, Jeong JW, Park JA, Kim SH, Bae MK, Choi SJ, Kim KW. Sumoylation increases HIF-1α stability and its transcriptional activity. Biochem Biophys Res Commun. 2004;324(1):394–400. doi: 10.1016/j.bbrc.2004.09.068. [DOI] [PubMed] [Google Scholar]