

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have poor efficacy in head and neck squamous cell carcinoma (HNSCC). Because the insulin-like growth factor-1 receptor (IGF1R) generates potent prosurvival signals and has been implicated in therapeutic resistance, its ability to induce resistance to EGFR-TKIs was studied in vitro. Five HNSCC cell lines demonstrated reduced sensitivity to the EGFR-TKI gefitinib when the IGF1R was activated. In SCC-25 and Cal27 cells, gefitinib inhibited basal and EGF-stimulated EGFR, Erk and Akt phosphorylation and reduced cell number. This correlated with initiation of apoptosis based on a 4-fold increase poly-(ADP-ribose) polymerase (PARP) cleavage and a 2.5-fold increase in Annexin V positivity. The apoptotic response and reduction in cell number were blocked by IGF1R activation, which resulted in phosphorylation of both Erk and Akt. In both cell lines, IGF1R-induced Erk but not Akt activation was eliminated by gefitinib. IGF1R-induced gefitinib resistance was unaffected by MEK inhibition with U0126, but was partially impaired by inhibition of phosphatidylinositol 3′-kinase (PI3K) with LY294002. The IGF1R-TKI PQ401 inhibited growth of SCC-25 and Cal27 cells alone and also acted synergistically with gefitinib. Thus the IGF1R can make HNSCC cells resistant to EGFR-TKI treatment via a prosurvival mechanism. Of 8 studied, all HNSCC tumor samples expressed the IGF1R and 5 demonstrated detectable IGF1R phosphorylation, suggesting that this receptor may be relevant in vivo, and thus combined EGFR/IGF1R inhibition may be necessary in some patients for effective targeted molecular therapy.
Keywords: targeted molecular therapy, head and neck squamous cell carcinoma, insulin-like growth factor-1 receptor, epidermal growth factor receptor, tyrosine kinase inhibitor, therapeutic resistance
INTRODUCTION
In head and neck squamous cell carcinoma (HNSCC), the epidermal growth factor receptor (EGFR) has emerged as a potential therapeutic target; more than 90% of HNSCCs overexpress the EGFR and elevated EGFR expression predicts decreased survival. Several targeted anti-EGFR agents have been developed, but their efficacy in HNSCC has been limited, showing both intrinsic (low initial response rate) and acquired (short duration of benefit) resistance. Phase II clinical trials with single agent EGFR-tyrosine kinase inhibitors (TKIs) showed response rates of only 5–15% (1, 2). Treatment failures do not result from lack of EGFR expression or inability to block receptor activation in vivo, and EGFR expression level does not predict effectiveness. To date, molecular markers that predict response or resistance in HNSCC have not been identified (3).
Insulin-like growth factor (IGF)-1 and IGF-2 are ubiquitously-produced protein hormones that interact with the IGF-1 receptor (IGF1R) to regulate growth, differentiation, and survival. The IGF1R activates both Ras/Erk-and phosphatidylinositol 3′-kinase (PI3K)/Akt-related signal transduction pathways, which act to promote proliferation and prevent apoptosis (4). The IGFs are regulated extracellularly by six IGF binding proteins (IGFBPs) that buffer receptor activation. Thus, dysregulation of IGF production, IGF1R expression or IGFBP secretion can alter growth regulation.
There is substantial evidence that the IGF1R plays a central role in cancer development and tumor cell growth. The IGF1R is expressed by nearly all tumor types studied, and is activated in an autocrine or paracrine fashion when tumor or stromal cells secrete IGFs (5, 6). In certain settings, the IGF1R is required for transformation by other agents (including the EGFR) (7), and the IGF1R encourages and supports properties of the transformed phenotype. In addition, IGF-1 is also involved in other aspects of cancer progression, such as angiogenesis and inflammation (8). Recent studies have demonstrated that elevated serum IGF-1 levels are associated with increased risk of a variety of epithelial cancers (9–12), and that reduced IGF-1 levels may be protective (13). Thus, it has been proposed that reduction of IGF signaling in some cancer types may have therapeutic benefit (4, 14). Augmenting this concept is the recent demonstration that the IGF1R can promote therapeutic resistance to multiple treatment approaches including radiation, cytotoxic chemotherapy, and molecular targeted therapy (15–18). The significance of these findings in HNSCC is, as yet, unknown.
In the present study, we demonstrate that activation of the IGF1R in HNSCC cells can overcome growth inhibition caused by EGFR-TKIs via a primarily anti-apoptotic mechanism. This validates the concept that, in the context of EGFR blockade, an alternate growth factor can continue to sustain tumor cell growth, and suggests that IGF1R signaling may be a mechanism of resistance to targeted anti-EGFR therapy in vivo. Thus, coinhibition of the EGFR and the IGF1R may lead to increased clinical response rates.
MATERIALS AND METHODS
Reagents
Recombinant human IGF-1 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA), des[1–3]IGF-1 from GroPep (Adelaide, Australia), EGF from Sigma (St. Louis, MO), and fetal bovine serum (FBS) from Invitrogen (Carlsbad, CA). U0126, PD158780 and LY294002 were obtained from EMD Biosciences (San Diego, CA), gefitinib and erlotinib from LC Laboratories (Woburn, MA), and PQ401 from Tocris Bioscience (Ellisville, MO). Anti-IGF1Rα was acquired from Santa Cruz Biotechnology, anti-p-Erk from Sigma, anti-p-Tyr and anti-PARP from BD Biosciences (San Jose, CA), anti-Akt, anti-p-Akt (S473), anti-Erk, anti-p-IGF1R, and anti-p-EGFR from Cell Signaling Technology (Beverly, MA).
Tissue Culture & Human Tissue Specimens
HNSCC cell lines included SCC-25, SCC-9, Cal27, and FaDu cells obtained from ATCC (Manassas, VA), and SCC-61 and UNC-7 cells kindly provided by Dr. Wendell Yarbrough (Vanderbilt University, Nashville, TN). These were selected to evaluate a variety of anatomic sites in the upper aerodigestive tract and because they exhibit a wide range of IGF1R expression. None of these cell lines had detectable basal IGF1R activation. They were grown in D-MEM/F12 supplemented with 400 ng/mL hydrocortisone and 5% FBS at 37°C and 5% CO2. In vitro, cells were histopathologically consistent with HNSCC on standard hematoxylin and eosin staining, and were positive for cytokeratin. RIG cells (Rat1 cells overexpressing the IGF1R) were kindly provided by Dr. Michael Weber (University of Virginia, Charlottesville, VA). They were grown in D-MEM and 5% FBS at 37°C and 5% CO2. Monolayers were starved in no or very low (0.5%) serum for 24 h before assays were performed. All cell lines were passaged for fewer than 6 months after resuscitation. Other than testing performed by the providers and the immunohistopathology described above, no additional authentication of these cell lines was performed by the authors. All cell lines were free of mycobacterial infection and exhibited behaviors similar to those previously published.
Human HNSCC tissue specimens were obtained with approval of the UVA Institutional Review Board. Small portions of surgically extirpated tumors were collected immediately after resection and snap frozen in liquid nitrogen. The specimens were stored at −80°C until protein extraction was performed on all specimens simultaneously. Standard clinical histopathology on the surgical specimens confirmed all tumors as squamous cell carcinoma.
Immunoblot
Cell monolayers were grown to 70% confluence and serum starved for 24 h, then washed with PBS and incubated with inhibitor or vehicle for 2 h followed by treatment with growth factor for 5–10 min. Stimulated monolayers were washed with ice cold PBS containing 2 mM sodium orthovanadate, collected, and resuspended in lysis buffer (50 mM HEPES (pH 8.0) containing 10 mM sodium pyrophosphate, 100 mM sodium fluoride, 4 mM EDTA, 1% Triton X-100, 1 mM phenylmethanesulphonylfluoride (PMSF), 2 mM sodium orthovanadate, 100 μM benzamidine, 1 μg/mL aprotinin, 2 μM pepstatin, and 25μM leupeptin), vortexed, and incubated at 4°C for 30 min. Insoluble material was removed by centrifugation at 13,000 g for 15 min at 4°C and sample buffer containing 0.1 M DTT was added. Proteins were resolved on an 8% SDS-polyacrylamide gel then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA). The PVDF was quenched, incubated with primary antibody, washed and incubated with secondary antibody according to the manufacturer’s instructions. Proteins were visualized by reaction with Immobilon Western Chemiluminescent Horseradish Peroxidase Substrate (Millipore).
Flow Cytometry
Monolayers were grown to 70% confluence and then incubated in 0.5% serum for 24 h. Monolayers were washed with PBS then incubated in medium containing inhibitor or vehicle for 2 h followed by treatment with growth factor for 48 h. The medium from each well was collected; cell monolayers were trypsinized, resuspended in the corresponding medium, centrifuged at 1,000 g for 7 min at 4°C, washed once with cold PBS, and resuspended in Annexin V-FITC and propidium iodide according to the manufacturer’s recommendations (Millipore). Flow cytometry was performed in the UVA Flow Cytometry Core Facility within 24 h of completion of the stimulation.
CellTiter 96® Aqueous Cell Proliferation Assay
Cells were plated at 5,000 cells/well in a 96-well plate, grown for 24 h and serum-starved for 24 h. Cells were washed with PBS, incubated for 2 h with inhibitor, and stimulated for 48 h with growth factor. MTS and PMS were added to each well according to the manufacturer’s protocol (Promega, Madison, WI). Cells were incubated for 4 h at 37°C and the absorbance at 490 nm was recorded, correcting for background absorbance. Control studies were performed to demonstrate the linearity of this assay at the cell densities used.
alamarBlue® Assay
Cells were prepared and treated as for the MTS assay above. alamarBlue (Invitrogen) was added to each well according to the manufacturer’s protocol. Cells were incubated for 3–4 h at 37°C and the fluorescence at 540 nm was recorded.
Drug Combination Analysis
Growth inhibition data resulting from coinhibition of both the EGFR and the IGF1R were assessed by median-effect analysis as described by Chou and Talalay (19) using CalcuSyn version 2.0 (Biosoft, Cambridge, UK). The combination index (CI) was calculated and an isobologram was plotted for multiple dose combinations. CI values of <1, 1, and >1 indicated synergism, additivity, and antagonism, respectively.
RESULTS
IGF1R-Induced Resistance to EGFR-TKIs
PD158780, gefitinib and erlotinib are well-characterized EGFR-TKIs; treatment of HNSCC cell lines with these compounds caused growth inhibition. Activation of the IGF1R was tested for its ability to alter HNSCC sensitivity to EGFR-TKIs (Fig. 1). IGFBP-2 and -3 are secreted at high levels by some HNSCC cells (20), and can interfere with activation of the IGF1R by exogenously added free IGF-1. Because of its dramatically reduced affinity for the IGFBPs, des[1–3]IGF-1 was used to activate the IGF1R.
Figure 1. Inhibition of HNSCC cell growth by EGFR-TKIs.

(A) Chemical structures of inhibitors. (B) After 24 h of serum starvation, SCC-25 cells were incubated for 48 h and MTS assay was performed. Graph shows average fold change compared to control (unstimulated, uninhibited) cells ± SEM for 4 independent quadruplicate experiments. *, p < 0.01 vs. unstimulated; +, p < 0.01 vs. uninhibited (Student’s t-test). Under the same conditions, cells were harvested 5 min after additions and whole cell lysates were immunoblotted. (C) SCC-25 cells were incubated for 48 h with an EGFR-TKI ± 10 nM des[1–3]IGF-1 and alamarBlue assay was performed. Graph shows average percent of control (unstimulated, uninhibited) cells ± SEM for 3 independent quadruplicate experiments. (D) Four additional SCCHN cell lines were treated with gefitinib ± 10 nM des[1–3]IGF-1 and assessed as in (C).
The original observation of IGF1R-induced resistance to EGFR inhibition was made in SCC-25 cells treated with PD158780 (Fig. 1A). PD158780 reduced cell number by 66% at 48 h. Addition of exogenous EGF caused a 40% increase in cell number; this effect was abolished by PD158780. Activation of the IGF1R with des[1–3]IGF-1 caused a similar increase in cell number which was only minimally affected by PD158780. Stimulation with FBS resulted in an 86% increase in cell number; this was partially blocked by PD158780 (Fig. 1A). These results demonstrate that activation of the IGF1R in SCC-25 cells confers resistance to the growth inhibitory effects of EGFR inhibition. The protective effect of IGF1R activation was measurable across a range of doses for multiple EGFR-TKIs, including PD158780, gefitinib, and erlotinib (Fig. 1B). Since it is most likely to have clinical applicability for HNSCC, gefitinib was tested in 4 additional HNSCC cell lines (Fig. 1C); activation of the IGF1R reduced gefitinib sensitivity in all 4 cell lines.
Erk and Akt Activation by the IGF1R
The effect of gefitinib and IGF1R activation on Erk and Akt phosphorylation was assessed in SCC-25 and Cal27 cells by immunoblot (Fig. 2). A small amount of Erk and Akt phosphorylation were present in unstimulated cells. Whereas EGF caused a large increase in p-Erk with little change in p-Akt, treatment with des[1–3]IGF-1 stimulated a modest increase in p-Erk and a large increase in p-Akt. Addition of gefitinib eliminated baseline p-Erk and p-Akt, confirming that their constitutive phosphorylation depends primarily on EGFR activity. Treatment with gefitinib eliminated EGFR- and des[1–3]IGF-1-induced Erk activation, suggesting that phosphorylation of Erk downstream of the IGF1R is EGFR dependent; this has been reported in other HNSCC cell lines (20, 21). Notably, des[1–3]IGF-1-induced Akt phosphorylation was unaffected by gefitinib in both cell lines, indicating that the IGF1R is capable of providing a persistent prosurvival signal in the presence of an EGFR-TKI.
Figure 2. Effect of gefitinib on Erk and Akt phosphorylation.

SCC-25 and Cal27 cells were incubated with gefitinib for 2 h then stimulated with des[1–3]IGF-1 or EGF for 5 min. Immunoblots were performed on whole cell lysates.
IGF1R Inhibition of PD158780-Induced Apoptosis
Cleavage of poly-(ADP-ribose) polymerase (PARP), a DNA repair enzyme, occurs in response to caspase-3 activation, serving as an early marker of apoptosis. Figure 3A shows an immunoblot analysis of SCC-25 cell lysates using an anti-PARP antibody that detects the initial PARP cleavage product (c-PARP). Treatment with gefitinib caused a dose-dependent increase in c-PARP that was completely blocked by IGF1R activation. Addition of exogenous EGF did not counteract gefitinib-induced PARP cleavage (Fig. 3B). The densitometric analysis in Figure 3B confirms that IGF1R activation in SCC-25 cells inhibits gefitinib-induced apoptosis. These data are in keeping with the understanding that the IGF1R can act as a potent prosurvival agent. A similar response to gefitinib and IGF1R activation was also noted in 3 other HNSCC cell lines (Fig. 3C).
Figure 3. Effect of gefitinib on apoptosis.

After 2 h of treatment with vehicle or gefitinib, cells were incubated for 6 h (A,B,C) or 48 h (D) with growth factors and harvested. (A,B,C) Cells were lysed and immunoblotted for PARP. In (B), PARP cleavage was quantified by densitometry (c-PARP density/total PARP density) and is shown as average percent of maximal (gefitinib alone) ± SEM for 3 independent experiments. (D) Cells were treated with Annexin V-FITC and PI and assessed by FACS. The average percentage of Annexin V-positive cells ± SEM is shown for 3 similar experiments. Early apoptosis = Annexin V-positive/PI-negative; late apoptosis/necrosis = Annexin V-positive/PI-positive; No Tx = no treatment; Gef = 5 μM gefitinib; IGF = 10 nM des[1–3]IGF-1; PQ401 = 5 μM PQ401. *, p < 0.05 vs. uninhibited (Student’s t-test).
Florescence-activated cell sorting (FACS) was used in conjunction with Annexin V-FITC and propidium iodide (PI) staining to identify cells in early and late apoptosis (Fig. 3D). As shown in the quantitative analysis in Figure 3D, the basal rate of early apoptosis plus late apoptosis (necrosis) was 11% at 48 h. Addition of gefitinib increased total apoptosis to 28%. While activation of the IGF1R had little effect on the basal apoptotic rate (10%), it dramatically inhibited the gefitinib-induced apoptosis rate to 13%. The effect of exogenous des[1–3]IGF-1 was blocked by addition of the IGF1R-TKI PQ401 (22, 23).
Prosurvival Signaling from the IGF1R
The MEK inhibitor U0126 and the PI3K inhibitor LY294002 were used to assess the roles of the Erk and Akt pathways in the anti-apoptotic response to IGF1R activation. U0126 and LY294002 treatment resulted in highly selective inhibition Erk and Akt, respectively (Fig. 4A). U0126 augmented gefitinib-induced PARP cleavage but des[1–3]IGF-1 still eliminated PARP cleavage in the presence of both inhibitors (gefitinib and U0126; Fig. 4B), demonstrating that IGF1R-induced resistance to gefitinib is not MEK/Erk dependent. Addition of LY294002 augmented gefitinib-induced PARP cleavage, and reduced but did not completely eliminate the ability of des[1–3]IGF-1 to block PARP cleavage (Fig. 4B). Although these data imply either incomplete inhibition of PI3K by LY294002, activation of Akt by an alternate (non-PI3K) pathway, or participation of additional signaling pathways in the prosurvival activity of the IGF1R, they strongly suggest involvement of the PI3K/Akt pathway rather than the MEK/Erk pathway in this process.
Figure 4. Role of MEK and PI3K in gefitinib-induced apoptosis and rescue by IGF1R activation.

After 24 h of serum starvation, SCC-25 cells were incubated for 5 min (A) or 48 h (B) with inhibitors ± growth factor. Cell lysates were immunoblotted. In (B), average PARP cleavage (c-PARP density/total PARP density) ± SEM is shown relative to gefitinib alone (100%) for 3 independent experiments.
EGFR and IGF1R Coinhibition
In order to determine the utility of IGF1R blockade, SCC-25 and Cal27 cells were treated with PQ401, an IGF1R-TKI (22, 23). PQ401 had an IC50 ~4–5 μM (Fig. 5A). Although p-IGF1R could not be detected in unstimulated cells, PQ401 inhibited des[1–3]IGF-1-stimulated IGF1R phosphorylation and downstream Akt phosphorylation in a dose-dependent manner (Fig. 5B). In SCC-25 cells, PQ401 was shown to reduce basal Akt phosphorylation at lower concentrations (Fig. 5C), suggesting that some basal Akt phosphorylation may be due to low level constitutive IGF1R activity. Figure 5D shows the effect of combined EGFR and IGF1R inhibition with gefitinib and PQ401. For these studies, PQ401 and gefitinib were combined in several ratios (1:1, 2:1, 1:2, and 1:4). Based on the combination indices and isobologram analyses, the two drugs are synergistic at IC50 levels.
Figure 5. IGF1R inhibition by PQ401 and synergism with gefitinib.

(A) SCC-25 and Cal27 cells were incubated for 48 h with PQ401 and alamarBlue assay performed. Graph shows average percent of control (unstimulated, uninhibited) cells ± SEM for 3 independent quadruplicate experiments. (B) SCC-25 and Cal27 cells were incubated for 2 h with PQ401 (0 = vehicle only) then for 5 min with des[1–3]IGF-1 or vehicle, lysed and immunoblotted. (C) Unstimulated SCC-25 cells were incubated for 2 h with PQ401 then lysed and immunoblotted. (D) SCC-25 and Cal27 cells were incubated for 48 h with gefitinib (Gef) and/or PQ401 (PQ) and alamarBlue assay performed. Growth inhibition was calculated and average results of 4 independent quadruplicate experiments (SEM < 5%) were analyzed for drug synergism using median-effect approach with CalcuSyn software. The graphs indicate dose response, combination index (CI) vs. fractional effect, and conservative ED50 isobologram curves for the individual drugs and fixed-ratio combinations as indicated.
IGF1R Expression in HNSCC Tumors
Figure 6 shows immunoblot analysis of human HNSCC tumor specimens. The tumor specimens used represent small biopsies of solid tumor tissue and the surrounding tissue was shown to be predominantly squamous carcinoma cells by standard clinical histopathology. When lysates of these specimens were immunoblotted, IGF1R was detectable in all tumors, and p-IGF1R was detectable in 5 of 8 specimens. An IGF-1-stimulated rat fibroblast cell line (Rat1) overexpressing the human IGF1R (RIG) (24) was used as a positive control. These data indicate that the IGF1R is present in HNSCC, and often constitutively phosphorylated, providing preliminary data that the IGF1R may have an important clinical role.
Figure 6. IGF1R in HNSCC tumor lysates.
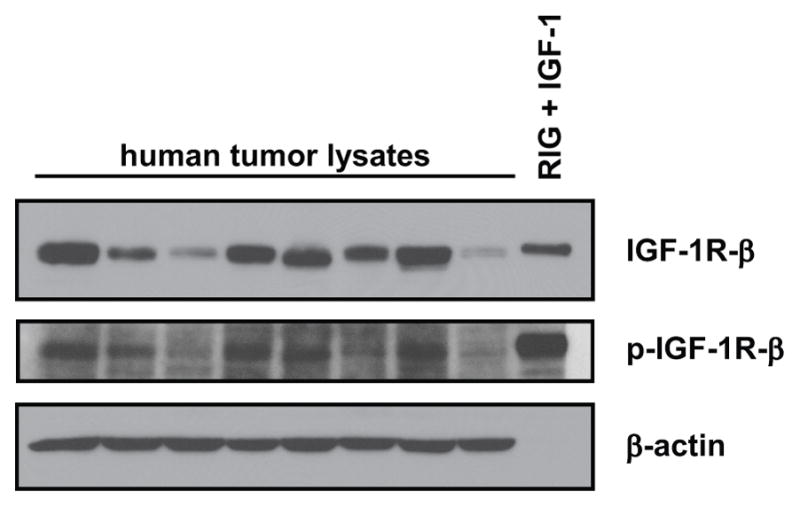
Tumor specimens were lysed and immunoblotted. RIG = Rat1 fibroblasts overexpressing human IGF1R
DISCUSSION
In HNSCC, EGFR-TKIs can induce tumor regression or stabilization and may augment the anti-tumor activity of radiotherapy and cytotoxic chemotherapy, but the proportion of responsive tumors is small, and the response is generally not sustained (2). Thus, clinical targeted anti-EGFR therapy yields disappointing results due to both intrinsic and acquired resistance. While EGFR mutations impact sensitivity to EGFR-TKIs in other tumor types, their role in HNSCC is not yet clear (25–27). Because EGFR-TKIs may impact their function, expression of other HER family members may also play an important role in sensitivity to EGFR inhibition. Gefitinib is more than 100-fold less potent at inhibiting HER2 (IC50 > 3.7 μM) compared to EGFR (IC50 = 0.033 μM) in in vitro kinase assays, but it is not known how important the effect of gefitinib on HER2 is in relation to growth inhibition. The expression and function of HER family members in HNSCC is even less well studied than in other epithelial tumors, but could be of significant consequence for EGFR-TKI therapy.
Signaling from other receptor tyrosine kinases (RTKs) can effectively substitute for EGFR activity; these may be constitutively active redundant pathways (intrinsic resistance) or compensatory responses (acquired resistance). The first well-described example of this was c-Met (28), and other RTKs, including the IGF1R, have since been implicated. In non-HNSCC cell lines, IGF1R activation has been shown to inhibit the apoptosis induced by cetuximab (29) and by the EGFR-TKI AG1478 (30). After long-term gefitinib exposure, HN11 HNSCC cells demonstrated elevated levels of activated IGF1R and Akt and increased sensitivity to IGFBP-3 (17). These findings support an important EGFR-IGF1R interaction, and, while the mechanism is not fully understood, the prosurvival activity of the IGF1R appears to be crucial. Chakravarti et al. (30) demonstrated that resistance of a glioblastoma cell line to EGFR-TKIs was mediated by increased IGF1R expression with persistent PI3K signaling. In multiple breast cancer cell lines, IGF1R inhibition synergistically increased apoptosis when combined with gefitinib; in these cell lines, inhibition of Akt activity required combined EGFR/IGF1R inhibition (31). Similarly, in A431 cells with induced resistance to gefitinib, Guix et al. (17) demonstrated that elimination of persistent IGF1R-induced Akt activity was required to reestablish gefitinib sensitivity. Based on these findings, persistent IGF1R activity may predict resistance to anti-EGFR therapy. More broadly, IGF1R inhibition also increases apoptosis in response to cytotoxic agents (32, 33), and thus IGF1R activity may also impact the effectiveness of non-targeted chemotherapeutics.
Constitutive activation of the IGF1R in human HNSCC tumor specimens (Fig. 6) suggests the existence of an autocrine or paracrine loop. This is consistent with other studies demonstrating the production of IGF-1 and/or IGF-2 by head and neck tumor cells (20). However, IGF production by epithelial tumor cells may not be necessary to activate the IGF1R; IGF-1 and/or IGF-2 produced by nearby stromal cells could serve as a paracrine growth stimulator. These data establish an important distinction between in vitro studies and the in vivo situation: while in vitro evaluation suggests no IGF1R phosphorylation in the absence of exogenous IGF, there is a high likelihood that HNSCC tumors in vivo are exposed to IGF-1 and/or IGF-2 and that the IGF1R exhibits some degree of constitutive activation. Combined with our findings of IGF1R-induced resistance to EGFR inhibition in multiple HNSCC cell lines in vitro, this would predict potential widespread resistance to EGFR-TKIs in vivo. Since HNSCC are highly heterogeneous tumors, a method for predicting IGF1R-based resistance to anti-EGFR therapy in the clinical setting will be crucial to evaluate the relevance of this mechanism in patients with HNSCC.
Accurate predictors of response to EGFR-TKIs in HNSCC have not been identified. A recent assessment by Rogers et al. (34) of a panel of 18 HNSCC cell lines demonstrated a correlation between sensitivity to gefitinib and degree of EGFR phosphorylation. While Met expression predicted response, Met knockdown had no effect on gefitinib sensitivity. In their study, neither IGF1R expression level nor degree of phosphorylation predicted response to gefitinib (34). However, there was no assessment of downstream targets in this study and thus no indication of the net impact of IGF1R level/activity. Unfortunately, it is unlikely that IGF1R-based resistance will correlate simply with IGF1R expression; in addition to the level and affinity of the IGF1R, the degree of IGF1R activation is dependent on the abundance of IGF ligands and IGFBPs, and the impact of IGF1R activity is highly context sensitive. For example, IGF1R activation can have a greater impact on cell growth when the EGFR is inhibited (Fig. 1C). Additionally, the mechanism(s) of interaction between the HER and IGF systems may be complex. A recent study demonstrated that, in breast cancer, trastuzumab regulates IGFBP-2 and -3 expression, which impacts IGF1R downstream signaling (35). Cytotoxic agents can also regulate IGFBP secretion: increased secretion of IGFBP-3 in prostate cancer cells treated with 5-fluorouracil plays an important role in its pro-apoptotic effect (36). Thus, due to the complexities of the IGF system and its regulation, further investigation will be required to identify a reliable marker of IGF1R-based resistance to therapy.
In general, the effectiveness of EGFR-TKIs in vivo has been based on assessment of EGFR phosphorylation. In SCC-25 and Cal27 cells treated with des[1–3]IGF-1, we detected persistent downstream signaling even when EGFR phosphorylation was undetectable (Fig. 2). Residual downstream target phosphorylation may be due to incomplete EGFR blockade or to the activity of other receptors such as the IGF1R. Janmaat et al. (37) have shown that failure of gefitinib to inhibit cell growth is correlated with persistent Erk and Akt activity. Therefore, complete assessment of the effectiveness of EGFR-TKIs both in vitro and in vivo requires analysis of phosphorylation of downstream targets.
SCC-25 and Cal27 cells exhibit high EGFR and IGF1R expression and constitutive EGFR, Erk, and Akt phosphorylation. Gefitinib induced apoptosis in a dose-dependent fashion; this was eliminated when the IGF1R was activated. Based on inhibitor studies, this effect is at least partially related to persistent Akt signaling, which is consistent with the well-established role of Akt as a prosurvival effector of the IGF1R. Addition of the PI3K inhibitor LY294002 reduced but did not eliminate IGF1R-based resistance to gefitinib-induced apoptosis suggesting incomplete PI3K inhibition, PI3K-independent Akt activation, or an alternate (Akt-independent) prosurvival pathway from the IGF1R (as suggested by other authors (24)). Activation of Erk, as observed in other contexts (20), is EGFR-dependent in SCC-25 and Cal27 cells, thus eliminating it as an anti-apoptotic agent when gefitinib is present. This was confirmed using the MEK inhibitor U0126. The remaining possibilities are incomplete PI3K blockade or an alternate non-Erk-, non-Akt-dependent prosurvival pathway.
It is important to note that the chemical inhibitors used in the present study are selective, and the results could be confounded by “off-target” effects. As mentioned, gefitinib inhibits HER2 with 100-fold less potency than EGFR; while this is unlikely to play an important role in our studies, cell lines with high HER2 expression might demonstrate altered responses when gefitinib is used at high concentrations. Similarly, PQ401 is a selective IGF1R inhibitor that undoubtedly also inhibits the insulin receptor (InsR), given the high homology between these two receptors. However, when stimulated with insulin, SCC-25 cells showed no response (i.e., no gefitinib resistance) at lower doses but showed similar behavior to IGF stimulation at higher doses (100-fold higher than IGF), indicating that the effect observed is due to IGF1R activation. Gable et al. (22) demonstrated similar findings using PQ401: it inhibited both basal and IGF-stimulated MCF-7 breast cancer cell growth, with inhibition of IGF1R and Akt phosphorylation at similar concentrations to those shown in Figure 5. As an inhibitor of IGF1R and Akt phosphorylation, PQ401 has also been shown to mimic IGFBP-3 and an IGF1R-blocking antibody that does not bind the InsR (23). The ability of low-dose exogenous IGF to attenuate the effects of gefitinib further substantiates the notion that PQ401 is acting on-target when it augments the effects of gefitinib. It is also noteworthy that inhibition of the InsR along with the IGF1R (i.e., non-selectivity) may be clinically desirable due to the potential for the InsR to substitute for the IGF1R when the IGF1R is selectively inhibited (38). In addition to these predictable off-target effects, there may be unexpected and as-yet unknown effects resulting from interactions with unrelated proteins. Future studies should more extensively evaluate the impact of these inhibitors on a broad range of molecular targets in HNSCC.
Our data showing augmented growth inhibition of SCC-25 and Cal27 cells with PQ401 provide preliminary evidence that combined EGFR/IGF1R inhibition may be an effective treatment approach to HNSCC. Other authors have used combined EGFR/IGF1R inhibition in limited in vivo models of HNSCC (39) but were unable to show a synergistic effect of the two inhibitors. Wilsbacher et al. (40) demonstrated that EGFR and IGF1R inhibition act synergistically in A431 cells; only the combination eliminated Akt phosphorylation and introduction of constitutively active Akt resulted in resistance to the drug combination. Erlotinib and the IGF1R-TKI PQIP were noted by Buck et al. (41) to be synergistic in a range of epithelial tumors. Their data suggested that coinhibition was necessary due, at least in part, to increased activation of the reciprocal receptor when either drug was used alone.
It seems clear that, in most cancers, effective targeted therapy will involve blockade of multiple targets. There is now mounting evidence that the IGF1R and/or its downstream signaling pathways will be of key importance in conjunction with the EGFR-TKIs. The most important element of attempting new combinations of targeted therapies will be tumor/patient selection. Thus, correlative evidence is needed that demonstrates biomolecular markers predictive of IGF1R-based EGFR-TKI resistance. It is currently unclear whether unique IGF system molecules (IGF1R, IGF-1 or -2, IGFBP-1 through -6) or downstream signaling molecules such as Akt or others will be the most significant markers for exploitation and continued evaluation is warranted.
CONCLUSION
The present study clearly demonstrates that the IGF1R can act as a mechanism of resistance to EGFR-TKIs in an in vitro model of HNSCC, and highlights the potential utility of coinhibition of the EGFR and IGF1R.
Acknowledgments
Financial Support: NIH grant K08-DE019477 (M. Jameson); American Head and Neck Society Pilot Grant (M. Jameson).
Abbreviation List
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- HNSCC
head and neck squamous cell carcinoma
- IGF
insulin-like growth factor
- IGF1R
IGF-1 receptor
- IGFBP
IGF binding protein
- InsR
insulin receptor
- MEK
mitogen-activated protein kinase (MAPK)/Erk kinase
- PARP
poly-(ADP-ribose) polymerase
- PI3K
phosphatidylinositol-3′-kinase
- RT
radiation therapy
- RTK
receptor tyrosine kinase
- TKI
tyrosine kinase inhibitor
References
- 1.Cohen EE, Rosen F, Stadler WM, Recant W, Stenson K, Huo D, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21:1980–7. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 2.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 3.Cohen EE. Role of epidermal growth factor receptor pathway-targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2006;24:2659–65. doi: 10.1200/JCO.2005.05.4577. [DOI] [PubMed] [Google Scholar]
- 4.Bohula EA, Playford MP, Macaulay VM. Targeting the type 1 insulin-like growth factor receptor as anti-cancer treatment. Anticancer Drugs. 2003;14:669–82. doi: 10.1097/00001813-200310000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Macaulay VM. Insulin-like growth factors and cancer. British Journal of Cancer. 1992;65:311–20. doi: 10.1038/bjc.1992.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouban A, Muraca P, Yeatman T, Coppola D. Expression and distribution of insulin-like growth factor-1 receptor in human carcinomas. Hum Pathol. 2003;34:803–8. doi: 10.1016/s0046-8177(03)00291-0. [DOI] [PubMed] [Google Scholar]
- 7.Coppola D, Ferber A, Miura M, Sell C, D’Ambrosio C, Rubin R, et al. A functional insulin-like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Molecular & Cellular Biology. 1994;14:4588–95. doi: 10.1128/mcb.14.7.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol. 2002;3:298–302. doi: 10.1016/s1470-2045(02)00731-3. [DOI] [PubMed] [Google Scholar]
- 9.Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279:563–6. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 10.Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst. 1999;91:151–6. doi: 10.1093/jnci/91.2.151. [DOI] [PubMed] [Google Scholar]
- 11.Toniolo P, Bruning PF, Akhmedkhanov A, Bonfrer JM, Koenig KL, Lukanova A, et al. Serum insulin-like growth factor-I and breast cancer. Int J Cancer. 2000;88:828–32. doi: 10.1002/1097-0215(20001201)88:5<828::aid-ijc22>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Zhao H, Do KA, Johnson MM, Dong Q, Hong WK, et al. Serum levels of insulin growth factor (IGF-I) and IGF-binding protein predict risk of second primary tumors in patients with head and neck cancer. Clin Cancer Res. 2004;10:3988–95. doi: 10.1158/1078-0432.CCR-03-0762. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y, Cui K, Miyoshi K, Hennighausen L, Green JE, Setser J, et al. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res. 2003;63:4384–8. [PubMed] [Google Scholar]
- 14.LeRoith D, Helman L. The new kid on the block(ade) of the IGF-1 receptor. Cancer Cell. 2004;5:201–2. doi: 10.1016/s1535-6108(04)00054-6. [DOI] [PubMed] [Google Scholar]
- 15.Jones HE, Goddard L, Gee JM, Hiscox S, Rubini M, Barrow D, et al. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer. 2004;11:793–814. doi: 10.1677/erc.1.00799. [DOI] [PubMed] [Google Scholar]
- 16.Warshamana-Greene GS, Litz J, Buchdunger E, Garcia-Echeverria C, Hofmann F, Krystal GW. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin Cancer Res. 2005;11:1563–71. doi: 10.1158/1078-0432.CCR-04-1544. [DOI] [PubMed] [Google Scholar]
- 17.Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–19. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan Z, Choy E, Harmon D, Yang C, Ryu K, Schwab J, et al. Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol Cancer Ther. 2009;8:2122–30. doi: 10.1158/1535-7163.MCT-09-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 20.Slomiany MG, Black LA, Kibbey MM, Tingler MA, Day TA, Rosenzweig SA. Insulin-like growth factor-1 receptor and ligand targeting in head and neck squamous cell carcinoma. Cancer Lett. 2007;248:269–79. doi: 10.1016/j.canlet.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Kuribayashi A, Kataoka K, Kurabayashi T, Miura M. Evidence that basal activity, but not transactivation, of the epidermal growth factor receptor tyrosine kinase is required for insulin-like growth factor I-induced activation of extracellular signal-regulated kinase in oral carcinoma cells. Endocrinology. 2004;145:4976–84. doi: 10.1210/en.2004-0713. [DOI] [PubMed] [Google Scholar]
- 22.Gable KL, Maddux BA, Penaranda C, Zavodovskaya M, Campbell MJ, Lobo M, et al. Diarylureas are small-molecule inhibitors of insulin-like growth factor I receptor signaling and breast cancer cell growth. Mol Cancer Ther. 2006;5:1079–86. doi: 10.1158/1535-7163.MCT-05-0397. [DOI] [PubMed] [Google Scholar]
- 23.Sivakumar R, Koga H, Selvendiran K, Maeyama M, Ueno T, Sata M. Autocrine loop for IGF-I receptor signaling in SLUG-mediated epithelial-mesenchymal transition. Int J Oncol. 2009;34:329–38. [PubMed] [Google Scholar]
- 24.Kulik G, Weber MJ. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Mol Cell Biol. 1998;18:6711–8. doi: 10.1128/mcb.18.11.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12:5064–73. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 26.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, Hilbe W, Schwentner I, Sprinzl GM, et al. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;42:109–11. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 27.Willmore-Payne C, Holden JA, Layfield LJ. Detection of EGFR- and HER2-activating mutations in squamous cell carcinoma involving the head and neck. Mod Pathol. 2006;19:634–40. doi: 10.1038/modpathol.3800552. [DOI] [PubMed] [Google Scholar]
- 28.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 29.Liu B, Fang M, Lu Y, Mendelsohn J, Fan Z. Fibroblast growth factor and insulin-like growth factor differentially modulate the apoptosis and G1 arrest induced by anti-epidermal growth factor receptor monoclonal antibody. Oncogene. 2001;20:1913–22. doi: 10.1038/sj.onc.1204277. [DOI] [PubMed] [Google Scholar]
- 30.Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002;62:200–7. [PubMed] [Google Scholar]
- 31.Camirand A, Zakikhani M, Young F, Pollak M. Inhibition of insulin-like growth factor-1 receptor signaling enhances growth-inhibitory and proapoptotic effects of gefitinib (Iressa) in human breast cancer cells. Breast Cancer Res. 2005;7:R570–R579. doi: 10.1186/bcr1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H, Rao J, Lin J, Yin B, Sheng H, Lin F, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-ADW742 sensitizes medulloblastoma to the effects of chemotherapy. Oncol Rep. 2011 doi: 10.3892/or.2011.1233. Epub- 2011 Mar 23. [DOI] [PubMed] [Google Scholar]
- 33.Shu S, Yang Y, Li X, Li T, Zhang Y, Xu C, et al. Down-regulation of IGF-1R expression inhibits growth and enhances chemosensitivity of endometrial carcinoma in vitro. Mol Cell Biochem. 2011 doi: 10.1007/s11010-011-0790-9. Epub- 2011 Mar 26. [DOI] [PubMed] [Google Scholar]
- 34.Rogers SJ, Box C, Chambers P, Barbachano Y, Nutting CM, Rhys-Evans P, et al. Determinants of response to epidermal growth factor receptor tyrosine kinase inhibition in squamous cell carcinoma of the head and neck. J Pathol. 2009;218:122–30. doi: 10.1002/path.2515. [DOI] [PubMed] [Google Scholar]
- 35.Dokmanovic M, Shen Y, Bonacci TM, Hirsch DS, Wu WJ. Trastuzumab regulates IGFBP-2 and IGFBP-3 to mediate growth inhibition: implications for the development of predictive biomarkers for trastuzumab-resistance. Mol Cancer Ther. 2011 doi: 10.1158/1535-7163.MCT-10-0980. Epub- 2011 Apr 12. [DOI] [PubMed] [Google Scholar]
- 36.Kawabata R, Oie S, Takahashi M, Kanayama H, Oka T, Itoh K. Up-regulation of insulin-like growth factor-binding protein 3 by 5-fluorouracil (5-FU) leads to the potent anti-proliferative effect of androgen deprivation therapy combined with 5-FU in human prostate cancer cell lines. Int J Oncol. 2011;38:1489–500. doi: 10.3892/ijo.2011.991. [DOI] [PubMed] [Google Scholar]
- 37.Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. 2003;9:2316–26. [PubMed] [Google Scholar]
- 38.Buck E, Mulvihill M. Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert Opin Investig Drugs. 2011;20:605–21. doi: 10.1517/13543784.2011.558501. [DOI] [PubMed] [Google Scholar]
- 39.Barnes CJ, Ohshiro K, Rayala SK, El-Naggar AK, Kumar R. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin Cancer Res. 2007;13:4291–9. doi: 10.1158/1078-0432.CCR-06-2040. [DOI] [PubMed] [Google Scholar]
- 40.Wilsbacher JL, Zhang Q, Tucker LA, Hubbard RD, Sheppard GS, Bamaung NY, et al. Insulin-like growth factor-1 receptor and ErbB kinase inhibitor combinations block proliferation and induce apoptosis through cyclin D1 reduction and Bax activation. J Biol Chem. 2008;283:23721–30. doi: 10.1074/jbc.M708360200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buck E, Eyzaguirre A, Rosenfeld-Franklin M, Thomson S, Mulvihill M, Barr S, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res. 2008;68:8322–32. doi: 10.1158/0008-5472.CAN-07-6720. [DOI] [PubMed] [Google Scholar]