

Abstract
Excess exogenous retinoic acid (RA) has been well documented to have teratogenic effects in the limb and craniofacial skeleton. Malformations that have been observed in this context include craniosynostosis, a common developmental defect of the skull that occurs in 1 in 2500 individuals and results from premature fusion of the cranial sutures. Despite these observations, a physiological role for RA during suture formation has not been demonstrated. Here, we present evidence that genetically based alterations in RA signaling interfere with human development. We have identified human null and hypomorphic mutations in the gene encoding the RA-degrading enzyme CYP26B1 that lead to skeletal and craniofacial anomalies, including fusions of long bones, calvarial bone hypoplasia, and craniosynostosis. Analyses of murine embryos exposed to a chemical inhibitor of Cyp26 enzymes and zebrafish lines with mutations in cyp26b1 suggest that the endochondral bone fusions are due to unrestricted chondrogenesis at the presumptive sites of joint formation within cartilaginous templates, whereas craniosynostosis is induced by a defect in osteoblastic differentiation. Ultrastructural analysis, in situ expression studies, and in vitro quantitative RT-PCR experiments of cellular markers of osseous differentiation indicate that the most likely cause for these phenomena is aberrant osteoblast-osteocyte transitioning. This work reveals a physiological role for RA in partitioning skeletal elements and in the maintenance of cranial suture patency.
Introduction
Both deficiency and excess of retinoic acid (RA) have broad and disparate effects during vertebrate embryogenesis1 and organogenesis and are indicative of finely tuned spatial and temporal regulation of its concentration during development. In particular, abnormalities of limb and craniofacial morphogenesis can result from exposure to exogenous RA at critical periods during fetal skeletal development.2, 3, 4 Despite these insights, the mechanistic basis for this teratogenic effect or the consequences of physiological upregulation of levels of RA during skeletogenesis has not been well characterized. During normal vertebrate development, the dose-dependent paracrine action of RA is regulated by synthetic (Raldh1-3) and degradative (Cyp26a1-c1) enzymes that establish RA tissue gradients by a source-and-sink mechanism.1
Cyp26 proteins belong to the cytochrome P450 family and oxidize all-trans-RA to biologically inactive metabolites.5, 6 A role for Cyp26-mediated RA catabolism in skeletogenesis is supported by animal models. Cyp26b1−/− knockout mice have foreshortened proximal limb segments, radiohumeral synostosis, oligodactyly, craniofacial defects, and reduced calvarial ossification,7, 8 whereas null and hypomorphic zebrafish cyp26b1 mutants, named dolphin (dol) and stocksteif (sst), display deficiencies in midline cartilaginous structures and hypermineralized facial and axial bones.9, 10
The malformations noted in these model organisms with Cyp26b1 mutations indicate that spatial and temporal deficits in RA degradation affect skeletogenesis by impacting on the function of several different cell types and morphogenic processes. For instance, the patterning defects in the limbs leading to oligodactyly are presumed to relate to the direct effect that RA has on the expression of key genes implicated in the morphogenesis of the autopod such as Hoxa13 and Hoxd13.7 Similarly, the failure of limb outgrowth could be attributable to analogous regulation of genes that dictate proximo-distal patterning by RA.7 The mechanism underlying the cartilaginous fusion of the radio-humeral joints noted in the Cyp26b1−/− mice, however, remains unclear. Similarly, the mechanism underlying the effect that Cyp26b1 insufficiency has on the mineralization of the skeleton is not satisfactorily explained. Although the hypermineralization and vertebral fusion observed in the zebrafish cyp26b1 mutants have been attributed to upregulation of osteoblastic activity in the absence of any effect of cellular proliferation,9, 10 the reason for the pronounced calvarial hypoplasia noted in the skulls of the knock-out mice is unclear.8 What is well demonstrated from the study of these model organisms is that RA affects both the development of structures such as the long bones that ossify via utilization of a cartilaginous template (endochondral ossification) and those that form bone by directly mineralizing mesenchymal anlagen (membranous ossification), the most notable example being the calvarium.
Here two overlapping human phenotypes are shown to be caused by mutations in the human gene encoding the RA-degrading cytochrome P450 enzyme CYP26B1 (MIM 605207). The phenotypes represent the effects of hypomorphic and null mutations that have radiohumeral fusions as a shared manifestation and extend to defects in calvarial and sutural ossification resulting in hypoplasia of the cranium and craniosynostosis. Through in vivo studies of treated or mutant mice or zebrafish and in vitro studies of cultured preosteoblasts, we propose a model in which RA promotes both chondrogenesis at the margins of cartilaginous structures, accounting for long-bone fusions, and the dose- and stage-dependent differentiation of matrix-producing osteoblasts to mineralizing osteocytes, accounting for the seemingly contradictory calvarial defects, namely hypoplasia and sutural fusion. This study emphasizes the importance of close temporal and spatial regulation of RA in the context of its multifarious functions in the patterning and mineralization of the vertebrate skeleton.
Material and Methods
Subjects
All subjects or their legal representatives gave written informed consent for the study. The study was performed in accordance to the Declaration of Helsinki protocols and approved by the Otago Ethics Committee and the University Hospital, Cologne local institutional review board. DNA from participating family members was extracted from peripheral blood lymphocytes by standard extraction procedures.
Genotyping, Mapping, and Modeling
SNP genotyping was performed with the Illumina OmniQuad platform and homozygous regions were identified with a sliding window and a tolerance for 2% residual apparent heterozygosity because of genotyping error. Database searches were performed to identify candidate genes within the critical region on chromosome 2p15-q11 (Ensemble Genome Server, UCSC Genome Bioinformatics) and genes were evaluated for candidacy on the basis of their expression in relevant tissues and phenotypic analysis of animal models. Primers to amplify all coding exons and adjacent splice sites of CYP26B1 were designed. All mutations and protein substitutions in the human are numbered according to the reference sequences NM_019885.2 and NP_063938.1. Primer sequences and conditions are available on request. The exons and intron-exon boundaries of CYP26B1 were amplified from genomic DNA of all affected members of the two families and sequenced with the BigDye Terminator method. All identified mutations were resequenced in independent experiments and tested for cosegregation within the families. Controls (190 for the p.Arg363Leu mutation; 200 healthy control individuals from Turkey for the p.Ser146Pro mutation) were examined with direct sequencing for the relevant mutation. The Turkish individual with Antley-Bixler syndrome was initially tested for mutations in FGFR2 (MIM 176943), POR (MIM 124015), and FGF9 (MIM 600921). No mutations were found. The structure of human CYP26B1 (residues 43-494) was modeled with the retinoic-acid-bound cyanobacterial (Synechocystis sp. PCC 6803) cytochrome P450 CYP120A1 structure (PDB 1VE3)11 as a template and the MODELER 912 and I-TASSER platforms13 independently calculating a similar solution. The quality of the model was externally assessed with MOLPROBITY.14
Zebrafish Lines
The following cyp26b1 alleles were used: sstt24295 (hypomorph10) and dolti230 g (amorph9).
Drug Treatments of Zebrafish and Mice
For zebrafish treatments, stock solutions of all-trans RA (100 μM, Sigma, St. Louis, MO) and R115866 (10 mM; Johnson and Johnson Pharmaceutical Research and Development, San Diego, CA) were prepared in dimethyl sulfoxide (DMSO) according to suppliers' instructions. Juvenile fish were incubated in the dark with a final concentration of 1 μM RA or 1 μM R115866 from 5 weeks to 6 postfertilization (wpf), followed by fixation at 7.5 wpf. For mouse experiments, R115866 was dissolved in PEG 200 (Sigma, 2.5 mg/ml) and administered to pregnant NMRI females by daily oral gavages, applying 10 mg per kilogram body weight, from E13–16.
Tissue-Labeling Procedures
Cartilage and mineralized bone from zebrafish and mice were stained with alcian blue or alizarin red, respectively, essentially as described.15, 16 Histological staining (periodic acid-Schiff reaction [PAS]; van Gieson, azan novum) were performed on 8 μM paraffin sections according to standard protocols (Merck, Whitehouse Station, NJ).
Whole-mount single17 and fluorescent-double in situ hybridizations18 and in situ hybridizations of paraffin sections19 were performed as described. A mouse Cyp26b1 cDNA fragment of 1.2 kb was amplified from tongue cDNA and cloned into pCRII (Invitrogen, Carlsbad, CA). For probe synthesis, the plasmid was linearized with HindIII and transcribed with T7 RNA polymerase. The riboprobe for mouse Gdf5 was synthesized as described.20 For zebrafish sost and phex, 1.2 kb cDNA fragments were excised from EST clones IMAGp998H1917181Q (sost) or IRALp962L1260Q (phex) and cloned into pBluescript. Resulting plasmids were linearized with HindIII (sost) or EcoRV (phex) and transcribed with T3 RNA polymerase. For zebrafish fgf23, a 0.8 cDNA fragment was amplified via RT-PCR and cloned into pCRII. The resulting plasmid was linearized with SpeI and transcribed with T7 RNA polymerase. Riboprobe for ankha was obtained from the full-length cDNA clone (IRBOp991C0761D; ImaGenes, Nottingham, UK), cloned via KpnI/SacI digest into pBSSK, linearized by SacI and transcribed with T7 RNA polymerase. The ankhb probe consisted of the full-length cDNA clone IRBOp991H0245D (ImaGenes) in pExpress1, linearized by StuI, and transcribed with T7 RNA polymerase. Probes for zebrafish col1a1,21 col10a,22 cyp26b1,9 opn,9 sox9a,23 and sparc24 were synthesized as described.
For phalloidin staining, 7 μM cryosections were cut, permeabilized for 45 min in 0.1% Triton X-100/0.1% sodium citrate, blocked in 10% calf serum in PBS plus Tween-20 (PBST), and stained overnight with Alexafluor 555-Phalloidin (1:200 Invitrogen A34055). Sections were embedded in Mowiol containing DAPI (1 μg/ml) and analyzed by confocal microscopy. For all histological, TUNEL, bromodeoxyuridine (BrdU), and in situ hybridization experiments sections of a minimum of three individual mutant and wild-type sibling fish were examined and representative results are depicted.
For transmission electron microscopy (TEM), juvenile zebrafish were fixed in 2% glutaraldehyde and 2% PFA in 0.1M cacodylate buffer (pH 7.4). Fixed specimens were washed several times in PBS, decalcified at room temperature for one week in 10% EDTA-Tris/HCl buffer (pH 7.35), postfixed with 2% osmium tetraoxide in 0.1 M PBS for 2 hr at 4°C, dehydrated in a graded series of ethanol, and embedded in araldite (Hundsman Advanced Materials, Derry, NH). Semithin sections (500 nm) and ultrathin sections (60 nm) were cut with a Reichert-Jung (Leica, Wetzlar, Germany) Ultra-cut ultra microtome. Semithin sections for light-microscopy were stained with methylene blue, whereas ultrathin sections were mounted on copper grids coated with formvar (SPI-Chem), stained with 0.2% uranyl acetate and lead citrate, and examined with a Zeiss EM 902 A electron microscope.
Cyp26b1 Luciferase Reporter Assay
HEK293 cells were seeded in 24-well plates at 0.75 × 105 cells/well. After 24 hr cells were transfected with RARE reporter plasmid (CCS-016L, SABiosciences) and control plasmid, wild-type hCYP26b1, or hCYP26b1 containing the Arg363Leu, Ser146Pro, or the zebrafish dolsa002 mutation,9 respectively. Mutations were introduced into hCYP26b1 by site-directed mutagenesis. Retinoic acid was administered 24 hr after transfection. A 1 mM RA stock solution (in ethanol) was diluted 1:1000 in cell culture medium to obtain a final RA concentration of 1 μM. After 24 hr, incubation cells were lysed and assayed with a Dual Luciferase Reporter Assay system (Promega) and a Glomax 96-microplate luminometer. The experiment was performed five times independently, each time in triplicate.
Osteoblast Cell Culture and qRT-PCR
MC3T3-E1 cells (subclone no. 4) were grown essentially as described.25 Cells were differentiated for indicated periods by adding 50 μg/ml ascorbic acid and 3 mM sodium phosphate to cell culture medium. 24 hr prior to treatment, cells were fasted in a-MEM 1% FBS, 50 μg/ml ascorbic acid, and 3 mM sodium phosphate. Cells were treated with different concentrations of retinoic acid (0.01−5 μM; Sigma R2625) or vehicle for 4, 8, or 24 hr and resuspended in Trizol for RNA and cDNA preparation according to standard protocols. Osteoblast and osteocyte gene expression was assayed by qPCR with SYBR green and normalized against 18S rRNA. Each experiment was performed at least three times independently. Primer sequences are available upon request.
Results
Human Individuals Homozygous for Loss-of-Function Mutations in CYP26B1 Display Multiple Skeletal Defects, Including Long-Bone Fusions and Craniosynostosis
A family consisting of three siblings born to first-cousin parents (Figure 1A) who exhibited combinations of severe craniofacial malformations, occipital encephalocele, radiohumeral fusions, oligodactyly, advanced osseous maturation, and calvarial mineralization defects were clinically characterized following presentation of the proband as a fetal death in utero at 35 weeks gestation (Figures 1B–1E; Figures S1 and S2, available online). The proband (Figure 1A; II:1) was partially macerated and the head and craniofacial region was distorted by the presence of a large occipital encephalocele that was the result of hypoplasia of the occipital and parietal bones. The brain had undergone liquefaction. There was arachnodactyly and terminal phalangeal aplasia of the thumbs. Both femora were grossly angulated (Figure 1B), the toes were elongated, and the great toes were absent bilaterally (Figure 1C). The upper limbs demonstrated ankylosis at the elbows because of complex radio-ulnar-humeral fusion (Figure 1D). In addition to the above anomalies, the thorax was narrowed, the scapulae and pelvis were hypoplastic, and the bone age was advanced. The second and third sibs (Figure 1A, II:2 and II:3) were identified as affected by ultrasonography in two separate subsequent pregnancies that were terminated at 15 and 12 weeks gestation, respectively. Both fetuses had occipital encephaloceles and shortened upper and lower limbs with pronounced angulation of the femora bilaterally. The great toes were absent in both siblings.
Figure 1.
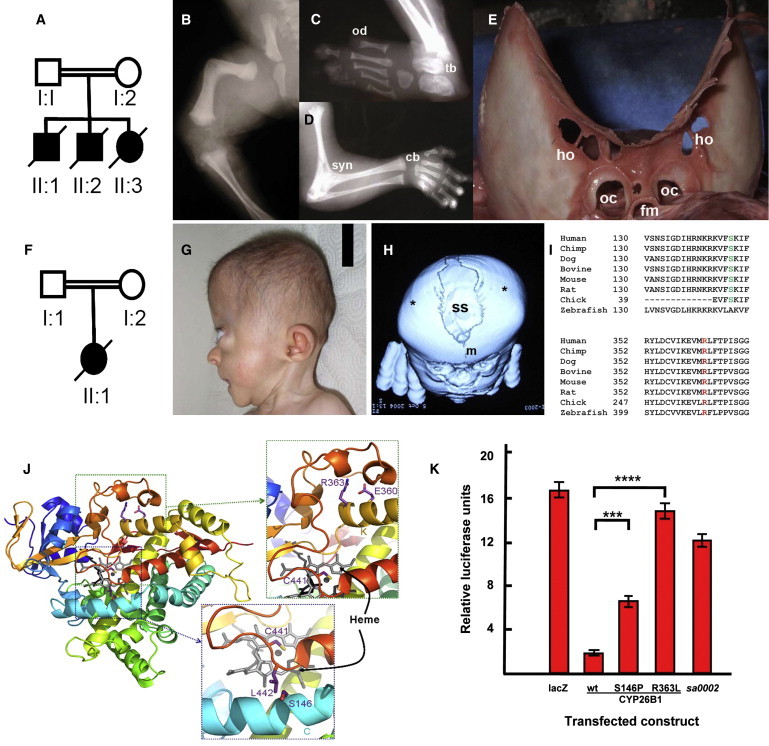
Humans with CYP26B1 Mutations
(A–E) Homozygosity for p.Arg363Leu. (A) Pedigree of family 1; (B–D) Sib 1 (36 weeks gestation). (B) Angulated femora, hypoplastic pelvis, (C) oligodactyly, and (D) elbow joint synostosis and advanced bone age; (E) skull of the proband at autopsy showing calvarial plate hypoplasia resulting in a large midline defect. Bone spicules at the margins of the calvarial plates, holes with the skull bones, and deficiency of the bases of the occipital condyles are all indicative of calvarial thinning.
(F–H) Homozygosity for p.Ser146Pro. (F) Pedigree of family 2 with the proband with a diagnosis of an Antley-Bixler-like syndrome; (G) exorbitism, maxillary retrusion, and brachycephaly. (H) 3D reconstructed cranial CT scan. Coronal suture craniosynostosis (∗) and a broadened sagittal suture (ss).
(I) Phylogenetic conservation of Ser146 and Arg363; (J) structural consequences of p.Arg363Leu and p.Ser146Pro substitutions. Heme (gray), RA (black), and residues as numbered. The p.Arg363Leu substitution ablates interactions with a loop essential for CYP450 catalytic function;30 p.Ser146Pro truncates a conserved helical element in P450 enzymes; (K) RARE-luciferase reporter assay of wild-type, p.Ser146Pro and p.Arg363Leu mutant CYP26B1 constructs in HEK293 cells compared to lacZ and sa0002, a zebrafish null allele.9 Relative luciferase activity units (RU ± standard deviation [SD]) are indicated (∗∗∗p = 5 × 10−5; ∗∗∗∗p = 4 × 10−8; student's t test). The following abbreviations are used: cb, carpal bones; fe, femur; fm, foramen magnum; ho, holes; hu, humerus; m, metopic suture; oc, occipital condyles; od, oligodactyly; ss, sagittal suture; syn, synostosis; tb, tarsal bones.
Two siblings (II:1 and II:2) were analyzed by autozygosity mapping under an assumption of homozygosity by descent for a single deleterious allele to identify the mutated gene. Each individual was genotyped with the Illumina Omniquad SNP array platform and these data sets were searched for tracts of homozygosity greater than 2 Mb in extent with IIlumina GenomeStudio (Illumina, San Diego, CA). Only one tract of shared homozygosity >2 Mb in extent was identified (chromosome 2; 61.24–103.62 Mb Figure S3; Table S1). This region contained 238 genes of which six (MEIS1 [MIM 601739], RMND5A, BMP10 [MIM 608748], MAP4K4 [MIM 604666], PPP3R1 [MIM 601302], and CYP26B1) were selected for sequencing to detect pathogenic mutations after a gene ontology analysis that implicated them in a role relating to skeletogenesis. Sequencing of CYP26B1 demonstrated homozygosity for a c.1088G>T transversion, predicting a p.Arg363Leu substitution, in CYP26B1 in all three siblings. Both parents were heterozygous for this sequence variant, and it was not observed in either 190 unrelated controls drawn from diverse ethnic backgrounds or in dbSNP.
To identify similarly affected subjects with mutations at the same locus, DNA samples obtained from a cohort of individuals with malformations of the skull, with or without associated skeletal anomalies, were subjected to CYP26B1 sequencing. In an unrelated individual with a likely diagnosis of Antley-Bixler syndrome (ABS [MIM 201750])26, 27 and born to consanguineous parents (Figures 1F–1H), a homozygous sequence variant, c.436T>C, predicting a p.Ser146Pro substitution in the same gene (Figure S4) was discovered. This female had a phenotype that included coronal and lambdoid craniosynostosis, a large sagittal skull defect, limited elbow extension, and arachnodactyly (Figures 1G–1H). She died from unknown causes at 5 months of age.
The p.Arg363Leu substitution occurs within an EXXR motif that is highly conserved across species and all human CYP450 proteins (Figure 1I; Figure S3). Substitutions of residues within this motif, including the arginine residue substituted in the family described here, are known to ablate the catalytic properties of these enzymes.28, 29 The p.Ser146Pro substitution is predicted to disrupt helix C, a broadly represented structural element in CYP450 enzymes30 (Figure 1J).
To demonstrate that these two substitutions impair the catalytic function of CYP26B1, we developed an in vitro assay designed to quantify enzymatic activity of mutation-bearing CYP26B1 constructs. Cellular cotransfection of CYP26B1-expression constructs encoding either the p.Arg363Leu or p.Ser146Pro substitutions alongside a RA-responsive luciferase reporter gene demonstrated a significantly attenuated ability to metabolize exogenously applied RA in HEK293 cells (Figure 1K; p.Arg363Leu 86% reduction; p.Ser146Pro 31% reduction). The reduction of enzymatic activity noted for the p.Arg363Leu substituted protein was comparable to that conferred by the truncating mutation underlying the zebrafish null allele sa00029, 10 (Figure 1K), indicating that the human mutation constitutes a null allele. In contrast, retention of activity by the p.Ser146Pro substitution was consistent with it constituting a hypomorphic mutation.
Complete Loss of Cyp26b1 Function in Mice and Zebrafish Results in Fusions or Overgrowth of Cartilaginous Structures
Similar to the p.Arg363Leu proband studied here (Figure 1D), Cyp26b1−/− mice display radiohumeral synostosis that manifests as a cartilaginous union before ossification initiates.7 We observed that in utero treatment of wild-type mice with the Cyp26 inhibitor R115866 results in a similar cartilaginous union of interphalangeal joints before the onset of ossification (Figures 2A and 2B). Notably Cyp26b1 is expressed close to developing tendons at the distal ends of phalangeal primordia in wild-type E15 embryos31 in a pattern identical to that of the nascent synovial joint marker gene Gdf5 (Figure 2C). Similarly, pectoral fins of the cyp26b1 null zebrafish mutant dolphin (dol) display outgrowths of the cartilaginous endochondral disc into distal-most fin regions (Figure 2E), regions that in wild-type siblings lack cartilage (Figure 2D). In situ hybridization experiments in wild-type zebrafish pectoral fins demonstrated cyp26b1 expression directly adjacent to sox9a-positive chondrocyte precursors at earlier time points,23 suggesting a role of this enzyme in defining this cartilaginous boundary (Figure 2F). Collectively, these data indicate that Cyp26b1 might establish and maintain synovial joint patency by restricting RA-mediated stimulation of chondrogenesis at borders of cartilaginous elements. Such a mechanism is consistent with in vitro data indicating that RA stimulates chondrogenesis by promoting chondrocyte maturation and transition to the hypertrophic state.32
Figure 2.
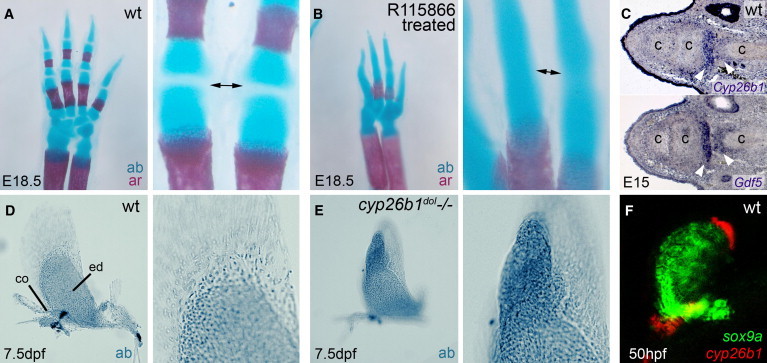
Cyp26b1 Inhibition Leads to Overgrowth or Fusion of Cartilaginous Elements in Developing Endochondral Bones of the Mouse Limb and Zebrafish Pectoral Fin
(A and B) Alcian blue (ab) and alizarin red (ar) staining of forelimbs of untreated E18.5 mice (A) and after treatment with the Cyp26 inhibitor R115866 from E13–16;9 (B) magnified views of the joint region between two phalanges (arrowed).
(C) Cyp26b1 and Gdf5 in situ hybridization on consecutive sections through developing digit of mouse forelimb at E15 indicating Cyp26b1 expression in the region corresponding to developing synovial joints.
(D and E) Alcian blue staining of cartilage of developing pectoral fin in wild-type (D) and cyp26b1 null mutant zebrafish, dolphin (dol) (E). In dol (E), the endochondral disc displays a massive outgrowth into distal fin regions, which normally lack cartilage (D).
(F) cyp26b1 (red) and sox9a (green) in the pectoral fin bud of wild-type zebrafish embryo. cyp26b1 is expressed adjacent to the distal border of the sox9a-expression domain. The following abbreviations are used: c, cartilage; co, scapulocoracoid; dpf, days postfertilization; ed, endochondral disc; hpf, hours postfertilization; wt, wild-type.
Partial Loss of Cyp26b1 Activity in Zebrafish Causes Coronal Craniosynostosis
Dysregulated chondrogenesis cannot be the mechanism underlying the craniosynostosis observed in the individual with the Ser146Pro mutation, however, because the calvarial plates are formed via dermal ossification without use of a cartilaginous intermediate. Because murine Cyp26b1−/− mutants lack a craniosynostosis phenotype and instead demonstrate calvarial hypoplasia8 (as did the p.Arg363Leu proband; see also Discussion), the human p.Ser146Pro phenotype is more comparable to the zebrafish hypomorphic mutant sst,10 which is semiviable and displays an abnormal craniofacial morphology analogous to this individual in addition to hyperossification of the skeleton.
Suture development in fish has only been described in outline.33 Normal zebrafish calvarial plate growth initiates laterally at ca. 4 weeks of age and proceeds rostromedially, forming the frontal, sagittal, and coronal sutures (Figures 3A–3D, 4C, and 4Q). The coronal suture is a wide, flexible joint formed by the frontal calvarial plate overlying the parietal plate at 6 weeks postfertilization that remains patent in fish throughout their lifetime (Figures 3E, 3F, and 4I, and 4O). As in mammals,34, 35 in situ hybridizations with col1a1, col10a, osteopontin (opn), and osteonectin (sparc)9, 21 labeled osteoblasts within the osteogenic fronts of growing zebrafish calvaria (Figure 3G) and later within the sutures (Figures 3H, 5I, 5K, and 5M). In contrast, cyp26b1 expression is only evident after suture formation and is restricted to the tips of the calvarial plates (Figures 3I and 3J). A similar expression at the tips of the calvarial plates was observed for Cyp26b1 in the coronal suture of newborn mice, pointing to a conserved function of this enzyme during vertebrate suture development (Figure S5).
Figure 3.
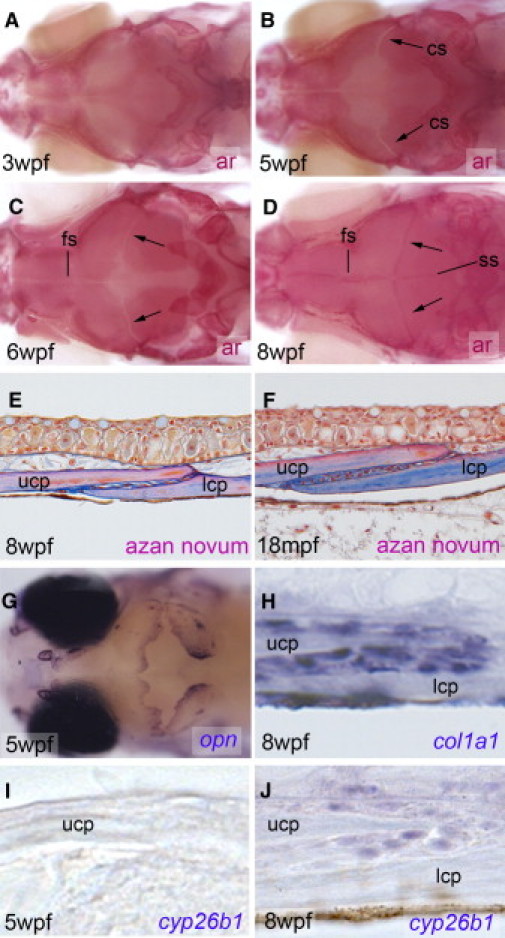
Cranial Suture Formation in Wild-Type Zebrafish
(A–D) Alizarin red (ar) staining of mineralized cranial bones. Dorsal views of the head. The calvarial plates grow toward the vertex of the head from anterior, posterior, and lateral aspects (A), and sutures are formed with a corresponding distal to proximal progression (C and D). At 8 weeks postfertilization (D), the interfrontal, sagittal, and coronal sutures are visible as dark red stripes because of the overlap of adjacent calvarial plates forming wide flexible joints.
(E and F) Azan novum staining of longitudinal sections through coronal suture, demonstrating the overlap of the lower and upper calvarial plates and the joint in the interzone. The flexible joint region also persists in aged fish (F), which is necessary because cranial growth persists over the entire zebrafish lifespan.
(G–J) in situ hybridization with indicated probes. (G) Dorsal view of the head, revealing strong opn expression at osteogenic fronts of calvarial plates as they grow toward each other. (H) Longitudinal section through coronal suture, revealing that col1a1 is expressed in a broader fashion than cyp26b1 (see below) in nearly all osteogenic cells (for even broader view at lower magnification see Figure 5I). (I and J) No cyp26b1 expression can be detected, whereas calvarial plates grow toward the cranial vertex and are still some distance from each other (I), whereas after suture formation, expression is restricted to the free edge of the calvarial plates (J). Ages of fish are indicated in lower left corners, stains, or in situ hybridization probes in lower right corners.
Figure 4.
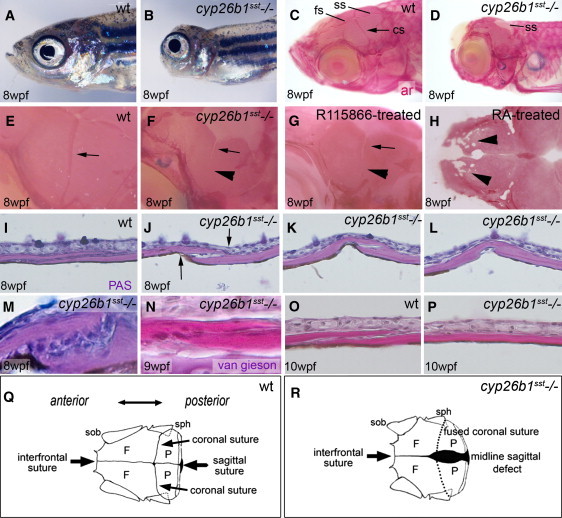
Coronal Craniosynostosis in Zebrafish Hypomorphic cyp26b1 Mutant sst Because of Elevated RA Levels
(A and B) Craniofacial morphology in (A) wild-type and (B) sst.
(C and D) Alizarin red (ar) staining of (C) wild-type and (D) sst mutant heads indicating sutures (arrows) and midline encephaloceles in sst (D).
(E and F) Magnified views of coronal sutures (cs; arrow) of animals in (C and D). (E) Normally forming cs; (F) fused lateral portion (arrowhead) of sst cs.
(G) Wild-type animal treated with R115866 from 5–6 wpf with coronal craniosynostosis identical to sst mutant.
(H) Wild-type animals treated with RA from 5–6 wpf. Note fragmented frontal bones (arrowhead).
(I–L) Longitudinal series of medial-to-lateral sections through wild-type (I) and sst (J–L) coronal sutures after PAS Reaction.
(M–P) Longitudinal sections through coronal sutures of wild-type (O) and sst mutant (M, N, and P) after PAS reaction (M) or van Gieson staining (N–P). (J–L) Suture fusion initiates at tips of calvarial plates (arrowheads) and progresses internally. (M and N) Magnified views revealing sutural cells progressively embedded in bony material. (O and P) Complete fusion of the coronal suture in the mutant without signs of embedded cells.
(Q) Diagram indicating rostral view of the zebrafish skull, outlining the position of the cranial sutures and calvarial bones; (R) diagram of a sst mutant skull showing a diminished anterior-posterior diameter, fused coronal sutures and a persistent midline sagittal defect that is most prominent posteriorly.
The following abbreviations are used: cs, coronal suture; F, frontal bone; fs, frontal suture; lcp, lower calvarial plate; P, parietal bone; PAS, periodic acid-Schiff reaction; sob, supraorbital bone; sph, sphenoid bone; ss, sagittal suture; ucp, upper calvarial plate, wpf, weeks postfertilization; wt, wild-type.
Figure 5.
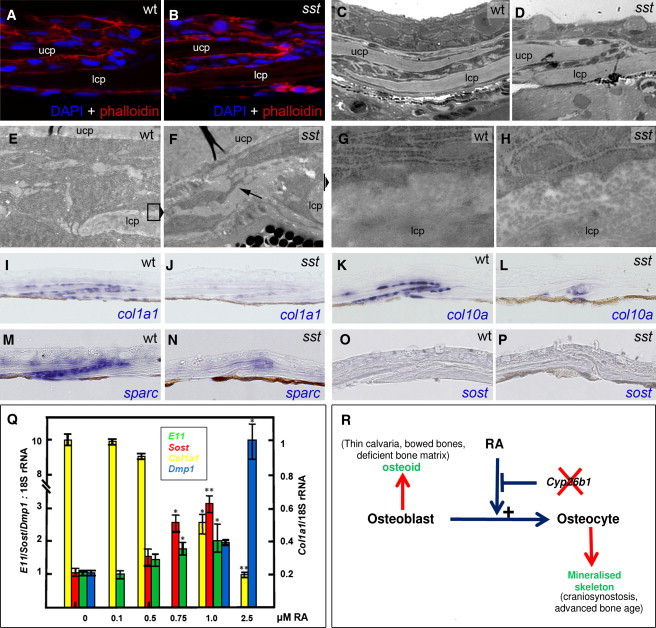
Coronal Sutural Cells in Zebrafish sst Mutants are Unaltered in Number but Demonstrate Aberrant Morphology and Gene Expression
(A–N) Sections through coronal sutures of wild-type siblings (A, C, E, G, I, K, M, and O) and sst mutants (B, D, F, H, J, L, N, and P) at 7.5 wpf, shortly before onset of sutural fusion. (A and B) DAPI and Alexa555-phalloidin staining revealing equivalent cell numbers in suture of (A) wild-type and (B) sst mutant fish. (C and D) Coronal suture (methylene blue) of individuals analyzed in (E–H). (E–H) TEM of the tip of the lower calvarial plate (lcp) in coronal suture; (E and F) 4400×. sst mutant cell with dendritic protrusions (F, arrow) compared to more globular wild-type cells (E). The positions of regions magnified in (g and h) are indicated. (G and H) 30,000×. Surface of upper side of lcp demonstrating reduced RER in osteogenic cell of sst mutant and reduced ECM/osteoid (H). (I–N) Downregulation of col1a1, col10a, and sparc expression in the suture of sst mutant.
(O and P) Lack of detectable sost expression in the suture of wild type and sst mutant via in situ hybridization.
(Q) RA treatment of mouse MC3T3 preosteoblasts causes downregulation of the osteoblast marker gene Col1a1 but upregulation of osteocyte markers Sost, E1 and Dmp1. The ratio of transcript abundance relative to 18S RNA ± SD is indicated; ∗p < 0.01; ∗∗p < 0.001.
(R) A model for the proposed effect of retinoic acid on osteoblast and osteocyte function in the vertebrate skeleton. Deleterious mutations in CYP26B1 accentuate the effect of RA on the promotion of differentiation of osteoblasts to osteocytes. Depending on the severity of the defect, phenotypic manifestations relating to deficient osteoblastic function (predominant effect of null alleles) or excessive osteocytic activity (more evident with hypomorphic alleles) are indicated (red).
Alizarin red bone staining of zebrafish sst mutants revealed a highly penetrant bilateral synostosis of the coronal suture at 10 weeks with fusion initiating laterally and progressing medially in a zipper-like fashion (Figures 4E, 4F, 4I–4L, and 4R), eventually resulting in closure of the entire suture (Figure 4M, 4N, and 4P). The sagittal suture remained widely open throughout (Figures 4C–4F, 4R), establishing cranial defects similar to those observed in the human p.Ser146Pro individual (Figure 1H). Coronal craniosynostosis was also obtained after treatment of wild-type fish with R115866 (Figure 4G) or with RA (data not shown). Escalation of RA treatment further induced porosity and thinning of the frontal bones (Figure 4H), similar to the human phenotype associated with the p.Arg363Leu mutation (Figure 1E). Serial longitudinal sections from medial-to-lateral revealed that the coronal synostosis of sst mutants begins at the two edges of the sutural plates (Figures 4J–4L), the same region that demonstrates maximal expression of cyp26b1 by osteogenic cells (Figure 3J). Magnified views further revealed that osteogenic sutural cells of sst mutants become embedded in bony material (Figures 4M and 4N). However, later, when fusion is complete, the former suture region is acellular and of normal calvarial morphology (Figures 4O and 4P), suggesting that embedded osteogenic cells have been lost and bone has been remodeled.
Sutural Cells of Cyp26b1 Hypomorphic Zebrafish Mutants and RA-Treated Preosteoblasts Manifest Accelerated Osteoblast-to-Osteocyte Transitioning
Analyses of suture sections, BrdU incorporation studies, and TUNEL staining of wild-type siblings and sst mutants shortly before the initiation of suture fusion revealed no significant alteration in the numbers, proliferative activity and apoptosis of sutural cells or aberrant morphology of the joint (Figures 5A–5D and data not shown). However, transmission electron microscopy showed striking differences in the shape and ultrastructure of sutural cells and in the organization of the extracellular matrix (ECM) at the free edges of the calvarial plates. Wild-type cells exhibited an osteoblastic morphology (globular, with massive rough endoplasmic reticulum [RER]) (Figures 5E and 5G), whereas sst sutures showed cells with a stellate shape, prominent cellular protrusions, and markedly reduced RER (Figures 5F and 5H) reminiscent of (pre)osteocytes.36, 37 The appearances of the ECM of sst mutants also suggested a marked reduction in matrix density (Figures 5G and 5H). Notably, the subject with the p.Arg363Leu substitution also demonstrated abnormalities of the osteoid matrix (Figure S2). Consistently, in situ hybridization revealed reduced expression of the osteoid collagen genes col1a1 and col10a (Figures 5I–5L) and the interstitial osteoid component sparc (osteonectin; Figures 5M and 5N) by sutural osteoblasts of sst mutants. In situ hybridizations performed in parallel with various osteocytic markers (sost, phex, fgf23, ankha, and ankhb)37 yielded inconclusive results; expression levels were below the detectable range in both sst mutants and wild-type siblings (Figures 5O and 5P and data not shown). These data are consistent with the notion that moderately elevated RA levels in sst mutants might be hastening the transition of sutural osteoblasts to preosteocytes. To test the potential of RA to promote osteoblast-osteocyte transitioning more directly, we employed an in vitro assay of osteoprogenitor cell differentiation. Treatment of murine MC3T3 preosteoblasts with escalating concentrations of RA not only led to a rapid and dose-dependent decline of Col1a1 expression but also to a progressive upregulation of osteocyte markers (Sost, E11/gp38, and Dmp1)37 (Figure 5Q). Collectively, these results suggest that the RA-induced sutural defect is characterized by a loss of osteoblastic and a gain of (pre)osteocyte-like characteristics of sutural cells and is accompanied by a reduction in osteoid production, the premature onset of mineralization, and the resultant craniosynostosis.
Discussion
Retinoic acid functions as a classical morphogen, establishing concentration gradients over developmental fields through regulation of its synthesis and degradation. Although animal models have been generated demonstrating that inactivation of synthetic and degradative enzymes have a drastic effect on morphogenesis,8, 9, 10, 38, 39, 40, 41 phenotypes that are known to be associated with abnormal regulation of RA metabolism in humans have not been described to date. Here, evidence is presented to show that deficiency in CYP26B1 action particularly impacts human skeletal development, both at the level of patterning and in the ossification of bone and the establishment of some synovial joints.
This work reinforces the observation that the roles that RA is known to play in skeletogenesis are multiple and include the regulation of genes that dictate antero-posterior and proximo-distal patterning of the limbs,7 chondrocyte proliferation, maturation and regulation of osteoblastic activity, and finally transitioning of osteoblasts to an osteocytic cell fate. Depending on the severity of the deficit conferred by the mutant alleles studied at this locus, the phenotypic effects can be variable.
A tentative model for how RA could mediate roles in mineralization is presented in Figure 5R. In this model, RA is proposed to promote osteoblastic differentiation to a preosteocytic/osteocytic cell fate. Surface osteoblasts mainly mediate the production of nonmineralized bone matrix (osteoid), whereas preosteocytes initiate and regulate matrix mineralization.42 The in vitro data presented here indicate that high levels of RA advance osteoblast-to-osteocyte transitioning to produce terminally differentiated osteocytes (Figure 5Q). Negative regulation of this effect by Cyp26b1 is necessary to promote osteoblast function, most notably the production of matrix osteoid at the growing edges of bones such as the calvarial plates or within the primary spongiosa immediately below the endochondral growth plate. Inappropriately accelerated maturation of osteoblasts could therefore conceivably result in the production of osteoid-deficient bone, whereas premature formation of osteocytic cells might result in accelerated mineralization of structures and manifest as advanced osseous maturation or craniosynostosis.
This model proposes that if the deficit in CYP26B1 activity is substantial, one of the consequences will be hypoplasia of the bones forming the calvarium. This might be the reason why both humans homozygous for the p.Arg363Leu mutation and Cyp26b1−/− mice do not display craniosynostosis, because frontal and parietal calvarial plates fail to grow to a size required for proper suture formation. However, Cyp26b1−/− mice do display fusions of other flat bones of the skull, such as the basioccipital and exoccipital bones of the skull base, that might not depend so much on lateral growth to form a sutural joint.8
In comparison, the effect in Cyp26b1 hypomorphic zebrafish mutants (sst) and in the individual with the p.Ser146Pro mutation is more attenuated. In this scenario, our model proposes that the residual Cyp26b1 activity appears sufficient to drive normal calvarial plate growth, whereas it is insufficient to maintain osteoblasts during later developmental stages in the coronal sutures themselves, structures that normally display highest Cyp26b1 expression (Figure 3J and Figure S5). Instead, moderately elevated concentrations of RA are proposed to lead to precocious osteoblast-osteocyte transitioning. Possibly this accelerated maturational effect advances cells only to a preosteocytic state37 because despite their (pre)osteocyte-characteristic morphology, we did not observe significantly increased osteocyte marker expression in sutural cells of zebrafish sst mutants. However, this effect is still sufficient to lead to increased bone mineralization and sutural synostosis.
It has been recently demonstrated that during early zebrafish bone development, RA has two distinct and seemingly contradictory effects on the osteoblast lineage: an early role to block the recruitment of osteoblasts from cephalic neural crest precursors and a later role in mature osteoblasts to promote bone matrix production,43 comparable to the described early negative and late positive effects of RA on chondroblast/chondrocyte differentiation.32 A similar negative effect of RA on the differentiation of postmigratory skeletogenic cephalic neural crest cells has also been discussed for mice.8 Interference mediated by Cyp26b1 mutations with this early role of RA would result in reduced numbers of osteoblasts formed from neural crest precursors and would provide an explanation for the bone deficiencies observed in Cyp26b1−/− mice8 and humans and in RA-treated zebrafish, whereas interference with the later role would result in premature osteoblast maturation, increased osteocytic activity, and the observed traits reflective of hyperossification. It is notable though that our previous observations that osteoblast numbers are not reduced in zebrafish cyp26b1 mutants9, 10 contradict this hypothesized negative role for RA in osteoprogenitor cell differentiation. More consistent with our data that demonstrate an ability of RA to promote the differentiation of preosteoblasts all the way to a mature osteocyte in a dose dependant fashion in vitro (Figure 5O) is a proposal that the different phenotypic traits observed under Cyp26b1 compromised conditions are all caused by an acceleration of osteoblast and osteocyte development and thus a general positive effect of elevated RA levels on bone-forming cells. Because osteoid is mainly generated by osteoblasts, whereas preosteocytes induce matrix mineralization,42 the hypoplasia of flat bones and developmental arrest of calvarial growth seen in Cyp26b1−/− mice8 and humans (Figure 1E) and the advanced skeletal maturation and craniosynostosis observed in individuals with hypomorphic mutations could all be attributed to a depletion of osteoblasts and an enrichment of (pre)osteocytic cells because of accelerated osteoblast-osteocyte transitioning.
Zebrafish cyp26b1 null mutant larvae (dol) also manifest fusions of centra, the vertebral precursors of the developing spine, most likely attributable to a precocious maturation and/or hyperactivity of osteoblasts affecting early processes in the life of their cellular precursors.9, 10 It remains puzzling why a corresponding spine defect does not occur in Cyp26b1−/− mice and humans. Cyp26b1−/− mice display fusions of the two first cervical vertebrae, the atlas, and the axis; these fusions have been attributed to shifts in anterior-posterior patterning, rather than hyperossification.8 Furthermore, treatment of mice with the Cyp26 inhibitor R115866 causes fusions of the neural arches of cervical vertebra, and premature fusions between the neural arches of thoracic and sacral vertebrae, but no fusions between the vertebral centra.9 Interspecies differences in cyp26 gene-expression patterns between fish and mammals might account for some of these differential responses. Another reason might be interspecies differences in the mode of centra formation. Mammalian vertebral centra form via endochondral ossification, whereas fish centra are formed via mineralization of the notochord sheath, a thick basement membrane-like structure formed by notochord cells that lacks a recognized contribution from chondrocytes.9, 10 Furthermore, there are data suggesting that osteoblasts involved in ossification of fish centra represent a distinct population of osteogenic cells, with a particular molecular signature,9, 10 and therefore possibly particularly sensitive to RA.
Functional relationships between RA/Cyp26b1 and other factors implicated in cranial suture patency are likely.35 Antley-Bixler syndrome (ABS [MIM 201750]) is an autosomal-recessive condition in humans with many similarities to the conditions described here. Craniosynostosis, radiohumeral joint fusions and arachnodactyly are all common manifestations of ABS in addition to genital anomalies associated with abnormal sex steroid profiles. ABS is caused by mutations in POR, which encodes P450 oxidoreductase, an electron donor/redox partner for all human type II CYP450 enzymes, including CYP26B1. Whereas the abnormalities of sex steroid metabolism are accounted for by functional insufficiency of CYP450 enzymes known to mediate metabolism of these compounds,27 the pathophysiology that underlies the skeletal anomalies in ABS remains obscure. Our data indicate that the joint fusions and craniosynostosis in ABS could be attributed to compromised CYP26B1 activity and elevated RA44 rather than impaired metabolism of other POR substrates, such as cholesterol, as has previously been suggested.27, 45
How deficits in RA degradation integrate with genes mutated to cause other human craniosynostosis syndromes, such as TWIST1 (MIM 601622) and the fibroblast growth factor (FGF) receptor genes FGFR1-3, is uncertain.35 At a morphological level, coronal craniosynostosis is the most common outcome of mutations at these loci46, 47 because it is in the condition we studied here in humans and zebrafish, and the sagittal suture remains widely patent. These observations suggest that the cell biologies of the sagittal and coronal sutures are distinct from one another. It is also notable that craniosynostosis-associated FGFR1-3 mutations render FGF signaling constitutively active, and recent in vitro studies have shown that Fgf2 induces an osteocyte-like phenotype,25 similar to the effect of RA described here. RA and FGFs could therefore interact to promote the osteoblast-to-osteocyte transition in a subset of cranial sutures, in contrast to their opposing effects in other developmental contexts.48
At a broader level our observations could have implications for bone health. Osteocytes are increasingly being implicated in the regulation of bone mineral density throughout the lifespan,49, 50 and our demonstration that RA can hasten the differentiation of osteoblasts to osteocytes, resulting in a bone matrix with reduced osteoid and accelerated mineralization, implicates RA as of potential relevance in the maintenance of bone-mineral homeostasis.
Acknowledgments
The families are thanked for their participation. Stefan Schulte-Merker is thanked for the zebrafish sst mutant and Christine Hartmann and Yingzi Yang are thanked for sending plasmids for the mouse probe synthesis. This work was supported by the German Research Foundation (DFG; SFB 572) (M.H.), the European Union (Seventh Framework Programme, Integrated Project ZF-HEALTH, EC Grant Agreement HEALTH-F4-2010-242048) (M.H.), the German Federal Ministry of Education and Research (BMBF; grant 01GM0801; E-RARE network CRANIRARE) (B.W.) and Curekids and the Marsden Fund (S.P.R.). Evelin Fahle, Christel Schenkel, Iris Riedl-Quinkertz, Mogdan Ghilav, Boidinh Chung, and Dan Mornin are thanked for their assistance. K.L. thanks Gerard Karsenty for support.
Published online: October 20, 2011
Footnotes
Supplemental Data include five figures and one table and can be found with this article online at http://www.cell.com/AJHG/.
Contributor Information
Matthias Hammerschmidt, Email: matthias.hammerschmidt@uni-koeln.de.
Stephen P. Robertson, Email: stephen.robertson@otago.ac.nz.
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC genome browser, http://www.genome.ucsc.edu/
Supplemental Data
References
- 1.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwasigroch T.E., Bullen M. Effects of isotretinoin (13-cis-retinoic acid) on the development of mouse limbs in vivo and in vitro. Teratology. 1991;44:605–616. doi: 10.1002/tera.1420440603. [DOI] [PubMed] [Google Scholar]
- 3.James A.W., Levi B., Xu Y., Carre A.L., Longaker M.T. Retinoic acid enhances osteogenesis in cranial suture-derived mesenchymal cells: Potential mechanisms of retinoid-induced craniosynostosis. Plast. Reconstr. Surg. 2010;125:1352–1361. doi: 10.1097/PRS.0b013e3181d62980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yip J.E., Kokich V.G., Shepard T.H. The effect of high doses of retinoic acid on prenatal craniofacial development in Macaca nemestrina. Teratology. 1980;21:29–38. doi: 10.1002/tera.1420210105. [DOI] [PubMed] [Google Scholar]
- 5.Fujii H., Sato T., Kaneko S., Gotoh O., Fujii-Kuriyama Y., Osawa K., Kato S., Hamada H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997;16:4163–4173. doi: 10.1093/emboj/16.14.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White J.A., Beckett-Jones B., Guo Y.D., Dilworth F.J., Bonasoro J., Jones G., Petkovich M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J. Biol. Chem. 1997;272:18538–18541. doi: 10.1074/jbc.272.30.18538. [DOI] [PubMed] [Google Scholar]
- 7.Yashiro K., Zhao X., Uehara M., Yamashita K., Nishijima M., Nishino J., Saijoh Y., Sakai Y., Hamada H. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell. 2004;6:411–422. doi: 10.1016/s1534-5807(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 8.Maclean G., Dollé P., Petkovich M. Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dyn. 2009;238:732–745. doi: 10.1002/dvdy.21878. [DOI] [PubMed] [Google Scholar]
- 9.Laue K., Jänicke M., Plaster N., Sonntag C., Hammerschmidt M. Restriction of retinoic acid activity by Cyp26b1 is required for proper timing and patterning of osteogenesis during zebrafish development. Development. 2008;135:3775–3787. doi: 10.1242/dev.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spoorendonk K.M., Peterson-Maduro J., Renn J., Trowe T., Kranenbarg S., Winkler C., Schulte-Merker S. Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development. 2008;135:3765–3774. doi: 10.1242/dev.024034. [DOI] [PubMed] [Google Scholar]
- 11.Kühnel K., Ke N., Cryle M.J., Sligar S.G., Schuler M.A., Schlichting I. Crystal structures of substrate-free and retinoic acid-bound cyanobacterial cytochrome P450 CYP120A1. Biochemistry. 2008;47:6552–6559. doi: 10.1021/bi800328s. [DOI] [PubMed] [Google Scholar]
- 12.Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007;Chapter 2 doi: 10.1002/0471140864.ps0209s50. Unit 2, 9. [DOI] [PubMed] [Google Scholar]
- 13.Roy A., Kucukural A., Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis I.W., Murray L.W., Richardson J.S., Richardson D.C. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32(Web Server issue) doi: 10.1093/nar/gkh398. W615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kessel M., Gruss P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell. 1991;67:89–104. doi: 10.1016/0092-8674(91)90574-i. [DOI] [PubMed] [Google Scholar]
- 16.Walker M.B., Kimmel C.B. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem. 2007;82:23–28. doi: 10.1080/10520290701333558. [DOI] [PubMed] [Google Scholar]
- 17.Hammerschmidt M., Pelegri F., Mullins M.C., Kane D.A., van Eeden F.J.M., Granato M., Brand M., Furutani-Seiki M., Haffter P., Heisenberg C.-P., et al. dino and mercedes, two genes regulating dorsal development in the zebrafish embryo. Development. 1996;123:95–102. doi: 10.1242/dev.123.1.95. [DOI] [PubMed] [Google Scholar]
- 18.Clay H., Ramakrishnan L. Multiplex fluorescent in situ hybridization in zebrafish embryos using tyramide signal amplification. Zebrafish. 2005;2:105–111. doi: 10.1089/zeb.2005.2.105. [DOI] [PubMed] [Google Scholar]
- 19.Lescher B., Haenig B., Kispert A. sFRP-2 is a target of the Wnt-4 signaling pathway in the developing metanephric kidney. Dev. Dyn. 1998;213:440–451. doi: 10.1002/(SICI)1097-0177(199812)213:4<440::AID-AJA9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Brunet L.J., McMahon J.A., McMahon A.P., Harland R.M. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- 21.Fisher S., Jagadeeswaran P., Halpern M.E. Radiographic analysis of zebrafish skeletal defects. Dev. Biol. 2003;264:64–76. doi: 10.1016/s0012-1606(03)00399-3. [DOI] [PubMed] [Google Scholar]
- 22.Nica G., Herzog W., Sonntag C., Nowak M., Schwarz H., Zapata A.G., Hammerschmidt M. Eya1 is required for lineage-specific differentiation, but not for cell survival in the zebrafish adenohypophysis. Dev. Biol. 2006;292:189–204. doi: 10.1016/j.ydbio.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 23.Yan Y.L., Willoughby J., Liu D., Crump J.G., Wilson C., Miller C.T., Singer A., Kimmel C., Westerfield M., Postlethwait J.H. A pair of Sox: Distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development. 2005;132:1069–1083. doi: 10.1242/dev.01674. [DOI] [PubMed] [Google Scholar]
- 24.Rotllant J., Liu D., Yan Y.L., Postlethwait J.H., Westerfield M., Du S.J. Sparc (Osteonectin) functions in morphogenesis of the pharyngeal skeleton and inner ear. Matrix Biol. 2008;27:561–572. doi: 10.1016/j.matbio.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta R.R., Yoo D.J., Hebert C., Niger C., Stains J.P. Induction of an osteocyte-like phenotype by fibroblast growth factor-2. Biochem. Biophys. Res. Commun. 2010;402:258–264. doi: 10.1016/j.bbrc.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flück C.E., Tajima T., Pandey A.V., Arlt W., Okuhara K., Verge C.F., Jabs E.W., Mendonça B.B., Fujieda K., Miller W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004;36:228–230. doi: 10.1038/ng1300. [DOI] [PubMed] [Google Scholar]
- 27.Scott R.R., Miller W.L. Genetic and clinical features of p450 oxidoreductase deficiency. Horm. Res. 2008;69:266–275. doi: 10.1159/000114857. [DOI] [PubMed] [Google Scholar]
- 28.Hatae T., Hara S., Yokoyama C., Yabuki T., Inoue H., Ullrich V., Tanabe T. Site-directed mutagenesis of human prostacyclin synthase: Alteration of Cys441 of the Cys-pocket, and Glu347 and Arg350 of the EXXR motif. FEBS Lett. 1996;389:268–272. doi: 10.1016/0014-5793(96)00600-x. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu T., Tateishi T., Hatano M., Fujii-Kuriyama Y. Probing the role of lysines and arginines in the catalytic function of cytochrome P450d by site-directed mutagenesis. Interaction with NADPH-cytochrome P450 reductase. J. Biol. Chem. 1991;266:3372–3375. [PubMed] [Google Scholar]
- 30.Hasemann C.A., Kurumbail R.G., Boddupalli S.S., Peterson J.A., Deisenhofer J. Structure and function of cytochromes P450: A comparative analysis of three crystal structures. Structure. 1995;3:41–62. doi: 10.1016/s0969-2126(01)00134-4. [DOI] [PubMed] [Google Scholar]
- 31.Abu-Abed S., MacLean G., Fraulob V., Chambon P., Petkovich M., Dollé P. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech. Dev. 2002;110:173–177. doi: 10.1016/s0925-4773(01)00572-x. [DOI] [PubMed] [Google Scholar]
- 32.Weston A.D., Hoffman L.M., Underhill T.M. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res. C Embryo Today. 2003;69:156–173. doi: 10.1002/bdrc.10010. [DOI] [PubMed] [Google Scholar]
- 33.Quarto N., Longaker M.T. The zebrafish (Danio rerio): A model system for cranial suture patterning. Cells Tissues Organs (Print) 2005;181:109–118. doi: 10.1159/000091100. [DOI] [PubMed] [Google Scholar]
- 34.Opperman L.A. Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 2000;219:472–485. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 35.Morriss-Kay G.M., Wilkie A.O. Growth of the normal skull vault and its alteration in craniosynostosis: Insights from human genetics and experimental studies. J. Anat. 2005;207:637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franz-Odendaal T.A., Hall B.K., Witten P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006;235:176–190. doi: 10.1002/dvdy.20603. [DOI] [PubMed] [Google Scholar]
- 37.Dallas S.L., Bonewald L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N Y Acad. Sci. 2010;1192:437–443. doi: 10.1111/j.1749-6632.2009.05246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niederreither K., Subbarayan V., Dollé P., Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999;21:444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- 39.Niederreither K., Vermot J., Schuhbaur B., Chambon P., Dollé P. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development. 2002;129:3563–3574. doi: 10.1242/dev.129.15.3563. [DOI] [PubMed] [Google Scholar]
- 40.Mic F.A., Sirbu I.O., Duester G. Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation. J. Biol. Chem. 2004;279:26698–26706. doi: 10.1074/jbc.M401920200. [DOI] [PubMed] [Google Scholar]
- 41.Abu-Abed S., Dollé P., Metzger D., Beckett B., Chambon P., Petkovich M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15:226–240. doi: 10.1101/gad.855001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonewald L.F., Dallas S.L., Gorski J.P. In: The skeletal system. Pourquié O., editor. Cold Spring Harbor Laboratory Press; New York: 2009. Bone Mineralization; pp. 277–295. [Google Scholar]
- 43.Li N., Kelsh R.N., Croucher P., Roehl H.H. Regulation of neural crest cell fate by the retinoic acid and Pparg signalling pathways. Development. 2010;137:389–394. doi: 10.1242/dev.044164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribes V., Otto D.M., Dickmann L., Schmidt K., Schuhbaur B., Henderson C., Blomhoff R., Wolf C.R., Tickle C., Dollé P. Rescue of cytochrome P450 oxidoreductase (Por) mouse mutants reveals functions in vasculogenesis, brain and limb patterning linked to retinoic acid homeostasis. Dev. Biol. 2007;303:66–81. doi: 10.1016/j.ydbio.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt K., Hughes C., Chudek J.A., Goodyear S.R., Aspden R.M., Talbot R., Gundersen T.E., Blomhoff R., Henderson C., Wolf C.R., Tickle C. Cholesterol metabolism: The main pathway acting downstream of cytochrome P450 oxidoreductase in skeletal development of the limb. Mol. Cell. Biol. 2009;29:2716–2729. doi: 10.1128/MCB.01638-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kan S.H., Elanko N., Johnson D., Cornejo-Roldan L., Cook J., Reich E.W., Tomkins S., Verloes A., Twigg S.R., Rannan-Eliya S., et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am. J. Hum. Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson D., Wilkie A.O. Craniosynostosis. Eur. J. Hum. Genet. 2011;19:369–376. doi: 10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diez del Corral R., Storey K.G. Opposing FGF and retinoid pathways: A signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays. 2004;26:857–869. doi: 10.1002/bies.20080. [DOI] [PubMed] [Google Scholar]
- 49.Kramer I., Halleux C., Keller H., Pegurri M., Gooi J.H., Weber P.B., Feng J.Q., Bonewald L.F., Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 2010;30:3071–3085. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tatsumi S., Ishii K., Amizuka N., Li M., Kobayashi T., Kohno K., Ito M., Takeshita S., Ikeda K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.