

Abstract
Abdominal aortic aneurysm (AAA) is a common cause of morbidity and mortality and has a significant heritability. We carried out a genome-wide association discovery study of 1866 patients with AAA and 5435 controls and replication of promising signals (lead SNP with a p value < 1 × 10−5) in 2871 additional cases and 32,687 controls and performed further follow-up in 1491 AAA and 11,060 controls. In the discovery study, nine loci demonstrated association with AAA (p < 1 × 10−5). In the replication sample, the lead SNP at one of these loci, rs1466535, located within intron 1 of low-density-lipoprotein receptor-related protein 1 (LRP1) demonstrated significant association (p = 0.0042). We confirmed the association of rs1466535 and AAA in our follow-up study (p = 0.035). In a combined analysis (6228 AAA and 49182 controls), rs1466535 had a consistent effect size and direction in all sample sets (combined p = 4.52 × 10−10, odds ratio 1.15 [1.10–1.21]). No associations were seen for either rs1466535 or the 12q13.3 locus in independent association studies of coronary artery disease, blood pressure, diabetes, or hyperlipidaemia, suggesting that this locus is specific to AAA. Gene-expression studies demonstrated a trend toward increased LRP1 expression for the rs1466535 CC genotype in arterial tissues; there was a significant (p = 0.029) 1.19-fold (1.04–1.36) increase in LRP1 expression in CC homozygotes compared to TT homozygotes in aortic adventitia. Functional studies demonstrated that rs1466535 might alter a SREBP-1 binding site and influence enhancer activity at the locus. In conclusion, this study has identified a biologically plausible genetic variant associated specifically with AAA, and we suggest that this variant has a possible functional role in LRP1 expression.
Introduction
Abdominal aortic aneurysm (AAA [MIM 100070]) is a common disease and is responsible for around 13,000 deaths in the USA per annum; it predominantly affects white populations. Family history of AAA is a strong risk-factor for AAA and individuals with a first-degree relative with AAA have between a 2- to 11-fold increased risk of developing an AAA themselves.1, 2 The heritability of AAA has been estimated in independent studies to be as high as 0.7.3, 4, 5 This strong genetic determination in the etiology of AAA has led to multiple candidate gene association studies of AAA, the majority of which have yielded findings that have not been replicated in independent cohorts, but with meta-analysis suggesting common variants in ACE (MIM 106180), MTHFR (MIM 607093), and MMP9 (MIM 120361) might all be contributory.6 Two replicated loci for AAA have been identified on 9p21.37 (which is also associated with coronary artery disease [MIM 611139] and intracranial aneurysm [MIM 611892]) and in DAB2IP (MIM 609205) on 9q33.2 (also associated with early-onset myocardial infarction, venous thromboembolism, and peripheral arterial disease).8 Other risk factors for AAA include age, male gender, and tobacco smoking.9, 10, 11 By contrast, diabetes has been shown to be a negative risk factor for AAA,11 whereas hypertension and hypercholesterolaemia, which are strong risk factors for other arterial diseases, are inconsistent risk factors for AAA.
The strong heritability of AAA and the low odds ratio for currently identified risk alleles suggests that other important risk loci for AAA remain to be identified. In an effort to identify new risk alleles for AAA, we undertook a GWAS in 1866 patients with AAA and 5435 controls and replication of promising signals in 2871 additional cases and 32,687 controls and performed a further follow-up study of 1491 AAA and 11,060 controls.
Material and Methods
Study Design and Subjects
We assembled cases of AAA together with available DNA recruited in eight centers in the UK, Australia, and New Zealand and also samples from the United Kingdom Small Aneurysm Trial (Table S1, available online) none of which had been used in any previous genome-wide association study (GWAS) of AAA cases were recruited from inpatient populations, outpatient clinics, or population screening programs in the participating centers. AAA ascertainment (infrarenal aortic diameter of >30 mm) was by either ultrasonography or by cross-sectional imaging except for patients who presented with acute rupture and for whom it was assumed that the AAA was >5.5 cm. Some of the centers also collected controls screened as being free of AAA by ultrasonography (Table S1). In addition to these assembled samples, control data from the Wellcome Trust Case Control Consortium (WTCCC) 2 study were obtained. The study flow is shown in Figure S1. All samples contributing to this study were collected under the approval of the ethics committee responsible for each center and informed consent was obtained from all individuals.
A genome-wide discovery study was performed by comparing data from 1866 cases with AAA and 5435 unscreened controls from the WTCCC2 study consisting of samples from the 1958 British Birth Cohort and from the UK National Blood Service. The lead SNP for each locus identified in the discovery study with a p value less than 1 × 10−5 was tested for replication by genotyping 1579 independent AAA cases and 2184 screened controls. In silico replication data for these lead SNPs from the discovery study was obtained from a previously published, independent GWAS of 1292 individuals with AAA and 30,503 controls.8 A follow-up study of SNPs demonstrating association with AAA in the replication studies was performed in independent samples (11,090 controls and 1491 AAA) from the Copenhagen City Heart Study and the Viborg Country AAA screening program and samples collected by two of the contributing centers during the conduct of the discovery and replication studies.
Genotyping
DNA samples for the discovery and replication studies were processed at the Wellcome Trust Sanger Institute. Genomic DNA was quantified by PicoGreen assay and quality control assured by both agarose gel electrophoresis and Sequenom iPLEX genotyping of 29 SNPs and four gender markers. Genotyping for the discovery study was performed with Illumina 1.2M (controls) or 670K (AAA) BeadChips. Raw intensity data were normalized with BeadStudio, and genotypes were called concurrently from the combined case control data set with the Illuminus algorithm.12 Replication study genotyping was performed with Sequenom's iPLEX assay. Follow-up studies of rs1466535 in the samples from Copenhagen, Leeds, Otago, and Viborg were performed with the TaqMan allelic discrimination method (with a predesigned, functionally tested assay). All genomic information was referenced to genome build 37.1/dbSNP build 132.
Quality Control
Quality control (QC) in the discovery study was first performed by exclusion of SNPs with call rates of less than 0.98 and those that demonstrated significant deviation from Hardy-Weinberg equilibrium in the control group (p < 0.0001). Duplicate samples and those that failed genotyping (sample call rate 0.98) were also excluded from further analysis. Genotyping cluster plots for all SNPs with p values less than 1 × 10−4 were visually inspected to exclude from further analysis positive associations generated by erroneous genotyping or calling. Checks for population stratification were first performed by PLINK13 identical-by-state clustering and extreme outliers were removed from the analysis. Further checks for population stratification were performed by multidimensional scaling and subsequent analyses were adjusted for the dimensions of this analysis. In total, 29,826 SNPs and 191 individuals (23 AAA and 168 controls) were excluded from the analysis as a result of quality control measures.
Association Analysis
Statistical analyses were performed with PLINK.13 The association between genotype and AAA was tested with a Cochran-Armitage test for trend in the discovery, replication, and follow-up studies, and Fishers method was used to combine p values in the replication studies. Two-tailed Cochran-Armitage tests were applied in all cases. The threshold chosen for statistical significance was adjusted with a Bonferroni correction in the replication analysis. Although this study was not powered to detect gene-environment interactions, we performed additional exploratory analyses to determine the independence of the SNPs identified from other risk factors for AAA. Adjusted analyses for SNPs of interest were carried out with a genotypic logistic regression model with the presence or absence of individual risk factors entered as covariates into separate models to examine each risk factor.
Imputation
Focused regional imputation was used to infer genotypes for SNPs previously associated with AAA where these SNPs (or any SNP in perfect linkage disequilibrium [LD]) were not directly typed or had failed to meet quality control thresholds (rs1333049). Imputation was conducted with MACH 1.014 run on the BC|SNPmax database platform (version 3.5, BC Platforms). The reference haplotypes were the 1000 Genomes Project (June 2010), CEU population (Utah residents with ancestry from Northern and Western European ancestry from the CEPH collection). Subsequent calls were filtered by quality score (>0.9) to identify high quality SNP's.
Association Analysis of rs1466535 with Cardiovascular Risk Factors and Coronary Disease
In order to determine whether rs1466535 was associated with cardiovascular risk factors or coronary disease, association results for rs1466525 were obtained from large-scale GWAS meta-analyses examining blood lipid levels,15 diabetes,16 hypertension,17 and coronary artery disease.18
Expression Analyses
To investigate whether there was any effect of rs1466535 on LRP1 expression in relevant tissues, we examined data from an existing genome-wide eQTL study of samples of ascending aorta, mammary artery, and liver taken from patients undergoing aortic valve surgery.19 Aortic biopsies were divided into intimal and adventitial halves. Periaortic fat was removed from the adventitial specimens where present. RNA from the tissue biopsies was hybridized to Affymetrix ST 1.0 Exon arrays and obtained scans were RMA normalized and log2 transformed. eQTL analysis was performed with genotypes from circulating blood DNA (Illumina 610w-Quad BeadArrays). Estimates of rs1466535 genotype effect on LRP1 expression was calculated with an additive linear model. No correction for multiple hypothesis testing was performed. Expression analyses were conducted with Bioconductor 2.7.20 Full methods for this study have been described previously.19
Potential Functional SNPs at 12q13.3
To assess whether rs1466535 is linked to any potential functional SNPs, we first identified all SNPs in LD with rs1466535 (r2 threshold > 0.2) with SNAP21 and the 1000 Genomes Project pilot 1 data set. Potential functionalilty of these variants (splice-site variation or peptide-shift) was then assessed by querying dbSNP 132 with Biomart.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assays (EMSAs) were performed to determine whether rs1466535 had any effect on localized DNA-protein interaction. Nuclear extracts were obtained from Huh7 cells with the NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Pierce, Northumberland, UK), with the addition of Complete Protease Inhibitor (Roche, West Sussex, UK) to buffers CER I and NER I. EMSA probes were designed with ∼15 bp on each side of the candidate SNP. Probes were labeled with the Biotin 3′ End DNA Labeling Kit (Pierce, UK). Probes were analyzed in isolation and bound to unlabelled probe competitors, a sterol regulatory element-binding protein 1 (SREBP-1)22 competitor and a random oligonucleotide competitor. Samples were loaded onto a 6% polyacrylamide gel and electrophoresed for 150 min at 120 V at 4°C. Transfer to positively charged nylon membrane was achieved through Southern transfer and detection with the Chemiluminescent Nucleic Acid Detection Module (Pierce, UK). Each binding reaction consisted of 2 μl of 10× binding buffer (100 mm Tris, 500 mm KCl [pH 7.5]), 1 μg of p[dI-dC], 5 μg of nuclear extract, and 200 fmol of biotin-labeled DNA made to a total of 20 μl with H2O and incubated at 25°C for 30 min, followed by the addition of 5× loading buffer. Primers for rs1466535 were designed based on genomic sequences surrounding the SNP (Table S2).
Luciferase Assay
A LRP1 promoter construct (previously described22) was kindly provided by Dr. Llorente-Cortés. The LRP1 promoter (+421 to −1350) was inserted into the restriction sites (NheI and HindIII) of the pGL3-basic vector polylinker site. A rs1466535 intron 2 construct (C genotype) was developed by PCR performed on 30 ng genomic DNA with 1× NH4 polmix (16 mM (NH4)2SO4, 67 mM Tris [pH 8.3], 0.01% Tween 20, 0.2 mM dNTPs each), 3 mM MgCl2, 0.1U Taq Polymase Bioline and 2 pmol forward and reverse primers (Tables S3 and S4). PCR conditions were as followed: 1 cycle at 95°C for 2 min; 30 cycles at 95°C for 30 s, at 62°C for 30 s, and at 72°C for 30 s; followed by 1 cycle at72°C for 5 min. This resulted in a 240 bp fragment, in the forward and reverse orientation. PCR products were ligated into the 3′ end of the LRP1 promoter clone at the SalI and BamHI sites with T4 ligase (NEB) according to manufacturer guidelines and then transformed into DH5α. To create the alternate variant (T genotype), we performed site directed mutagenesis by using the Quick Change Lightning Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer. All new clones were checked by direct sequencing.
Huh 7 immortal hepatocellular carcinoma cells were chosen for the luciferase assay because these cells contain large quantities of SREBP-1. Huh7 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) High Glucose (4.5 g/l), supplemented with 10% fetal bovine serum. Cultures were grown and maintained at 37°C containing 5% CO2. Cells were plated out into a 96-well plate at a concentration of 2 × 104 per well the day before transfection to be 70% confluent at the time of transfection. Transfection was carried out with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were lysed 48 hr after transfection. Luciferase assays were carried out with the Dual Luciferase Reporter Assay System from Promega according to the manufacturer's instructions. Each construct was replicated 12 times in five different experiments. Luciferase activity was normalized by the Renilla values and is expressed in normalized fold change activity to the LRP1 promoter construct, which does not include the intron 2 variant. Mean relative expression differences were calculated with a t test.
Results
Discovery Stage
Following stringent quality control, we tested 535,296 SNPs for association with AAA in 1866 cases (mean AAA size, 4.8 cm and median age, 72 years) and 5435 unscreened controls (median age in the UK Blood Transfusion Service samples was 45 years and all individuals in the 1958 Birth Cohort were 52 years old at the time of analysis). Identical-by-state clustering identified 23 genomic outliers in the AAA group and 168 in the control group that were excluded from further analyses. Further checks for population stratification were performed by multidimensional scaling (Figure S2) that revealed potential stratification and the analysis was therefore adjusted for the first six dimensions from this analysis. The genomic inflation factor (λ) was calculated as 1.096 following these exclusions (Figure S3), and an adjustment for this was applied in subsequent analyses. Following these adjustments, nine loci demonstrated association with AAA at a p value threshold of 1 × 10−5 (Figure 1 and Table S5) and were taken forward for replication.
Figure 1.
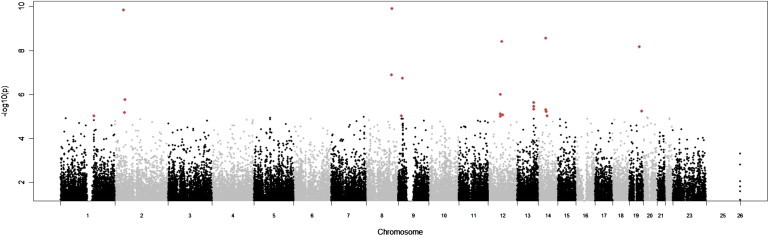
Whole-Genome Manhattan Plot of the Results of the Discovery Study
p values expressed as a negative logarithm are plotted on the vertical axis and chromosomal positions of each SNP are plotted on the horizontal axis. SNPs with p values < 1 × 10−5 are shown in red, and represent the loci taken forward into the replication study.
Replication Stage
The discovery genome-wide association analysis identified nine loci that demonstrated an association with AAA with a p value < 1 × 10−5, and one lead SNP from each of these loci was taken forward for replication. Replication was performed in 2871 cases and 32,687 controls, comprising 1579 cases (mean AAA size, 5.4 cm and median age, 72 years) and 2184 screened controls (median age, 68 years) who underwent wet-lab replication and 1292 cases and 30,503 controls with in silico data8 (Table S5). rs1466535 (risk-allele frequency [C] = 0.68 and discovery p value 9.99 × 10−7) at 12q13.3 demonstrated significant association in the replication study (p = 4.2 × 10−3) allowing for multiple testing (Bonferroni corrected replication study p value threshold 0.0056) (Table 1). The regional discovery study association plot and LD plot for 12q13.3 are shown in Figure 2.
Table 1.
Association of rs1466535 in LRP1 on Chromosomal Region 12q13.3 with AAA
|
Controls |
AAA |
OR (95% CI) | p value | Phet | |||
|---|---|---|---|---|---|---|---|
| N | AF | n | AF | ||||
| Genome-wide analysis | |||||||
| Discovery | 5435 | 0.63 | 1866 | 0.68 | 1.22 (1.13–1.32) | 9.99 × 10−7 | |
| Replication | |||||||
| Iceland | 27,712 | 0.58 | 452 | 0.61 | 1.12 (0.98–1.28) | 0.10 | |
| Netherlands | 2791 | 0.65 | 840 | 0.68 | 1.14 (1.02–1.28) | 0.026 | |
| Laboratory | 2184 | 0.65 | 1579 | 0.68 | 1.12 (1.02–1.23) | 0.02 | |
| Combined replication studies | 1.13 (1.06–1.19) | 0.0042 | 0.97 | ||||
| Combined discovery and replication studies | 1.16 (1.11–1.23) | 2.86 × 10−9 | 0.49 | ||||
| Follow-up studies | |||||||
| Leeds | 254 | 0.64 | 216 | 0.67 | 1.12 (0.95–1.33) | 0.38 | |
| Viborg | 196 | 0.64 | 503 | 0.66 | 1.07 (0.94–1.22) | 0.60 | |
| Otago | 430 | 0.61 | 579 | 0.66 | 1.23 (1.04–1.45) | 0.028 | |
| Copenhagen | 10,180 | 0.63 | 193 | 0.65 | 1.04 (0.96–1.12) | 0.64 | |
| Combined follow-up studies | 1.11 (1.01–1.23) | 0.04 | 0.60 | ||||
| Overall combined analysis (6228 AAA, 49,182 controls) | 1.15 (1.10–1.21) | 4.52 × 10−10 | 0.67 | ||||
Results for the genome-wide analysis, replication, and follow-up studies of the SNP rs1466535 in LRP1 associated with AAA. Allele frequencies (AF) shown are for the risk (C) allele. Association testing was performed with the Cochran-Armitage trend test (two-tailed) analysis in the discovery study corrected for a genomic inflation factor of 1.096 and the first six components of a multidimensional scaling analysis. Two-tailed Cochran-Armitage trend tests were used in the replication and follow-up studies. The genome-wide discovery study was performed comparing a pooled AAA case set from contributing centers with the WTCCC2 unscreened controls. The laboratory replication study was performed with a pooled case and control set (not all centers contributed both cases and controls to the analysis). The control samples used in the genome-wide analysis, and the studies from Iceland and Copenhagen were not screened for the presence of AAA. The combined analysis was performed by generic inverse variance weighted meta-analysis with a fixed effects model (Phet is the p value for Cochrans Q test for heterogeneity). The overall combined analysis of the discovery, replication and follow-up studies is shown in bold type.
Figure 2.
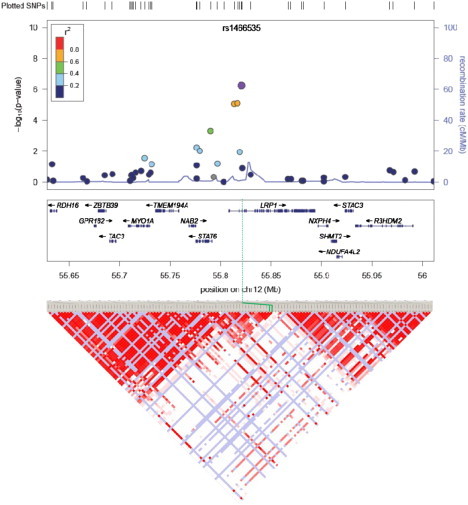
GWAS Discovery Study Associations for the Region around rs1466535 Showing All Genes in the Region
rs1466535 is indicated by the purple circle, the other SNPs typed in the discovery study are shown as filled circles with the color corresponding to their linkage with rs1466535. The solid blue line shows the regional recombination rate (right axis). Below the regional association plot is the LD plot for the same region with the standard D'/LOD Haploview color scheme. rs1466535 is highlighted by green lines.
None of the other variants taken forward to replication showed significant association (Table S5) In particular SNP rs3019885 (risk-allele frequency [T] = 0.51) at 8q24.11 reached genome-wide significance in the discovery study (p value 1.24 × 10−10), but there was no consistent evidence for association in the replication samples (Table S5). Notably for this variant, the observed effect direction in the in silico data from Iceland was reversed compared to the other replication data sets and the discovery study. To ensure that this observation was not due to erroneous calling, we checked the frequencies of a SNP in LD with rs3019885 (rs2938864) in the Iceland data set and found them to be consistent. Because of the lack of replication, this locus was not carried forward for any further analyses.
rs1466535 genotypes were determined in a further follow-up sample comprising 1491 AAA and 11060 controls, and this again confirmed the association of rs1466535 and AAA (p = 0.035) (Table 1). When all the samples genotyped for rs1466535 were analyzed together (6228 AAA, 49,182 controls), the combined p value was 4.52 × 10−10 (odds ratio [OR] 1.15 [1.10–1.21]) with a consistent effect direction in all sample sets (Table 1). Little change in effect size was seen for rs1466535 when the combined discovery and wet-lab replication samples (those samples that had AAA size data available, 2871 cases and 7619 controls) were stratified by AAA size. In those with AAA < 4.5 cm, the OR for rs1466535[C] was 1.18 (1.07 to 1.30, p = 7.6 × 10−4), and in those AAA ≥ 4.5 cm, the rs1466535[C] OR was 1.21 (1.12 to 1.31, p = 2.7 × 10−6). Aortic size was not assessed as a quantitative trait because the method of measuring aortic size was not standardized between or within the centers contributing to the study. Because the method used to assess aortic size has a considerable effect on the measured size, this heterogeneity would result in a lack of power to detect any association. This effect is limited by the dichotomization of the data as adopted here. rs1466535[C] frequencies for each center contributing to the discovery and replication studies are shown in Table S6. Those for the studies contributing to the in silico replication and follow-up studies are shown in Table 1.
Two further SNPs previously reported to be associated with AAA were also genotyped in the replication samples (chr9p21 rs1333049 and DAB2IP rs10818583 [a proxy for rs7025486]).7, 8 rs1333049 demonstrated highly significant association in both the discovery data set (p = 8.91 × 10−5, based on imputed data) and the replication data set (p = 3.3 × 10−5), combined odds ratio of 1.15 (95% confidence [CI]: 1.09 to 1.22, p = 4.4 × 10−7). rs7025486 (DAB2IP) showed a borderline association in the discovery stage (p = 0.027). In the replication stage, rs7025486 again showed borderline association (p = 0.048), resulting in a combined p value of 9.9 × 10−3 (OR 1.09, 95% CI: 1.02 to 1.16). When samples contributing to the GWAS that identified this association were removed from the replication analysis, the effect size for rs7025486 remained the same but at a reduced level of significance (p = 0.017, OR 1.098 [0.89–1.35]).
To assess whether the association of the 12q13.3 locus with AAA is driven by the presence of clinical factors associated with AAA, we performed an analysis with adjustment for self-reported history of hyperlipidaemia; smoking; or diabetes, which has a negative association with AAA, in subjects where this information was available and found that the association was independent of each of these factors (Table S7). By examining previous genome-wide meta-analyses of lipid traits, diabetes, and blood pressure, we also confirmed that there is no association of this locus with these traits (Table S8). Because AAA and coronary artery disease (CAD [MIM 608320]) often coexist, we also carried out an analysis adjusted for the presence of overt CAD in our subjects and showed that the association with AAA was still significant (Table S7), and by an examination of the locus in the CARDIoGRAM study18 we showed that the locus does not show a signal for CAD (Figure S4). These data indicate that the observed association of the locus with AAA is independent of conventional risk factors for AAA and not due to confounding by the concomitant presence of CAD in the cases.
Functional Assessment of rs1466535
SNP rs1466535 is located in the second intron of the low-density-lipoprotein receptor-related protein 1 (LRP1 [MIM 107770]) gene (Figure 2). There are no known nonsynonymous SNPs or splice-site variants in linkage disequilibrium (r2 > 0.2) with rs1466535. We therefore examined whether rs1466535 was associated with expression of any of the genes at the locus. Expression QTL data sets were available for liver and two arterial tissues (the internal mammary artery and ascending aorta). There was a trend toward increased LRP1 expression with the rs1466535 CC genotype in all arterial tissues with a significant (p = 0.029) 1.19-fold (1.04–1.36) increase in LRP1 expression level in CC homozygotes compared to TT homozygotes in aortic adventitia (Figure S5 and Table 2).
Table 2.
Effect of rs1466535 Genotype on LRP1 Expression
| Tissue | Number | CC versus TT Fold-Change (95% CI) |
|---|---|---|
| Aortic adventitia | 133 | 1.19 (1.04–1.36) |
| Aortic intima media | 138 | 1.17 (0.96–1.42) |
| Mammary artery intima media | 89 | 1.24 (0.99–1.56) |
| Liver | 212 | 0.99 (0.91–1.07) |
Tissue samples were obtained from 316 patients undergoing aortic valve surgery; 572 unique tissue samples were analyzed. Extracted RNA was hybridized to Affymetrix ST 1.0 Exon arrays and obtained scans were RMA normalized and log2 transformed. DNA was extracted from circulating blood cells and hybridized to Illumina 610w-Quad BeadArrays. rs1466535 genotype was directly measured. Estimates of genotype effect on expression of genes were calculated with an additive linear model. The fold-change is calculated as the difference between the mean expression levels in the CC homozygotes compared to the TT homozygotes. Values greater than one correspond to higher expression in CC homozygous individuals. The results for aortic adventitia are shown in Figure S5.
SREBP-1 is known to play a role in transcriptional regulation of LRP1.22 We noted that there was sequence homology between the SREBP-1-binding sequence and the region containing rs1466535. To assess whether rs1466535 affects SREBP-1 binding, we performed an electrophoretic mobility-shift assay (EMSA) and found selective binding of nuclear extract from Huh7 cells to an oligonucleotide representing the C allele that was competed out by excess amount of a probe competitor for SREBP-1 (Figure 3). These results indicate that the rs1466535 T allele might disrupt a binding site for SREBP-1.
Figure 3.
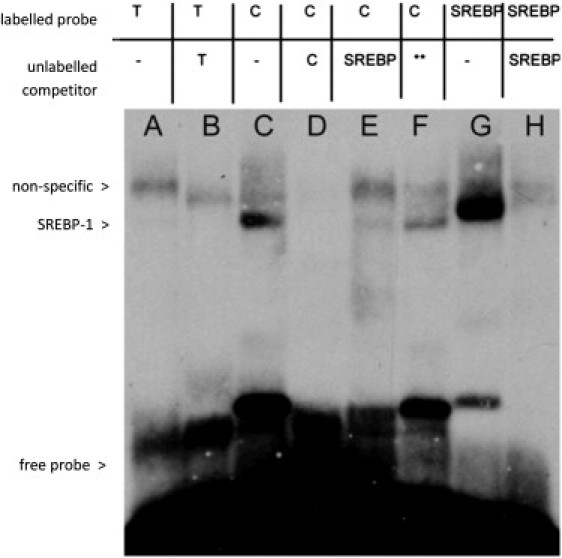
Electrophoretic Mobility Shift Assay Demonstrating Allele-Specific Transcription Factor Binding to rs1466535
Lane A: binding of nuclear extract to labeled oligonucleotide containing rs1466535 T allele; lane B: T-allele binding with addition of excess unlabeled T-allele DNA competitor. The remaining band indicates a nonspecific protein-DNA interaction. Lane C: binding of nuclear extract to labeled oligonucleotide containing C allele. Lane D: C-allele binding with addition of excess unlabeled C-allele probe. Elimination of bands present in lane C indicates the binding of a specific protein. Lane E: C-allele-labeled oligonucleotide with addition of excess unlabeled fSREBP-1 (a transcription factor known to play an important role in LRP1 transcription) DNA competitor. The reduction in intensity of the lower band indicates that this binding is likely to be due to SREBP-1. Lane F: C-allele-labeled oligonucleotide with addition of excess non-SREBP-1-binding oligonucleotide competitor (sequence available on request) as a negative control. Lane G: binding of nuclear extract to labeled SREBP-1 consensus sequence, the lower band migrates with the putative SREBP-1 bound to the C allele. Lane H: SREBP-1 consensus sequence with the addition of excess SREBP-1 unlabeled competitor DNA. The lower band is eliminated, indicating this band represents the DNA-SREBP-1 complex. Together, these results suggest that rs1466535[T] removes a binding site for SREBP-1 in vitro.
In order to determine the possible transcriptional effect of the rs1466535 variant, a luciferase reporter system was created whereby the LRP1 promoter was inserted upstream of luciferase and the sequence containing the two different genotypes of rs1466535 were placed downstream, in both reverse and forward directions. Figure S6 shows the relative changes in luciferase activity (compared with the promoter-only construct) between the two different alleles of rs1466535. In both the forward and reverse directions, the T (nonrisk) allele significantly reduced luciferase activity. This finding is consistent with the disruption of SREP-1 binding observed in the EMS assay as well as the LRP1 gene-expression findings in arterial tissue.
Discussion
We report an association of a common and potentially functional variant in LRP1 with risk of AAA. The association with LRP1 with AAA remained after adjustment for cardiovascular risk factors and furthermore, no association of rs1466535 was seen in data from association studies of coronary artery disease, blood pressure, diabetes, or hyperlipidaemia, suggesting that this association is AAA specific rather than associated with cardiovascular disease in general. The risk allele is common (MAF = 0.62) such that roughly 38% of individuals of white European origin will carry two risk alleles and have an approximately 14% higher risk of developing AAA than noncarriers. Interestingly, in South Asian populations, who rarely develop AAA, the allele frequency of the rs1466535 C (risk) allele is lower (0.54), whereas in African American populations (who have a risk of AAA in between that of South Asians and white Europeans) the risk-allele frequency is higher (0.83).23
Although the association study by itself cannot implicate LRP1 as the causal gene at the locus, other data support its candidacy. Early studies on aneurysmal aortic tissue identified MMP9 as having a prime role in the aneurysmal degradation of the aortic wall.24, 25, 26 LRP1 has been shown to have a role in the regulation of MMP9 expression in response to tPA binding to the LRP1 receptor,27 as well as a direct effect on MMP9 cellular processing.28 In addition to this, LRP1 knockout mice are prone to aneurysm formation.29 These murine models have demonstrated that LRP1 is essential for the maintenance of vascular wall integrity, particularly in the presence of high plasma lipid levels,30 and that this effect is mediated via PDGF receptor beta and Smad signaling.29 Paradoxically, we observed higher LRP1 expression in aortic tissue in those carrying the risk allele for rs1466535, data supported by the findings from the luciferase assay; therefore, whether the risk of AAA relates to higher or lower expression of LRP1 remains to be established. The magnitude of the observed difference in gene expression between genotype groups was small, and therefore caution needs to be exercised in its interpretation. It is possible that this degree of change in expression over a lifetime could have an impact on AAA risk; alternately, interaction of the genotype with other AAA risk factors could result in a much larger change in LRP1 expression in the aneurysmal wall in susceptible individuals. There are no immunohistochemical or expression studies reporting the identification, location, or cellular colocalization of LRP1 in the normal abdominal aortic wall or in aneurysmal aortic tissue, and further investigation of the role of LRP1 in aneurysmal disease is necessary. Similarly, although we found evidence that rs1466535 C allele might create a binding site for SREBP-1, whether this is responsible for the association of the locus with risk of AAA requires further elucidation and more investigation into the determination of the function of this and other SNPs in the region is warranted.
LRP1 is a large endocytic transmembrane receptor named because it contains three separate clusters of ligand-binding domains that bear structural similarity to that of the other LDL receptors. It is, however, far larger than the other LDL receptors and has a more diverse range of functions. This variant (rs1466535) has not been associated with circulating lipid levels in large cohort studies,31 but unrelated SNPs in this region have demonstrated association with triglyceride and HDL levels but without evidence of an increased risk of CAD.15 The fact that rs1466535 alters expression levels in aortic rather than in hepatic tissue suggests that its effect might not be via circulating lipid levels, although the mechanism whereby the higher expression levels of LRP1 in rs1466535 C-allele carriers leads to an increased risk of AAA is unclear.
Previous large-scale genetic association studies for AAA have discovered variants in 9p21.3 and DAB2IP.7,8 These loci are involved in modulating cellular senescence and proliferation32, 33, 34, 35 and inflammation.36 LRP1 knockout mice develop aortic aneurysms secondary to a proliferative smooth-muscle phenotype29 and mutations in ACTA2 [MIM 102620] and TGFBR2 [MIM 190182], causative for thoracic aortic aneurysms, also alter vascular smooth-muscle cells (VSMCs) in favor of excessive proliferation.37, 38, 39 Taken together, the data from GWAS so far point to pathways involved in the differentiation of VSMCs as important in the development of AAA.
Currently, there are no specific medical treatments available for AAA, and repair (endovascular or open surgery) when the aneurysms reaches a size of more than 5.5 cm, becomes symptomatic, or presents acutely following rupture is the only therapeutic option. Our findings suggest a mechanism contributing to AAA formation via the LRP1 pathway, and exploration of this mechanism could provide future therapeutic approaches to preventing the development and/or progression of AAA.
Acknowledgments
This study was funded by the Wellcome Trust (number 084695). This study makes use of data generated by the WTCCC. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the WTCCC project was provided by the Wellcome Trust under award 076113 and 085475. Further funding was provided by the Higher Education Funding Council for England (M.J.B.), Health Research Council of New Zealand (08/075) (Vascular Research Consortium of New Zealand), National Health Medical Research Council (NHMRC) and the Office of Health and Medical Research (J.G.); Swedish Research Council, Swedish Heart-Lung foundation, the European Commission (FAD, Health-F2-2008-200647), a donation by Fredrik Lundberg (for the expression study); National Institute for Health Research (NIHR) Biomedical Research Centre at Guy's and St Thomas' National Health Service (NHS) Foundation Trust (R.C.), NHMRC (Australia), The Dunhill Medical Trust (Leicester), and the Garfield Weston Trust for Medical Research into Heart Surgery (Leeds) (provided additional funding for source cohorts); the European Community's Seventh Framework Programme (FP7/2007-2013); Fighting Aneurysmal Disease Project grant agreement HEALTH-F2-2008-200647 (provided replication data sets from Iceland and Netherlands); St George's Hospital NHS Trust Special Trustees and the Peter and Sonia Field Charitable Trust (A.H.C.); British Heart Foundation Clinical Training Fellowship (FS/11/16/28696) (S.E.H.); British Heart Foundation (RG2008/08) (S.E.H., A.J.S., J.P.); Netherlands Heart Foundation (2009T001) (A.F.B.); British Heart Foundation chairs (S.E.H and N.J.S.); and Leicester NIHR Biomedical Research Unit in Cardiovascular Disease (M.J.B., R.D.S. and N.J.S.). deCODE genetics is a biotechnology company, and authors employed by deCODE own stock or stock options in the company.
Published online: November 3, 2011
Footnotes
Supplemental Data include six figures, eight tables, and a full list of CARDIoGRAM Consortium, Global BPgen Consortium, DIAGRAM Consortium, and VRCNZ Consortium members and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Biomart, www.biomart.org
Genome Wide Associations Scans for Total Cholesterol, HDL-C, LDL-C and triglycerides, http://www.sph.umich.edu/csg/abecasis/public/lipids2010/
Locuszoom, http://csg.sph.umich.edu/locuszoom/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Supplemental Data
References
- 1.Larsson E., Granath F., Swedenborg J., Hultgren R. A population-based case-control study of the familial risk of abdominal aortic aneurysm. J. Vasc. Surg. 2009;49:47–50. doi: 10.1016/j.jvs.2008.08.012. discussion 51. [DOI] [PubMed] [Google Scholar]
- 2.Johansen K., Koepsell T. Familial tendency for abdominal aortic aneurysms. JAMA. 1986;256:1934–1936. [PubMed] [Google Scholar]
- 3.Majumder P.P., St Jean P.L., Ferrell R.E., Webster M.W., Steed D.L. On the inheritance of abdominal aortic aneurysm. Am. J. Hum. Genet. 1991;48:164–170. [PMC free article] [PubMed] [Google Scholar]
- 4.Powell J.T., Greenhalgh R.M. Multifactorial inheritance of abdominal aortic aneurysm. Eur. J. Vasc. Surg. 1987;1:29–31. doi: 10.1016/s0950-821x(87)80020-8. [DOI] [PubMed] [Google Scholar]
- 5.Wahlgren C.M., Larsson E., Magnusson P.K., Hultgren R., Swedenborg J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J. Vasc. Surg. 2010;51:3–7. doi: 10.1016/j.jvs.2009.08.036. discussion 7. [DOI] [PubMed] [Google Scholar]
- 6.Thompson A.R., Drenos F., Hafez H., Humphries S.E. Candidate gene association studies in abdominal aortic aneurysm disease: A review and meta-analysis. Eur. J. Vasc. Endovasc. Surg. 2008;35:19–30. doi: 10.1016/j.ejvs.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 7.Helgadottir A., Thorleifsson G., Magnusson K.P., Grétarsdottir S., Steinthorsdottir V., Manolescu A., Jones G.T., Rinkel G.J., Blankensteijn J.D., Ronkainen A., et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat. Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 8.Gretarsdottir S., Baas A.F., Thorleifsson G., Holm H., den Heijer M., de Vries J.P., Kranendonk S.E., Zeebregts C.J., van Sterkenburg S.M., Geelkerken R.H., et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat. Genet. 2010;42:692–697. doi: 10.1038/ng.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilmink A.B., Quick C.R. Epidemiology and potential for prevention of abdominal aortic aneurysm. Br. J. Surg. 1998;85:155–162. doi: 10.1046/j.1365-2168.1998.00714.x. [DOI] [PubMed] [Google Scholar]
- 10.Chichester Aneurysm Screening Group. Viborg Aneurysm Screening Study. Western Australian Abdominal Aortic Aneurysm Program. Multicentre Aneurysm Screening Study A comparative study of the prevalence of abdominal aortic aneurysms in the United Kingdom, Denmark, and Australia. J. Med. Screen. 2001;8:46–50. doi: 10.1136/jms.8.1.46. [DOI] [PubMed] [Google Scholar]
- 11.Lederle F.A., Johnson G.R., Wilson S.E., Chute E.P., Littooy F.N., Bandyk D., Krupski W.C., Barone G.W., Acher C.W., Ballard D.J., Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Group Prevalence and associations of abdominal aortic aneurysm detected through screening. Ann. Intern. Med. 1997;126:441–449. doi: 10.7326/0003-4819-126-6-199703150-00004. [DOI] [PubMed] [Google Scholar]
- 12.Teo Y.Y., Inouye M., Small K.S., Gwilliam R., Deloukas P., Kwiatkowski D.P., Clark T.G. A genotype calling algorithm for the Illumina BeadArray platform. Bioinformatics. 2007;23:2741–2746. doi: 10.1093/bioinformatics/btm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J., et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeggini E., Scott L.J., Saxena R., Voight B.F., Marchini J.L., Hu T., de Bakker P.I., Abecasis G.R., Almgren P., Andersen G., et al. Wellcome Trust Case Control Consortium Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newton-Cheh C., Johnson T., Gateva V., Tobin M.D., Bochud M., Coin L., Najjar S.S., Zhao J.H., Heath S.C., Eyheramendy S., et al. Wellcome Trust Case Control Consortium Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schunkert H., König I.R., Kathiresan S., Reilly M.P., Assimes T.L., Holm H., Preuss M., Stewart A.F., Barbalic M., Gieger C., et al. Cardiogenics. CARDIoGRAM Consortium Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Folkersen L., van't Hooft F., Chernogubova E., Agardh H.E., Hansson G.K., Hedin U., Liska J., Syvänen A.C., Paulsson-Berne G., Franco-Cereceda A., et al. BiKE and ASAP study groups Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ Cardiovasc Genet. 2010;3:365–373. doi: 10.1161/CIRCGENETICS.110.948935. [DOI] [PubMed] [Google Scholar]
- 20.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O'Donnell C.J., de Bakker P.I. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costales P., Aledo R., Vérnia S., Das A., Shah V.H., Casado M., Badimon L., Llorente-Cortés V. Selective role of sterol regulatory element binding protein isoforms in aggregated LDL-induced vascular low density lipoprotein receptor-related protein-1 expression. Atherosclerosis. 2010;213:458–468. doi: 10.1016/j.atherosclerosis.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 23.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., et al. International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman K.M., Ogata Y., Malon A.M., Irizarry E., Gandhi R.H., Nagase H., Tilson M.D. Identification of matrix metalloproteinases 3 (stromelysin-1) and 9 (gelatinase B) in abdominal aortic aneurysm. Arterioscler. Thromb. 1994;14:1315–1320. doi: 10.1161/01.atv.14.8.1315. [DOI] [PubMed] [Google Scholar]
- 25.Newman K.M., Malon A.M., Shin R.D., Scholes J.V., Ramey W.G., Tilson M.D. Matrix metalloproteinases in abdominal aortic aneurysm: Characterization, purification, and their possible sources. Connect. Tissue Res. 1994;30:265–276. doi: 10.3109/03008209409015042. [DOI] [PubMed] [Google Scholar]
- 26.Thompson R.W., Holmes D.R., Mertens R.A., Liao S., Botney M.D., Mecham R.P., Welgus H.G., Parks W.C. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J. Clin. Invest. 1995;96:318–326. doi: 10.1172/JCI118037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X., Lee S.R., Arai K., Lee S.R., Tsuji K., Rebeck G.W., Lo E.H. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat. Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 28.Hahn-Dantona E., Ruiz J.F., Bornstein P., Strickland D.K. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J. Biol. Chem. 2001;276:15498–15503. doi: 10.1074/jbc.M100121200. [DOI] [PubMed] [Google Scholar]
- 29.Boucher P., Gotthardt M., Li W.P., Anderson R.G., Herz J. LRP: Role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 30.Zhou L., Takayama Y., Boucher P., Tallquist M.D., Herz J. LRP1 regulates architecture of the vascular wall by controlling PDGFRbeta-dependent phosphatidylinositol 3-kinase activation. PLoS ONE. 2009;4:e6922. doi: 10.1371/journal.pone.0006922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talmud P.J., Drenos F., Shah S., Shah T., Palmen J., Verzilli C., Gaunt T.R., Pallas J., Lovering R., Li K., et al. ASCOT investigators. NORDIL investigators. BRIGHT Consortium Gene-centric association signals for lipids and apolipoproteins identified via the HumanCVD BeadChip. Am. J. Hum. Genet. 2009;85:628–642. doi: 10.1016/j.ajhg.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Visel A., Zhu Y., May D., Afzal V., Gong E., Attanasio C., Blow M.J., Cohen J.C., Rubin E.M., Pennacchio L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie D., Gore C., Zhou J., Pong R.C., Zhang H., Yu L., Vessella R.L., Min W., Hsieh J.T. DAB2IP coordinates both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc. Natl. Acad. Sci. USA. 2009;106:19878–19883. doi: 10.1073/pnas.0908458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarinova O., Stewart A.F., Roberts R., Wells G., Lau P., Naing T., Buerki C., McLean B.W., Cook R.C., Parker J.S., McPherson R. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler. Thromb. Vasc. Biol. 2009;29:1671–1677. doi: 10.1161/ATVBAHA.109.189522. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y., Sanoff H.K., Cho H., Burd C.E., Torrice C., Mohlke K.L., Ibrahim J.G., Thomas N.E., Sharpless N.E. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS ONE. 2009;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harismendy O., Notani D., Song X., Rahim N.G., Tanasa B., Heintzman N., Ren B., Fu X.D., Topol E.J., Rosenfeld M.G., Frazer K.A. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inamoto S., Kwartler C.S., Lafont A.L., Liang Y.Y., Fadulu V.T., Duraisamy S., Willing M., Estrera A., Safi H., Hannibal M.C., et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc. Res. 2010;88:520–529. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milewicz D.M., Kwartler C.S., Papke C.L., Regalado E.S., Cao J., Reid A.J. Genetic variants promoting smooth muscle cell proliferation can result in diffuse and diverse vascular diseases: Evidence for a hyperplastic vasculomyopathy. Genet. Med. 2010;12:196–203. doi: 10.1097/GIM.0b013e3181cdd687. [DOI] [PubMed] [Google Scholar]
- 39.Guo D.C., Pannu H., Tran-Fadulu V., Papke C.L., Yu R.K., Avidan N., Bourgeois S., Estrera A.L., Safi H.J., Sparks E., et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.