

Abstract
A subset of ciliopathies, including Sensenbrenner, Jeune, and short-rib polydactyly syndromes are characterized by skeletal anomalies accompanied by multiorgan defects such as chronic renal failure and retinitis pigmentosa. Through exome sequencing we identified compound heterozygous mutations in WDR19 in a Norwegian family with Sensenbrenner syndrome. In a Dutch family with the clinically overlapping Jeune syndrome, a homozygous missense mutation in the same gene was found. Both families displayed a nephronophthisis-like nephropathy. Independently, we also identified compound heterozygous WDR19 mutations by exome sequencing in a Moroccan family with isolated nephronophthisis. WDR19 encodes IFT144, a member of the intraflagellar transport (IFT) complex A that drives retrograde ciliary transport. We show that IFT144 is absent from the cilia of fibroblasts from one of the Sensenbrenner patients and that ciliary abundance and morphology is perturbed, demonstrating the ciliary pathogenesis. Our results suggest that isolated nephronophthisis, Jeune, and Sensenbrenner syndromes are clinically overlapping disorders that can result from a similar molecular cause.
Main Text
The cilium is an antenna-like structure that protrudes out of the apical membrane of most vertebrate cells. Dysfunction of this organelle has been shown to result in a number of inherited diseases, ranging from isolated disorders, such as cystic kidney disease and retinitis pigmentosa to more complex disorders such as Bardet-Biedl (MIM 209900) and Meckel (MIM 249000) syndromes.1
Recently, it has been demonstrated that the genetically heterogeneous asphyxiating thoracic dysplasia, also called Jeune syndrome (MIM 611263, MIM 613091, and MIM 613819); short-rib polydactyly (MIM 263510, MIM 263530, MIM 263520, and MIM 269860); and cranioectodermal dysplasia, also known as Sensenbrenner syndrome (MIM 218330, MIM 613610, MIM 614099) are also caused by disruption of cilia.1, 2 This group of disorders is characterized by abnormal development of the bones, that is short ribs, shortening of the long bones, short fingers, and polydactyly. Extraskeletal anomalies such as renal insufficiency, hepatic fibrosis, heart anomalies, and retinitis pigmentosa are also often part of the phenotype. Patients with Sensenbrenner syndrome may also present with craniosynostosis and ectodermal abnormalities such as malformed teeth, sparse hair, and skin laxity.3, 4 Jeune syndrome is less complex and is primarily characterized by a narrow rib cage and respiratory insufficiency.5, 6 Although Jeune and Sensenbrenner syndromes are considered to be rather mild forms of the same phenotypic spectrum, the embryonically lethal short-rib polydactyly is thought to be at the severe end of this spectrum.7, 8, 9, 10 Renal disease has been reported in all of these syndromes and involves nephronophthisis, a chronic tubulointerstitial nephropathy in most cases leading to end-stage renal failure during childhood or young adulthood. The kidneys in juvenile and adolescent nephronophthisis are of normal or even reduced size and are characterized histologically by disruption as well as focal thickening and replication of basement membranes in nonatrophic tubules, associated with interstitial fibrosis and tubular atrophy. Cysts may develop late in the course of the disease, typically at the corticomedullary junction. Nephronophthisis (NPHP [MIM 256100]) is considered a ciliopathy since the mutations that have been associated with this disorder are nearly all located in genes that encode proteins that have a role in the cilium.11
Intraflagellar transport (IFT) is an important transport process that occurs in the cilium. Transport towards the ciliary tip is regulated by the IFT complex B (IFT-B), consisting of at least 15 IFT proteins, in association with kinesin motors, whereas transport from the ciliary tip back to the base is executed by a dynein motor in association with the IFT complex A (IFT-A), currently known to be composed of six IFT proteins.12, 13, 14 Nearly all mutations that have been associated with skeletal ciliopathies are located in genes that encode proteins that are part of the IFT-A complex and the IFT-A-associated motor protein. Specifically, mutations were found in IFT122 (mutated in patients with Sensenbrenner syndrome; MIM 606045),15 WDR35 (associated with Sensenbrenner and short-rib polydactyly syndromes; MIM 613602),10, 16 TTC21B (mutated in Jeune syndrome and nephronophthisis; MIM 612014),17 IFT43 (previously called C14ORF179, associated with Sensenbrenner syndrome; MIM 614068),18 and DYNC2H1 (associated with Jeune and short-rib polydactyly syndromes; MIM 603297).8 IFT80 (MIM 611177) is the only known gene encoding an IFT-B particle subunit that is involved in ciliopathies that affect the skeleton.7, 19 In addition, mutations in NEK1 (MIM 604588), which encodes a serine/threonine kinase involved in cell-cycle regulation, have recently been described in short-rib polydactyly patients.20 Still, there is an emerging theme that mutations in genes encoding IFT proteins, and predominantly the IFT-A particle subunits, are associated with the etiology of skeletal ciliopathies.
In this report we applied exome sequencing to identify the genetic cause of Sensenbrenner syndrome in a Norwegian family with two affected children and their healthy sibling. The clinical findings of these patients are illustrated in Figure 1, Figure 2 and Figure S1, available online, and a summary is provided in Table S1. Patient II-1, a 21-year-old female, is the second child of unrelated, healthy parents. At birth, developmental dysplasia of both hips and general hypotonia were observed. Ophthalmoscopy at the age of 11 years revealed attenuated arteries and bone-spicule-shaped deposits in the periphery of the retina (Figure 1A). An electroretinogram showed completely extinguished signals, indicative of tapetoretinal dystrophy. She had recurrent pneumonia and asthma, consistent with respiratory function tests that indicated reduced lung capacity. At the age of 11 years she was investigated for retardation of statural growth with height 2–3 cm under the 2.5th centile, but no underlying cause was found. After treatment with growth hormone therapy, her height attained the 10th centile. The mid-parental height was 175 cm. When she was 11 years old, she presented with a moderately increased serum creatinine and hypertension. Ultrasound showed hyperechoic kidneys, and histological examination of a renal biopsy revealed unspecific changes that were compatible with a nephronophthisis-like disease (Figures 1D and 1E). The nephropathy progressed rapidly, and a successful renal transplantation was performed when she was 14 years old. She also had idiopathic bone marrow hypoplasia. The diagnosis of Sensenbrenner syndrome was made based on the combination of clinical findings of the patient and her brother. Patient II-2, the 16-year-old brother of patient II-1, presented with developmental dysplasia of both hips and craniosynostosis of the sagittal suture. Although he did not have ocular complaints at the age of 7, ophthalmoscopy revealed attenuated retinal vessels and an electroretinogram showed extinguished rod signals and severely reduced cone signals compatible with a rod-cone dystrophy (Figure 1C). An ultrasound showed hyperechoic kidneys, but there were no clinical or biological signs of renal disease at follow-up. Both siblings had multiple dental anomalies (Figure 2 and Table S1) that are different from the tooth phenotypes from their parents (Figure S2).
Figure 1.
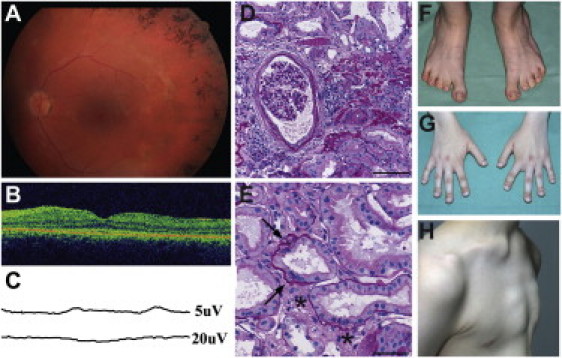
Clinical Features and Renal Pathology of Sensenbrenner Patients
(A) Fundus image of Sensenbrenner patient II-1 showing bone-spicule-shaped pigment deposits in the midperiphery. Slight attenuation of retinal vessels is also seen.
(B) Optical coherence tomography of the same patient showing disrupted photoreceptor inner and outer segment layer most clearly seen outside of the foveal area.
(C) Patient II-2 was examined by electroretinogram at the age of 7. Rod signals were extinguished. Cone function was barely recordable as illustrated by photopic flicker recording seen at 5 μV (top). At the age of 14, no cone response could be detected; shown here at 20 μV (bottom).
(D and E) Renal biopsy from patient II-1 (periodic acid-Schiff staining). The patient had nephron loss with 40% globally sclerosed glomeruli and focal tubular atrophy and interstitial sclerosis. (D) Some of the remaining open glomeruli are enlarged and show ischemic changes with slightly retracted capillary convolute and fibrosis of the Bowman's capsule. (E) Nonatrophic tubules show focal lamellation of the basement membrane (arrows) and interstitial fibrosis (indicated by an asterisk [∗]). The scale bars are 100 μm and 50 μm in (D) and (E), respectively.
(F) Feet of patient II-1. Pes planus with short 2nd to 5th toes.
(G) The hands of patient II-1 have short and broad distal phalanges and short 2nd and 5th second phalanges.
(H) Patient II-2 has a pectus excavatum.
Figure 2.
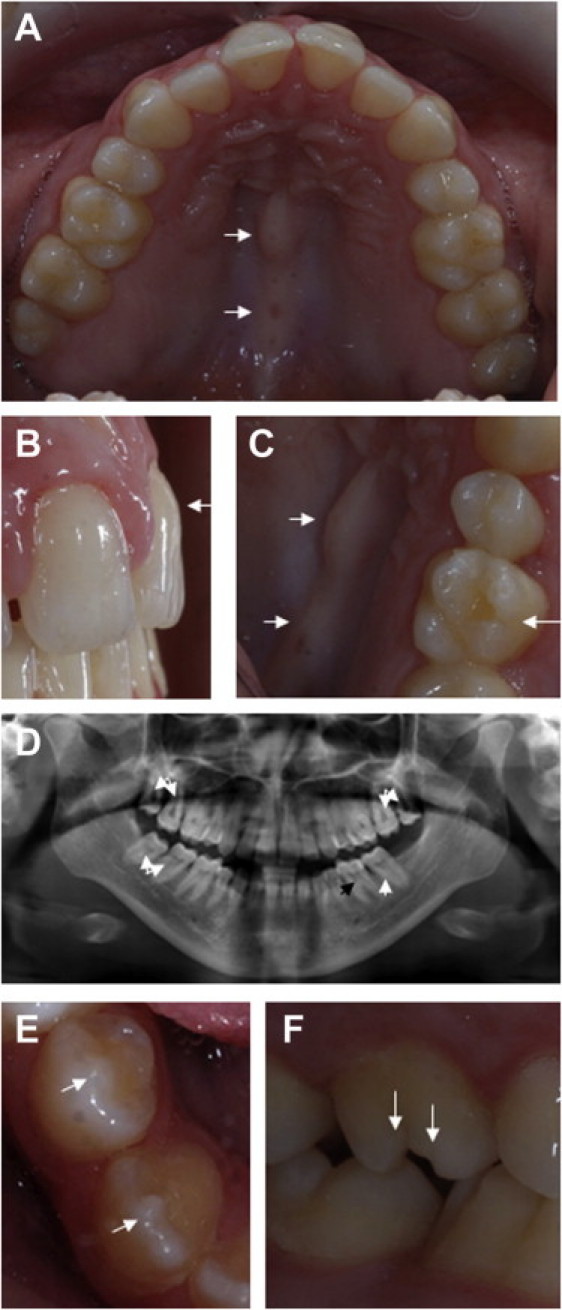
Dental Defects of Sensenbrenner Patients
(A–D) Dental and palatal defects of patient II-1. (A) Patient II-1 has a bony protrusion of the hard palate (torus palatinus, indicated by the arrows in A and C) and decreased maximum width of the teeth with diastemata between maxillary incisors. (B) The morphology of her central incisors is atypical. The arrow points at excessive bulging of the gingival aspect of the buccal surface. (C) The conical first maxillary molars with atypical cusps is also pointed out (right arrow). (D) The panoramic radiograph shows taurodontism of second and third molars (white arrows). The black arrow points out normal pulp of the first molar.
All first premolars are extracted, and there is hypodontia of the lower left third molar (E–F) Tooth anomalies of patient II-2. Arrows point out nipple cusps of mandibular premolar and canine (E) and atypical cusps of maxillary first molar (F).
The parents, both affected siblings, and their healthy brother were screened using Affymetrix 6.0 SNP arrays. No pathogenic copy-number variations were found. Neither did we find any significant homozygous regions (>2 Mb) that were shared by the two affected siblings after homozygosity mapping by using the PLINK program.21 Haplotype analysis of the chromosomal regions encompassing the loci for IFT122, WDR35, and IFT43 did not show segregation with the disease within this family, thus excluding the genes that previously had been shown to be mutated in Sensenbrenner patients. We therefore decided to proceed with exome sequencing to identify the causative genetic defect in this family. Our study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre and by the Regional Committee for Medical and Health Research Ethics, Western Norway (IRB no. 00001872). We obtained written informed consent to participate in the study from all family members as well as informed consent for publication of the clinical photographs.
The exome of patient II-2 was sequenced, and 96.47% of all genes from the NCBI RNA reference sequences collection (RefSeq) were targeted. This was done using the 50 Mb SureSelect human exome kit (Agilent, Santa Clara, CA, USA). We applied posthybridization barcoding of the library, which was subsequently sequenced on a quarter of a SOLiD slide. This resulted in 3.48 Gb of mappable sequence data. The sequence reads were mapped to the hg19 reference genome by using SOLiD BioScope software version 1.3. The median coverage of the targeted exome was 69 fold. In the coding regions or splice sites, 19,053 genetic variants were identified. After mapping and calling the genetic variations, we used a prioritization scheme to identify the pathogenic mutations as we did in our previous work.16, 22 Known dbSNP132 variants were excluded as well as variants from our in-house database. The latter consists of 359,220 variants identified in more than 220 Nijmegen exome-resequencing samples from healthy individuals or patients with other rare diseases.
We determined which genetic private nonsynonymous and splice site variations matched with a recessive inheritance model and identified seven candidate genes; two with compound heterozygous variations (WDR19 and MICALCL [MIM 612355]) and five with homozygous variations (PAQR6, TFDP3 [MIM 300772], PRB1 [MIM 180989], PLXNB3 [MIM 300214] and PPIL2 [MIM 607588]). These variations were subsequently validated with Sanger sequencing and the distribution of the variants in the family was determined (Table S2). The only gene in which variants could be validated that were shown to cosegregate with the disease was WDR19 (RefSeq NM_025132.3) encoding IFT144, a gene containing 37 exons (Figures 3A and 3B and Figure S3A and Table S2). Both affected siblings carry compound heterozygous variations (c.2129T>C [p.Leu710Ser] and c.3307C>T [p.Arg1103∗]) in WDR19, while each of the parents is heterozygous for one of the changes, which is consistent with an autosomal-recessive inheritance model (Figure 3B, Table S2, and Figures S3A and S4). Haplotype analysis of the SNP array data that had previously been obtained indicated that WDR19 is indeed located in a 26.4 Mb haplotype block (from rs2286540 to rs3890818) that is shared by both affected siblings, further confirming our results. We did not detect the identified p.Leu710Ser and p.Arg1103∗ variations in WDR19 in 422 control alleles from the same population as the patients. The genetic association of WDR19 with the phenotype in this family fully matches the recently established connection of the IFT-A particle with ciliopathies that are characterized by skeletal anomalies10, 15, 16, 17, 18 because the WDR19-encoded IFT144 is one of the six core subunits of this particle.
Figure 3.
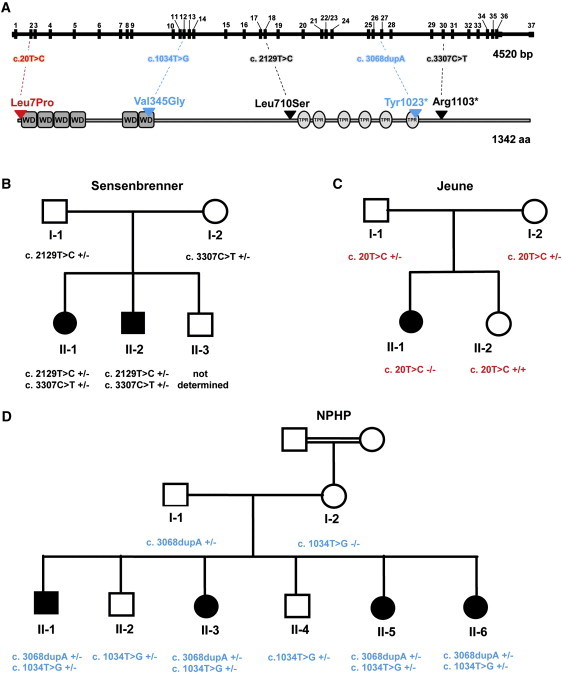
Variants in WDR19
(A) Gene and protein structure of WDR19 encoding IFT144. WD domains are indicated with gray boxes and tetratricopeptide repeat (TPR) motifs with light gray ovals. The nature and position of the identified mutations in patients with Sensenbrenner syndrome (black), Jeune syndrome (red), and isolated nephronophthisis (blue) are indicated.
(B) The pedigree shows that both siblings with Sensenbrenner syndrome (II-1 and II-2) carry compound heterozygous mutations in WDR19. The missense mutation (p.Leu710Ser) was inherited from the father (I-1), whereas the stop mutation (p.Arg1103∗) was transferred by the mother (I-2).
(C) A homozygous missense mutation (p.Leu7Pro) was identified in a patient with Jeune syndrome (II-1). The parents (I-1 and I-2) are both heterozygous for this variant. The unaffected sister (II-2) of the patient has wild-type alleles.
(D) All four affected siblings (II-1, II-3, II-5, and II-6) from a large nonconsanguineous Morrocan family with isolated nephronophthisis carry compound heterozygous variants in WDR19 (p.Val345Gly and p.Tyr1023∗). Their two unaffected siblings are heterozygous for the p.Val345Gly mutation. The nonsense mutation was inherited from the father (I-1), who carries this variant in a heterozygous state. The missense mutation is inherited from the mother (I-2). Notably, the unaffected mother (I-2), who is a child from a consanguineous family, is homozygous for the p.Val345Gly mutation.
We obtained fibroblasts from Sensenbrenner patient II-1, a carrier (I-2), and a healthy individual and stained these cells with antibodies against IFT144 (mouse polyclonal, Abnova, H00057728-B01P, Taipei, Taiwan; 1:100), acetylated α-tubulin (a mouse monoclonal from Life Technologies, Bleiswijk, The Netherlands; 1:1000) and RPGRIP1L (SNC040,23 guinea pig polyclonal; 1:500) to analyze the localization and abundance of IFT144 in the cilia and to study cilia number and morphology as previously described.18 Alexa Fluor 568- and 488-conjugated antibodies against mouse or guinea pig immunoglobulins (Life Technologies, Bleiswijk, The Netherlands; 1:300) were used as secondary antibodies. In control fibroblasts IFT144 was detected throughout the cilium, with prominent signals at its base, where it colocalizes with RPGRIP1L, and the ciliary tip (Figure 4A and Figure S5). Stainings of cilia from fibroblasts from carrier I-2, show a similar pattern but with a significantly lower intensity of the signals. We did not detect any IFT144 in cilia from patient II-1, indicating a complete loss of function of IFT144. We also studied IFT144 in cilia from fibroblasts from a recently described Sensenbrenner patient with mutated IFT43.18 Interestingly, we found that IFT144 was less abundantly present in the cilia, and its localization was restricted to the (distal part of the) ciliary axoneme and the ciliary tip (Figure 4B). Mutations in other IFT complex A proteins such as IFT43 can thus also result in IFT144 mislocalization, emphasizing that mutations causing Sensenbrenner syndrome are affecting the function of the entire IFT-A particle.
Figure 4.
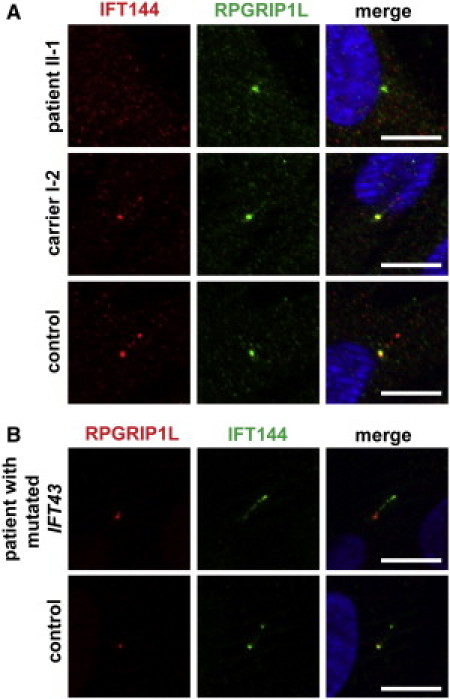
IFT144 Is Absent or Mislocalized in Cilia from Sensenbrenner Patients with Mutations in Genes that Encode IFT Complex A Proteins
(A) Serum-starved fibroblasts were stained with antibodies against IFT144 (red) and RPGRIP1L (green). The latter marks the base of the cilium. DAPI (blue) stains nuclei and is shown in the merged image. IFT144 is not detected in cilia from Sensenbrenner patient II-1. The distribution of IFT144 in primary cilia in cells from the heterozygous mother of patient II-1 is normal, but IFT144 is present in decreased amounts compared to the amount in cilia from the control individual. The scale bars represent 10 mm.
(B) Aberrant ciliary localization of IFT144 in cilia in fibroblasts from a patient with a mutation in IFT43. IFT144 (green) is mainly present in the ciliary base and tip in control cilia; however, it is not detected in the base of cilia in cells from the patient. Instead, IFT144 was detected primarily at the ciliary tip and also occurred as a smear along the ciliary axoneme in the cilia of the patient. DAPI stains the nuclei (in merged images). The scale bars represent 10 μm.
Acetylated alpha-tubulin staining of the ciliary axonemes of fibroblasts from patient II-1 revealed a significant reduction of the number of ciliated cells as well as a reduction in cilium length of most of the cilia that were detected compared to cilia from fibroblasts from two control cell lines (Figures 5A–5C). This points at defects in ciliogenesis and/or cilium maintenance. Similar ciliary defects have been described in fibroblasts of patients with IFT122 mutations.15
Figure 5.
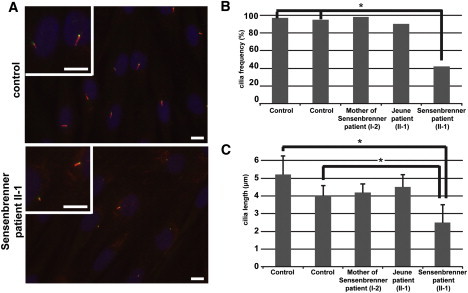
Aberrant Ciliary Morphology in Fibroblasts from Sensenbrenner Patient II-1
(A) Primary cilia in fibroblasts from a control individual (upper panel) and Sensenbrenner patient II-1 (lower panel). The control cells are from an individual with comparable age, sex, and ethnicity as the patient. Fibroblasts are stained with anti-acetylated alpha-tubulin (red) and RPGRIP1L (a marker of the base of the cilium, green). Nuclei are stained with DAPI (blue). High magnifications of cilia are shown. The scale bars all represent 10 μm.
(B) The cilia frequency is significantly lower (p < 0.0001) in cells from Sensenbrenner patient II-1 compared to cells from two control cell lines as was determined with a Fisher's exact test (indicated with an asterisk). The abundance of cilia in cells from the mother of the Sensenbrenner patient and the patient with Jeune syndrome is comparable to the cilia frequency in control cell lines.
(C) The length of cilia in Sensenbrenner patient II-1 and two control individuals differ significantly. Significance was calculated with a Student's t test (p < 0.0001). Error bars represent the standard error of the mean. The average ciliary length of cells from the mother from the Sensenbrenner patient and the patient with Jeune syndrome are comparable to the average ciliary length in cells from two healthy controls.
To investigate whether WDR19 mutations are a frequent cause of Sensenbrenner syndrome, and to examine whether WDR19 is also associated with syndromes that clinically overlap with Sensenbrenner syndrome, we screened all coding exons of WDR19 for mutations in nine Sensenbrenner, 14 Jeune and two short-rib polydactyly patients by Sanger sequencing. Thirty-six protein-coding exons were amplified by standard PCR and analyzed with dye-termination chemistry (BigDye Terminator, version 3 on a 3730 DNA analyzer; Applied Biosystems, Foster City, CA, USA). We did not identify mutations in the other Sensenbrenner patients. However, in one of our patients with Jeune syndrome (previously described as case 1 in de Vries et al.,6 Table S1 and Figures S6A and S6B), we found a homozygous missense variation in exon 2, c.20T>C [p.Leu7Pro] (Figures 3A and 3C and Figures S3B and S4). Aside from the typical skeletal features that characterize Jeune syndrome, this patient also had chronic renal disease and several eye abnormalities, that is cataracts, attenuated arteries, and macular abnormalities for which she is currently under investigation. Both parents are heterozygous for the missense variation, and the unaffected sister of the patient has wild-type alleles. The altered base is highly conserved in vertebrates (phylo-P24 is 4.4 for 46 vertebrate species) and is not described in dbSNP132 or the 1000 Genomes Project or in our in-house database. Diagnostic screening with Affymetrix 250K SNP microarrays did not reveal any uncommon copy-number variants or large homozygous regions, and no mutations were found in genes that have previously been shown to be mutated in patients with Jeune syndrome, which further supports that mutations in WDR19 can result in the clinically overlapping Sensenbrenner and Jeune syndromes.
In order to functionally assess the effect of the p.Leu7Pro mutation identified in the Jeune patient, we also studied the cilia in fibroblasts from this patient. In contrast to the results from the Sensenbrenner patient, we did not observe major differences in cilia number and length in cells from the Jeune patient compared to the cells in two controls (Figures 5B and 5C), and IFT144 could still localize to the cilium although the overall level seemed reduced (data not shown). A common ciliary phenotype associated with IFT-A dysfunction is the accumulation of IFT-B complex proteins in ciliary tips; this accumulation has previously been reported in human cells and several model organisms.18, 25, 26 By staining the Jeune fibroblasts with anti-IFT57 (kindly provided by Greg Pazour), ciliary markers GT335 (a gift from Carsten Janke), and anti-RPGRIP1L, we indeed observed this defect in a low but significant number of cells, whereas it was virtually absent in the controls (Figures S7A and S7B). The clinical and ciliary phenotypes from the patient with Jeune syndrome are thus less dramatic compared to the phenotypes in the patient(s) with Sensenbrenner syndrome; this difference corresponds to the identification of a milder genetic defect in this patient.
In an independent study in which a combined approach of linkage analysis and exome sequencing was used for gene hunting, we identified compound heterozygous mutations in WDR19 in a large nonconsanguineous Moroccan family with isolated nephronophthisis (Figure 3). The unrelated parents of this family have three affected children, two healthy children, and one child with an unknown status. The clinical findings of the patients are illustrated in Figures S6C and S6D, and a summary of the clinical information is provided in Table S3. The three affected siblings II-1, II-3, and II-5 all presented with small kidneys and reached end-stage renal disease (ESRD) at the ages of 19, 20, and 13 years, respectively (Table S3). Histological examination of the renal biopsies from patients II-1 and II-5 revealed interstitial fibrosis with atrophic tubules and thickening of the tubular basement membrane, indicative of nephronophthisis (Figures S6C and S6D). The youngest sister, II-6, only displayed mild features of renal disease (Table S3), whereas the 31- and 26-year-old siblings and both parents were asymptomatic. Once our study was approved by the Comité de Protection des Personnes “Ile-De-France II” and informed consent was obtained, all family members were genotyped with Affymetrix 250k SNP arrays. Haplotype analysis, performed with MERLIN software, revealed that the affected siblings (II-1, II-3, and II-5) share 11 chromosomal regions; however, none of these encompassed known NPHP loci. We subsequently performed exome sequencing for patient II-3 with a comparable, yet slightly different, technical and bioinformatic approach as described previously. We used a 50 Mb Agilent SureSelect assay and a SOLiD4 sequencer (50 bases fragment reads). This resulted in 1.9 Gb of mappable sequence data that were aligned to the human genome reference sequence (hg19 build) with a BWA aligner.27 The variants were annotated with an in-house pipeline based on the Ensembl database (release 61). Known variants from dbSNP132, the 1000 Genomes Project, and in-house exome data were excluded from the 330 genetic variants identified in the coding regions or splice sites. Finally (after comparison with linkage data), the only gene in which two variants could be validated that cosegregated with the disease in the family was WDR19 (Table S4). All affected siblings carry compound heterozygous variations (c.1034T>G [p.Val345Gly] and c.3068dupA [p.Tyr1023∗]) (Figure 3D and Figure S3C). The father is heterozygous for the p.Tyr1023∗ mutation, whereas the mother (whose parents are consanguineous) carries the p.Val345Gly change at a homozygous state (Figure 3D). The two unaffected children are both heterozygous for the p.Val345Gly mutation but do not carry the nonsense mutation (Figure 3D). We did not detect the p.Val345Gly variant in 200 alleles from the same ethnic origin. While in silico analysis of p.Val345Gly in PolyPhen2 indicates that this variant is benign, SIFT predicts that this missense mutation does alter protein function. Based on our bioinformatic analyses and the location of the mutation (in the end of the WD40 repeat stretch, Figure 3A), we propose that this variation only mildly affects IFT144 function. This is consistent with the fact that the mother, who carries the p.Val345Gly variant in a homozygous state, is asymptomatic. However, the association of this hypomorphic variant with the nonsense mutation p.Tyr1023∗ in the affected siblings could enhance the mild effect of the p.Val345Gly variant and lead to a kidney phenotype. This is reminiscent to what has been described for the c.686G>A (p.Arg229Gln) variant in nephrotic syndrome (NPHS2, nephrotic syndrome type 2 [MIM 600995]): whereas homozygosity for this variant is nonpathogenic,28 compound heterozygosity for c.686G>A and another NPHS2 (MIM 604766) mutation is associated with adult-onset steroid resistant nephrotic syndrome.29 WDR19 mutations are thus likely also associated with isolated nephronophthisis. We further screened by Sanger sequencing all coding exons of WDR19 for mutations in 22 additional patients with nephronophthisis and skeletal defects (cone-shaped epiphysis, craniosynostosis, or narrow thorax) and 30 patients with isolated nephronophthisis; however, this did not result in the identification of additional homozygous or compound heterozygous WDR19 mutations. This is not surprising considering our cohort size and the marked genetic heterogeneity in nephronophthisis. Although we cannot exclude that compound pathogenic mutations in noncoding regions have been missed in our exome analysis or that additional modifying mutation(s) in another ciliary related gene (accompanied with the WDR19 defects) contribute to disease, we conclude that the phenotypic spectrum associated with WDR19 mutations most likely extends from Jeune and Sensenbrenner syndromes to isolated nephronophthisis. The phenotypic variability in patients with genetic defects in the genes encoding IFT-associated proteins that have been identified thus far ranges from isolated nephronophthisis to the embryonically lethal short-rib polydactyly.8, 10, 15, 16, 17, 18, 19 Because mutation type may be one of the factors that influence the phenotype in these disorders, the phenotypic spectrum associated with WDR19 mutations may be broader than reported in this paper (i.e., extending beyond isolated nephronophthisis and Sensenbrenner and Jeune syndromes). We did not detect combinations of two truncating mutations in any of our families. It is conceivable that a combination of two mutations that severely disrupt the protein function (e.g., truncating mutations or deletions) may result in more complex and/or embryonically lethal phenotypes such as short-rib polydactyly. Yet, as the IFT144 protein was completely absent from cilia in fibroblasts from one of our Sensenbrenner patients (indicative of a loss of function), it could also be that Sensenbrenner syndrome is at the end of the phenotypic spectrum that is associated with WDR19.
In addition to mutation type, genetic load, modifier effects, and oligogenic inheritance, which all refer to the possibility that mutations in more than one gene affect the phenotype, have also been proposed as explanations for the clinical variability within ciliopathies and within families suffering from these disorders.17, 20, 30, 31 Notably, the two siblings with Sensenbrenner syndrome described in this paper also display several different clinical features, for example patient II-2 has a craniosynostosis, whereas the skull of patient II-1 is normal (Table S1). In this sense, it is of interest that recent studies in Dync2h1 knockout mice indicate that additional mutations in Ift172 and Ift122 can rescue the cilia and hedgehog phenotype of these mice.32 Modifier effects of genes encoding IFT-associated proteins could influence the phenotypes in humans. In our analysis of the exome data of Sensenbrenner patient II-2, we did not make observations that could further explain the intrafamilial phenotypic variability. No additional private variants in any of these genes, nor in any of the genes that were previously reported to be mutated in patients with ciliopathies, were identified aside from the WDR19 mutations. As new mutations in IFT and ciliary genes are continuously being discovered, future studies may reveal whether the factors that cause the different clinical manifestations of Sensenbrenner syndrome in this family are of genetic and/or environmental origin.
It is interesting that all IFT-A genes (including the WDR19 findings from this paper) except for IFT140 have been found to be mutated in skeletal ciliopathies such as Sensenbrenner, Jeune, and/or short-rib polydactyly syndromes.10, 15, 16, 17, 18 Although, we did not find any pathogenic genetic defects in this gene in any of our patients (data not shown), we still hypothesize that mutations in IFT140 might cause ciliopathies that are characterized by skeletal defects.
Dysfunction of IFT-A proteins have frequently been associated with chronic renal disease.10, 15, 17, 18 All the families with WDR19 mutations discussed in this paper present with a nephronophthisis-like nephropathy. TTC21B, encoding IFT139/THM1/TTC21B, is thus far the only other gene encoding an IFT protein in which mutations have been found both in association with a syndrome (Jeune) and isolated nephronophthisis.17 The exact role of the IFT proteins in the kidney remains to be investigated although hypotheses that they play a role in the regulation of ciliary signaling cascades (e.g., wnt and hedgehog) have been put forward. One of the suggested mechanisms is that IFT proteins, by contributing to the ciliary localization of Inversin, regulate the noncanonical wnt pathway involved in establishing planar cell polarity.33 Because Inversin is a central protein mutated in infantile nephronophthisis and known to associate with other Nephrocystin proteins, this could explain why mutations of IFT genes also cause nephronophthisis.
Retinal dystrophy is a feature that accompanies several ciliopathies, including Sensenbrenner and Jeune syndromes.1, 3 The two siblings with Sensenbrenner syndrome in our report were both diagnosed with retinitis pigmentosa. This demonstrates that mutations in WDR19 can be associated with tapetoretinal degeneration in humans. The Jeune patient also had retinal abnormalities; however, although the changes could be compatible with a retinal dystrophy, no such diagnosis had been made. None of the Sensenbrenner, Jeune, or nephronophthisis patients with mutations in other IFT-A genes were reported to present signs of retinal dystrophy.15, 16, 17, 18 However, the majority of these patients are still young, and they may develop retinal problems later in life. It has been shown that multiple IFT proteins (several IFT-B proteins and IFT140 and Ttc21b) localize in the retina.17, 34 LCA5 (MIM 611408), encoding Lebercilin, is associated with Leber congenital amaurosis (MIM 604537), a severe hereditary tapetoretinal degeneration. Although Lebercilin is not an integral component of the IFT machinery, it associates to the IFT particles, and mutations in LCA5 have been found to lead to disrupted IFT, confirming the importance of IFT for retinal function.35 It is thus conceivable that more genes encoding IFT-A subunits, or proteins closely connected to its function, are associated with retinal disease.
Dental anomalies were seen in both of our Sensenbrenner patients; however, no major abnormalities were reported in our Jeune and nephronophthisis families. The dental phenotypes of our Sensenbrenner patients are similar to the tooth anomalies (e.g., reduced enamel thickness, hypodontia, microdontia, taurodontism, and malformations of the cusps) that have been described in other Sensenbrenner patients4, 36, 37 and in patients with ciliopathies such as Ellis-van Creveld syndrome (MIM 225500), orofaciodigital syndrome (MIM 311200), and Bardet-Biedl syndrome.38, 39 Tooth morphogenesis is a complex process in which cilia seem to play an important role. This is, for example, indicated by the fact that transgenic mice with loss of function of FoxJ1, a major regulator of cilia development, have a reduced and defective ameloblast layer.40 In addition, there is emerging evidence that human odontoblasts express important primary cilium components, including IFT proteins.41 With respect to IFT, it has been shown that loss of Polaris (the murine IFT88 ortholog) results in supernumerary teeth because of ectopic hedgehog signaling in mice.42, 43 This tooth phenotype is opposite to the hypo- and oligodontia that is described in Sensenbrenner syndrome. In this respect, it is at least remarkable that IFT88 and IFT144 also have contrasting ciliary transport functions; whereas IFT88 is an IFT-B protein that regulates anterograde ciliary transport, IFT144 is part of the IFT-A machinery that regulates retrograde ciliary transport. As such, we hypothesize that these contrasting dental phenotypes might be due to different imbalances of the Hedgehog pathway associated with dysfunction of either the IFT-A or the IFT-B complex (as has previously been suggested for neural tube and limb development).44 It is thus clear that there is a link between cilia dysfunction and aberrant tooth development; however, the molecular mechanisms that underlie these dental defects are still poorly understood.
In conclusion, we identified through exome-sequencing compound heterozygous mutations in WDR19 in a Norwegian family with Sensenbrenner syndrome as well as in a Moroccan family with isolated nephronophthisis. A homozygous missense mutation in WDR19 in a Dutch family with Jeune syndrome was also found. By demonstrating aberrant ciliogenesis and IFT protein localization in the cilia of fibroblasts from a Sensenbrenner patient, we validated the ciliary dysfunction. Our results suggest that there is a large phenotypic spectrum associated with WDR19 mutations, as previously reported for other IFT genes. While mutations in Sensenbrenner syndrome can result from defects in retrograde transport, Jeune syndrome and isolated nephronophthisis could be due to less severe mutations in the same gene with milder effects on the same molecular mechanism.
Acknowledgments
We thank the families with Sensenbrenner and Jeune syndromes and nephronophthisis for their participation in this study. We thank H.G. Brunner, K. Rosendahl, E.M.H.F. Bongers, M.C. Gubler, N. Aitsahlia, and C.M.A. van Ravenswaaij for useful discussions and communications and B. Kjersem, T. Peters, C. Arrondel, N. Prince, C. Masson, M. Zarhrate, and S. Pruvost for technical assistance. We also thank P.L. Beales, J. de Vries, J.L. Yntema, and A.F.M. Hoogeboom and other clinicians for supplying additional patients with Sensenbrenner and Jeune syndromes. This study was supported by grants from the Dutch Kidney Foundation (KJPB09.009 and IP11.58 to H.H.A.), the European Community's Seventh Framework Programme FP7/2009 under grant agreement number 241955, SYSCILIA (to R.R.), grants from the Fondation pour la Recherche Médicale (FRM DEQ20071210558 to S.S.), the Agence National de la Recherche (grant RPV11012KK to S.S.), a grant from the Netherlands Organization for Scientific Research (NWO Vidi-91786396; to R.R.), and grants from the Western Norway Regional Health Authority (911466 and 911688 to C.B.). None of the authors have any conflicts of interests.
Published online: October 20, 2011
Footnotes
Supplemental Data include seven figures and four tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes project, http://www.1000genomes.org/
IGV browser, http://www.broadinstitute.org/igv
Online Mendelian Inheritance in Man, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
Supplemental Data
References
- 1.Waters A.M., Beales P.L. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gascue C., Katsanis N., Badano J.L. Cystic diseases of the kidney: ciliary dysfunction and cystogenic mechanisms. Pediatr. Nephrol. 2011;26:1181–1195. doi: 10.1007/s00467-010-1697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fry A.E., Klingenberg C., Matthes J., Heimdal K., Hennekam R.C., Pilz D.T. Connective tissue involvement in two patients with features of cranioectodermal dysplasia. Am. J. Med. Genet. A. 2009;149A:2212–2215. doi: 10.1002/ajmg.a.33027. [DOI] [PubMed] [Google Scholar]
- 4.Sensenbrenner J.A., Dorst J.P., Owens R.P. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig. Artic. Ser. 1975;11:372–379. [PubMed] [Google Scholar]
- 5.Jeune M., Beraud C., Carron R. Asphyxiating thoracic dystrophy with familial characteristics. Arch. Fr. Pediatr. 1955;12:886–891. [PubMed] [Google Scholar]
- 6.de Vries J., Yntema J.L., van Die C.E., Crama N., Cornelissen E.A., Hamel B.C. Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. Eur. J. Pediatr. 2010;169:77–88. doi: 10.1007/s00431-009-0991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavalcanti D.P., Huber C., Sang K.H., Baujat G., Collins F., Delezoide A.L., Dagoneau N., Le Merrer M., Martinovic J., Mello M.F., et al. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J. Med. Genet. 2011;48:88–92. doi: 10.1136/jmg.2009.069468. [DOI] [PubMed] [Google Scholar]
- 8.Dagoneau N., Goulet M., Geneviève D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L., et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho N.C., Francomano C.A., van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am. J. Med. Genet. 2000;90:310–314. doi: 10.1002/(sici)1096-8628(20000214)90:4<310::aid-ajmg9>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 10.Mill P., Lockhart P.J., Fitzpatrick E., Mountford H.S., Hall E.A., Reijns M.A., Keighren M., Bahlo M., Bromhead C.J., Budd P., et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 2011;88:508–515. doi: 10.1016/j.ajhg.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurd T.W., Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron, Exp. Nephrol. 2011;118:e9–e14. doi: 10.1159/000320888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cole D.G. The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic. 2003;4:435–442. doi: 10.1034/j.1600-0854.2003.t01-1-00103.x. [DOI] [PubMed] [Google Scholar]
- 13.Cole D.G., Snell W.J. SnapShot: Intraflagellar transport. Cell. 2009;137:784. doi: 10.1016/j.cell.2009.04.053. e1. [DOI] [PubMed] [Google Scholar]
- 14.Piperno G., Siuda E., Henderson S., Segil M., Vaananen H., Sassaroli M. Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects. J. Cell Biol. 1998;143:1591–1601. doi: 10.1083/jcb.143.6.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A., et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G., et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., et al. NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arts H.H., Bongers E.M., Mans D.A., van Beersum S.E., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A., Schuurs-Hoeijmakers J.H., de Leeuw N., et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 19.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J., et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 20.Thiel C., Kessler K., Giessl A., Dimmler A., Shalev S.A., von der Haar S., Zenker M., Zahnleiter D., Stöss H., Beinder E., et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becker J., Semler O., Gilissen C., Li Y., Bolz H.J., Giunta C., Bergmann C., Rohrbach M., Koerber F., Zimmermann K., et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Märker T., Voesenek K., Kartono A., et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 24.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Absalon S., Blisnick T., Kohl L., Toutirais G., Doré G., Julkowska D., Tavenet A., Bastin P. Intraflagellar transport and functional analysis of genes required for flagellum formation in trypanosomes. Mol. Biol. Cell. 2008;19:929–944. doi: 10.1091/mbc.E07-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iomini C., Li L., Esparza J.M., Dutcher S.K. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics. 2009;183:885–896. doi: 10.1534/genetics.109.101915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tonna S.J., Needham A., Polu K., Uscinski A., Appel G.B., Falk R.J., Katz A., Al-Waheeb S., Kaplan B.S., Jerums G., et al. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol. 2008;9:13. doi: 10.1186/1471-2369-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Machuca E., Hummel A., Nevo F., Dantal J., Martinez F., Al-Sabban E., Baudouin V., Abel L., Grünfeld J.P., Antignac C. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009;75:727–735. doi: 10.1038/ki.2008.650. [DOI] [PubMed] [Google Scholar]
- 30.Badano J.L., Leitch C.C., Ansley S.J., May-Simera H., Lawson S., Lewis R.A., Beales P.L., Dietz H.C., Fisher S., Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 31.Hoefele J., Wolf M.T., O'Toole J.F., Otto E.A., Schultheiss U., Dêschenes G., Attanasio M., Utsch B., Antignac C., Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J. Am. Soc. Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 32.Ocbina P.J., Eggenschwiler J.T., Moskowitz I., Anderson K.V. Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao C., Malicki J. Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 2011;30:2532–2544. doi: 10.1038/emboj.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sedmak T., Wolfrum U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J. Cell Biol. 2010;189:171–186. doi: 10.1083/jcb.200911095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boldt K., Mans D.A., Won J., van Reeuwijk J., Vogt A., Kinkl N., Letteboer S.J., Hicks W.L., Hurd R.E., Naggert J.K., et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J. Clin. Invest. 2011;121:2169–2180. doi: 10.1172/JCI45627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amar M.J., Sutphen R., Kousseff B.G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome) Am. J. Med. Genet. 1997;70:349–352. doi: 10.1002/(sici)1096-8628(19970627)70:4<349::aid-ajmg3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 37.Young I.D. Cranioectodermal dysplasia (Sensenbrenner's syndrome) J. Med. Genet. 1989;26:393–396. doi: 10.1136/jmg.26.6.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borgström M.K., Riise R., Tornqvist K., Granath L. Anomalies in the permanent dentition and other oral findings in 29 individuals with Laurence-Moon-Bardet-Biedl syndrome. J. Oral Pathol. Med. 1996;25:86–89. doi: 10.1111/j.1600-0714.1996.tb00198.x. [DOI] [PubMed] [Google Scholar]
- 39.Hanemann J.A., de Carvalho B.C., Franco E.C. Oral manifestations in Ellis-van Creveld syndrome: report of a case and review of the literature. J. Oral Maxillofac. Surg. 2010;68:456–460. doi: 10.1016/j.joms.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 40.Venugopalan S.R., Li X., Amen M.A., Florez S., Gutierrez D., Cao H., Wang J., Amendt B.A. Hierarchical interactions of homeodomain and forkhead transcription factors in regulating odontogenic gene expression. J. Biol. Chem. 2011;286:21372–21383. doi: 10.1074/jbc.M111.252031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thivichon-Prince B., Couble M.L., Giamarchi A., Delmas P., Franco B., Romio L., Struys T., Lambrichts I., Ressnikoff D., Magloire H., Bleicher F. Primary cilia of odontoblasts: possible role in molar morphogenesis. J. Dent. Res. 2009;88:910–915. doi: 10.1177/0022034509345822. [DOI] [PubMed] [Google Scholar]
- 42.Ohazama A., Haycraft C.J., Seppala M., Blackburn J., Ghafoor S., Cobourne M., Martinelli D.C., Fan C.M., Peterkova R., Lesot H., et al. Primary cilia regulate Shh activity in the control of molar tooth number. Development. 2009;136:897–903. doi: 10.1242/dev.027979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Q., Murcia N.S., Chittenden L.R., Richards W.G., Michaud E.J., Woychik R.P., Yoder B.K. Loss of the Tg737 protein results in skeletal patterning defects. Dev. Dyn. 2003;227:78–90. doi: 10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]
- 44.Ko H.W., Liu A., Eggenschwiler J.T. Analysis of hedgehog signaling in mouse intraflagellar transport mutants. Methods Cell Biol. 2009;93:347–369. doi: 10.1016/S0091-679X(08)93017-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.