

Abstract
Say-Barber-Biesecker-Young-Simpson syndrome (SBBYSS or Ohdo syndrome) is a multiple anomaly syndrome characterized by severe intellectual disability, blepharophimosis, and a mask-like facial appearance. A number of individuals with SBBYSS also have thyroid abnormalities and cleft palate. The condition usually occurs sporadically and is therefore presumed to be due in most cases to new dominant mutations. In individuals with SBBYSS, a whole-exome sequencing approach was used to demonstrate de novo protein-truncating mutations in the highly conserved histone acetyltransferase gene KAT6B (MYST4/MORF)) in three out of four individuals sequenced. Sanger sequencing was used to confirm truncating mutations of KAT6B, clustering in the final exon of the gene in all four individuals and in a further nine persons with typical SBBYSS. Where parental samples were available, the mutations were shown to have occurred de novo. During mammalian development KAT6B is upregulated specifically in the developing central nervous system, facial structures, and limb buds. The phenotypic features seen in the Qkf mouse, a hypomorphic Kat6b mutant, include small eyes, ventrally placed ears and long first digits that mirror the human phenotype. This is a further example of how perturbation of a protein involved in chromatin modification might give rise to a multisystem developmental disorder.
Main Text
Blepharophimosis, an abnormal narrowing of the palpebral fissures, is seen in association with intellectual disability in the group of disorders collectively known as the blepharophimosis-mental retardation syndromes.1 Although the first of these was described by Ohdo et al.2 [MIM 249620], the most distinctive phenotype is the Say-Barber-Biesecker-Young-Simpson syndrome (SBBYSS),3, 4, 5 which is characterized by a distinctive facial appearance with severe blepharophimosis, an immobile mask-like face, a bulbous nasal tip, and a small mouth with a thin upper lip. (Figure 1A). The condition presents in infancy with severe hypotonia and feeding problems. Associated skeletal problems include joint laxity, abnormally long thumbs and great toes, and dislocated or hypoplastic patellae. Structural cardiac defects are present in around 50% of cases, and dental anomalies, including small and pointed teeth, are common. Many affected individuals have abnormalities of thyroid structure or function.5 SBBYSS is usually associated with severe mental retardation, delayed motor milestones, and significantly impaired speech. The majority of individuals with SBBYSS are simplex cases. Microarray analyses of a previously reported clinical cohort6 failed to identify novel variants to explain the severe phenotype of typical SBBYSS, and it was postulated that this condition was most likely due to new dominant mutations within a single gene. Here, we demonstrate that de novo protein-truncating mutations within KAT6B [MIM 605880] on chromosomal region 10q22, which encodes a highly conserved histone acetyltransferase involved in chromatin modification,7 give rise to the SBBYSS phenotype.
Figure 1.
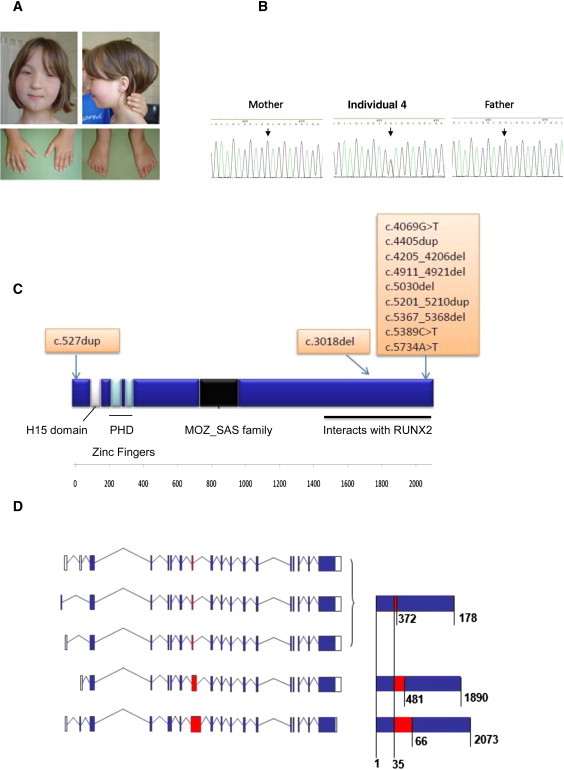
Clinical Phenotype, Structure, and Truncating Mutation of KAT6B
(A) Photos showing clinical features of individual 10 with typical SBBYSS. Facial features include severe blepharophimosis, a bulbous nasal tip, and a mask-like appearance. The thumbs and great toes are abnormally long and straight.
(B) Sanger sequencing of KAT6B in individual 4. The c.4069G>T (p.Glu1357X) mutation was absent in both parents and had arisen de novo.
(C) Schematic of KAT6B including the conserved domains and position of truncating mutations. The catalytic MYST domain of the MOZ-SAS family is the histoneacetyltransferase domain and contains a specific C2HC zinc finger together with an acetyl-coenzyme A binding domain. There is a conserved N-terminal domain ([NEMM] the N-terminal domain conserved in Enok) and a highly conserved C-terminal SM-rich domain. Mutations in typical SBBYSS individuals cluster in exon 18, the final exon, and leave the catalytic domain intact.
(D) KAT6B isoforms. Alternative splicing of KAT6B produces five protein coding transcripts. Three transcripts vary only in the 5′ UTR and produce the same mature protein. In-frame splicing within exon 8 results in three protein isoforms of 1781, 1890, and 2013 amino acids in length. As amino acid composition before and after exon 8 is identical in each isoform, the mutations found in exons 15 and 18 are assumed to have the same effect. White boxes indicate exons in UTR; blue boxes indicate translated exons; red box indicates exon 8; blue lines indicate introns. Mature protein contributed by exon 8 is shown in red; values indicate amino acid positions.
A cohort of 19 individuals with a presumed diagnosis of SBBYSS were referred to an ethically approved research study. Informed consent was obtained from each family and clinical evaluation and genetic testing were carried out in accordance with the ethics approval granted (04/MRE10/1). Four of the families were approached to participate in an extension study involving whole-exome sequencing (MREC 10/H1016/12) and informed consent was again obtained. On clinical evaluation, 12 individuals were considered to have typical features of SBBYSS syndrome, two had suggestive but milder features, and five were classified as atypical. The clinical features of each person are documented in detail in Table S1, available online.
Microarray analysis was carried out in 9 out of 17 individuals with the Affymetrix Genome-Wide Human SNP6.0 Assay following the manufacturer's protocol. Copy-number data processing and visualization were performed with Affymetrix Genotyping Console (version 3.0.1) set at a 50 kb threshold for genomic segment size with a minimum of 25 markers per segment. External resources included the Database of Genomic Variants, Ensembl, and Online Mendelian Inheritance in Man. Seven out of nine individuals (numbered 1, 2, 4, 5, 6, 10, and 14) had a normal array result. Individual 3 was shown to have a de novo duplication of approximately 2 Mb within the long arm of chromosome 1 at band q21.1 (Figure S1A), similar to the recurrent duplications of this region reported by Mefford et al.8 Because this duplication is usually associated with a very mild clinical phenotype, it was not considered to be the cause of the severe SBBYSS phenotype in this individual. Individual 17 had two significant imbalances of chromosome 1, a deletion of ∼8.3 Mb (1p36.12 to 1p36.21 nucleotides chromosome 1: 13339380-21692325 based on Ensembl Build 36) and a duplication of ∼1.3 Mb (1p34, nucleotides chromosome 1: 34572912-35949096, Figures S1B and S1C). Although maternal fluorescence in situ hybridization studies were normal, a paternal sample was not available, and a de novo origin could not be confirmed.
For exome sequencing, targeted enrichment and sequencing were performed on DNA extracted from the peripheral blood of individuals 1, 2, 3, and 4, all of whom were considered to have typical features of SBBYSS. Enrichment was performed with SureSelect Human All Exon 50MB Kit (Agilent), and the four samples were run indexed on a SOLiD 4 sequencer (Life Technologies). Reads were aligned to the human reference genome sequence (USCS hg19 February 2009 Genome Reference Consortium GRCh37). Bioinformatic analysis of results obtained is summarized in Table 1. Assuming a de novo mutation model, we filtered for variants on the same gene across multiple individuals. This identified eight single nucleotide variants (SNVs) of which only two were nonsense mutations. Individual 4 had a heterozygous c.4069G>T (p.Glu1357X) variant in KAT6B (RefSeq NM_012330.2). Two heterozygous indels (a 1 bp insertion and a 2 bp deletion) were found in the same gene in individuals 1 and 2, respectively. All three variants were confirmed with Sanger sequencing. The c.4069G>T mutation was absent in both parents and had arisen de novo (Figure 1B).
Table 1.
Filtering Procedure for Bioinformatics Analysis
| Filtering for Minimum 20× Novel Allele Depth | Total SNVa | Variants not in dbSNP132b | Total Nonsynonymous and Splice-Site SNV | Nonsynonymous and Splice-Site SNV not in dbSNP132c | Nonsense SNV not in dbSNP132 | Nonsense SNV not in dbSNP132 | Indels not in dbSNP132 |
|---|---|---|---|---|---|---|---|
| Person 1 | 29972 | 7478 | 8755 | 3533 | 174 | 0 | 1d |
| Person 2 | 28883 | 6775 | 8331 | 3229 | 136 | 1 | 1d |
| Person 3 | 29061 | 6806 | 8286 | 3119 | 143 | 0 | 0 |
| Person 4 | 31467 | 7000 | 8828 | 3224 | 148 | 1d | 0 |
The total number of SNVs seen in each individual is shown in column 1. Of the nonsynonymous and splice-site variants, those not in dbSNP132 were identified. Filtering for genes where either nonsense variants or small indels were shared by the individuals studied revealed that three individuals had either a nonsense variant or a small indel predicted to cause protein truncation in the KAT6B gene.
SNV.
SNP database.
Unless seen in Human Gene Mutation Database.
In KAT6B.
Subsequently, all 19 individuals with a SBBYSS or a SBBYSS-like phenotype were sequenced for the entire KAT6B coding region by classical Sanger sequencing. Details of primer sequences are included in the supplemental information and PCR methods are available on request. Results are shown in Table 2. Truncating mutations were confirmed in 13 individuals, including individual 3 who had a heterozygous frameshift mutation, c.3018del (p.Glu1007ArgfsX5) in exon 15, not initially detected on whole-exome sequencing. Individual 14, who had a milder phenotype with less severe facial features and milder intellectual disability, had a missense variant, c.1078G>A (p.Glu360Lys) in exon 8. His parents were not available for study. This same missense mutation was subsequently identified in a normal control and was therefore deemed to be nonpathogenic. None of the other variants were found in dbSNP132 or in 401 exomes of the ClinSeq cohort9 of healthy individuals recruited to a pilot study of exome sequencing (Table 3).
Table 2.
KAT6B Sequence Variants in 19 Individuals Tested
| Individual | Mutation | Exon | Parents | Comments |
|---|---|---|---|---|
| 1 | NM_012330.2(KAT6B):c.4405dup (p.Ser1469Phefs∗18) | 18 | not tested | typical; case 322 |
| 2 | NM_012330.2(KAT6B):c.5370_5373dup (p.Ile1792GlnfsX12) | 18 | not tested | typical |
| 3 | NM_012330.2(KAT6B):c.3018del (p.Glu1007Argfs∗5) | 15 | negative | typical; also has 1q21.1 microdup |
| 4 | NM_012330.2(KAT6B):c.4069G>T (p.Glu1357∗) | 18 | negative | typical. Figure 3 in Day et al.6 |
| 5 | NM_012330.2(KAT6B):c.4205_4206del (p.Ser1402Cysfs∗5) | 18 | negative | typical |
| 6 | NM_012330.2(KAT6B):c.5734A>T (p.Arg1912∗) | 18 | not tested | typical case 222 |
| 7 | NM_012330.2(KAT6B):c.5030del (p.Thr1677Metfs∗38) | 18 | not tested | typical |
| 8 | NM_012330.2(KAT6B):c.5201_5210dup (p.Gln1737Hisfs∗41) | 18 | not tested | typical |
| 9 | NM_012330.2(KAT6B):c.5201_5210dup (p.Gln1737Hisfs∗41) | 18 | negative | typical |
| 10 | NM_012330.2(KAT6B):c.4205_4206del (p.Ser1402Cysfs∗5) | 18 | not tested | typical case 422 |
| 11 | NM_012330.2(KAT6B):c.527dup (p.Tyr176∗) | 3 | not tested | SBBYSS-like, not typical |
| 12 | NM_012330.2(KAT6B):c.4911_4921del (p.Val1638Alafs∗27) | 18 | negative | typical |
| 13 | NM_012330.2(KAT6B):c.5389C>T (p.Arg1797∗) | 18 | negative | typical |
| 14 | NM_012330.2(KAT6B):c.1078G>A (p.Glu360Lys) | 8 | not tested | milder phenotype |
| 15 | negative | atypical | ||
| 16 | negative | atypical | ||
| 17 | negative | atypical; has 1p36 microdeletion | ||
| 18 | negative | atypical | ||
| 19 | negative | atypical |
This table shows the Sanger sequencing results from the cohort of individuals tested. Individuals 1–4 are those where exome sequencing was carried out. Twelve of the individuals were judged to have a typical clinical presentation, and sequence variants predicted to cause protein truncation were identified in all of these. Eleven of these twelve variants were in exon 18. In addition, a truncating mutation was identified in exon 3 in a less typical individual and a missense variant was identified in exon 8 in an individual with a much milder phenotype. This boy had only a moderate learning disability and significantly less speech delay. This missense variant has subsequently been identified in a normal control and is now considered to be unrelated to the clinical presentation. The chance of finding a KAT6B mutation in our series of clinically typical individuals was therefore high.
Table 3.
Variants in KAT6B NM_12330 from the ClinSeq Cohort
| Chromosome | Start | End | Human Genome Variation Society | Type of Variant | RS | Homozygous Frequency | Heterozygous Frequency |
|---|---|---|---|---|---|---|---|
| 10 | 76784992 | 76784993 | c.3649G>T (p.Ala1217Ser) | nonsynonymous | rs57372986 | 0.9975 | 0.0025 |
| 10 | 76781027 | 76781028 | c.3005G>A (p.Arg1002Gln) | nonsynonymous | – | 0.9975 | 0.0025 |
| 10 | 76781906 | 76781930 | c.3289_3291del3 | DIV-c | rs111976843 | 0.7292 | 0.2708 |
| 10 | 76789968 | 76789969 | c.5386G>A (p.Glu1796Lys) | nonsynonymous | – | 0.9975 | 0.0025 |
| 10 | 76789304 | 76789305 | c.4722C>G (p.Asn1574Lys) | nonsynonymous | – | 0.9975 | 0.0025 |
| 10 | 76784747 | 76784748 | c.3404G>A (p.Arg1135His) | nonsynonymous | – | 0.9975 | 0.0025 |
| 10 | 76789417 | 76789418 | c.4835G>A (p.His1612Arg) | nonsynonymous | rs72803461 | 0.9925 | 0.0075 |
| 10 | 76735369 | 76735379 | c.1274_1279del (p.Lys426_Val427del) | DIV-c | – | 0.9975 | 0.0025 |
| 10 | 76735563 | 76735564 | c.1469C>T (p.Pro490Leu) | nonsynonymous | – | 0.995 | 0.005 |
| 10 | 76781875 | 76781876 | c.3258A>C (p.Glu1086Asp) | nonsynonymous | – | 0.9973 | 0.0027 |
| 10 | 76789092 | 76789093 | c.4510G>A (p.Glu1504Lys) | nonsynonymous | – | 0.9975 | 0.0025 |
| 10 | 76790184 | 76790185 | c.5602G>A (p.Val1868Ile) | nonsynonymous | – | 0.9975 | 0.0025 |
| 10 | 76789077 | 76789078 | c.4495G>A (p.Val1499Ile) | nonsynonymous | rs3740321 | 0.9551 | 0.0424 |
| 10 | 76790331 | 76790332 | c.5749A>G (p.Ile1917Val) | nonsynonymous | – | 0.9975 | 0.0025 |
Mice carrying a gene trap insertion in the mouse ortholog Kat6b (Qkf/Myst4/Morf) that produces approximately 5% of the normal amount of Kat6b mRNA10 were examined for phenotypic characteristics seen in SBBYSS. All experiments in mice were carried out according to the Australian National Health and Medical Research Council (NHMRC) code of conduct and approval of the Melbourne Health Animal Ethics Committee. The Qkfgt/gt hypomorphic mutant displayed a number of defects that mirror SBBYS syndrome, though the phenotype in the mice is milder. Mice are of normal size at birth but fail to thrive and have brain developmental defects as well as craniofacial defects.11 Observed abnormalities include short and narrow palpebral fissures, low set ears (Figures 2A and 2B), and malocclusion. Qkf mRNA is strongly expressed in the eyelids and teeth primordia during development. (Figures 2C and 2D) Moreover, β-galactosidase expressed from a lacZ reporter gene inserted into the Kat6b locus in the Qkfgt allele, which reflects the high-level expression domain of the Qkf locus, indicates that Qkf is activated in the medial aspect of the hand and footplate at embryonic day 10 in the mouse and marks all digits as they arise (Figure 2E). Similar to individuals with SBBYSS, the Qkfgt/gt mice have long, slender feet and disproportionally long first digits (Figure 2F). The thyroid gland in neonatal Qkf mice is morphologically and histologically normal, though the possibility of a functional defect of the thyroid has not been explored.
Figure 2.
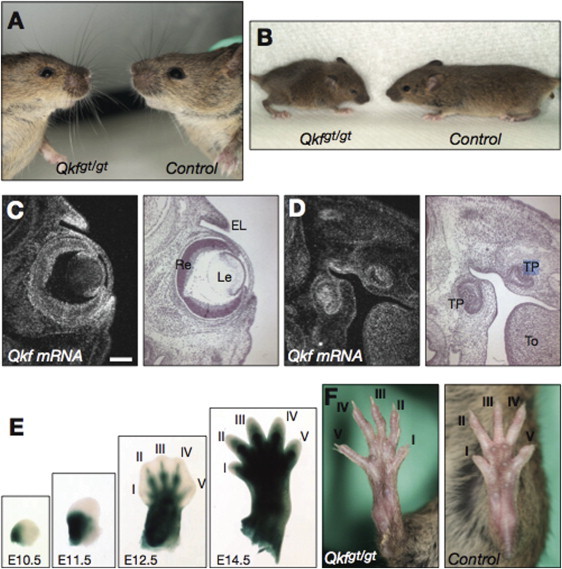
Morphology of Qkfgt/gt Mutant Mice and Expression of Kat6b mRNA in Tissues Affected in SBBYSS Individuals
(A) Facial features including small eyes and upturned nose in a 10-month-old male Qkfgt/gt mutant mouse compared to a same-sex wild-type littermate control.
(B) External appearance of a 3-week-old male Qkfgt/gt mutant mouse and a male littermate control.
(C) Qkf mRNA distribution in E15.5 eyes and eyelids. Precipitated silver grains representing Qkf mRNA appear white in the darkfield image; the brightfield image is in the adjacent panel.
(D) Qkf mRNA distribution in E15.5 tooth primordia.
(E) β-galactosidase reporter staining (blue) indicates high-expression activity of the Qkf locus in the developing hindlimb at the stages indicated.
(F) Feet of a 3-week-old male Qkfgt/gt mutant mouse and a male littermate control. Note the long and slender appearance of the Qkfgt/gt foot and digits. The following abbreviations are used: I to V, digits I to V of the hindlimb; EL, eyelids; Le, lens; To, tongue; TP, tooth primordium; Re, retina. The scale bars (C and D) equal 133 μm. Embryo recovery, histological processing and sectioning, radioactive in situ hybridization, and β-galactosidase staining were performed as previously published.10, 20, 21
Clinical diagnosis of blepharophimosis-learning disability syndromes is challenging. Autosomal-recessive, autosomal-dominant, and X-linked inheritance have all been proposed,1 making risk assessment for future pregnancies difficult. Our findings of KAT6B mutations in all individuals with typical features of SBBYSS suggest that this particular phenotype is a homogeneous disorder and underlines the overlap between the phenotypes described by Say and Barber,3 Biesecker,4 and Young and Simpson5 (YSS [MIM 603736]); the latter authors emphasize the additional features of polydactyly and thyroid abnormalities. Our data suggest that the likelihood of identifying a pathogenic KAT6B mutation in individuals with the typical SBBYSS phenotype is high. Where the presentation is atypical, the phenotype might have a separate genetic etiology as evidenced by the likely pathogenic microarray abnormality in Individual 17. We consider the 1q21 duplication in Individual 3 to be a coincidental occurrence.
The MYST proteins including KAT6B are the largest group of histone acetyltransferases.7 Studies of the hypomorphic Qkfgt/gt mutant have demonstrated that Kat6b is upregulated in developing cerebral cortex. It is also expressed in adult neural stem cells, osteoblasts, and germ cells.11, 12 Structural brain abnormalities have not been studied extensively in individuals with SBBYSS, although two individuals have been documented to have abnormalities of the corpus callosum,6 and white matter changes and delayed myelination have also been observed.13
Individuals with deletions of 10q22 involving KAT6B are rare. Tzschach et al.14 reported three individuals with deletions encompassing KAT6B, and all had a severe learning disability. Their facial features were not, however, typical of SBBYSS. Translocations through intron 16 of KAT6B have been implicated in acute myeloid leukemia15 where the gene forms a fusion protein with Creb-binding protein. Another translocation between the C terminus of KAT6B and KAT2A (GCN5L2) [MIM 602301] occurs in benign uterine leiomyomata.16 So far, no one with the SBBYSS has developed a malignancy or uterine leiomyomata, but follow-up into adulthood has been limited.
KAT6B has a highly conserved C-terminal serine and methionine (SM)-rich domain encompassing exon 18 where the KAT6B mutations causing typical SBBYSS cluster. This C-terminal region is known to bind to the transcription factor RUNX2 (CBFA1) [MIM 600211] in vitro and in vivo.17 Heterozygous loss-of-function mutations of RUNX2 have been shown to cause cleidocranial dysplasia (CCD [MIM 119600]), a disorder of membranous bone ossification that affects the skull and clavicles. Endogenous KAT6B is required for RUNX2-dependent transcriptional activation.
Recently, a boy diagnosed with a phenotype that overlapped with Noonan syndrome (NS1 [MIM 163950]) was reported to be haploinsufficient for KAT6B as a result of a de novo t(10;13)(q22.3;q34) translocation with a breakpoint in intron 3.18 This individual shared some clinical features with those we have described who have heterozygous point mutations and SBBYSS, including a learning disability, blepharophimosis, ptosis, and ear anomalies; however, the facial characteristics of the typical SBBYSS individuals are more distinctive than his are. In this regard, the Qkfgt mice more closely resemble the milder phenotype that overlaps with Noonan syndrome than the SBBYSS phenotype associated with heterozygous point mutations. These observations suggest that mutations of protein-protein interaction domains in exon 18 result in a more complex phenotype that is not due to simple haploinsufficiency of KAT6B and raises the possibility that exon 18 mutations are activating or have a dominant negative effect. Alternatively, haploinsufficiency for just a single important domain could be responsible for the phenotype. This situation is somewhat analogous to that seen in Hajdu-Cheney syndrome (HJCYS [MIM 102500]), where truncating mutations are confined to the C-terminal region of NOTCH219 [MIM 600275] that contains a proline-glutamate-serine-threonine-rich region important in proteolytic recognition. If this region is missing, NOTCH-2 signaling is increased. One argument against this hypothesis might be the finding of an exon 15 mutation in individual 3. However, assessing this child clinically was difficult because she also has a copy number variation at chromosomal region 1q21.1 that is likely to have also contributed to her phenotype, and it is possible that this exon 15 variant would produce a milder phenotype in the absence of the 1q21.1 duplication.
The finding of de novo KAT6B mutations in individuals with the SBBYS phenotype confirms the condition as a distinct clinical entity. Although the possibility of gonadal mosaicism must be considered, and most likely explains the rare familial recurrence of the condition,6 other individuals with SBBYSS are simplex cases, and unaffected parents who do not carry the same mutation can now be counseled with some reassurance that the risk of recurrence is low, likely to be 1% or less on the basis of clinical observations. This observation confirms the important role of chromatin modifying genes in human development.
Acknowledgments
Clinical and Laboratory work carried out within the Genetic Medicine Department at St Mary's Hospital was supported by the Manchester Biomedical Research Centre funded by the National Institute for Health Research. Research into clefting syndromes in Manchester is supported by the Healing Foundation. The Clinseq project was funded by the National Institute of Health. A.K.V. and T.T. were supported by project grants and research fellowships from the Australian NHMRC as well as through infrastructure funding from the Victorian government.
Published online: November 10, 2011
Footnotes
Supplemental Data include two figures and two tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
dbSNP132, http://www.ncbi.nlm.nih.gov/snp
Elements of Morphology, http://elementsofmorphology.nih.gov/index.cgi
Ensembl Genome Browser, http://www.ensemble.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Supplemental Data
References
- 1.Verloes A., Bremond-Gignac C., Isidor B., David A., Baumann C., Leroy M.A., Stevens R., Gillerot Y., Héron D., Héron B., et al. Blepharophimosis-mental retardation syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am. J. Med. Genet. A. 2006;140:1285–1296. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- 2.Ohdo S., Madokoro H., Sonoda T., Hayakawa K. Mental retardation associated with congenital heart disease, blepharophimosis, blepharoptosis, and hypoplastic teeth. J. Med. Genet. 1986;23:242–244. doi: 10.1136/jmg.23.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Say B., Barber N. Mental retardation with blepharophimosis. J. Med. Genet. 1987;24:511. doi: 10.1136/jmg.24.8.511-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biesecker L.G. The Ohdo blepharophimosis syndrome: a third case. J. Med. Genet. 1991;28:131–134. doi: 10.1136/jmg.28.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young I.D., Simpson K. Unknown syndrome: abnormal facies, congenital heart defects, hypothyroidism, and severe retardation. J. Med. Genet. 1987;24:715–716. doi: 10.1136/jmg.24.11.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Day R., Beckett B., Donnai D., Fryer A., Heidenblad M., Howard P., Kerr B., Mansour S., Maye U., McKee S., et al. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin. Genet. 2008;74:434–444. doi: 10.1111/j.1399-0004.2008.01087.x. [DOI] [PubMed] [Google Scholar]
- 7.Voss A.K., Thomas T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays. 2009;31:1050–1061. doi: 10.1002/bies.200900051. [DOI] [PubMed] [Google Scholar]
- 8.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D., et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., et al. NISC Comparative Sequencing Program The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas T., Voss A.K., Chowdhury K., Gruss P. Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development. 2000;127:2537–2548. doi: 10.1242/dev.127.12.2537. [DOI] [PubMed] [Google Scholar]
- 11.Merson T.D., Dixon M.P., Collin C., Rietze R.L., Bartlett P.F., Thomas T., Voss A.K. The transcriptional coactivator Querkopf controls adult neurogenesis. J. Neurosci. 2006;26:11359–11370. doi: 10.1523/JNEUROSCI.2247-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGraw S., Morin G., Vigneault C., Leclerc P., Sirard M.A. Investigation of MYST4 histone acetyltransferase and its involvement in mammalian gametogenesis. BMC Dev. Biol. 2007;7:123. doi: 10.1186/1471-213X-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szakszon K., Berényi E., Jakab A., Bessenyei B., Balogh E., Köbling T., Szilvássy J., Knegt A.C., Oláh E. Blepharophimosis mental retardation syndrome Say-Barber/Biesecker/Young-Simpson type - new findings with neuroimaging. Am. J. Med. Genet. A. 2011;155A:634–637. doi: 10.1002/ajmg.a.33837. [DOI] [PubMed] [Google Scholar]
- 14.Tzschach A., Bisgaard A.M., Kirchhoff M., Graul-Neumann L.M., Neitzel H., Page S., Ahmed A., Müller I., Erdogan F., Ropers H.H., et al. Chromosome aberrations involving 10q22: report of three overlapping interstitial deletions and a balanced translocation disrupting C10orf11. Eur. J. Hum. Genet. 2010;18:291–295. doi: 10.1038/ejhg.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panagopoulos I., Fioretos T., Isaksson M., Samuelsson U., Billström R., Strömbeck B., Mitelman F., Johansson B. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13) Hum. Mol. Genet. 2001;10:395–404. doi: 10.1093/hmg/10.4.395. [DOI] [PubMed] [Google Scholar]
- 16.Moore S.D., Herrick S.R., Ince T.A., Kleinman M.S., Dal Cin P., Morton C.C., Quade B.J. Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res. 2004;64:5570–5577. doi: 10.1158/0008-5472.CAN-04-0050. [DOI] [PubMed] [Google Scholar]
- 17.Pelletier N., Champagne N., Stifani S., Yang X.J. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene. 2002;21:2729–2740. doi: 10.1038/sj.onc.1205367. [DOI] [PubMed] [Google Scholar]
- 18.Kraft M., Cirstea I.C., Voss A.K., Thomas T., Goehring I., Sheikh B.N., Gordon L., Scott H., Smyth G.K., Ahmadian M.R., et al. Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J. Clin. Invest. 2011;121:3479–3491. doi: 10.1172/JCI43428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K., et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 20.Voss A.K., Thomas T., Gruss P. Efficiency assessment of the gene trap approach. Dev. Dyn. 1998;212:171–180. doi: 10.1002/(SICI)1097-0177(199806)212:2<171::AID-AJA3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 21.Voss A.K., Thomas T., Gruss P. Mice lacking HSP90beta fail to develop a placental labyrinth. Development. 2000;127:1–11. doi: 10.1242/dev.127.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Clayton-Smith J., Krajewska-Walasek M., Fryer A., Donnai D. Ohdo-like blepharophimosis syndrome with distinctive facies, neonatal hypotonia, mental retardation and hypoplastic teeth. Clin. Dysmorphol. 1994;3:115–120. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.