

Abstract
Mutations in POLR3A encoding the largest subunit of RNA polymerase III (Pol III) were found to be responsible for the majority of cases presenting with three clinically overlapping hypomyelinating leukodystrophy phenotypes. We uncovered in three cases without POLR3A mutation recessive mutations in POLR3B, which codes for the second largest subunit of Pol III. Mutations in genes coding for Pol III subunits are a major cause of childhood-onset hypomyelinating leukodystrophies with prominent cerebellar dysfunction, oligodontia, and hypogonadotropic hypogonadism.
Main Text
Leukodystrophies are a heterogeneous group of neurodegenerative disorders characterized by abnormal central nervous system white matter.1 It is estimated that at least 30% to 40% of patients with leukodystrophies remain without a precise diagnosis despite extensive investigations.2 We recently demonstrated that the majority of cases affected by tremor-ataxia with central hypomyelination (TACH),3, 4 hypomyelination, hypodontia, and hypogonadotropic hypogonadism (4H syndrome [MIM 612440]),5, 6, 7, 8, 9 and leukodystrophy with oligodontia (LO [MIM 607694])10, 11 are caused by mutations in POLR3A (MIM 610210).12 POLR3A is the largest of the 17 subunits that constitute RNA polymerase III (Pol III). It forms, together with the second largest subunit (POLR3B), the catalytic center of the enzyme. Pol III transcribes small untranslated RNAs (e.g., tRNAs, 5S RNA, 7SK RNA, and U6 RNA) involved in the regulation of essential cellular processes, including transcription, RNA processing, and translation.13 We suggested that these allelic conditions be referred to as Pol III-related hypomyelinating leukodystrophies. Having not identified POLR3A mutations in four of nine 4H syndrome cases,12 we searched for mutations in POLR3B, which codes for the other subunit that forms Pol III's catalytic site.
The research project was approved by the institutional ethics committee of the Centre de Recherche du CHUM (CRCHUM), Montreal, Canada; the institutional review board of the National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA; and the Children's National Medical Center, Washington D.C., USA. Informed consent was obtained from all participants. We sequenced all exons, exon-intron boundaries and 3′ and 5′ UTR of POLR3B (NM_018082, hg19) on available genomic DNA from two of the four 4H cases not found previously to carry POLR3A mutations (individuals 1 and 2)12 and one other case of 4H syndrome (individual 3) (Table S1, available online).
All three cases were found to be compound heterozygote for mutations in POLR3B (Figure 1A). One missense mutation in exon 15, c.1568T>A (p.Val523Glu), is common to all three individuals, who are from different ethnic backgrounds; two of the individuals reside in the USA and are of mixed European descent (individuals 1 and 2), and one individual is Italian (individual 3). This common mutation could result from an ancestral haplotype shared by all three individuals. However, we do not have access to genotyping data on these individuals to validate this hypothesis. Individual 1 carries another missense mutation, also in exon 15, that changes a threonine for a lysine at position 503 (c.1508C>A [p.Thr503Lys]). The impact of these two missense mutations on POLR3B was assessed with AlignGVGD, PhD-SNP, PolyPhen and SIFT. All bioinformatics programs predicted the mutations to be pathogenic. In addition to the common mutation, individual 2 was carrying a deletion of one base pair (c.1533delT [p.Ile511MetfsX513]) in exon 15 causing a frameshift and leading to a premature stop codon at position 513. The second mutation in individual 3 is a nonsense variant in exon 23 (c.2686A>T) creating a stop codon at position 896 (p.Lys896X). As seen in POLR3A mutations, none of the participants is homozygous for null mutations.12 All parents were unaffected and found to carry one of their affected child's mutations. All variants appear to be conserved across species (Figure 1A) and were absent in more than 340 control chromosomes except for the common mutation, c.1568T>A, which was found in 2/374 (0.5%) of control chromosomes. All control individuals were of European descent. This variant has never been reported in the SNPs database (dbSNP132).
Figure 1.
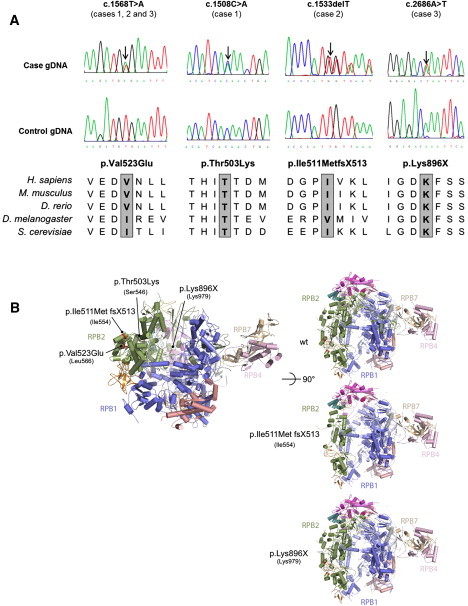
POLR3B Mutations
(A) Genomic sequence chromatograms and amino acid conservation across species is shown for all missense and nonsense POLR3B mutations. Individual 1 is compound heterozygote for mutations c.1508C>A and c.1568T>A; individual 2 is compound heterozygote for mutations c.1533delT and c.1568T>A, and individual 3 is compound heterozygote for mutations c.1568T>A and c.2686A>T. The amino acid conservation across species is shown for all mutations. The following abbreviations are used: D. melanogaster, Drosophila melanogaster; D. rerio, Danio rerio; H. sapiens, Homo sapiens; M. musculus, Mus musculus; S. cerevisiae, Saccharomyces cerevisiae.
(B) 3D representations of POLR3B missense, deletion (frameshift), and nonsense mutations. Point mutations identified in POLR3B are displayed according to their equivalent positions in the yeast RNA Polymerase II RPB2 subunit.24 Numbers in brackets refer to the positions in yeast RPB2. Images have been generated with PyMol software (Schrödinger). RPB1 is shown in blue, RPB2 in green, RPB7 in brown, RPB4 in pink, RPB8 in orange, and RPB11 in magenta.
The locations of the different mutations are shown in Figure 1B. POLR3B (RPC2) and POLR2B (RPB2) subunits of RNA polymerase III and RNA polymerase II, respectively, are highly conserved proteins with a similar structure.14 Based on the extrapolation of POLR3B (RPC2) from the yeast Pol II structure, the common mutation c.1568T>A (p.Val523Glu) corresponds to Leu566 in POLR2B (RPB2) that is, across species, always an aliphatic or aromatic residue. The mutation c.1508C>A (p.Thr503lys) corresponds to Ser546 in RPB2, which is highly conserved across species. The null mutation c.2686A>T (p.Lys896X), which corresponds to Lys979 in POLR2B (RPB2), and the deletion mutation leading to a frameshift and to a premature stop codon, c.1533delT (p.Ile511MetX513), which corresponds to Ile554 in POLR2B (RPB2), are predicted to generate, if the mRNA is stable enough, a truncated POLR3B subunit leading to a nonfunctional polymerase because the active site is expected to be largely affected in its function. Based on the Pol III electron microscopy structure and photocross-linking experiments,15, 16 the p.Val523Glu and p.Thr503Lys residues are located near the “jaw,” an area of Pol III where POLR3D (RPC4) and POLR3E (RPC5) are localized (counterpart of yeast RPC53 and RPC37). These two point mutations are predicted to affect the POLR3B structure locally and thus impair the proper function of RPC4 and RPC5 in transcription.
All three cases had characteristic clinical phenotypes of 4H syndrome.5, 8, 17 They all presented in early childhood with developmental delays and developed dysarthria as well as progressive motor difficulties, including cerebellar ataxia and, in individuals 1 and 2, progressive spasticity. Individuals 1 and 3 both developed hypogonadotropic hypogonadism, whereas individual 2 was too young to evaluate for endocrine dysfunction. All three individuals had teeth abnormalities, which are presented in Figure 2 for individuals 1 and 2 and consisted of neonatal upper incisors, delayed eruption of deciduous teeth and permanent teeth, abnormal sequence of eruption, and malposition in individual 3. The clinical features of individual 3 have been published previously.18 MRI features of the three individuals were also characteristic of previous descriptions of 4H syndrome19 (Figure 3).
Figure 2.
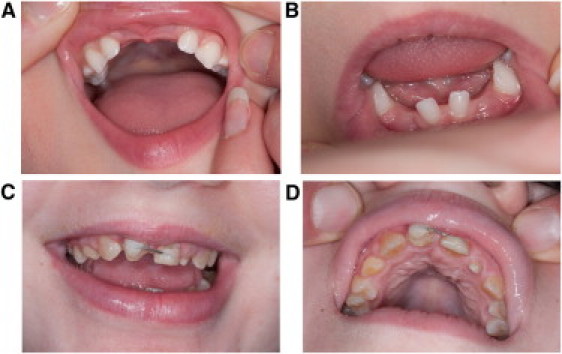
Teeth Abnormalities
(A and B) The teeth abnormalities seen in individual 1: unspecified age at tooth eruption and the absence of permanent mandibular second premolars with abnormal shape of the permanent maxillary central and lateral incisors.
(C and D) The teeth abnormalities of individual 2: delayed tooth eruption with complete retention of the primary maxillary central incisors and the mandibular lateral incisors.
Figure 3.
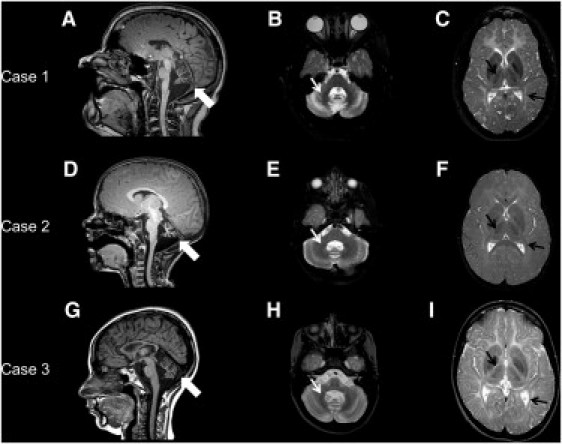
MRI Characteristics
(A, D, and G) Sagittal T1-weighted images of the brain for individuals 1 (A), 2 (D), and 3 (G) at the level of the midline demonstrating the thin corpus callosum and vermian cerebellar atrophy (thick white arrows).
(B, E, and H) Axial T2-weighted images of the brain for individuals 1 (B), 2 (E), and 3 (H) at the level of the pons and the dentate nucleus demonstrating a mild T2 hyperintensity of the cerebellar white matter associated with the typical hypointensity of the dentate nucleus (small white arrows).19
(C, F, and I) Axial T2-weighted images of the brain for individuals 1 (C), 2 (F), and 3 (I) at the level of the basal ganglia showing the mild hyperintense white matter characteristic of hypomyelination; there is relative sparing of the optic radiations and anterolateral nucleus of the thalamus (small black arrows), which appear hypointense.19
Pol III-related hypomyelinating leukodystrophies are a genetically and clinically heterogeneous group of disorders. We demonstrated previously that POLR3A mutations account for the majority of our relatively small group of affected individuals and are reporting that POLR3B mutations account for the remaining cases for which DNA was available (2/4), as well as one other case of 4H syndrome (individual 3). It is, however, likely that genes encoding for the other Pol III subunits are mutated in cases of hypomyelinating leukodystrophies. As previously suggested, because Pol III is responsible for the transcription of transfer RNAs (tRNAs) and other essential small RNAs, it is possible that mutations in POLR3A and POLR3B lead to an abnormal Pol III function that might affect the levels of certain tRNAs important for the development of the central nervous system white matter,20 leading to abnormal protein production. This pathophysiological mechanism is also attributed to leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL [MIM 611105]) caused by mutations in the nuclear-coded mitochondrial aspartyl-tRNA synthetase (DARS2 [MIM 610956]) and hypomyelinating leukodystrophy-3 (MIM 260600) caused by a mutation in aminoacyl-tRNA synthetase complex-interacting-multifunctional protein 1 (AIMP1/p43 [MIM 603605]).21, 22, 23 We believe that genes coding for other Pol III subunits and possibly genes coding for proteins interacting with Pol III could be mutated in cases of genetically uncharacterized hypomyelinating leukodystrophies.
Acknowledgments
We would like to thank all participants and the clinicians who referred patients to us. We would like to thank the “Fondation sur les Leucodystrophies” and the European Leukodystrophy Association for financing this research project. G.B. received fellowship scholarships from the Réseau de Médecine Génétique Appliquée and Fonds de Recherche en Santé du Québec. M. Tétreault received the Frederick Banting and Charles Best Doctoral scholarship from the Canadian Institute of Health Research. A.V.'s contribution is supported in part by the Intramural Research Program of the National Human Genome Research Institute and by the Myelin Disorders Bioregistry Project. M. Teichmann has received grants from the Agence Nationale de Recherche, the National Cancer Institute, the regional government of Aquitaine, and the Ligue Contre le Cancer (équipe labellisée). S.F. has received grants from La Ligue Contre le Cancer (Comité Dordogne), the regional government of Aquitaine, and the Inserm. The authors wish to thank all collaborators (including William McClintock) and patients participating in the Myelin Bioregistry Project.
Published online October 27, 2011
Footnotes
Supplemental data include one table and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for the data presented herein are as follows:
AlignGVGD, http://agvgd.iarc.fr/agvgd_input.php
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PhD-SNP, http://gpcr.biocomp.unibo.it/∼emidio/PhD-SNP/PhD-SNP.htm
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SIFT, http://sift.jcvi.org/
Supplemental Data
References
- 1.Schiffmann R., van der Knaap M.S. The latest on leukodystrophies. Curr. Opin. Neurol. 2004;17:187–192. doi: 10.1097/00019052-200404000-00017. [DOI] [PubMed] [Google Scholar]
- 2.Schiffmann R., van der Knaap M.S. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernard G., Thiffault I., Tetreault M., Putorti M.L., Bouchard I., Sylvain M., Melançon S., Laframboise R., Langevin P., Bouchard J.P., et al. Tremor-ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3-10q23.31. Neurogenetics. 2010;11:457–464. doi: 10.1007/s10048-010-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tetreault, M., Putorti, M.L., Thiffault, I., Sylvain, M., Vanderver, A., Schiffmann, R., Brais, B., and Bernard, G. Refinement of the locus responsible for Tremor-Ataxia with Central Hypomyelination (TACH) leukodystrophy to chromosome 10q22.3-23.1. Can. J. Neurol. Sci., in press.
- 5.Timmons M., Tsokos M., Asab M.A., Seminara S.B., Zirzow G.C., Kaneski C.R., Heiss J.D., van der Knaap M.S., Vanier M.T., Schiffmann R., Wong K. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. 2006;67:2066–2069. doi: 10.1212/01.wnl.0000247666.28904.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vázquez-López M., Ruiz-Martín Y., de Castro-Castro P., Garzo-Fernández C., Martín-del Valle F., Márquez-de la Plata L. Central hypomyelination, hypogonadotrophic hypogonadism and hypodontia: A new leukodystrophy. Rev. Neurol. 2008;47:204–208. [PubMed] [Google Scholar]
- 7.Wolf N.I., Harting I., Boltshauser E., Wiegand G., Koch M.J., Schmitt-Mechelke T., Martin E., Zschocke J., Uhlenberg B., Hoffmann G.F., et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology. 2005;64:1461–1464. doi: 10.1212/01.WNL.0000158615.56071.E3. [DOI] [PubMed] [Google Scholar]
- 8.Wolf N.I., Harting I., Innes A.M., Patzer S., Zeitler P., Schneider A., Wolff A., Baier K., Zschocke J., Ebinger F., et al. Ataxia, delayed dentition and hypomyelination: A novel leukoencephalopathy. Neuropediatrics. 2007;38:64–70. doi: 10.1055/s-2007-985137. [DOI] [PubMed] [Google Scholar]
- 9.Bekiesinska-Figatowska M., Mierzewska H., Kuczynska-Zardzewialy A., Szczepanik E., Obersztyn E. Hypomyelination, hypogonadotropic hypogonadism, hypodontia - First Polish patient. Brain Dev. 2010;32:574–578. doi: 10.1016/j.braindev.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Atrouni S., Darazé A., Tamraz J., Cassia A., Caillaud C., Mégarbané A. Leukodystrophy associated with oligodontia in a large inbred family: Fortuitous association or new entity? Am. J. Med. Genet. A. 2003;118A:76–81. doi: 10.1002/ajmg.a.10019. [DOI] [PubMed] [Google Scholar]
- 11.Chouery E., Delague V., Jalkh N., Salem N., Kfoury J., Rodriguez D., Chabrol B., Boespflug-Tanguy O., Lévy N., Serre J.L., Mégarbané A. A whole-genome scan in a large family with leukodystrophy and oligodontia reveals linkage to 10q22. Neurogenetics. 2011;12:73–78. doi: 10.1007/s10048-010-0256-3. [DOI] [PubMed] [Google Scholar]
- 12.Bernard G., Chouery E., Putorti M.L., Tétreault M., Takanohashi A., Carosso G., Clément I., Boespflug-Tanguy O., Rodriguez D., Delague V., et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011;89:415–423. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumay-Odelot H., Durrieu-Gaillard S., Da Silva D., Roeder R.G., Teichmann M. Cell growth- and differentiation-dependent regulation of RNA polymerase III transcription. Cell Cycle. 2010;9:3687–3699. doi: 10.4161/cc.9.18.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jasiak A.J., Armache K.J., Martens B., Jansen R.P., Cramer P. Structural biology of RNA polymerase III: Subcomplex C17/25 X-ray structure and 11 subunit enzyme model. Mol. Cell. 2006;23:71–81. doi: 10.1016/j.molcel.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Fernández-Tornero C., Böttcher B., Rashid U.J., Steuerwald U., Flörchinger B., Devos D.P., Lindner D., Müller C.W. Conformational flexibility of RNA polymerase III during transcriptional elongation. EMBO J. 2010;29:3762–3772. doi: 10.1038/emboj.2010.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C.C., Lin Y.C., Chen H.T. The TFIIF-like Rpc37/53 dimer lies at the center of a protein network to connect TFIIIC, Bdp1, and the RNA polymerase III active center. Mol. Cell. Biol. 2011;31:2715–2728. doi: 10.1128/MCB.05151-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolff A., Koch M.J., Benzinger S., van Waes H., Wolf N.I., Boltshauser E., Luder H.U. Rare dental peculiarities associated with the hypomyelinating leukoencephalopathy 4H syndrome/ADDH. Pediatr. Dent. 2010;32:386–392. [PubMed] [Google Scholar]
- 18.Orcesi S., Tonduti D., Uggetti C., Larizza D., Fazzi E., Balottin U. New case of 4H syndrome and a review of the literature. Pediatr. Neurol. 2010;42:359–364. doi: 10.1016/j.pediatrneurol.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 19.Steenweg M.E., Vanderver A., Blaser S., Bizzi A., de Koning T.J., Mancini G.M., van Wieringen W.N., Barkhof F., Wolf N.I., van der Knaap M.S. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–2982. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmar K.A., Goodenbour J.M., Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinstein M., Markus B., Noyman I., Shalev H., Flusser H., Shelef I., Liani-Leibson K., Shorer Z., Cohen I., Khateeb S., et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010;87:820–828. doi: 10.1016/j.ajhg.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheper G.C., van der Klok T., van Andel R.J., van Berkel C.G., Sissler M., Smet J., Muravina T.I., Serkov S.V., Uziel G., Bugiani M., et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 23.Boespflug-Tanguy O., Aubourg P., Dorboz I., Bégou M., Giraud G., Sarret C., Vaurs-Barrière C. Neurodegenerative disorder related to AIMP1/p43 mutation is not a PMLD. Am. J. Hum. Genet. 2011;88:392–393. doi: 10.1016/j.ajhg.2010.12.015. author reply 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Armache K.J., Mitterweger S., Meinhart A., Cramer P. Structures of complete RNA polymerase II and its subcomplex, Rpb4/7. J. Biol. Chem. 2005;280:7131–7134. doi: 10.1074/jbc.M413038200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.