

Non-technical summary
Cardiac glycosides (CGs) have been routinely used in the treatment of congestive heart failure (HF). Unfortunately, the therapeutic use of CGs in treating HF is limited by their adverse side effects, including cardiac arrhythmias. The arrhythmic side effects of CGs have been traditionally ascribed to excessive cellular Ca2+ retention (Ca2+ overload) leading to spontaneous discharges of intracellular Ca2+ stores, or Ca2+ waves, in turn causing oscillations of the cardiac membrane potential. In the present study, we demonstrate that the proarrhythmic effects of CGs on Ca2+ cycling in cardiac myocytes involve alterations in the function of ryanodine receptor calcium channels caused by oxidative changes in the channel structure by reactive oxygen species. Our findings reveal a new mechanism for CG-induced Ca2+ waves and suggest a potential target for antiarrhythmic therapy in HF patients treated with CGs.
Abstract
Abstract
The therapeutic use of cardiac glycosides (CGs), agents commonly used in treating heart failure (HF), is limited by arrhythmic toxicity. The adverse effects of CGs have been attributed to excessive accumulation of intracellular Ca2+ resulting from inhibition of Na+/K+-ATPase ion transport activity. However, CGs are also known to increase intracellular reactive oxygen species (ROS), which could contribute to arrhythmogenesis through redox modification of cardiac ryanodine receptors (RyR2s). Here we sought to determine whether modification of RyR2s by ROS contributes to CG-dependent arrhythmogenesis and examine the relevant sources of ROS. In isolated rat ventricular myocytes, the CG digitoxin (DGT) increased the incidence of arrhythmogenic spontaneous Ca2+ waves, decreased the sarcoplasmic reticulum (SR) Ca2+ load, and increased both ROS and RyR2 thiol oxidation. Additionally, pretreatment with DGT increased spark frequency in permeabilized myocytes. These effects on Ca2+ waves and sparks were prevented by the antioxidant N-(2-mercaptopropionyl) glycine (MPG). The CG-dependent increases in ROS, RyR2 oxidation and arrhythmogenic propensity were reversed by inhibitors of NADPH oxidase, mitochondrial ATP-dependent K+ channels (mito-KATP) or permeability transition pore (PTP), but not by inhibition of xanthine oxidase. These results suggest that the arrhythmogenic adverse effects of CGs involve alterations in RyR2 function caused by oxidative changes in the channel structure by ROS. These CG-dependent effects probably involve release of ROS from mitochondria possibly mediated by NADPH oxidase.
Introduction
Cardiac glycosides (CGs), whose primary target is the Na+/K+-ATPase (NKA), have been commonly used in the treatment of congestive heart failure (HF) (Gheorghiade et al. 2004). The beneficial impact of CGs has been attributed to positive inotropic effects due to improvements of myocyte Ca2+ handling by these reagents. A common view is that inhibition of NKA by CGs results in elevated intracellular [Na+] which reduces Ca2+ extrusion via Na+/Ca2+ exchanger (NCX), thus leading to increased gain of cellular and SR Ca2+ and increased myocyte contractility (Bers, 2001; Reuter et al. 2002; Altamirano et al. 2006). Unfortunately, the therapeutic use of CGs in treating HF is limited by their adverse side effects, including cardiac arrhythmias (Ferrier, 1977). The arrhythmic side effects of CGs have been traditionally ascribed to excessive cellular Ca2+ retention (Ca2+ overload) leading to spontaneous discharges of intracellular Ca2+ stores, or Ca2+ waves, in turn causing oscillations of the membrane potential, known as delayed afterdepolarizations (DADs), extra-systolic action potentials and triggered activity (Wier & Hess, 1984; Fujiwara et al. 2008; Eisner et al. 2009; Weiss et al. 2011).
Recently, several groups of investigators have demonstrated that in addition to leading to physiological and pathological changes associated with inhibition of NKA ion transport activity and alterations in ionic balance, binding of CGs to NKA initiates a chain of signalling events that is independent of changes in intracellular [Na+] and [Ca2+] (Liu et al. 2000; Tian & Xie, 2008). In particular, conformational changes on binding of CGs have been reported to initiate a signalling cascade involving the activation of Src kinase and tyrosine phosphorylation of the epidermal growth factor receptor (EGFR) with the concomitant increase in production of reactive oxygen species (ROS) (Tian et al. 2003, 2006; Pasdois et al. 2007) via mechanisms yet to be fully defined. ROS have been shown to contribute to cardiac arrhythmogenesis and contractile dysfunction through redox modifications of the cardiac Ca2+ release channels, or ryanodine receptors (RyR2s), rendering them hyperactive and ‘leaky’ in various disease settings (Györke & Carnes, 2008; Terentyev et al. 2008; Belevych et al. 2009; Xie et al. 2009). Consequently, the goal of the present study was to test the hypothesis that the arrhythmogenic adverse effects of CGs involve redox modification of RyR2s resulting from increased production of ROS in cardiac myocytes. In particular, we sought to determine whether exposure of cells to antioxidants reverses the arrhythmogenic effects of CGs on myocyte Ca2+ handling and examine the mechanisms of ROS generation by CGs. Our results show for the first time that the arrhythmogenic toxicity of CGs indeed involves alterations in RyR2 function caused by ROS derived from mitochondria.
Methods
Ventricular myocytes from 50 adult LBNF1 male rats (250–300 g) were isolated following standard procedures (Györke et al. 1997). Rats were anaesthetized with Nembutal (75 mg kg−1, intraperitoneal injection) and euthanized by exsanguination. All animal procedures were approved by The Ohio State University Institutional Animal Care and Use Committee and conforpmed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The authors have read, and the experiments comply with, the policies and regulations of The Journal of Physiology given by Drummond (2009).
Ca2+ imaging and ROS production measurements
Intracellular Ca2+ cycling and ROS production in isolated rat ventricular myocytes were monitored by an Olympus Fluoview 1000 confocal microscope using the Ca2+- and ROS-sensitive indicators Fluo-3 and CM-H2DCFDA, respectively. The fluorescent probes were excited with the 488 nm line of an argon laser and emission was collected at 500–600 nm for Fluo-3 and 500–560 nm for CM-H2DCFDA. Fluo-3 fluorescence was recorded in the line scan mode of the confocal microscope (0.414 μm per pixel, 2 to 5 ms per line). The external, Tyrode solution contained (mm): 140 NaCl, 5.4 KCl, 1 CaCl2, 0.5 MgCl2, 10 Hepes and 5.6 glucose (pH 7.3). Myocytes were paced at 0.3–2 Hz using extracellular platinum electrodes. For Ca2+ spark recordings, the cardiac myocytes were permeabilized with saponin (0.01% for 20–30 s). The internal solution contained (mm): 120 potassium aspartate, 20 KCl, 0.81 MgCl2, 1 KH2PO4, 0.5 EGTA (free [Ca2+]∼50 nm), 3 MgATP, 10 phosphocreatine, 0.03 Fluo-3 pento potassium salt, 20 Hepes (pH 7.2) and 5 U ml−1 creatine phosphokinase. To assess the SR Ca2+ load, 20 mm caffeine was applied at the end of the experiments.
Myocyte arrhythmic potential was measured as the number of spontaneous Ca2+ waves per second. To test the proarrhythmic effects of CGs, myocytes were exposed to digitoxin (25–100 nm) or ouabain (100 μm). In some experiments, myocytes were pretreated with the reducing reagent and ROS scavenger N-(2-mercaptopropionyl) glycine (MPG, 750 μm). To examine the contribution of xanthine oxidase and NADPH oxidase to ROS generation by CGs, we used allopurinol (ALLO, 250 μm) and diphenyliodonium chloride (DPI, 50 μm), respectively. To identify the digitoxin-induced Na+/K+-ATPase cascade, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2, Src family kinase inhibitor), 5-hydroxydecanoate (5-HD, a mito-KATP channel blocker) and cyclosporin A (CsA, a potent inhibitor of the permeability transition pore in mitochondria) were applied.
RyR2 free thiol content
The content of free thiols in RyR2s was determined using the monobromobimane (mBB) fluorescence method (Terentyev et al. 2008). Briefly, samples prepared from isolated myocytes were incubated with 20 mm mBB for 1 h in the dark at room temperature. Subsequently proteins were acetone precipitated and subjected to SDS-PAGE. To examine the effects of DGT on RyR2 redox status, some of the cells were treated with digitoxin (100 nm, 30 min, 37°C). To minimize RyR2 thiol oxidation, some of the samples were incubated with 10 mm dithiothreitol (DTT, reducing agent). As a positive control to maximize RyR2 oxidation, we included 200 μm 2,2′-dithiodipyridine (DTDP, oxidizing agent). mBB fluorescence was normalized to the RyR2 levels quantified using Coomassie Blue staining of gels run in parallel.
Mitochondrial membrane potential measurement
Mitochondrial membrane potential was monitored with a voltage-sensitive fluorescent indicator, tetramethylrhodamine ethyl ester (TMRE). Freshly isolated rat ventricular myocytes were loaded with TMRE (75 nm, 20 min, 37°C) and TMRE fluorescence was measured as a series of X–Y confocal images. TMRE was excited at 543 nm with a helium–neon laser, and the emission signals were collected at 570–650 nm. TMRE fluorescence was normalized to the fluorescence signal obtained in the presence of the mitochondrial uncoupler carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP, 3 μm).
Reagents
All chemicals were obtained from Sigma (St Louis, MO, USA) or Calbiochem (La Jolla, CA, USA) and fluorescent dyes were purchased from Molecular Probes (Eugene, OR, USA).
Statistical analysis
Data are presented as means ± SEM. Statistical analyses were performed using ANOVA. A level of P < 0.05 was accepted as statistically significant.
Results
Disruption of normal Ca2+ cycling by DGT
Intracellular Ca2+ cycling in paced (at 0.3 Hz) isolated rat ventricular myocytes was monitored in the line scan mode of a confocal microscope using the Ca2+ indicator Fluo-3. The effects of DGT at 70 and 100 nm on myocyte Ca2+ cycling are illustrated in Fig. 1A and summarized in Fig. 1B–F. Exposure to DGT resulted in significant increases in the amplitude of the Ca2+ transients at both 70 and 100 nm (Fig. 1B) without a significant change in the rate of decay of the Ca2+ transients (Fig. 1D). At the same time, DGT caused a marked increase in the incidence of arrhythmogenic spontaneous Ca2+ waves (SCWs) at 100 nm, although Ca2+ wave frequency was unaffected at 70 nm (Fig. 1A and B). The maximal effect of DGT at 100 nm on Ca2+ wave frequency was attained within 3–5 min (Supplemental Fig. S1A) and wave frequency was highest at 0.3 Hz pacing rate in the range of 0.3–2 Hz (Supplemental Fig. S1B), conditions used throughout this study. Thus, whereas at 70 nm DGT increased Ca2+ transient amplitude without affecting the frequency of Ca2+ waves, at 100 nm DGT enhanced both Ca2+ transient size and arrhythmogenic propensity.
Figure 1. Concentration-, frequency-, and time- dependencies of pro-arrhythmic effects of digitoxin.
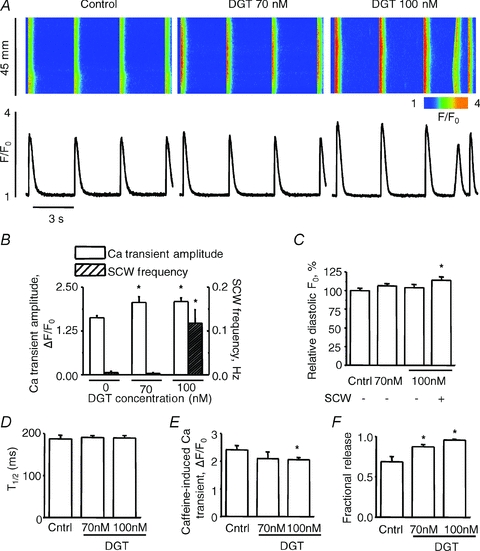
A, representative line-scan images (top) and time-dependent profiles (bottom) of spontaneous Ca2+ waves (SCWs) under control condition and in the presence of 70 nm digitoxin (DTG) or 100 nm digitoxin, as indicated. B, pooled data for the Ca2+ transient amplitude with the frequency of SCWs in control, 70 nm or 100 nm digitoxin. C, bar graph of relative diastolic Fluo-3 fluorescence (F0) under control condition and in the presence of 70 nm or 100 nm digitoxin. For 100 nm digitoxin, the bars present either total averaged diastolic F0 or averaged diastolic F0 excluding image areas containing SCWs as indicated. The values of F0 represent the mean F0 obtained during the last 1.5 s of each recording. D, pooled data for the duration of Ca2+ transients. E and F, pooled data for the averaged amplitude of caffeine-induced Ca2+ transients and fractional SR Ca2+ release, calculated as the ratio between the Ca2+ transient amplitude and SR Ca2+ content under each condition. Data are means ± SEM from 5 to 43 cells from 2 to 3 heart preparations. *P < 0.05 vs. control.
The positive inotropic and proarrhythmic effects of CGs are generally associated with increased cytosolic and SR Ca2+ content. In our experiments, DGT caused no detectable changes in baseline Fluo-3 fluorescence (indexing diastolic [Ca2+]) in myocytes lacking Ca2+ waves (at 70 nm) or when assessed in areas of line scan images outside of Ca2+ waves (at 100 nm) (Fig. 1C). The SR Ca2+ content assessed by application of caffeine was significantly reduced by 100 nm DGT, and 70 nm DGT tended to decrease the SR Ca2+ content (Fig. 1E). Notably, fractional SR Ca2+ release, defined as the ratio of Ca2+ transient amplitude to the SR Ca2+ content, was increased by DGT at both concentrations (Fig. 1F). Since fractional release is an indicator of the functional status of the Ca2+ release mechanism (Bers, 2001), both the positive inotropic and proarrhythmic effects of DGT in our study appear to arise as a consequence of enhanced functional activity of the SR Ca2+ release channels rather than by an increase in the SR Ca2+ content.
Clearly, the SR Ca2+ content depends on timing of caffeine application with respect to the occurrence of SCWs as Ca2+ waves deplete the SR of Ca2+. To examine whether the reduction of the SR Ca2+ content by 100 nm DGT was merely due to SR Ca2+ depletion by Ca2+ waves, we measured the SR Ca2+ content following periods with and without diastolic Ca2+ waves. Myocytes were paced at 0.3 Hz. After cessation of pacing, the SR Ca2+ content was measured by caffeine application within 10 s during which SCWs occurred in some myocytes but not in others. As expected, the SR Ca2+ content was lower following a diastolic Ca2+ wave than following an idle diastolic period (Fig. 2A). However, even in the absence of Ca2+ waves the SR Ca2+ content was lower in DGT-treated myocytes than in control myocytes (Fig. 2C). Of note, the amplitude of Ca2+ transients was increased in both DGT-treated groups (Fig. 2B) despite the reduced Ca2+ loading state of the SR, thus indicating an increase of fractional release (Fig. 2D), similar to the experiments in Fig. 1.
Figure 2. SR Ca2+ content with and without a preceding spontaneous Ca2+ wave (SCW) in 100 nm digitoxin-treated myocytes vs. control.
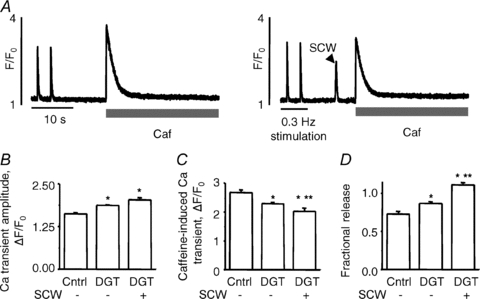
A, representative recordings of SR Ca2+ content measured by application of Caffeine (Caf, 20 mM) following (after pacing at 0.3 Hz) either an idle period (left-hand trace) or a SCW (right-hand trace) in the presence of 100 nM DGT. B and C, pooled data for the Ca2+ transient amplitude and SR Ca2+ content in each condition, as previously described. D, fractional SR Ca2+ release. Data are means ± SEM from 5 to 13 cells from 2 to 6 heart preparations. *P < 0.05 vs. control, **P < 0.05 vs. DGT without waves.
Arrhythmogenic effects of DGT are prevented by antioxidant pretreatment
To test the possibility that the increased propensity for Ca2+ wave generation in DGT-treated myocytes involves ROS, we examined the effects of the antioxidant N-(2-mercaptopropionyl) glycine (MPG) on Ca2+ cycling. Again, DGT (100 nm) increased the amplitude of Ca2+ transients and enhanced the frequency of Ca2+ waves (Fig. 3A). Exposure of myocytes to MPG by itself did not result in significant changes in the properties of Ca2+ transients, frequency of SCWs or SR Ca2+ content (Fig. 3B, C and F). However, as shown in Fig. 3A and B, pretreatment with MPG prevented the increase in SCW frequency by DGT. Notably, MPG did not prevent the increase in Ca2+ transient amplitude observed in response to DGT; however, it did avert the decline in the SR Ca2+ content caused by DGT, thereby partly normalizing fractional SR Ca2+ release (Fig. 3G). These results suggest that the positive inotropic and proarrhythmic effects of DGT in cardiac myocytes involve ROS. Similar results were obtained with another CG, ouabain (Supplemental Fig. S2).
Figure 3. Alterations in Ca2+ handling caused by digitoxin and their reversal by the antioxidant, MPG.
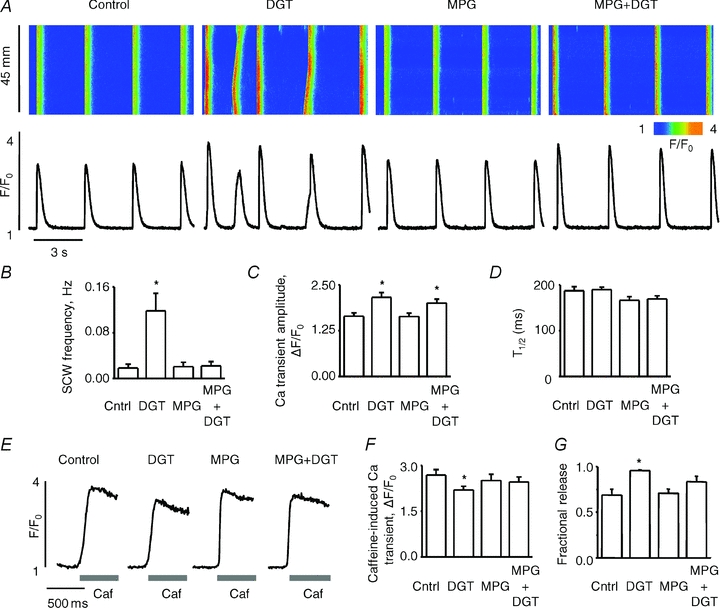
A, representative line-scan images (top) and time-dependent profiles (bottom) of spontaneous Ca2+ waves (SCWs) under control condition and in the presence of 100 nm digitoxin, 750 μm MPG or 100 nm digitoxin + 750 μm MPG, as indicated. B, pooled data for the frequency of SCWs. C and D, pooled data for the amplitude and duration, respectively, of Ca2+ transients in control, 100 nm digitoxin, 750 μm MPG or 100 nm digitoxin + 750 μm MPG, as indicated. E, traces of caffeine-induced Ca2+ transients under different experimental conditions as indicated at the top. F, pooled data for the averaged amplitude of caffeine-induced Ca2+ transients under each condition. G, bar graph of fractional SR Ca2+ release. Data are means ± SEM from 9 to 31 cells from 6 to 10 heart preparations. *P < 0.05 vs. control.
Effects of DGT on Ca2+ sparks
To more closely examine the effects of DGT on SR Ca2+ release, we performed measurements of Ca2+ sparks in saponin-permeabilized myocytes at fixed cytosolic [Ca2+]. In these experiments, myocytes were first exposed to DGT and then permeabilized with saponin. As shown in Fig. 4A–C, pretreatment with DGT increased the frequency of Ca2+ sparks by ∼30% and reduced their amplitude by ∼15% compared to control; these effects were prevented by MPG. Similar to our observations in intact cells, DGT significantly reduced the SR Ca2+ content (by ∼15%), consistent with the reduced amplitude of Ca2+ sparks; this effect was also prevented by MPG (Fig. 4D and E). Thus, as in intact cells, DGT stimulated Ca2+ release channel activity in permeabilized cells, in a manner preventable by treatment with the antioxidant MPG. The effects of DGT pretreatment on the properties of Ca2+ sparks and SR Ca2+ content were mimicked by the oxidizing agent 2,2′-dithiodipyridine (DTDP) (Supplemental Fig. S3). These results further support the notion that DGT affects SR Ca2+ release by redox modification of RyR2s.
Figure 4. Pretreatment with digitoxin increases the frequency of Ca2+ sparks in permeabilized myocytes.
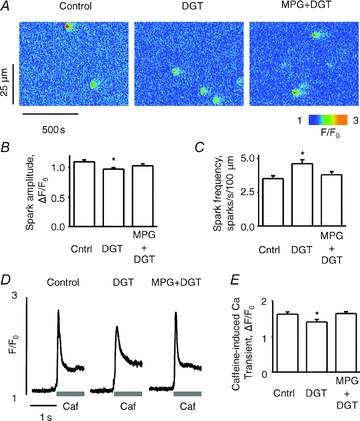
A, representative line-scan images of Ca2+ sparks in saponin-permeabilized myocytes under control condition and following pretreatment (before permeabilization) with 100 nm digitoxin (DGT) or 100 nm digitoxin + 750 μm MPG, as indicated. B and C, average amplitude and frequency of Ca2+ sparks, respectively, for each condition. D, traces of caffeine-induced Ca2+ transients under different experimental conditions as indicated at the top. E, pooled data for the averaged amplitude of caffeine-induced Ca2+ transients for each condition. Data are means ± SEM from 5 to 384 cells from 5 heart preparations.*P < 0.05 vs. control.
Increased ROS and RyR2 redox modification caused by DGT
The effects of digitoxin (DGT) on myocyte ROS production were examined using the ROS-sensitive indicator CM-H2DCFDA. In agreement with previous reports (Xie et al. 1999; Liu et al. 2000; Tian et al. 2003), exposure of myocytes to DGT resulted in a significant increase in the rate of ROS production (Fig. 5). RyR2 contains multiple thiols (Xu et al. 1998) that could become modified when myocyte ROS levels are increased. We directly examined the extent of RyR2 modification in myocytes treated with DGT using a mBB fluorescence labelling assay. Based on mBB fluorescence, the fraction of free thiols was indeed decreased significantly in myocytes treated with DGT, indicating increased levels of redox modifications of RyR2 (Fig. 6). The observed DGT-dependent changes in both ROS and RyR2 redox status were prevented by DPI, an inhibitor of NADPH oxidase, an important ROS generating system in cardiac myocytes (Figs 5 and 6).
Figure 5. Digitoxin increases ROS in myocytes.
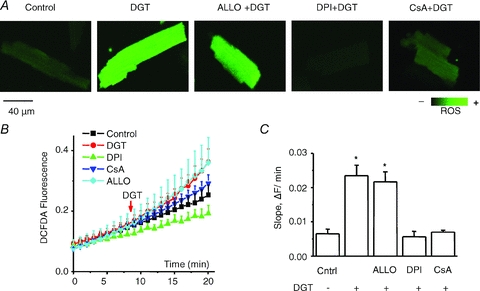
A, representative images of ROS generation measured by using the ROS-sensitive dye, CM-H2DCFDA, for control, 100 nm digitoxin (DGT), 100 nm digitoxin + 250 μm allopurinol (ALLO), 100 nm digitoxin + 50 μm DPI and 100 nm digitoxin + 10 μm CsA, as indicated. B, averaged traces of ROS production in the different groups. C, pooled data for ROS accumulation rates obtained from the slopes in the presence or absence of digitoxin. Data are means ± SEM from 10 to 28 cells from 4 to 8 heart preparations. *P < 0.05 vs. control.
Figure 6. RyR2 thiol oxidation in myocytes by digitoxin.
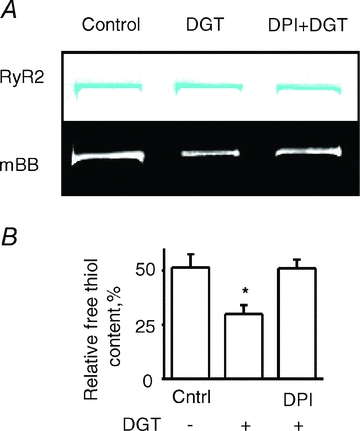
A, representative Coomassie Blue-stained gels (upper panels) and corresponding mBB fluorescence intensity signals (lower panels) of RyR2 from normal, digitoxin-pretreated, and digitoxin-pretreated with DPI myocytes. B, pooled data for free thiol contents normalized by DTT and DTDP. Data are means ± SEM from 6 heart preparations. *P < 0.05 vs. control.
Mitochondrial ROS release is involved in DGT-dependent arrhythmogenesis
In addition to NADPH oxidase, the main sources of ROS in myocytes include xanthine oxidase (XO) and mitochondria (Giordano, 2005). To evaluate the potential contribution of these systems to increased ROS production by DGT, we used inhibitors of XO (allopurinol) and mitochondrial ROS release (cyclosporin A, CsA). Whereas allopurinol failed to produce a significant effect, CsA significantly reduced ROS in DGT-treated myocytes (Fig. 5). In addition, consistent with previous reports (Liu et al. 2000; Garlid et al. 2003; Tian et al. 2003; Pasdois et al. 2007), DGT-dependent ROS was significantly inhibited by an inhibitor of mito-KATP channels, 5-HD, and by an inhibitor of Src kinase, PP2 (not shown). Importantly, these compounds as well as DPI, at the same concentrations at which ROS production was inhibited, also significantly decreased the frequency of DGT-induced SCWs (Fig. 7). These results further support the notion that the increased arrhythmogenic propensity in cardiomyocytes induced by DGT is mediated by ROS. Moreover, they suggest that the increased generation of ROS by DGT may involve NADPH oxidase and mitochondria but not XO.
Figure 7. Analysis of the signalling pathways involved in digitoxin-induced increase in myocyte arrhythmic potential.
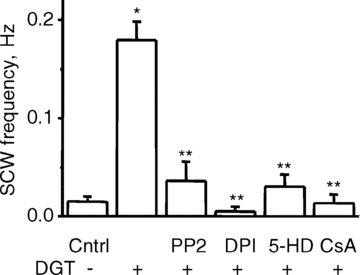
Myocyte arrhythmic potential is assessed as the number of spontaneous Ca2+ waves per second in myocytes paced at 0.3 Hz. Pooled data for the frequency of spontaneous Ca2+ waves for control, 100 nm digitoxin, and 100 nm digitoxin in the presence of 1 μm PP2, 50 μm DPI, 200 μm 5-HD or 10 μm CsA, as indicated. Data are means ± SEM from 8 to 94 cells from 4 to 10 heart preparations. *P < 0.05 vs. control, **P < 0.05 vs. DGT.
To further examine the mechanism of ROS production by CGs, we performed mitochondrial potential measurements using tetramethylrhodamine ethyl ester. As shown in Fig. 8, DGT caused a significant depolarization of the mitochondrial potential, which was prevented by inhibitors of mito-KATP channels, permeability transition pore and NADPH oxidase, 5-HD, CsA and DPI, respectively. These results provide further evidence for the role of mitochondria in DGT-dependent ROS production and arrhythmogenesis.
Figure 8. Mitochondrial membrane depolarization by digitoxin and its prevention by inhibitors of NADPH oxidase, mitochondrial KATP channels and permeability transition pore.
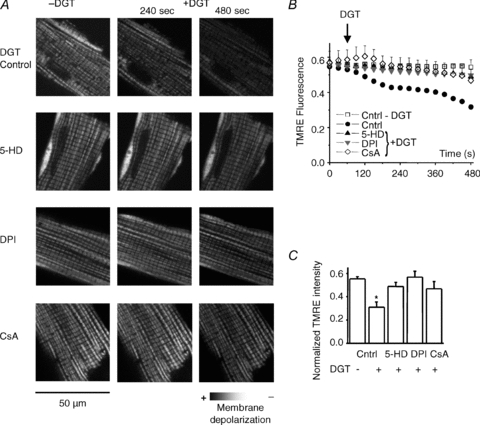
A, representative images of mitochondrial membrane potential at 0, 240 and 480 s measured by using TMRE, for control, 100 nm digitoxin (DGT), 100 nm digitoxin in the presence of 50 μm DPI, 200 μm 5-HD and 10 μm CsA. B, traces of normalized TMRE fluorescence. Data are normalized to the minimum TMRE fluorescence in the presence of 3 μm FCCP. (Fx,y,t–Fmin)/Fmin, where Fx,y,t is the average fluorescence at the X–Y scan at time t, Fmin is the minimal fluorescence signal during FCCP application. C, bar graphs of mitochondrial membrane potential 7 min after digitoxin application. Data are means ± SEM from 3 to 5 cells from 2 to 5 heart preparations. *P < 0.05 vs. control.
Discussion
The main finding of the present study is that the proarrhythmic effects of CGs on Ca2+ cycling in cardiac myocytes involve modifications in the RyR2 channel structure by ROS. Furthermore, our results suggest that the CG-induced generation of ROS and proarrhythmic Ca2+ waves involve release of ROS from mitochondria which may be mediated by NADPH oxidase. Commonly, spontaneous Ca2+ waves in the presence of CGs are attributed to increased cellular accumulation of Ca2+ secondary to the inhibition of the ion transport activity of Na+/K+ ATPase. Our findings reveal an alternative mechanism for digitoxin-induced Ca2+ waves and suggest a potential target for antiarrhythmic therapy in HF patients treated with CGs.
CG-dependent alterations in myocyte Ca2+ signalling involve ROS
Consistent with previous studies of the effects of CGs (Wier & Hess, 1984), DGT increased the amplitude of Ca2+ transients without promoting Ca2+ waves at relatively low concentrations and increased Ca2+ wave frequency (along with enhancing Ca2+ transients) at a higher concentration (Fig. 1). Importantly, these stimulatory effects on release were associated with either no change or a decrease in the SR Ca2+ content, suggesting that DGT acted on myocyte Ca2+ cycling by stimulating RyR2 functional activity rather than through increasing the SR Ca2+ load. Increased RyR2 activity was further indicated by increased frequency of Ca2+ sparks observed on the backdrop of reduced SR Ca2+ content in permeabilized myocytes treated with DGT (Fig. 4). The role of redox modification as a cause of DGT-induced changes in RyR2 function was shown by the following set of results: (1) DGT increased myocyte ROS and decreased the level of free, unmodified thiols in RyR2 (Figs 5 and 6); (2) the potentiatory effects of DGT on fractional release and Ca2+ waves were reversed by the antioxidant MPG and inhibitors of cellular ROS generating systems (Fig. 3); and (3) the increased frequency of Ca2+ sparks by DGT was observed following the pretreatment and permeabilization of myocytes with the glycoside and was reversed by MPG (Fig. 4); moreover, the effects of DGT on sparks and SR Ca2+ content were mimicked by the oxidizing agent DTDP (Supplemental Fig. S3).
Our present findings are consistent with previous studies that demonstrated that modification of RyR2s by ROS enhances RyR2 functional activity (Boraso & Williams, 1994; Xu et al. 1998; Zima & Blatter, 2006; Donoso et al., 2011) and results in increased frequency of spontaneous Ca2+ waves and arrhythmogenic electrical events including early or delayed afterdepolarizations (EADs or DADs) in cardiac myocytes (Terentyev et al. 2008; Belevych et al. 2009; Xie et al. 2009). RyR2 thiol oxidation reportedly increases the sensitivity of the channel to luminal Ca2+, thus lowering the critical SR Ca2+ content at which spontaneous Ca2+ waves occur (Terentyev et al. 2008). Spontaneous Ca2+ waves and DADs are considered to be the basis of the triggered activity characteristic of CG-induced arrhythmias (Ferrier, 1977; Bers, 2001; Eisner et al. 2009).
According to recent evidence, synchronization of Ca2+ waves and DADs between individual cardiac cells required for the generation of ectopic action potentials involves temporally aligned SR Ca2+ refilling during the Ca2+ release–uptake cycle across the myocardium (Wasserstrom et al. 2010). Collectively, these results suggest that increased RyR2 function due to redox modification could provide a common mechanism for both the positive inotropic and proarrhythmic effects of CGs in myocytes. Moreover, an excessive SR Ca2+ leak could also contribute to the negative inotropic effects of CGs at higher concentrations as hyperoxidation of RyR2 by ROS has been shown to contribute to the increased SR Ca2+ leak and depressed cytosolic Ca2+ transients in myocytes from failing hearts (Terentyev et al. 2008; Belevych et al. 2011).
Previously, it has been suggested that CGs enhance SR Ca2+ release by acting directly on RyR2 (Rardon & Wasserstrom, 1990; McGarry & Williams, 1993; Sagawa et al. 2002; Nishio et al. 2004). We believe, however, such direct effects of DGT on RyR2 could not account for most of our results because the DGT-induced increases in the frequency of Ca2+ waves and sparks were reversed by an antioxidant treatment (Figs 3 and 4). Nevertheless, our study does not completely rule out the possibility of a direct modulation of RyR2s by CGs which would be expected to complement the stimulatory effects of RyR2 redox modification. Thus, direct action upon RyR2 could explain, at least in part, the residual potentiation of Ca2+ transients by DGT in the presence of MPG (Fig. 3). At the same time, it is also possible that RyR2 oxidation contributed to the stimulation of SR Ca2+ release by CGs under Na+-free conditions, which precludes a mechanism involving alterations of intracellular ionic balances, observed by Nishio et al. (2004). Previously, Altamirano et al. (2006) reported that ouabain has no effects on frequency and amplitude characteristics of Ca2+ sparks when applied directly to permeabilized myocytes. Although this result appears to conflict with direct effects of CGs on RyR2s, the differences between Altamirano et al and our studie could be attributed to myocyte permeabilization disrupting the ROS-mediated signalling mechanisms revealed by our experiments. Finally, increased RyR2 phosphorylation by CaMKII could also contribute to the arrhythmogenic effects of CGs (Sapia et al. 2010) as a consequence of potential activation of CaMKII either by elevated [Ca2+] or ROS (Xie et al. 2009). However, this possibility seems unlikely for this particular study as RyR2 phosphorylation at the CaMKII phosphorylation site Ser 2814 was not changed following incubation with DGT under our experiments condition (not shown).
Although glycoside-induced Ca2+ waves are commonly attributed to increased Ca2+ levels in both extra- and intra-SR compartments, there is a lack of direct experimental evidence for increased SR Ca2+ content for either therapeutic or toxic concentrations of CGs, and an elevation in diastolic [Ca2+] has been shown only at relatively high CG concentrations (Wasserstrom & Aistrup, 2005). In the present study, we found no detectable increases in diastolic Ca2+ in areas outside of the propagating Ca2+ waves in DGT-treated myocytes (Fig. 1), suggesting that the baseline cytosolic Ca2+ was not considerably increased by DGT when assessed independently of SR-derived Ca2+. Moreover, the SR Ca2+ content was, in fact, reduced in the presence of the drug (Fig. 1E and F) (or unchanged at lower, purely positive inotropic concentrations of DGT). The reduction in the SR Ca2+ content by proarrhythmic concentrations of DGT is attributable to increased diastolic SR Ca2+ release in the form of spontaneous Ca2+ waves and sparks. Additionally, oxidation-dependent inhibition of SERCA2 activity (Kennedy et al. 2006) could contribute to the decreased SR Ca2+ by CGs.
Mechanism of CG-dependent ROS generation
Our findings are also supported by previous reports (Liu et al. 2000; Tian et al. 2003; Pasdois et al. 2007) showing that CGs increase ROS production in cardiac myocytes. While the mechanisms mediating increased ROS remain to be clarified, several laboratories have demonstrated that they involve signalling cascades that are independent of the changes in intracellular Na+ or Ca2+ concentrations (expected as a consequence of inhibition of the activity of NKA) (Tian & Xie, 2008). In particular, it has been established that binding of ouabain to NKA results in the initiation of signalling cascades that involve the activation of Src kinase and opening of the mito-KATP channels, ultimately leading to increased myocyte ROS (Tian et al. 2003, 2006; Pasdois et al. 2007). Consistent with these studies, inhibitors of Src and mito-KATP channels reversed arrhythmogenesis in myocytes in the presence of DGT (Fig. 7). Moreover, DGT caused mitochondrial membrane depolarization, which was prevented by inhibition of mito-KATP channels and mitochondrial permeability transition pore (Fig. 8), thus further supporting the role of mitochondria in DGT-dependent ROS increase and arrhythmogenesis.
Considering the nearly complete prevention of DGT-dependent ROS, mitochondrial depolarization and arrhythmogenesis by inhibitors of mitochondrial PTP and mito-KATP, it appears that most of the bioactive ROS under our experimental conditions is derived from the mitochondria. The main potential sources of ROS in cardiac myocytes in addition to mitochondria include XO and NADPH oxidase (Giordano, 2005). While inhibition of XO had no significant effects on ROS, the general NADPH oxidase inhibitor DPI markedly inhibited both ROS and SCWs in DGT-treated cells (Figs 5, 6 and 7). One possible explanation for this result is that mitochondrial ROS release is caused by a relatively small level of ROS supplied by NADPH oxidase. Thus, DPI in addition to inhibiting NADPH oxidase would also prevent the release of ROS from mitochondria. This interpretation is compatible with the concept of ROS-induced ROS generation (Brady et al. 2006; Zorov et al. 2006) according to which ROS can induce mitochondrial depolarization and subsequent ROS release through mitochondrial PTP in cardiac myocytes. In support of this possibility, NADPH oxidase has been demonstrated to play a role in angiotensin II-mediated cardiac protection by causing mitochondrial ROS release that was sensitive to 5-HD (Kimura et al. 2005).
While suggesting a new mechanism for glycoside-dependent arrhythmogenesis, our results do not rule out a potential role for the classical mechanisms involving changes in cytosolic Na+ and Ca2+ concentrations brought about by inhibition of NKA ion transport activity. Indeed, although not observed in our study, increased diastolic [Ca2+] resulting from inhibition of the NKA by CGs is likely to contribute to arrhythmogenesis by stimulating RyR2s at high drug concentrations. Additionally, CG-induced elevations in cytosolic [Ca2+] could facilitate ROS generation by activation of PKC with subsequent stimulation of NADPH oxidase. Thus, PKC could present a site for integration of Ca2+-dependent and Ca2+-independent pathways in activation of NADPH oxidase and ROS production. Additionally, mitochondrial ROS production by CGs could be facilitated by increased mitochondrial Ca2+ load and subsequent effects on electron transport chain and mitochondrial NOS (Dedkova & Blatter, 2009). Alternatively, elevated cytosolic [Na+] caused by CGs could contribute to increased ROS accumulation by impairing mitochondrial energetic and redox balance via activation of mitochondrial Na+/Ca2+ exchanger and blunting mitochondrial Ca2+ accumulation (Liu et al. 2010).
Limitations
The widely used pharmacological agents DPI, 5-HD and CsA were employed to inhibit NADPH oxidase, mito-KATP and PTP, respectively. Characteristic of experimentation with pharmacological inhibitors, we cannot rule out the possibility of non-specific and secondary effects in the present study, including inhibition by DPI of mitochondrial complex I (Li & Trush, 1998).
Conclusions
In conclusion, our study shows that the arrhythmogenic effects of CGs on Ca2+ cycling in cardiac myocytes involve alterations in RyR2 function caused by oxidative changes in the channel structure by ROS. Furthermore, our data suggest that the CG-dependent ROS is likely to involve release of ROS from the mitochondria possibly mediated by NADPH oxidase. Targeting ROS-dependent alterations in RyR2 function may present a strategy for enhancing the utility of CGs in treating HF.
Acknowledgments
This work was supported by National Institutes of Health grants (HL074045 and HL063043 to S.G.).
Glossary
Abbreviations
- 5-HD
5-hydroxydecanoate
- ALLO
allopurinol
- CGs
cardiac glycosides
- CsA
cyclosporin A
- DADs
delayed afterdepolarizations
- DGT
digitoxin
- DPI
diphenyliodonium chloride
- DTDP
2,2′-dithiodipyridine
- DTT
dithiothreitol
- EADs
early afterdepolarizations
- EGFR
epideral growth factor receptor
- FCCP
carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone
- HF
heart failure
- mBB
monobromobimane
- mito-KATP
mitochondrial ATP-dependent K+ channels
- MPG
N-(2-mercaptopropionyl) glycine
- NCX
Na+/Ca2+ exchanger
- NKA
Na+/K+-ATPase
- PP2
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- PTP
permeability transition pore
- ROS
reactive oxygen species
- RyR2s
ryanodine receptors
- SR
sarcoplasmic reticulum
- TMRE
tetramethylrhodamine ethyl ester
- XO
xanthine oxidase
Author contributions
H.-T.H, C.A.C, D.T. and S.G. contributed to the conception, design, data interpretation and manuscript preparation. H.-T.H, S.C.W.S and R.T were responsible for experimental work. H.-T.H contributed to data collection and analysis. All authors approved the final version of the manuscript.
Supplementary material
Supplemental Fig. S1
Supplemental Fig. S2
Supplemental Fig. S3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Altamirano J, Li Y, Desantiago J, Piacentino V, III, Houser SR, Bers DM. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J Physiol. 2006;575:845–854. doi: 10.1113/jphysiol.2006.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Györke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Györke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd edn. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- Boraso A, Williams AJ. Modification of the gating of the cardiac sarcoplasmic reticulum Ca2+-release channel by H2O2 and dithiothreitol. Am J Physiol Heart Circ Physiol. 1994;267:H1010–H1016. doi: 10.1152/ajpheart.1994.267.3.H1010. [DOI] [PubMed] [Google Scholar]
- Brady NR, Hamacher-Brady A, Westerhoff HV, Gottlieb RA. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid Redox Signal. 2006;8:1651–1665. doi: 10.1089/ars.2006.8.1651. [DOI] [PubMed] [Google Scholar]
- Dedkova EN, Blatter LA. Characteristics and function of cardiac mitochondrial nitric oxide synthase. J Physiol. 2009;587:851–872. doi: 10.1113/jphysiol.2008.165423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Frontiers in Bioscience-Landmark. 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ J. 2009;73:1561–1567. doi: 10.1253/circj.cj-09-0478. [DOI] [PubMed] [Google Scholar]
- Ferrier GR. Digitalis arrhythmias: role of oscillatory afterpotentials. Prog Cardiovasc Dis. 1977;19:459–474. doi: 10.1016/0033-0620(77)90010-x. [DOI] [PubMed] [Google Scholar]
- Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–518. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Dos SP, Xie ZJ, Costa AD, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim Biophys Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Adams KF, Jr, Colucci WS. Digoxin in the management of cardiovascular disorders. Circulation. 2004;109:2959–2964. doi: 10.1161/01.CIR.0000132482.95686.87. [DOI] [PubMed] [Google Scholar]
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke S, Carnes C. Dysregulated sarcoplasmic reticulum calcium release: potential pharmacological target in cardiac disease. Pharmacol Ther. 2008;119:340–354. doi: 10.1016/j.pharmthera.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke S, Lukyanenko V, Györke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol. 1997;500:297–309. doi: 10.1113/jphysiol.1997.sp022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DJ, Vetteth S, Xie M, Periyasamy SM, Xie Z, Han C, Basrur V, Mutgi K, Fedorov V, Malhotra D, Shapiro JI. Ouabain decreases sarco(endo)plasmic reticulum calcium ATPase activity in rat hearts by a process involving protein oxidation. Am J Physiol Heart Circ Physiol. 2006;291:H3003–H3011. doi: 10.1152/ajpheart.00603.2006. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem. 2000;275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- Liu T, Brown DA, O'Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–736. doi: 10.1016/j.yjmcc.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry SJ, Williams AJ. Digoxin activates sarcoplasmic reticulum Ca2+-release channels: a possible role in cardiac inotropy. Br J Pharmacol. 1993;108:1043–1050. doi: 10.1111/j.1476-5381.1993.tb13503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Ruch SW, Kelly JE, Aistrup GL, Sheehan K, Wasserstrom JA. Ouabain increases sarcoplasmic reticulum calcium release in cardiac myocytes. J Pharmacol Exp Ther. 2004;308:1181–1190. doi: 10.1124/jpet.103.060004. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Quinlan CL, Rissa A, Tariosse L, Vinassa B, Costa AD, Pierre SV, Dos SP, Garlid KD. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am J Physiol Heart Circ Physiol. 2007;292:H1470–H1478. doi: 10.1152/ajpheart.00877.2006. [DOI] [PubMed] [Google Scholar]
- Rardon DP, Wasserstrom JA. Cardiotonic steroids activate cardiac sarcoplasmic-reticulum calcium release channels. Circulation. 1990;82:342. [Google Scholar]
- Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res. 2002;90:305–308. doi: 10.1161/hh0302.104562. [DOI] [PubMed] [Google Scholar]
- Sagawa T, Sagawa K, Kelly JE, Tsushima RG, Wasserstrom JA. Activation of cardiac ryanodine receptors by cardiac glycosides. Am J Physiol Heart Circ Physiol. 2002;282:H1118–H1126. doi: 10.1152/ajpheart.00700.2001. [DOI] [PubMed] [Google Scholar]
- Sapia L, Palomeque J, Mattiazzi A, Petroff MV. Na+/K+-ATPase inhibition by ouabain induces CaMKII-dependent apoptosis in adult rat cardiac myocytes. J Mol Cell Cardiol. 2010;49:459–468. doi: 10.1016/j.yjmcc.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY, Xie ZJ. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell. 2006;17:317–326. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Liu J, Garlid KD, Shapiro JI, Xie Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial KATP channels. Mol Cell Biochem. 2003;242:181–187. [PubMed] [Google Scholar]
- Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology (Bethesda.) 2008;23:205–211. doi: 10.1152/physiol.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstrom JA, Aistrup GL. Digitalis: new actions for an old drug. Am J Physiol Heart Circ Physiol. 2005;289:H1781–H1793. doi: 10.1152/ajpheart.00707.2004. [DOI] [PubMed] [Google Scholar]
- Wasserstrom JA, Shiferaw Y, Chen W, Ramakrishna S, Patel H, Kelly JE, O'Toole MJ, Pappas A, Chirayil N, Bassi N, Akintilo L, Wu M, Arora R, Aistrup GL. Variability in timing of spontaneous calcium release in the intact rat heart is determined by the time course of sarcoplasmic reticulum calcium load. Circ Res. 2010;107:1117–1126. doi: 10.1161/CIRCRESAHA.110.229294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Nivala M, Garfinkel A, Qu Z. Alternans and arrhythmias: from cell to heart. Circ Res. 2011;108:98–112. doi: 10.1161/CIRCRESAHA.110.223586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier WG, Hess P. Excitation-contraction coupling in cardiac Purkinje fibers. Effects of cardiotonic steroids on the intracellular [Ca2+] transient, membrane potential, and contraction. J Gen Physiol. 1984;83:395–415. doi: 10.1085/jgp.83.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Kometiani P, Liu J, Li J, Shapiro JI, Askari A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J Biol Chem. 1999;274:19323–19328. doi: 10.1074/jbc.274.27.19323. [DOI] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.