

Abstract
The asymmetric unit of the title compound, C17H17ClN2O2, contains one half-molecule with a center of inversion at the mid-point of the central C—C bond. The amide N—H group is anti to the meta-chloro/methyl groups in the adjacent benzene rings. The dihedral angle between the benzene ring and the NH—C(O)—CH2 segment is 43.5 (1)°. In the crystal, intermolecular N—H⋯O hydrogen bonds link the molecules into chains along the a axis. The methyl group and the Cl atom occupy the same position and were treated in a disorder model with site-occupation factors of 0.5 each.
Related literature
For our studies on the effects of substituents on the structures of N-(aryl)-amides, see: Bhat & Gowda (2000 ▶); Gowda et al. (2007 ▶); Saraswathi et al. (2011a ▶,b
▶) and on the structures of N-(aryl)-methanesulfonamides, see: Jayalakshmi & Gowda (2004 ▶). For similar structures, see: Pierrot et al. (1984 ▶). For restrained geometry, see: Nardelli (1999 ▶).
Experimental
Crystal data
C17H17ClN2O2
M r = 316.78
Triclinic,
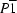
a = 4.840 (1) Å
b = 5.560 (1) Å
c = 14.752 (3) Å
α = 93.47 (2)°
β = 91.39 (2)°
γ = 97.71 (2)°
V = 392.46 (13) Å3
Z = 1
Mo Kα radiation
μ = 0.25 mm−1
T = 293 K
0.44 × 0.20 × 0.08 mm
Data collection
Oxford Diffraction Xcalibur diffractometer with Sapphire CCD detector
Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009 ▶) T min = 0.897, T max = 0.980
2451 measured reflections
1567 independent reflections
1249 reflections with I > 2σ(I)
R int = 0.010
Refinement
R[F 2 > 2σ(F 2)] = 0.050
wR(F 2) = 0.130
S = 0.99
1567 reflections
110 parameters
14 restraints
H-atom parameters constrained
Δρmax = 0.17 e Å−3
Δρmin = −0.17 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2009 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2009 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2009 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811028121/wm2508sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811028121/wm2508Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811028121/wm2508Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O1i | 0.86 | 2.05 | 2.894 (2) | 168 |
Symmetry code: (i) 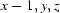 .
.
Acknowledgments
BSS thanks the University Grants Commission, Government of India, New Delhi, for the award of a research fellowship under its faculty improvement program.
supplementary crystallographic information
Comment
The amide and sulfonamide moieties are important constituents of many biologically significant compounds. As part of our studies on the substituent effects on the structures of this class of compounds (Bhat & Gowda, 2000; Gowda et al., 2007; Jayalakshmi & Gowda, 2004; Saraswathi et al., 2011a,b), in the present work, the structure of N-(3-chlorophenyl),N-(3-methylphenyl)- succinamide, (I), has been determined (Fig.1). The asymmetric unit of (I) contains half a molecule with a center of inversion at the mid-point of the central C—C bond, similar to that observed in bis(2-chlorophenylaminocarbonylmethyl)disulfide, (II), (Pierrot et al., 1984), N,N-bis(3-chlorophenyl)-succinamide, (III), (Saraswathi et al., 2011a) and N,N-bis(3-methylphenyl)-succinamide dihydrate, (IV), (Saraswathi et al., 2011b).
The conformations of the amide O atoms are anti to the H atoms attached to the adjacent C atoms. Further, the conformations of the N—H bonds in the amide fragments are anti to the meta-chloro/methyl groups in the adjacent benzene rings, similar to the anti conformations observed with respect to the meta-chloro groups in (III) and meta-methyl groups in (IV).
Further, the C1—N1—C7—C8 and C1a—N1a—C7a—C8a segments in (I) are nearly planar and so also the C1—N1—C7—O1 and C1a—N1a—C7a—O1a segments, similar to those observed in (III) and (IV). The torsion angles of C2—C1—N1—C7 and C6—C1—N1—C7 are -43.2 (4)° and 138.6 (3)°, in contrast to the values of -35.0 (3)° and 147.5 (2)° in (III), and 5.4 (9)° and -173.6 (6)° in (IV).
The dihedral angle between the benzene ring and the NH—C(O)—CH2 segment is 43.5 (1)°, compared to the values of 62.1 (2)° in (III) and 5.6 (4)° in (IV).
The packing of the molecules in the crystal is accomplished by N—H···O hydrogen bonds (Table 1) that lead is shown in Fig. 2.
Experimental
Succinic anhydride (0.01 mol) in toluene (25 ml) was treated dropwise with 3-chloroaniline (0.01 mol) also in toluene (20 ml) with constant stirring. The resulting mixture was stirred for one hour and set aside for an additional hour at room temperature for completion of the reaction. The mixture was then treated with dilute hydrochloric acid to remove unreacted 3-chloroaniline. The resultant solid N-(3-chlorophenyl)-succinamic acid was filtered under suction and washed thoroughly with water to remove the unreacted succinic anhydride and succinic acid. The compound was recrystallized to a constant melting point from ethanol. The purity of the compound was checked by elemental analysis and characterized by its infrared and NMR spectra.
The N-(3-chlorophenyl)succinamic acid obtained was then treated with phosphorous oxychloride and excess of 3-methylaniline at room temperature with constant stirring. The resultant mixture was stirred for 4 h, kept aside for additional 6 h for completion of the reaction and poured slowly into crushed ice with constant stirring. It was kept aside for a day. The resultant solid, N-(3-chlorophenyl), N-(3-methylphenyl)-succinamide was filtered under suction, washed thoroughly with water, dilute sodium hydroxide solution and finally with water. It was recrystallized to a constant melting point from a mixture of acetone and toluene (3:1 v/v). The compound was characterized by its infrared and NMR spectra.
Prism-like colorless single crystals used in X-ray diffraction studies were grown in a mixture of acetone and toluene (3:1 v/v) at room temperature.
Refinement
The H atoms were positioned with idealized geometry using a riding model with C—H = 0.93 Å for aromatic, C—H = 0.97 Å for methylene and N—H = 0.86 Å for amide H atoms and were refined with isotropic displacement parameters, set to 1.2×Ueq of the parent atom. Atoms C9 and Cl1 occupy the same position. The disorder was treated by using a split-atom model. The corresponding site-occupation factors were fixed to 0.50:0.50. The Uij components of these atoms were restrained to approximate isotropic behavior (Nardelli, 1999), the bond lenghts C3—C9 and C3—Cl1 were restrained.
Figures
Fig. 1.

Molecular structure of the title compound, showing the atom labelling scheme and displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

Molecular packing of the title compound with hydrogen bonding shown as dashed lines.
Crystal data
| C17H17ClN2O2 | Z = 1 |
| Mr = 316.78 | F(000) = 166 |
| Triclinic, P1 | Dx = 1.340 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 4.840 (1) Å | Cell parameters from 1094 reflections |
| b = 5.560 (1) Å | θ = 2.8–27.7° |
| c = 14.752 (3) Å | µ = 0.25 mm−1 |
| α = 93.47 (2)° | T = 293 K |
| β = 91.39 (2)° | Prism, colourless |
| γ = 97.71 (2)° | 0.44 × 0.20 × 0.08 mm |
| V = 392.46 (13) Å3 |
Data collection
| Oxford Diffraction Xcalibur diffractometer with Sapphire CCD detector | 1567 independent reflections |
| Radiation source: fine-focus sealed tube | 1249 reflections with I > 2σ(I) |
| graphite | Rint = 0.010 |
| Rotation method data acquisition using ω scans | θmax = 26.3°, θmin = 2.8° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009) | h = −6→5 |
| Tmin = 0.897, Tmax = 0.980 | k = −6→6 |
| 2451 measured reflections | l = −18→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.050 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.130 | H-atom parameters constrained |
| S = 0.99 | w = 1/[σ2(Fo2) + (0.0501P)2 + 0.2642P] where P = (Fo2 + 2Fc2)/3 |
| 1567 reflections | (Δ/σ)max < 0.001 |
| 110 parameters | Δρmax = 0.17 e Å−3 |
| 14 restraints | Δρmin = −0.17 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| O1 | 0.2418 (3) | 0.7102 (3) | −0.09593 (13) | 0.0651 (6) | |
| N1 | −0.2109 (3) | 0.5789 (3) | −0.13211 (12) | 0.0417 (5) | |
| H1 | −0.3807 | 0.5949 | −0.1204 | 0.050* | |
| C1 | −0.1722 (4) | 0.4107 (4) | −0.20578 (14) | 0.0384 (5) | |
| C2 | 0.0217 (4) | 0.4712 (4) | −0.27063 (14) | 0.0445 (5) | |
| H2 | 0.1310 | 0.6226 | −0.2657 | 0.053* | |
| C3 | 0.0541 (5) | 0.3084 (5) | −0.34254 (15) | 0.0520 (6) | |
| C4 | −0.1059 (6) | 0.0831 (5) | −0.34971 (18) | 0.0630 (7) | |
| H4A | −0.0831 | −0.0283 | −0.3977 | 0.076* | |
| C5 | −0.2988 (6) | 0.0250 (5) | −0.2854 (2) | 0.0659 (7) | |
| H5A | −0.4070 | −0.1269 | −0.2904 | 0.079* | |
| C6 | −0.3364 (5) | 0.1866 (4) | −0.21353 (16) | 0.0502 (6) | |
| H6 | −0.4703 | 0.1453 | −0.1709 | 0.060* | |
| C7 | −0.0058 (4) | 0.7190 (4) | −0.08329 (14) | 0.0414 (5) | |
| C8 | −0.0999 (4) | 0.8842 (4) | −0.00851 (15) | 0.0441 (5) | |
| H8A | −0.1160 | 0.7990 | 0.0470 | 0.053* | |
| H8B | −0.2828 | 0.9240 | −0.0249 | 0.053* | |
| C9 | 0.251 (3) | 0.364 (4) | −0.4221 (10) | 0.155 (8) | 0.50 |
| H9A | 0.3567 | 0.5224 | −0.4105 | 0.185* | 0.50 |
| H9B | 0.1426 | 0.3602 | −0.4776 | 0.185* | 0.50 |
| H9C | 0.3763 | 0.2442 | −0.4274 | 0.185* | 0.50 |
| Cl1 | 0.2883 (5) | 0.3904 (5) | −0.42162 (12) | 0.0742 (6) | 0.50 |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0252 (8) | 0.0862 (13) | 0.0794 (12) | 0.0103 (7) | 0.0031 (7) | −0.0380 (10) |
| N1 | 0.0249 (8) | 0.0507 (11) | 0.0484 (10) | 0.0059 (7) | 0.0052 (7) | −0.0101 (8) |
| C1 | 0.0295 (9) | 0.0447 (12) | 0.0415 (11) | 0.0094 (8) | −0.0013 (8) | −0.0030 (9) |
| C2 | 0.0366 (11) | 0.0497 (13) | 0.0457 (12) | 0.0028 (9) | 0.0031 (9) | −0.0039 (9) |
| C3 | 0.0458 (12) | 0.0695 (16) | 0.0415 (12) | 0.0152 (11) | 0.0031 (10) | −0.0051 (11) |
| C4 | 0.0707 (17) | 0.0637 (17) | 0.0532 (14) | 0.0147 (13) | 0.0009 (12) | −0.0188 (12) |
| C5 | 0.0738 (18) | 0.0468 (14) | 0.0717 (17) | −0.0039 (12) | −0.0025 (14) | −0.0111 (12) |
| C6 | 0.0462 (12) | 0.0493 (13) | 0.0526 (13) | −0.0017 (10) | 0.0059 (10) | −0.0003 (10) |
| C7 | 0.0283 (10) | 0.0485 (12) | 0.0471 (12) | 0.0073 (8) | 0.0046 (8) | −0.0065 (10) |
| C8 | 0.0302 (10) | 0.0550 (13) | 0.0458 (11) | 0.0071 (9) | 0.0048 (8) | −0.0114 (10) |
| C9 | 0.149 (9) | 0.162 (9) | 0.152 (9) | 0.019 (5) | 0.009 (5) | 0.006 (5) |
| Cl1 | 0.0639 (9) | 0.1109 (15) | 0.0466 (7) | 0.0093 (9) | 0.0242 (7) | −0.0080 (8) |
Geometric parameters (Å, °)
| O1—C7 | 1.224 (2) | C4—H4A | 0.9300 |
| N1—C7 | 1.342 (3) | C5—C6 | 1.380 (3) |
| N1—C1 | 1.423 (3) | C5—H5A | 0.9300 |
| N1—H1 | 0.8591 | C6—H6 | 0.9300 |
| C1—C6 | 1.382 (3) | C7—C8 | 1.510 (3) |
| C1—C2 | 1.381 (3) | C8—C8i | 1.507 (4) |
| C2—C3 | 1.378 (3) | C8—H8A | 0.9700 |
| C2—H2 | 0.9300 | C8—H8B | 0.9700 |
| C3—C4 | 1.379 (4) | C9—H9A | 0.9600 |
| C3—Cl1 | 1.686 (3) | C9—H9B | 0.9600 |
| C3—C9 | 1.550 (9) | C9—H9C | 0.9600 |
| C4—C5 | 1.369 (4) | ||
| C7—N1—C1 | 125.39 (16) | C6—C5—H5A | 119.2 |
| C7—N1—H1 | 118.5 | C5—C6—C1 | 119.0 (2) |
| C1—N1—H1 | 116.1 | C5—C6—H6 | 120.5 |
| C6—C1—C2 | 119.8 (2) | C1—C6—H6 | 120.5 |
| C6—C1—N1 | 119.33 (19) | O1—C7—N1 | 122.90 (18) |
| C2—C1—N1 | 120.85 (19) | O1—C7—C8 | 121.54 (18) |
| C3—C2—C1 | 120.4 (2) | N1—C7—C8 | 115.51 (16) |
| C3—C2—H2 | 119.8 | C7—C8—C8i | 112.1 (2) |
| C1—C2—H2 | 119.8 | C7—C8—H8A | 109.2 |
| C2—C3—C4 | 120.0 (2) | C8i—C8—H8A | 109.2 |
| C2—C3—Cl1 | 119.1 (2) | C7—C8—H8B | 109.2 |
| C4—C3—Cl1 | 120.9 (2) | C8i—C8—H8B | 109.2 |
| C2—C3—C9 | 124.4 (7) | H8A—C8—H8B | 107.9 |
| C4—C3—C9 | 115.4 (7) | C3—C9—H9A | 109.5 |
| Cl1—C3—C9 | 5.9 (7) | C3—C9—H9B | 109.5 |
| C5—C4—C3 | 119.2 (2) | H9A—C9—H9B | 109.5 |
| C5—C4—H4A | 120.4 | C3—C9—H9C | 109.5 |
| C3—C4—H4A | 120.4 | H9A—C9—H9C | 109.5 |
| C4—C5—C6 | 121.6 (2) | H9B—C9—H9C | 109.5 |
| C4—C5—H5A | 119.2 | ||
| C7—N1—C1—C6 | 138.7 (2) | C9—C3—C4—C5 | −175.8 (9) |
| C7—N1—C1—C2 | −43.0 (3) | C3—C4—C5—C6 | −0.1 (4) |
| C6—C1—C2—C3 | −0.6 (3) | C4—C5—C6—C1 | −1.0 (4) |
| N1—C1—C2—C3 | −178.9 (2) | C2—C1—C6—C5 | 1.3 (3) |
| C1—C2—C3—C4 | −0.5 (4) | N1—C1—C6—C5 | 179.6 (2) |
| C1—C2—C3—Cl1 | 178.8 (2) | C1—N1—C7—O1 | −2.4 (4) |
| C1—C2—C3—C9 | 175.8 (9) | C1—N1—C7—C8 | 179.8 (2) |
| C2—C3—C4—C5 | 0.8 (4) | O1—C7—C8—C8i | 32.9 (4) |
| Cl1—C3—C4—C5 | −178.4 (2) | N1—C7—C8—C8i | −149.2 (2) |
Symmetry codes: (i) −x, −y+2, −z.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O1ii | 0.86 | 2.05 | 2.894 (2) | 168. |
Symmetry codes: (ii) x−1, y, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: WM2508).
References
- Bhat, D. K. & Gowda, B. T. (2000). J. Indian Chem. Soc. 77, 279–284.
- Gowda, B. T., Foro, S. & Fuess, H. (2007). Acta Cryst. E63, o1975–o1976.
- Jayalakshmi, K. L. & Gowda, B. T. (2004). Z. Naturforsch. Teil A, 55, 491–500.
- Nardelli, M. (1999). J. Appl. Cryst. 32, 563–571.
- Oxford Diffraction (2009). CrysAlis CCD and CrysAlis RED Oxford Diffraction Ltd, Yarnton, England.
- Pierrot, M., Baldy, A., Maire, J. C., Mehrotra, R. C., Kapoor, T. S. & Bachlas, B. P. (1984). Acta Cryst. C40, 1931–1934.
- Saraswathi, B. S., Foro, S. & Gowda, B. T. (2011a). Acta Cryst. E67, o966. [DOI] [PMC free article] [PubMed]
- Saraswathi, B. S., Foro, S. & Gowda, B. T. (2011b). Acta Cryst. E67, o1591. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811028121/wm2508sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811028121/wm2508Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811028121/wm2508Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report