

Abstract
In the crystal structure of the title compound, C17H18N2O4S3, molecules are connected into centrosymmetric dimers via weak intermolecular C—H⋯π interactions. These dimers are further connected through a series of weak C—H⋯O hydrogen bonds, while futher C—H⋯π interactions involving the phenyl and thiazoline rings are also observed. The thiazolidine ring is twisted from the benzene rings rings by dihedral angles of 79.1 (1) and 85.0 (1)°, while the dihedral angle between two benzene rings is 76.0 (1)°.
Related literature
For background to N-heterocyclic sulfanilamide derivatives, see: Kuz’mina et al. (1962 ▶); Jensen & Thorsteinsson (1941 ▶); Hunter & Kolloff (1943 ▶); Hultquist et al. (1951 ▶). For a related synthesis, see: Razvodovskaya et al. (1990 ▶). 
Experimental
Crystal data
C17H18N2O4S3
M r = 410.51
Monoclinic,
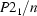
a = 9.3825 (2) Å
b = 14.4047 (2) Å
c = 14.2279 (3) Å
β = 102.666 (1)°
V = 1876.14 (6) Å3
Z = 4
Mo Kα radiation
μ = 0.42 mm−1
T = 296 K
0.40 × 0.40 × 0.40 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2001 ▶) T min = 0.845, T max = 0.845
17749 measured reflections
4652 independent reflections
3756 reflections with I > 2σ(I)
R int = 0.025
Refinement
R[F 2 > 2σ(F 2)] = 0.037
wR(F 2) = 0.107
S = 1.04
4652 reflections
236 parameters
H-atom parameters constrained
Δρmax = 0.29 e Å−3
Δρmin = −0.29 e Å−3
Data collection: APEX2 (Bruker, 2010 ▶); cell refinement: SAINT (Bruker, 2010 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811028807/fj2445sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811028807/fj2445Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811028807/fj2445Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg1 and Cg2 are the centroids of the C4–C9 and C11–C16 benzene rings, respectively.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C10—H10B⋯Cg1i | 0.97 | 2.91 | 3.567 (1) | 127 |
| C2—H2B⋯Cg2ii | 0.97 | 3.09 | 3.821 (1) | 134 |
| C1—H1B⋯O1ii | 0.97 | 2.59 | 3.394 (3) | 141 |
| C12—H12A⋯O3iii | 0.93 | 2.47 | 3.318 (2) | 151 |
Symmetry codes: (i) 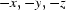 ; (ii)
; (ii) 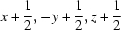 ; (iii)
; (iii)  .
.
Acknowledgments
We are grateful to the National Science Council of the Republic of China and the Nanya Institute of Technology for support.
supplementary crystallographic information
Comment
In a series of N-heterocyclic sulfanilamide derivatives which prepared and are investigating biologically one of the compounds, 2-sulfanilyl-aminothiazoline, proved to be of particular interest, both chemically and therapeutically. (Kuz'mina et al., 1962; Jensen et al., 1941; Hunter et al., 1943; Hultquist et al., 1951). The synthesis and character the 3-substituted 2-(thiophosphorylimino)thiazolidine compounds are also reported (Razvodovskaya et al., 1990). Within this project the crystal structure of the title compound was determined. The crystal structure features inversion-related dimers linked by the weak intermolecular C—H···pi interactions in the solid state, while Cg1 and Cg2 are the centers of C4—C9 and C11—C16 and these carbon atoms of mean devition from plane are 0.0008 and 0.0043 Å. Weak C—H···O hydrogen bonds among the molecules are also observed in the solid state. The thiazolidine and the phenyl rings are not coplanar but twisted with each other by an interplanar angles of 79.1 (1) and 85.0 (1)°, respectively, while the dihedral angle between two phenyl groups is 76.0 (1)°.
Experimental
The title compound was prepared according to a published procedure (Razvodovskaya et al., 1990). Block like crystals suitable for X-ray crystallography were obtained by slow evaporization of the solvent from a solution of the title compound in methanol.
Refinement
All the hydrogen atoms were discernible in the difference Fourier maps. However, they were situated into the idealized positions and constrained by the riding atom approximation: C—Hmethyl = 0.96 Å and C—Hmethylene = 0.97 Å while the methyls and methylenes were allowed to rotate about their respective axes; C—Haryl = 0.93 Å; Uiso(Hmethyl) = 1.5Ueq(Cmethyl); Uiso(Haryl or methylene) = 1.2Ueq(Caryl or methylene).
Figures
Fig. 1.

Crystal structure of the title compound with atom labeling and displacement ellipsoids drawn at the 30% probability level.
Crystal data
| C17H18N2O4S3 | F(000) = 856 |
| Mr = 410.51 | Dx = 1.453 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 8638 reflections |
| a = 9.3825 (2) Å | θ = 2.4–28.2° |
| b = 14.4047 (2) Å | µ = 0.42 mm−1 |
| c = 14.2279 (3) Å | T = 296 K |
| β = 102.666 (1)° | Block, colourless |
| V = 1876.14 (6) Å3 | 0.40 × 0.40 × 0.40 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 4652 independent reflections |
| Radiation source: fine-focus sealed tube | 3756 reflections with I > 2σ(I) |
| graphite | Rint = 0.025 |
| phi and ω scans | θmax = 28.3°, θmin = 2.0° |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | h = −12→12 |
| Tmin = 0.845, Tmax = 0.845 | k = −15→19 |
| 17749 measured reflections | l = −17→18 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.037 | H-atom parameters constrained |
| wR(F2) = 0.107 | w = 1/[σ2(Fo2) + (0.0497P)2 + 0.6237P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.04 | (Δ/σ)max < 0.001 |
| 4652 reflections | Δρmax = 0.29 e Å−3 |
| 236 parameters | Δρmin = −0.29 e Å−3 |
| 0 restraints | Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0091 (9) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.16502 (15) | 0.17067 (10) | 0.38640 (10) | 0.0446 (3) | |
| N2 | 0.08736 (15) | 0.26664 (10) | 0.25505 (10) | 0.0448 (3) | |
| S1 | 0.34585 (5) | 0.17353 (4) | 0.27357 (4) | 0.06158 (16) | |
| S2 | 0.10549 (5) | 0.31474 (3) | 0.15432 (3) | 0.04983 (13) | |
| S3 | 0.01808 (5) | 0.19186 (3) | 0.43270 (3) | 0.04805 (13) | |
| O1 | −0.00727 (19) | 0.38361 (9) | 0.13422 (10) | 0.0693 (4) | |
| O2 | 0.25206 (18) | 0.34381 (12) | 0.15775 (11) | 0.0739 (4) | |
| O3 | 0.04496 (18) | 0.13807 (11) | 0.51854 (10) | 0.0702 (4) | |
| O4 | 0.00217 (15) | 0.28956 (9) | 0.43817 (10) | 0.0580 (3) | |
| C1 | 0.2585 (2) | 0.08847 (14) | 0.41762 (15) | 0.0590 (5) | |
| H1A | 0.2091 | 0.0321 | 0.3909 | 0.071* | |
| H1B | 0.2818 | 0.0836 | 0.4873 | 0.071* | |
| C2 | 0.3947 (2) | 0.10280 (16) | 0.38077 (16) | 0.0664 (6) | |
| H2A | 0.4335 | 0.0436 | 0.3656 | 0.080* | |
| H2B | 0.4684 | 0.1338 | 0.4290 | 0.080* | |
| C3 | 0.18420 (17) | 0.21042 (11) | 0.30207 (11) | 0.0407 (3) | |
| C4 | 0.06041 (19) | 0.22754 (11) | 0.06650 (12) | 0.0431 (4) | |
| C5 | −0.08309 (19) | 0.19554 (12) | 0.04084 (13) | 0.0481 (4) | |
| H5A | −0.1533 | 0.2177 | 0.0723 | 0.058* | |
| C6 | −0.1198 (2) | 0.13072 (13) | −0.03153 (14) | 0.0534 (4) | |
| H6A | −0.2156 | 0.1094 | −0.0486 | 0.064* | |
| C7 | −0.0173 (2) | 0.09647 (13) | −0.07954 (13) | 0.0542 (4) | |
| C8 | 0.1244 (2) | 0.12907 (15) | −0.05287 (14) | 0.0607 (5) | |
| H8A | 0.1943 | 0.1068 | −0.0844 | 0.073* | |
| C9 | 0.1647 (2) | 0.19422 (14) | 0.01986 (14) | 0.0538 (4) | |
| H9A | 0.2606 | 0.2152 | 0.0370 | 0.065* | |
| C10 | −0.0606 (3) | 0.02670 (16) | −0.15964 (16) | 0.0787 (7) | |
| H10A | −0.1623 | 0.0119 | −0.1675 | 0.118* | |
| H10B | −0.0034 | −0.0287 | −0.1439 | 0.118* | |
| H10C | −0.0438 | 0.0524 | −0.2185 | 0.118* | |
| C11 | −0.12963 (18) | 0.14496 (12) | 0.34887 (12) | 0.0460 (4) | |
| C12 | −0.1544 (2) | 0.04982 (13) | 0.35063 (15) | 0.0578 (5) | |
| H12A | −0.0965 | 0.0124 | 0.3971 | 0.069* | |
| C13 | −0.2660 (2) | 0.01228 (15) | 0.28240 (17) | 0.0652 (5) | |
| H13A | −0.2837 | −0.0512 | 0.2836 | 0.078* | |
| C14 | −0.3526 (2) | 0.06605 (15) | 0.21206 (15) | 0.0579 (5) | |
| C15 | −0.3267 (2) | 0.16119 (15) | 0.21243 (15) | 0.0557 (5) | |
| H15A | −0.3850 | 0.1986 | 0.1661 | 0.067* | |
| C16 | −0.21635 (19) | 0.20075 (13) | 0.28026 (14) | 0.0507 (4) | |
| H16A | −0.2002 | 0.2644 | 0.2800 | 0.061* | |
| C17 | −0.4719 (3) | 0.0231 (2) | 0.13607 (19) | 0.0864 (8) | |
| H17A | −0.4738 | −0.0427 | 0.1461 | 0.130* | |
| H17B | −0.4536 | 0.0354 | 0.0735 | 0.130* | |
| H17C | −0.5644 | 0.0494 | 0.1403 | 0.130* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0461 (7) | 0.0435 (7) | 0.0418 (7) | 0.0049 (6) | 0.0045 (6) | −0.0002 (6) |
| N2 | 0.0453 (7) | 0.0459 (8) | 0.0429 (7) | 0.0017 (6) | 0.0095 (6) | 0.0013 (6) |
| S1 | 0.0508 (3) | 0.0734 (3) | 0.0623 (3) | 0.0140 (2) | 0.0163 (2) | −0.0073 (2) |
| S2 | 0.0627 (3) | 0.0401 (2) | 0.0470 (2) | −0.00536 (18) | 0.0128 (2) | 0.00129 (17) |
| S3 | 0.0555 (3) | 0.0491 (3) | 0.0406 (2) | −0.00057 (18) | 0.01281 (18) | −0.00071 (17) |
| O1 | 0.1039 (12) | 0.0425 (7) | 0.0589 (8) | 0.0189 (7) | 0.0122 (8) | 0.0043 (6) |
| O2 | 0.0802 (10) | 0.0758 (10) | 0.0684 (9) | −0.0367 (8) | 0.0217 (8) | −0.0044 (8) |
| O3 | 0.0878 (11) | 0.0805 (10) | 0.0427 (7) | 0.0010 (8) | 0.0149 (7) | 0.0108 (7) |
| O4 | 0.0645 (8) | 0.0504 (7) | 0.0619 (8) | 0.0006 (6) | 0.0197 (7) | −0.0130 (6) |
| C1 | 0.0684 (12) | 0.0496 (10) | 0.0521 (10) | 0.0146 (9) | −0.0017 (9) | 0.0011 (8) |
| C2 | 0.0637 (12) | 0.0655 (13) | 0.0631 (12) | 0.0251 (10) | −0.0009 (10) | −0.0143 (10) |
| C3 | 0.0399 (8) | 0.0402 (8) | 0.0402 (8) | −0.0027 (6) | 0.0052 (6) | −0.0082 (6) |
| C4 | 0.0484 (9) | 0.0396 (8) | 0.0416 (8) | 0.0018 (7) | 0.0110 (7) | 0.0055 (7) |
| C5 | 0.0472 (9) | 0.0468 (9) | 0.0520 (10) | 0.0050 (7) | 0.0144 (7) | 0.0031 (8) |
| C6 | 0.0529 (10) | 0.0479 (10) | 0.0546 (10) | −0.0025 (8) | 0.0014 (8) | 0.0027 (8) |
| C7 | 0.0726 (12) | 0.0458 (10) | 0.0403 (9) | 0.0106 (9) | 0.0041 (8) | 0.0038 (7) |
| C8 | 0.0659 (12) | 0.0680 (13) | 0.0519 (11) | 0.0153 (10) | 0.0210 (9) | −0.0008 (9) |
| C9 | 0.0484 (9) | 0.0636 (11) | 0.0512 (10) | 0.0007 (8) | 0.0153 (8) | 0.0020 (8) |
| C10 | 0.1124 (19) | 0.0631 (13) | 0.0513 (12) | 0.0143 (13) | −0.0025 (12) | −0.0083 (10) |
| C11 | 0.0473 (9) | 0.0452 (9) | 0.0478 (9) | −0.0052 (7) | 0.0155 (7) | 0.0038 (7) |
| C12 | 0.0651 (12) | 0.0470 (10) | 0.0626 (12) | −0.0066 (9) | 0.0171 (9) | 0.0100 (9) |
| C13 | 0.0710 (13) | 0.0470 (11) | 0.0826 (15) | −0.0152 (9) | 0.0279 (11) | −0.0052 (10) |
| C14 | 0.0491 (10) | 0.0687 (12) | 0.0607 (11) | −0.0102 (9) | 0.0226 (9) | −0.0147 (10) |
| C15 | 0.0441 (9) | 0.0665 (12) | 0.0578 (11) | −0.0001 (8) | 0.0139 (8) | 0.0053 (9) |
| C16 | 0.0467 (9) | 0.0451 (9) | 0.0622 (11) | −0.0022 (7) | 0.0157 (8) | 0.0064 (8) |
| C17 | 0.0689 (14) | 0.0996 (19) | 0.0894 (18) | −0.0144 (13) | 0.0148 (13) | −0.0413 (15) |
Geometric parameters (Å, °)
| N1—C3 | 1.376 (2) | C6—H6A | 0.9300 |
| N1—C1 | 1.483 (2) | C7—C8 | 1.382 (3) |
| N1—S3 | 1.6811 (15) | C7—C10 | 1.507 (3) |
| N2—C3 | 1.289 (2) | C8—C9 | 1.387 (3) |
| N2—S2 | 1.6340 (15) | C8—H8A | 0.9300 |
| S1—C3 | 1.7368 (16) | C9—H9A | 0.9300 |
| S1—C2 | 1.808 (2) | C10—H10A | 0.9600 |
| S2—O2 | 1.4282 (15) | C10—H10B | 0.9600 |
| S2—O1 | 1.4327 (15) | C10—H10C | 0.9600 |
| S2—C4 | 1.7566 (17) | C11—C16 | 1.383 (3) |
| S3—O4 | 1.4192 (14) | C11—C12 | 1.391 (3) |
| S3—O3 | 1.4215 (14) | C12—C13 | 1.373 (3) |
| S3—C11 | 1.7536 (18) | C12—H12A | 0.9300 |
| C1—C2 | 1.498 (3) | C13—C14 | 1.380 (3) |
| C1—H1A | 0.9700 | C13—H13A | 0.9300 |
| C1—H1B | 0.9700 | C14—C15 | 1.392 (3) |
| C2—H2A | 0.9700 | C14—C17 | 1.508 (3) |
| C2—H2B | 0.9700 | C15—C16 | 1.375 (3) |
| C4—C9 | 1.383 (2) | C15—H15A | 0.9300 |
| C4—C5 | 1.394 (2) | C16—H16A | 0.9300 |
| C5—C6 | 1.377 (3) | C17—H17A | 0.9600 |
| C5—H5A | 0.9300 | C17—H17B | 0.9600 |
| C6—C7 | 1.386 (3) | C17—H17C | 0.9600 |
| C3—N1—C1 | 114.30 (15) | C7—C6—H6A | 119.2 |
| C3—N1—S3 | 122.77 (11) | C8—C7—C6 | 118.23 (17) |
| C1—N1—S3 | 120.66 (13) | C8—C7—C10 | 121.2 (2) |
| C3—N2—S2 | 121.62 (12) | C6—C7—C10 | 120.6 (2) |
| C3—S1—C2 | 92.76 (9) | C7—C8—C9 | 121.55 (18) |
| O2—S2—O1 | 117.85 (10) | C7—C8—H8A | 119.2 |
| O2—S2—N2 | 112.27 (9) | C9—C8—H8A | 119.2 |
| O1—S2—N2 | 104.73 (8) | C4—C9—C8 | 119.15 (18) |
| O2—S2—C4 | 108.29 (9) | C4—C9—H9A | 120.4 |
| O1—S2—C4 | 107.52 (9) | C8—C9—H9A | 120.4 |
| N2—S2—C4 | 105.40 (8) | C7—C10—H10A | 109.5 |
| O4—S3—O3 | 119.63 (9) | C7—C10—H10B | 109.5 |
| O4—S3—N1 | 107.86 (8) | H10A—C10—H10B | 109.5 |
| O3—S3—N1 | 103.39 (8) | C7—C10—H10C | 109.5 |
| O4—S3—C11 | 110.02 (9) | H10A—C10—H10C | 109.5 |
| O3—S3—C11 | 109.83 (9) | H10B—C10—H10C | 109.5 |
| N1—S3—C11 | 104.90 (8) | C16—C11—C12 | 120.69 (18) |
| N1—C1—C2 | 106.21 (17) | C16—C11—S3 | 120.74 (14) |
| N1—C1—H1A | 110.5 | C12—C11—S3 | 118.52 (15) |
| C2—C1—H1A | 110.5 | C13—C12—C11 | 118.67 (19) |
| N1—C1—H1B | 110.5 | C13—C12—H12A | 120.7 |
| C2—C1—H1B | 110.5 | C11—C12—H12A | 120.7 |
| H1A—C1—H1B | 108.7 | C12—C13—C14 | 121.87 (19) |
| C1—C2—S1 | 107.17 (13) | C12—C13—H13A | 119.1 |
| C1—C2—H2A | 110.3 | C14—C13—H13A | 119.1 |
| S1—C2—H2A | 110.3 | C13—C14—C15 | 118.41 (19) |
| C1—C2—H2B | 110.3 | C13—C14—C17 | 121.1 (2) |
| S1—C2—H2B | 110.3 | C15—C14—C17 | 120.5 (2) |
| H2A—C2—H2B | 108.5 | C16—C15—C14 | 120.96 (19) |
| N2—C3—N1 | 120.10 (15) | C16—C15—H15A | 119.5 |
| N2—C3—S1 | 128.55 (13) | C14—C15—H15A | 119.5 |
| N1—C3—S1 | 111.35 (12) | C15—C16—C11 | 119.39 (17) |
| C9—C4—C5 | 120.25 (17) | C15—C16—H16A | 120.3 |
| C9—C4—S2 | 120.28 (14) | C11—C16—H16A | 120.3 |
| C5—C4—S2 | 119.40 (13) | C14—C17—H17A | 109.5 |
| C6—C5—C4 | 119.28 (17) | C14—C17—H17B | 109.5 |
| C6—C5—H5A | 120.4 | H17A—C17—H17B | 109.5 |
| C4—C5—H5A | 120.4 | C14—C17—H17C | 109.5 |
| C5—C6—C7 | 121.53 (18) | H17A—C17—H17C | 109.5 |
| C5—C6—H6A | 119.2 | H17B—C17—H17C | 109.5 |
Hydrogen-bond geometry (Å, °)
| Cg1 and Cg2 are the centroids of the C4–C9 and C11–C16 benzene rings, respectively. |
| D—H···A | D—H | H···A | D···A | D—H···A |
| C10—H10B···Cg1i | 0.97 | 2.91 | 3.567 (1) | 127 |
| C2—H2B···Cg2ii | 0.97 | 3.09 | 3.821 (1) | 134 |
| C1—H1B···O1ii | 0.97 | 2.59 | 3.394 (3) | 141 |
| C12—H12A···O3iii | 0.93 | 2.47 | 3.318 (2) | 151 |
Symmetry codes: (i) −x, −y, −z; (ii) x+1/2, −y+1/2, z+1/2; (iii) −x, −y, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2445).
References
- Bruker (2001). SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2010). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Hultquist, M. E., Germann, R. P., Webb, J. S., Wright, W. B. Jr, Roth, B., Smith, J. M. Jr & SubbaRow, Y. (1951). J. Am. Chem. Soc. 73, 2558–2566.
- Hunter, J. H. & Kolloff, H. G. (1943). J. Am. Chem. Soc. 65, 156–159.
- Jensen, K. A. & Thorsteinsson, T. (1941). Dansk Tidsskrift Farmaci, 15, 41–77.
- Kuz’mina, K. K., Ostroumova, N. G., Markova, Yu. V. & Shchukina, M. N. (1962). Zhurnal Obshchei Khimii. 32, 3390–3393.
- Razvodovskaya, L. V., Vorob’eva, N. N., Grapov, A. F. & Mel’nikov, N. N. (1990). Zhurnal Obshchei Khimii, 60, 1518–1525.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811028807/fj2445sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811028807/fj2445Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811028807/fj2445Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report