

Abstract
The molecule of the title compound, C12H16O5, has crystallographically imposed mirror symmetry with the mirror plane passing through the endocyclic O atom and the mid-point of the double bond. In the crystal, molecules are linked by C—H⋯O hydrogen bonds, forming chains running along the a axis.
Related literature
Compounds containing the 8-oxabicyclo[3.2.1]octane framework have shown broad utility as chiral building blocks for synthesis of polyketides, see: Coste & Gerber-Lemaire (2005 ▶); Meilert et al. (2003 ▶); Schwenter & Vogel (2001 ▶); Gerber-Lemaire & Vogel (2003 ▶); Gerber & Vogel (1999 ▶, 2001 ▶); Re et al. (2009 ▶); Pascual et al. (2004 ▶); Derwick (1998 ▶). For the inhibitory activity of calystegines and other tropane alkaloids against several glycosidase enzymes, see: Asano et al. (2000 ▶); Drager (2004 ▶). Several 8-oxabicyclo[3.2.1] octane derivatives possess moderate anti-HIV activity, see: Montana et al. (2009 ▶). For the syntheses of a full set of hybrid d- and l-C-glycosides and thymine polyoxin C starting with the unsaturated 8-oxabicyclo[3.2.1]octane framework, see: Gethin & Simpkins (1997 ▶); Hoffmann et al. (2001 ▶). For the synthesis of an 8-oxabicyclo[3.2.1]octane from tetrachlorocyclopropene and furan, see: Batson et al. (2004 ▶). For a synthetic approach to 8-oxabicyclo[3.2.1]octane derivatives based on the reaction of tetrachlorocyclopropene with furan, see: Law & Tobey (1968 ▶). For structures of related 8-oxabicyclo[3.2.1]octanes, see: Kreiselmeier et al. (2006 ▶); Hoffmann et al. (2001 ▶). For a report of prior research, see: Tafeenko et al. (2009 ▶). 
Experimental
Crystal data
C12H16O5
M r = 240.25
Orthorhombic,
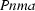
a = 6.8680 (12) Å
b = 12.295 (4) Å
c = 14.120 (3) Å
V = 1192.3 (5) Å3
Z = 4
Ag Kα radiation
λ = 0.56085 Å
μ = 0.06 mm−1
T = 296 K
0.1 × 0.07 × 0.05 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
1974 measured reflections
1974 independent reflections
1085 reflections with I > 2s(I)
2 standard reflections every 120 min intensity decay: none
Refinement
R[F 2 > 2σ(F 2)] = 0.055
wR(F 2) = 0.138
S = 1.02
1974 reflections
91 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.24 e Å−3
Δρmin = −0.17 e Å−3
Data collection: CAD-4 Software (Enraf–Nonius, 1989 ▶); cell refinement: CAD-4 Software; data reduction: XCAD4 (Harms & Wocadlo, 1995 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg, 2000 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811027292/mw2015sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811027292/mw2015Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C6—H6⋯O2i | 0.93 | 2.55 | 3.482 (2) | 178 |
Symmetry code: (i)  .
.
supplementary crystallographic information
Comment
Compounds containing the 8-oxabicyclo[3.2.1]octane framework are important precursors in the field of biologically active compounds. They have shown broad utility as chiral building blocks for synthesis of polyketides (Coste & Gerber-Lemaire, 2005; Meilert et al., 2003; Schwenter & Vogel, 2001; Gerber-Lemaire & Vogel, 2003), C-linked disaccharides (Gerber & Vogel, 1999; Gerber & Vogel, 2001), calystegines (Re et al., 2009; Pascual et al., 2004; Derwick, 1998) and other natural products. Calystegines and other tropane alkaloids show remarkable inhibitory activities against several glycosidase enzymes, in comparison with other alkaloidal glycosidase inhibitors (Asano et al., 2000; Drager, 2004). They are used medicinally, e.g., as anticholinergics, competing with acetylcholine for the muscarinic receptor site of the parasympathetic nervous system (Derwick, 1998). De novo syntheses of a full set of hybrid d- and l-C-glycosides and thymine polyoxin C starting with the unsaturated 8-oxabicyclo[3.2.1]octane framework have been reported (Hoffmann et al., 2001; Gethin & Simpkins, 1997). Moreover, recent research showed that several 8-oxabicyclo[3.2.1] octane derivatives possess moderate anti-HIV activity (Montana et al., 2009). In studies of novel biologically active homoinositol compounds, including selective glycosidase inhibitors, we have investigated a new synthetic approach to 8-oxabicyclo[3.2.1]octane derivatives based on the reaction of tetrachlorocyclopropene with furan (Law et al., 1968) for the preparation of (1R,2R,3r,4S,5S)-3-methyl-8-oxabicyclo[3.2.1]oct-6-ene- 2,4-diol (4). Several structural results in this field have been previously reported (Tafeenko et al., 2009; Batson et al., 2004; Kreiselmeier et al., 2006; Hoffmann et al. 2001).
Determination of the relative stereochemistry of compound (4) (Fig. 2) by NMR methods was ambiguous so recourse was made to X-ray crystallography for which purpose the crystalline diacetate (I) was synthesized.
Molecule (I) has crystallographically-imposed mirror symmetry with the mirror plane, m, passing through atoms C3, C8, the endocyclic oxygen O8 and the midpoint of the double bond C6/C6i (i: x,1.5 - y, z). The 6-membered ring of the molecule adopts a chair conformation, with atoms O8 and C3 displaced out of plane defined by the atoms C2/C2i/C1/C1i (plane 1) by 0.856 (2) and -0.525 (2) Å, respectively. The carbon atom of the methyl-group and atoms C6,C6i (double bond) are displaced out of plane 1 by -2.028 (2) Å and -1.326 (2) Å respectively. The molecules (I) are linked by means of weak C—H···O hydrogen bonds to form chains running along a axis.
Experimental
(1R,5S)-3-Chloro-3-methyl-8-oxabicyclo[3.2.1]oct-6-ene-2,4-dione (see Fig.2) (2).
To a solution of 0.5 g (2.9 mmol) of (1) (Law & Tobey, 1968) in acetone (10 ml) 0.83 g (5.8 mmol) K2CO3 and 1.38 g (8.7 mmol) MeI were added. The mixture was stirred at 298 K for 36 h, then concentrated under reduced pressure. The residue was purified via silica gel flash chromatography (10% EtOAc in CH2Cl2), to give a pale yellow crystalline solid; yield: 0.45 g, (83%) (compound 2), mp 370–372 K.
(1R,5S)-3-methyl-8-oxabicyclo[3.2.1]oct-6-ene-2,4-dione (see Fig.2) (3).
To a solution of 0.4 g (2.1 mmol) of diketone (2) in 4 ml of glacial acetic acid was added 0.23 g of Zn-powder. After the beginning of heating-up the mixture was stirred at 298 K for 0.5 h and 20% aqueous NaOH was added dropwise until solid formed. The solid was dissolved by addition of 1 ml 0.1 N HCl, the mixture was extracted with CH2Cl2 (5*20 ml), the combined organic layers were dried over Na2SO4 and evaporated under reduced pressure to yield 0.26 g of compound (3) as a pale yellow crystalline solid, (compound 3) mp 359–361 K.
(1R,2R,4S,5S)-3-methyl-8-oxabicyclo[3.2.1]oct-6-ene-2,4 -diol (see Fig.2) (4).
To a solution of 200 mg (1.3 mmol) of diketone (3) in 15 ml of MeOH 340 mg (8.9 mmol) of NaBH4 was added portionwise for 3 h, during which time the reaction temperature was kept between 293–303 K. The mixture was stirred at 298 K for 2 h, then 0.4 g of crystalline NH4Cl was added followed by 0.5 ml of 1 N HCl. The solvents were evaporated under reduced pressure, the residue was flashed by EtOAc/Me2CO (1:1) through a silica gel column to give 0.2 g of a yellow oil. The 1H and 13C NMR spectra showed that the oil contained several isomers so it was purified by column chromatography on silica gel eluting with a EtOAc/Me2CO (5:2) mixture to afford 95 mg of the major isomer (4) as a colorless oil, yield 46%; Rf = 0.5 (Et2O/Me2CO = 3:1).
(1R,2R,3r,4S,5S)-3-methyl-8-oxabicyclo[3.2.1]oct-6-ene- 2,4-diyl diacetate (see Fig.2) (I).
Compound (4) (95 mg, 0.61 mmol) was dissolved in 5 ml of py and 3 ml of Ac2O was added. The mixture was stirred for 24 h at room temperature and then concentrated under reduced pressure (1 mm Hg) to give a thick brown oil. Diacetate (I) was separated by flash chromatography on silica gel (CH2Cl2) as colorless crystals, yield 134 mg (92%), mp 428–430 K, Rf = 0.4 (CH2Cl2). Crystals suitable for diffraction analysis were obtained by slow evaporation of solvents from a dichloromethane-hexane solution.
Refinement
The positions of the H atoms were determined from Fourier difference maps; H atoms attached to carbons were then placed in calculated positions and allowed to ride on their parent atoms [C—H = 0.93–0.98 Å. Uiso(H) = xUeq(parent atom), where x = 1.2.] Hydrogen (H8, H81) atoms at C8 are refined freely.
Figures
Fig. 1.

The molecular structure of (I) with displacement ellipsoids drawn at the 50% probability level. Atoms C,N,O, and Ci,Ni,Oi are related by symmetry code: (i) x,1.5 - y, z; The H atoms at C8 are not shown for clarity.
Fig. 2.

How the title compound was obtained.
Crystal data
| C12H16O5 | Dx = 1.338 Mg m−3 |
| Mr = 240.25 | Melting point: 428 K |
| Orthorhombic, Pnma | Ag Kα radiation, λ = 0.56085 Å |
| Hall symbol: -P 2ac 2n | Cell parameters from 25 reflections |
| a = 6.8680 (12) Å | θ = 11–14° |
| b = 12.295 (4) Å | µ = 0.06 mm−1 |
| c = 14.120 (3) Å | T = 296 K |
| V = 1192.3 (5) Å3 | Prism, colorless |
| Z = 4 | 0.1 × 0.07 × 0.05 mm |
| F(000) = 512 |
Data collection
| Enraf–Nonius CAD-4 diffractometer | Rint = 0.000 |
| Radiation source: fine-focus sealed tube | θmax = 24.0°, θmin = 1.7° |
| graphite | h = −9→0 |
| non–profiled ω scans | k = −17→0 |
| 1974 measured reflections | l = −20→0 |
| 1974 independent reflections | 2 standard reflections every 120 min |
| 1085 reflections with I > 2s(I) | intensity decay: none |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.055 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.138 | w = 1/[σ2(Fo2) + (0.0492P)2 + 0.2946P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 1974 reflections | Δρmax = 0.24 e Å−3 |
| 91 parameters | Δρmin = −0.17 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.031 (4) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.74430 (17) | 0.55206 (10) | 0.09878 (9) | 0.0459 (4) | |
| O2 | 1.06414 (18) | 0.53353 (12) | 0.12399 (11) | 0.0604 (4) | |
| O8 | 0.6552 (3) | 0.7500 | −0.08400 (12) | 0.0550 (5) | |
| C1 | 0.6076 (3) | 0.65913 (15) | −0.02489 (13) | 0.0479 (5) | |
| H1 | 0.5863 | 0.5928 | −0.0619 | 0.057* | |
| C2 | 0.7802 (2) | 0.64791 (14) | 0.04233 (12) | 0.0409 (4) | |
| H2 | 0.8972 | 0.6349 | 0.0042 | 0.049* | |
| C3 | 0.8160 (3) | 0.7500 | 0.10391 (17) | 0.0382 (6) | |
| H3 | 0.9552 | 0.7500 | 0.1193 | 0.046* | |
| C6 | 0.4243 (3) | 0.69639 (16) | 0.02399 (13) | 0.0510 (5) | |
| H6 | 0.3285 | 0.6518 | 0.0496 | 0.061* | |
| C8 | 0.7082 (5) | 0.7500 | 0.19833 (19) | 0.0494 (7) | |
| C9 | 0.9005 (3) | 0.50142 (15) | 0.13532 (13) | 0.0451 (4) | |
| C10 | 0.8443 (3) | 0.40349 (17) | 0.18986 (17) | 0.0647 (6) | |
| H10A | 0.9539 | 0.3554 | 0.1947 | 0.097* | |
| H10B | 0.7395 | 0.3669 | 0.1581 | 0.097* | |
| H10C | 0.8029 | 0.4246 | 0.2521 | 0.097* | |
| H8 | 0.749 (3) | 0.8125 (16) | 0.2358 (16) | 0.071 (7)* | |
| H81 | 0.575 (5) | 0.7500 | 0.194 (3) | 0.092 (12)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0411 (6) | 0.0396 (7) | 0.0569 (8) | −0.0008 (6) | 0.0002 (6) | 0.0067 (6) |
| O2 | 0.0422 (8) | 0.0569 (9) | 0.0820 (10) | 0.0018 (7) | 0.0016 (7) | 0.0071 (8) |
| O8 | 0.0746 (13) | 0.0542 (11) | 0.0362 (9) | 0.000 | −0.0022 (10) | 0.000 |
| C1 | 0.0580 (11) | 0.0423 (10) | 0.0434 (9) | −0.0048 (9) | −0.0061 (9) | −0.0032 (8) |
| C2 | 0.0420 (9) | 0.0387 (9) | 0.0420 (9) | −0.0002 (7) | 0.0056 (8) | 0.0006 (8) |
| C3 | 0.0330 (11) | 0.0392 (13) | 0.0425 (13) | 0.000 | 0.0001 (10) | 0.000 |
| C6 | 0.0403 (9) | 0.0602 (11) | 0.0524 (11) | −0.0044 (8) | −0.0128 (8) | 0.0003 (10) |
| C8 | 0.0507 (17) | 0.0601 (18) | 0.0374 (14) | 0.000 | 0.0003 (13) | 0.000 |
| C9 | 0.0494 (10) | 0.0372 (9) | 0.0487 (10) | 0.0021 (9) | 0.0002 (8) | −0.0051 (9) |
| C10 | 0.0650 (13) | 0.0516 (12) | 0.0775 (15) | −0.0036 (11) | −0.0095 (12) | 0.0119 (11) |
Geometric parameters (Å, °)
| O1—C9 | 1.344 (2) | C3—C2i | 1.547 (2) |
| O1—C2 | 1.444 (2) | C3—H3 | 0.9800 |
| O2—C9 | 1.202 (2) | C6—C6i | 1.318 (4) |
| O8—C1 | 1.432 (2) | C6—H6 | 0.9300 |
| C1—C6 | 1.507 (2) | C8—H8 | 0.97 (2) |
| C1—C2 | 1.525 (2) | C8—H81 | 0.92 (4) |
| C1—H1 | 0.9800 | C9—C10 | 1.481 (3) |
| C2—C3 | 1.547 (2) | C10—H10A | 0.9600 |
| C2—H2 | 0.9800 | C10—H10B | 0.9600 |
| C3—C8 | 1.525 (4) | C10—H10C | 0.9600 |
| C9—O1—C2 | 116.98 (13) | C2—C3—H3 | 106.2 |
| C1i—O8—C1 | 102.51 (19) | C2i—C3—H3 | 106.2 |
| O8—C1—C6 | 102.74 (16) | C6i—C6—C1 | 107.70 (10) |
| O8—C1—C2 | 104.80 (15) | C6i—C6—H6 | 126.1 |
| C6—C1—C2 | 113.07 (14) | C1—C6—H6 | 126.1 |
| O8—C1—H1 | 111.9 | C3—C8—H8 | 109.7 (13) |
| C6—C1—H1 | 111.9 | C3—C8—H81 | 115 (2) |
| C2—C1—H1 | 111.9 | H8—C8—H81 | 108.8 (18) |
| O1—C2—C1 | 106.53 (14) | O2—C9—O1 | 122.91 (17) |
| O1—C2—C3 | 112.28 (14) | O2—C9—C10 | 125.48 (18) |
| C1—C2—C3 | 113.59 (15) | O1—C9—C10 | 111.60 (16) |
| O1—C2—H2 | 108.1 | C9—C10—H10A | 109.5 |
| C1—C2—H2 | 108.1 | C9—C10—H10B | 109.5 |
| C3—C2—H2 | 108.1 | H10A—C10—H10B | 109.5 |
| C8—C3—C2 | 114.47 (13) | C9—C10—H10C | 109.5 |
| C8—C3—C2i | 114.47 (13) | H10A—C10—H10C | 109.5 |
| C2—C3—C2i | 108.50 (19) | H10B—C10—H10C | 109.5 |
| C8—C3—H3 | 106.2 | ||
| C1i—O8—C1—C6 | 38.7 (2) | O1—C2—C3—C8 | −30.9 (2) |
| C1i—O8—C1—C2 | −79.64 (19) | C1—C2—C3—C8 | 90.0 (2) |
| C9—O1—C2—C1 | 154.76 (15) | O1—C2—C3—C2i | −160.12 (11) |
| C9—O1—C2—C3 | −80.30 (19) | C1—C2—C3—C2i | −39.2 (2) |
| O8—C1—C2—O1 | −175.93 (13) | O8—C1—C6—C6i | −24.28 (13) |
| C6—C1—C2—O1 | 72.93 (19) | C2—C1—C6—C6i | 88.12 (14) |
| O8—C1—C2—C3 | 59.93 (19) | C2—O1—C9—O2 | 1.2 (3) |
| C6—C1—C2—C3 | −51.2 (2) | C2—O1—C9—C10 | −178.85 (15) |
Symmetry codes: (i) x, −y+3/2, z.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C6—H6···O2ii | 0.93 | 2.55 | 3.482 (2) | 178 |
Symmetry codes: (ii) x−1, y, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: MW2015).
References
- Asano, N., Nash, R. J., Molyneux, R. J. & Fleet, G. W. J. (2000). Tetrahedron Asymmetry, 11, 1645–1680.
- Batson, W. A., Abboud, K. A., Battiste, M. A. & Wright, D. L. (2004). Tetrahedron Lett. 45, 2093–2096.
- Brandenburg, K. (2000). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Coste, G. & Gerber-Lemaire, S. (2005). Tetrahedron Asymmetry, 16, 2277–2283.
- Derwick, P. M. (1998). In Medicinal Natural Products Chichester: Wiley.
- Drager, B. (2004). Nat. Prod. Rep. 21, 211–223. [DOI] [PubMed]
- Enraf–Nonius (1989). CAD-4 Software. Enraf–Nonius, Delft, The Netherlands.
- Farrugia, L. J. (1999). J. Appl. Cryst. 32, 837–838.
- Gerber, P. & Vogel, P. (1999). Tetrahedron Lett. 40, 3165–3168.
- Gerber, P. & Vogel, P. (2001). Helv. Chim. Acta, 84, 1363–1395.
- Gerber-Lemaire, S. & Vogel, P. (2003). Eur. J. Org. Chem. pp. 2959–2963.
- Gethin, D. M. & Simpkins, N. S. (1997). Tetrahedron, 53, 14417–14436.
- Harms, K. & Wocadlo, S. (1995). XCAD4 University of Marburg, Germany.
- Hoffmann, H. M. R., Dunkel, R., Mentzel, M., Reuter, H. & Stark, C. B. W. (2001). Chem. Eur. J. 7, 4771–4789. [DOI] [PubMed]
- Kreiselmeier, G., Frey, W. & Fohlisch, B. (2006). Tetrahedron, 62, 6029–6035.
- Law, D. C. F. & Tobey, S. W. (1968). J. Am. Chem. Soc., 90, 2376–2386.
- Meilert, K. M., Schwenter, M. E., Shatz, Y., Dubbaka, S. R. & Vogel, P. (2003). J. Org. Chem. 68, 2964–2967. [DOI] [PubMed]
- Montana, A. M., Barcia, J. A., Kociok-Kohn, G. & Font-Bardia, M. (2009). Tetrahedron, 65, 5308–5321.
- Pascual, M. V., Proemmel, S., Beil, W., Wartchow, R. & Hoffmann, H. M. R. (2004). Org. Lett. 6, 4155–4158. [DOI] [PubMed]
- Re, D. L., Franco, F., Sanchez-Cantalejo, F. & Tamayo, J. A. (2009). Eur. J. Org. Chem. pp. 1984–1993. [DOI] [PubMed]
- Schwenter, M. E. & Vogel, P. (2001). J. Org. Chem. 66, 7869–7872. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Tafeenko, V. A., Aslanov, L. A., Proskurnina, M. V., Sosonyuk, S. E. & Khlevin, D. A. (2009). Acta Cryst. E65, o1580. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811027292/mw2015sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811027292/mw2015Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report