

Abstract
The title compound, C6H10N2O, is a zwitterionic pyrazole derivative. The crystal packing is predominantly governed by a three-center iminium–amine N+—H⋯O−⋯H—N interaction, leading to an undulating sheet-like structure lying parallel to (100).
Related literature
For related structures and the preparation of similar compounds, see: Ragavan et al. (2009 ▶, 2010 ▶) and references therein. For related salt-bridge-mediated sheet structures, see: Shylaja et al. (2008 ▶).
Experimental
Crystal data
C6H10N2O
M r = 126.16
Monoclinic,
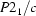
a = 9.1299 (15) Å
b = 7.1600 (11) Å
c = 11.374 (2) Å
β = 113.232 (9)°
V = 683.2 (2) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 296 K
0.21 × 0.19 × 0.11 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2001 ▶) T min = 0.64, T max = 0.83
12120 measured reflections
1332 independent reflections
961 reflections with I > 2σ(I)
R int = 0.034
Refinement
R[F 2 > 2σ(F 2)] = 0.049
wR(F 2) = 0.136
S = 1.03
1332 reflections
92 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.17 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT-Plus (Bruker, 2007 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and PLATON (Spek, 2009 ▶); software used to prepare material for publication: SHELXL97 and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S160053681102808X/su2287sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S160053681102808X/su2287Isup2.hkl
Supplementary material file. DOI: 10.1107/S160053681102808X/su2287Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O5i | 0.91 (2) | 1.82 (2) | 2.730 (2) | 175 (2) |
| N2—H2⋯O5ii | 0.96 (2) | 1.75 (2) | 2.693 (2) | 168 (2) |
Symmetry codes: (i) 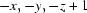 ; (ii)
; (ii) 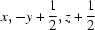 .
.
Acknowledgments
We acknowledge the CCD facility, set up under the IRHPA–DST program at the IISc, Bangalore. VV thanks the DST for financial assistance under the Fast-Track young scientist scheme, and RSR acknowledges the CSIR for funding under the scientist’s pool scheme.
supplementary crystallographic information
Comment
As a part of our interest in antimicrobial compounds, we have synthesized the title pyrazole derivative using the procedure described earlier by (Ragavan et al., 2009, and references therein; 2010, and references therein).
The molecular structure of the title molecule is shown in Fig 1. The methyl atom (C3B) of the 3-ethyl substituent lies out of the mean plane of the pyrazole moiety (N1,N2,C3-C5) by 1.366 (4) Å.
The crystal packing is a fine balance of strong N—H···O hydrogen bonds (Table 1) and salt bridges, which normally tend to promote the formation of a planar structure and compact packing (Shylaja et al., 2008). In the title compound all the hydrogen bonding donors, iminium N+H (N1) and amine NH (N2), and the O-(O1) acceptor, are in the plane of the pyrazole moiety, which would normally yield a planar hydrogen-bonded structure. However, in order to accommodate the out-of-plane methyl group, (C3B), an undulating hydrogen bonded sheet-like structure, lieing paralallel to (100), is formed (Fig. 2).
Experimental
The title compound was synthesized using the method described earlier by (Ragavan et al., 2009, 2010). It was crystallized using an ethanol-chloroform (1:1) mixture. Yield, 74%; m.p. 779-780 K.
Refinement
The NH atoms were located in a difference Fourier map and were freely refined: N2—H2 = 0.92 (2) Å and N1+—H1 = 0.95 (3) Å. The methylene and methyl hydrogen atoms were placed in calculated positions and refined as riding atoms: C-H = 0.97 and 0.96 Å, for CH and CH3 H-atoms, respectively, with Uiso(H) = k × Ueq(C,) where k = 1.5 for CH3 H-atoms and 1.2 for the CH H-atoms.
Figures
Fig. 1.

A view of the molecular structure of the title molecule, with labelling scheme and displacement ellipsoids drawn at the 30% probability level.
Fig. 2.

A view of the N—H···O hydrogen bonded (dashed cyan lines) sheet structure in the crystal structure of the title compound (see Table 1 for details).
Crystal data
| C6H10N2O | F(000) = 272 |
| Mr = 126.16 | Dx = 1.227 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 3015 reflections |
| a = 9.1299 (15) Å | θ = 2.4–22.9° |
| b = 7.1600 (11) Å | µ = 0.09 mm−1 |
| c = 11.374 (2) Å | T = 296 K |
| β = 113.232 (9)° | Plate, colourless |
| V = 683.2 (2) Å3 | 0.21 × 0.19 × 0.11 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 1332 independent reflections |
| Radiation source: fine-focus sealed tube | 961 reflections with I > 2σ(I) |
| graphite | Rint = 0.034 |
| φ and ω scans | θmax = 26.0°, θmin = 2.4° |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | h = −11→11 |
| Tmin = 0.64, Tmax = 0.83 | k = −8→8 |
| 12120 measured reflections | l = −13→13 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.049 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.136 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.0674P)2 + 0.2195P] where P = (Fo2 + 2Fc2)/3 |
| 1332 reflections | (Δ/σ)max < 0.001 |
| 92 parameters | Δρmax = 0.17 e Å−3 |
| 0 restraints | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.0756 (2) | 0.1929 (2) | 0.58699 (14) | 0.0427 (5) | |
| H1 | 0.029 (3) | 0.087 (3) | 0.601 (2) | 0.051 (6)* | |
| N2 | 0.1373 (2) | 0.3275 (2) | 0.67795 (15) | 0.0462 (5) | |
| H2 | 0.116 (3) | 0.326 (3) | 0.754 (2) | 0.062 (6)* | |
| O5 | 0.07244 (17) | 0.12652 (19) | 0.38750 (11) | 0.0489 (4) | |
| C3 | 0.2107 (2) | 0.4552 (3) | 0.63369 (17) | 0.0402 (5) | |
| C3A | 0.2903 (3) | 0.6189 (3) | 0.7141 (2) | 0.0573 (6) | |
| H3A1 | 0.2994 | 0.7177 | 0.6591 | 0.069* | |
| H3A2 | 0.2238 | 0.6648 | 0.7566 | 0.069* | |
| C3B | 0.4512 (4) | 0.5766 (4) | 0.8123 (3) | 0.1014 (12) | |
| H3B1 | 0.4424 | 0.4859 | 0.8714 | 0.152* | |
| H3B2 | 0.4982 | 0.6889 | 0.8577 | 0.152* | |
| H3B3 | 0.5171 | 0.5277 | 0.7714 | 0.152* | |
| C4 | 0.1999 (2) | 0.4015 (2) | 0.51474 (17) | 0.0367 (5) | |
| C4A | 0.2643 (3) | 0.4995 (3) | 0.4291 (2) | 0.0544 (6) | |
| H41 | 0.3131 | 0.6149 | 0.4681 | 0.082* | |
| H42 | 0.1789 | 0.5248 | 0.3482 | 0.082* | |
| H43 | 0.3422 | 0.4216 | 0.4162 | 0.082* | |
| C5 | 0.1135 (2) | 0.2330 (3) | 0.48611 (16) | 0.0359 (5) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0661 (11) | 0.0389 (9) | 0.0308 (8) | −0.0145 (8) | 0.0275 (8) | −0.0061 (7) |
| N2 | 0.0698 (12) | 0.0444 (10) | 0.0313 (9) | −0.0109 (8) | 0.0275 (8) | −0.0098 (7) |
| O5 | 0.0759 (10) | 0.0484 (8) | 0.0304 (7) | −0.0197 (7) | 0.0295 (7) | −0.0092 (6) |
| C3 | 0.0470 (11) | 0.0362 (10) | 0.0369 (10) | −0.0008 (8) | 0.0162 (9) | −0.0008 (8) |
| C3A | 0.0725 (15) | 0.0467 (12) | 0.0519 (13) | −0.0103 (11) | 0.0237 (12) | −0.0149 (10) |
| C3B | 0.084 (2) | 0.082 (2) | 0.097 (2) | −0.0121 (16) | −0.0077 (17) | −0.0341 (18) |
| C4 | 0.0428 (10) | 0.0361 (10) | 0.0322 (9) | −0.0018 (8) | 0.0159 (8) | 0.0016 (8) |
| C4A | 0.0632 (14) | 0.0566 (13) | 0.0485 (12) | −0.0143 (11) | 0.0274 (11) | 0.0035 (10) |
| C5 | 0.0455 (10) | 0.0378 (10) | 0.0266 (9) | −0.0018 (8) | 0.0165 (8) | 0.0003 (8) |
Geometric parameters (Å, °)
| N1—C5 | 1.354 (2) | C3A—H3A2 | 0.9700 |
| N1—N2 | 1.363 (2) | C3B—H3B1 | 0.9600 |
| N1—H1 | 0.92 (2) | C3B—H3B2 | 0.9600 |
| N2—C3 | 1.343 (3) | C3B—H3B3 | 0.9600 |
| N2—H2 | 0.95 (3) | C4—C5 | 1.408 (3) |
| O5—C5 | 1.284 (2) | C4—C4A | 1.495 (3) |
| C3—C4 | 1.372 (3) | C4A—H41 | 0.9600 |
| C3—C3A | 1.488 (3) | C4A—H42 | 0.9600 |
| C3A—C3B | 1.484 (4) | C4A—H43 | 0.9600 |
| C3A—H3A1 | 0.9700 | ||
| C5—N1—N2 | 109.01 (16) | H3B1—C3B—H3B2 | 109.5 |
| C5—N1—H1 | 128.2 (13) | C3A—C3B—H3B3 | 109.5 |
| N2—N1—H1 | 122.3 (13) | H3B1—C3B—H3B3 | 109.5 |
| C3—N2—N1 | 108.38 (16) | H3B2—C3B—H3B3 | 109.5 |
| C3—N2—H2 | 130.8 (14) | C3—C4—C5 | 106.50 (16) |
| N1—N2—H2 | 120.5 (14) | C3—C4—C4A | 128.08 (17) |
| N2—C3—C4 | 109.04 (16) | C5—C4—C4A | 125.42 (17) |
| N2—C3—C3A | 120.03 (18) | C4—C4A—H41 | 109.5 |
| C4—C3—C3A | 130.90 (18) | C4—C4A—H42 | 109.5 |
| C3B—C3A—C3 | 113.6 (2) | H41—C4A—H42 | 109.5 |
| C3B—C3A—H3A1 | 108.8 | C4—C4A—H43 | 109.5 |
| C3—C3A—H3A1 | 108.8 | H41—C4A—H43 | 109.5 |
| C3B—C3A—H3A2 | 108.8 | H42—C4A—H43 | 109.5 |
| C3—C3A—H3A2 | 108.8 | O5—C5—N1 | 122.03 (16) |
| H3A1—C3A—H3A2 | 107.7 | O5—C5—C4 | 130.92 (17) |
| C3A—C3B—H3B1 | 109.5 | N1—C5—C4 | 107.05 (15) |
| C3A—C3B—H3B2 | 109.5 | ||
| C5—N1—N2—C3 | 1.6 (2) | C3A—C3—C4—C4A | −1.7 (3) |
| N1—N2—C3—C4 | −1.4 (2) | N2—N1—C5—O5 | 178.68 (17) |
| N1—N2—C3—C3A | −179.69 (17) | N2—N1—C5—C4 | −1.2 (2) |
| N2—C3—C3A—C3B | 80.6 (3) | C3—C4—C5—O5 | −179.5 (2) |
| C4—C3—C3A—C3B | −97.3 (3) | C4A—C4—C5—O5 | 0.9 (3) |
| N2—C3—C4—C5 | 0.7 (2) | C3—C4—C5—N1 | 0.3 (2) |
| C3A—C3—C4—C5 | 178.7 (2) | C4A—C4—C5—N1 | −179.25 (18) |
| N2—C3—C4—C4A | −179.80 (19) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O5i | 0.91 (2) | 1.82 (2) | 2.730 (2) | 175 (2) |
| N2—H2···O5ii | 0.96 (2) | 1.75 (2) | 2.693 (2) | 168 (2) |
Symmetry codes: (i) −x, −y, −z+1; (ii) x, −y+1/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: SU2287).
References
- Bruker (2001). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2007). APEX2 and SAINT-Plus Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Ragavan, R. V., Vijayakumar, V. & Kumari, N. S. (2009). Eur. J. Med. Chem. 44, 3852–3857.
- Ragavan, R. V., Vijayakumar, V. & Kumari, N. S. (2010). Eur. J. Med. Chem. 45, 1173–1180. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shylaja, S., Mahendra, K. N., Varma, K. B. R., Narasimhamurthy, T. & Rathore, R. S. (2008). Acta Cryst. C64, o361–o363. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148-155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S160053681102808X/su2287sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S160053681102808X/su2287Isup2.hkl
Supplementary material file. DOI: 10.1107/S160053681102808X/su2287Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report