

Abstract
Periapical granulomas are induced by bacterial infection of the dental pulp and result in destruction of the surrounding alveolar bone. In previous studies we have reported that the bone resorption in this model is primarily mediated by macrophage-expressed interleukin-1 (IL-1). The expression and activity of IL-1 is in turn modulated by a network of Th1 and Th2 regulatory cytokines. In the present study, the functional roles of the Th1 cytokine gamma interferon (IFN-γ) and IFN-γ-inducing cytokines IL-12 and IL-18 were determined in a murine model of periapical bone destruction. IL-12−/−, IL-18−/−, and IFN-γ−/− mice were subjected to surgical pulp exposure and infection with a mixture of four endodontic pathogens, and bone destruction was determined by microcomputed tomography on day 21. The results indicated that all IL-12−/−, IL-18−/−, and IFN-γ−/− mice had similar infection-stimulated bone resorption in vivo as wild-type control mice. Mice infused with recombinant IL-12 also had resorption similar to controls. IFN-γ−/− mice exhibited significant elevations in IL-6, IL-10, IL-12, and tumor necrosis factor alpha in lesions compared to wild-type mice, but these modulations had no net effect on IL-1α levels. Recombinant IL-12, IL-18, and IFN-γ individually failed to consistently modulate macrophage IL-1α production in vitro. We conclude that, at least individually, endogenous IL-12, IL-18, and IFN-γ do not have a significant effect on the pathogenesis of infection-stimulated bone resorption in vivo, suggesting possible functional redundancy in proinflammatory pathways.
The periapical granuloma is an inflammatory and immune response that is elicited by anaerobic infection of the dental pulp as a consequence of caries, tooth fracture, and traumatic operative dental procedures. This inflammatory process ultimately results in destruction of the alveolar bone surrounding the tooth.
Interleukin-1 (IL-1) is a potent bone-resorptive cytokine that is strongly up-regulated following pulpal infection (20). IL-1 mRNA and protein are markedly increased in infiltrating macrophages and polymorphonuclear leukocytes (23), and IL-1 levels generally correlate with the extent of bone destruction (15, 22). Most bone-resorptive activity present in periapical granulomas is neutralized by anti-IL-1 antibodies, and bone resorption is significantly inhibited in vivo with IL-1 receptor antagonist (16, 22).
Both proinflammatory Th1 and antiinflammatory Th2 cytokines are also induced by pulpal infection and may modulate IL-1 expression and activity by macrophages (10). Th1 cytokines (IL-12 and gamma interferon [IFN-γ]) and bone-resorptive cytokines (IL-1 and tumor necrosis factor alpha [TNF-α]) were up-regulated in a linear fashion over 4 weeks following infection, and resorptive cytokines were positively correlated with Th1 cytokine expression. In contrast, Th2 cytokines exhibited increased expression up to 2 weeks after infection, with a decline in levels thereafter. These data suggest that Th1 cytokine-mediated proinflammatory pathways generally predominate in inflammatory bone lesions and are therefore expected to potentiate inflammation and bone resorption adjacent to sites of infection.
At the same time, the central Th1 cytokine IFN-γ as well as the IFN-γ-inducing cytokines IL-12 and IL-18 also possess osteoclast-inhibitory properties, at least in vitro. These three cytokines have been reported to directly or indirectly reduce osteoclast differentiation and bone resorption (7, 8, 18, 21, 25). As a consequence of these opposing activities, the overall role of Th1 cytokines in inflammatory bone resorption in vivo is difficult to predict.
In this study, we therefore determined the individual function of IL-12, IL-18, and IFN-γ in the pathogenesis of infection-stimulated bone destruction, using a well-established in vivo periapical lesion model and appropriate knockout mice. The modulatory effect of these three cytokines on endodontic pathogen-stimulated IL-1 expression by macrophages was also assessed in vitro.
MATERIALS AND METHODS
Animals.
IL-12 knockout (IL-12−/−), IL-18−/−, and IFN-γ−/− animals were used in this study. IL-12−/− (C57BL/6 background) mice were purchased from Jackson Laboratory, Bar Harbor, Maine. IL-18−/− mice (C57BL/6 background) (19) were kindly provided by S. Akira. IFN-γ−/− mice (BALB/c background) (5) were the generous gift of F. W. van Ginkel and J. R. McGhee, Department of Microbiology, University of Alabama, Birmingham, Ala. IL-18−/− and IFN-γ−/− mice were bred in the animal facility of the Forsyth Institute, and all animals were maintained under pathogen-free conditions. Age-matched C57BL/6J and BALB/cJ mice were purchased from Jackson Laboratory and were used as wild-type controls.
Induction of periapical lesions.
Infection-stimulated periapical bone destruction was induced following the regimen previously described (2, 10, 15). In brief, mice were anesthetized with ketamine HCl (62.5 mg/kg of body weight) and xylazine (12.5 mg/kg). Mandibular first molar pulps were exposed using a variable-speed electric dental handpiece (Osada Electric, Los Angeles, Calif.) with a no. 1/4 dental round bur, under a surgical microscope (MC-M92; Seiler, St. Louis, Mo.). The exposure site was approximately 1.5 times the diameter of the bur.
Exposed pulps were infected with a mixture of four common endodontic pathogens, Prevotella intermedia (ATCC 25611; American Type Culture Collection [ATCC], Manassas, Va.), Fusobacterium nucleatum (ATCC 25586), Peptostreptococcus micros (ATCC 33270), and Streptococcus intermedius (ATCC 27335). A bacterial mixture containing approximately 1010 cells of each bacterial species/ml in methylcellulose (10 mg/ml) was prepared as previously described (15). At the time of pulp exposure (day zero), exposed dental pulps were directly inoculated with 10 μl of the bacterial mixture. Mice without pulp exposures and infection were established as negative controls.
rIL-12 infusion.
Animals were continuously infused with recombinant mouse IL-12 (rIL-12; generous gift of the Genetics Institute, Cambridge, Mass.), 0.3 μg (approximately 710 U)/mouse/day in sterile phosphate-buffered saline (PBS), using a microosmotic pump (model 1007D; ALZET Corporation, Palo Alto, Calif.) (17). The osmotic pump was subcutaneously implanted on day zero just after surgical pulp exposure and was replaced on days 7 and 14 under general anesthesia. Infusions of sterile PBS served as a negative control.
Tissue sample preparation.
Animals were killed on day 21 after pulp exposure, and mandibles were isolated. After removal of soft tissue, one hemi-mandible was fixed in fresh 4% paraformaldehyde in PBS for microcomputed tomography (micro-CT). The periapical tissues surrounding the mesial and distal root apices were carefully extracted from the other hemi-mandible, together with surrounding bone in a block specimen under a surgical microscope for cytokine enzyme-linked immunosorbent assays (ELISAs). Periapical tissues were rinsed in cold PBS, freed of clots, weighed, and immediately frozen at −70°C.
Micro-CT analysis.
Hemi-mandibles were scanned using a compact fan-beam-type tomograph (Micro-CT 20; Scanco Medical AG, Bassersdorf, Switzerland) providing a 17-μm nominal resolution as previously described (1). The most centrally located section that included the distal root of the mandibular first molar and its root canal apex was chosen from the stack of images for measurement. The cross-sectional area of periapical lesions was defined using Adobe Photoshop 5.5 (Adobe Systems, San Jose, Calif.) and measured with NIH Image 1.62 (Wayne Rasband, National Institutes of Health, Bethesda, Md.). Results were expressed as square millimeters of resorbed area.
Protein extraction and cytokine ELISA.
For protein extraction, frozen periapical tissue samples were ground using a precooled sterile mortar and pestle, and the tissue fragments were dispersed in 1 ml of lysis buffer as previously described (10, 15). The mixture was incubated at 4°C for 1 h, and the supernatant was collected after centrifugation and stored at −70°C until assay.
For the IL-1α assay, a mouse capture-ELISA system was used (detection range, 7.8 to 500 pg/ml). In brief, 100 μl of sample with or without dilution was added to immunomodules (Nunc, Naperville, Ill.) coated with mouse IL-1α capture antibody (R&D Systems, Minneapolis, Minn.). Captured IL-1α was detected with 100 μl of biotinylated mouse IL-1α-specific polyclonal antibody (R&D Systems), which was reacted with 100 μl of streptavidin-horseradish peroxidase (Zymed, San Francisco, Calif.). One hundred microliters of substrate solution (R&D Systems) was added and incubated for 1 h, and the reaction was stopped by adding 50 μl of 1 N H2SO4. The optical density of the reaction was read using an ELISA plate reader at 450 nm. Standards for the assay were prepared with rIL-1α (R&D Systems).
ELISAs for other mediators in extracts employed commercially available kits (all from BioSource International, Camarillo, Calif.) as follows: IL-4 (sensitivity, >5 pg/ml), IL-6 (>3 pg/ml), IL-10 (>13 pg/ml), IL-12 (>2 pg/ml), IFN-γ (>1 pg/ml) and TNF-α (>3 pg/ml). All assays using commercial kits were carried out according to the manufacturer's instructions. Results were expressed as picograms of cytokine per milligram of periapical tissue.
Macrophage cultures.
Resident unstimulated peritoneal macrophages were isolated from IL-12−/−, IL-18−/−, and IFN-γ−/− mice following the regimen previously described (15). In brief, a total of 5 ml of cold culture medium, consisting of RPMI 1640 (BioWhittaker, Walkersville, Md.) supplemented with l-glutamine (2 mM), 1% penicillin-streptomycin (Invitrogen, Carlsbad, Calif.), and 10% heat-inactivated fetal bovine serum (Sigma, St. Louis, Mo.), was injected into the peritoneal cavity and collected with constituent peritoneal cells under sterile conditions. After washing three times with cold medium, the cells were resuspended at 106 cells/ml. Aliquots (160 μl) were dispensed into 96-well culture plates (Corning Inc., Corning, N.Y.) and incubated for 2 h at 37°C in an atmosphere of 5% CO2-95% air. Nonadherent cells were removed by washing three times with warm medium, and adherent cells (>90% macrophages) were subjected to stimulation experiments.
Isolated macrophages were preincubated with or without regulatory cytokines for 1 h. Regulatory cytokines included rIL-12 (Genetics Institute), rIL-18 (Medical & Biological Laboratories, Nagoya, Japan), and rIFN-γ (R&D Systems). Cytokines were used at 0 (control), 0.1, 1, and 10 U/ml. Following preincubation, macrophages were stimulated with each bacterial pathogen for 24 h in 37°C in an atmosphere of 5% CO2-95% air. The four pathogens described above were grown and prepared as previously described, fixed in 0.5% Formol-PBS, and used at 1.6 × 106 cells/well (15). Escherichia coli lipopolysaccharide (LPS; serotype 026:B6; Sigma) was used as a positive control (160 ng/well). After 24 h of stimulation, the culture supernatants were collected and stored at −70°C until assay.
Statistical analysis.
Effects of genotype and pulp exposure on bone resorption and cytokine expression were compared using a two-way fractorial analysis of variance. Differences in IL-1α production stimulated by pathogens in vitro were analyzed by Dunnett's two-tailed t test and Duncan's multiple range test.
RESULTS
Effect of IL-12, IL-18, and IFN-γ on periapical bone destruction.
As noted, IL-12, IL-18, and IFN-γ exert opposing regulatory functions with respect to inflammatory bone destruction, i.e., they up-regulate inflammatory responses but have also been reported to inhibit osteoclast formation. We therefore determined the functional role of these three cytokines in infection-stimulated bone destruction in vivo. Experimental periapical lesions were induced by surgical pulp exposure and infection of the mandibular first molars of groups of IL-12−/−, IL-18−/−, IFN-γ−/−, and appropriate wild-type mice in three separate experiments (background strains, C57BL/6J for IL-12−/− and IL-18−/− and BALB/cJ for IFN-γ−/− animals). The teeth of negative controls of all groups remained uninfected. After 21 days, the extent of periapical bone resorption was determined by micro-CT (1, 2, 15). As shown in Fig. 1, unexposed and uninfected controls exhibited a minimal area of radiolucency surrounding the distal root, representing the intact periodontal ligament space that anchors teeth to the mandible. In contrast, all exposed and infected animals showed significant increases in periapical bone resorption compared to uninfected controls. Importantly, infected IL-12−/−, IL-18−/−, and IFN-γ−/− mice all had periapical bone resorption that was similar in extent to their respective wild-type infected controls.
FIG. 1.

Effect of Th1 cytokine deficiency on periapical bone destruction. Hemi-mandibles were scanned by micro-CT on day 21, and the area of the normal periodontal ligament space or infection-stimulated bone resorption was measured. Open columns, normal periodontal ligament space; closed columns, periapical bone destruction; vertical bars, standard deviations. No significant differences were present in knockout mice versus wild-type controls.
We also attempted to modulate periapical resorption by infusion of rIL-12, which is known to enhance Th1-type responses in vivo. As shown in Fig. 2, rIL-12 failed to up-regulate bone resorption compared to PBS. Taken together, these data indicate that endogenous IL-12, IL-18, and IFN-γ do not exert a major effect upon the pathogenesis of infection-stimulated bone destruction, at least individually, suggesting that there may be considerable functional redundancy among these cytokines.
FIG. 2.

Effect of IL-12 infusion on periapical bone destruction. The area of the normal periodontal ligament space or infection-stimulated bone resorption was determined by micro-CT. Open column, uninfected control with normal periodontal ligament space; closed columns, periapical bone destruction; vertical bars, standard deviations. No significant difference was observed between the IL-12- and PBS-infused groups.
Cytokine responses in inflammatory lesions.
IL-1α has previously been shown to be a primary mediator of bone resorption in this model (22). In addition, the elimination of either IL-10 or IL-6 potently increases IL-1α production in periapical lesions in vivo, which correlates with increased bone destruction (2, 15). Therefore, the levels of IL-1α and other regulatory cytokines in inflammatory periapical tissues were assessed by ELISA.
As shown in Fig. 3, bone-resorptive cytokines were up-regulated in lesions after pulp exposure and infection compared to uninfected controls. In IFN-γ−/− mice, the major bone-resorptive cytokine, IL-1α, was unchanged, whereas less-potent resorptive mediators, TNF-α and IL-6, were significantly increased (P < 0.05). IL-12 and IL-10 production was also increased (P < 0.05). IL-18 deficiency did not affect bone-resorptive cytokine expression in the periapical tissues. Both IFN-γ and IL-10 production were significantly increased in IL-18−/− mice (P < 0.05). Thus, although there was a significant modulation of IFN-γ, IFN-γ-inducing cytokines, and Th2 cytokines in IL-18−/− and IFN-γ−/− mice, the expression of IL-1α, thought to be the primary mediator of periapical bone destruction, was relatively unaffected.
FIG. 3.

Profile of cytokine expression in periapical inflammatory lesions. Cytokine concentrations in inflammatory tissues were determined by ELISA on day 21, and values were normalized to the weight of periapical tissue. Vertical bar, standard deviation by two-way fractorial analysis of variance. *, P < 0.05 for exposure effect; †, P < 0.05 for genotype effect.
Modulation of IL-1α production by IL-12, IL-18, and IFN-γ in vitro.
The ability of IL-12, IL-18, and IFN-γ to modulate macrophage IL-1α production in vitro was determined using resident peritoneal macrophages as previously described (15). As shown in Fig. 4, the effects of rIL-12, rIL-18, and rIFN-γ on macrophage IL-1 production in response to all pathogens were somewhat inconsistent, although IFN-γ clearly up-regulated IL-1α production at the lowest concentration tested (0.1 U/ml; P < 0.01 in response to LPS and P. intermedia). These data suggest that macrophage IL-1α production appears to be IL-12-, IL-18-, and IFN-γ independent, at least when these cytokines are assessed separately.
FIG. 4.

Modulation of pathogen-stimulated macrophage IL-1α production by Th1 cytokines. Resident peritoneal macrophages were harvested from IL-12−/−, IL-18−/−, and IFNγ−/− mice and challenged with bacterial pathogens in the presence or absence of recombinant Th1 cytokines. IL-1α concentrations were determined by ELISA, and values were normalized to the volume of culture supernatant. Each plot indicates the bacterial stimulant, and the x axis indicates the modulating cytokine. Vertical bar, standard deviation. Statistical differences were determined by Dunnett's two-tailed t test versus controls preincubated without cytokines. *, P < 0.01.
DISCUSSION
Anaerobic infections of the dental pulp result in infrabony inflammation, cytokine expression, and resorption of the surrounding periapical bone. Macrophages are prominent in the cellular infiltrate early after pulpal infection and have been shown to express IL-12 and IL-18, as well as bone-resorptive cytokines IL-1 and TNF-α (10, 13). IL-12 induces Th1 cell differentiation from naive T cells and their subsequent IFN-γ production (24). Although IL-18 alone cannot induce Th1 cell commitment, IL-18 synergizes with IL-12 (11). IL-12 and IL-18 also induce IFN-γ production in NK cells (4, 13). IFN-γ activates macrophages, reduces macrophage-suppressive activity, and induces IL-1, NO synthesis, and 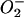 (3, 12, 14). There is thus strong evidence that Th1-mediated pathways may be involved in increasing inflammation following infection.
(3, 12, 14). There is thus strong evidence that Th1-mediated pathways may be involved in increasing inflammation following infection.
In the present study, we tested the hypothesis that the central Th1 cytokine, IFN-γ, and the IFN-γ-inducing cytokines, IL-12 and IL-18, enhance periapical inflammation and bone resorption in vivo. Somewhat surprisingly, our findings demonstrated that IL-12−/−, IL-18−/−, and IFN-γ−/− mice did not have significantly reduced infection-stimulated bone resorption compared to wild-type animals. Furthermore, rIL-12-infused mice failed to significantly up-regulate bone destruction compared to control mice. The local level of IL-1 in inflammatory lesions, previously found to be a primary mediator of infection-stimulated bone resorption (22, 23), was significantly lower only in IL-12−/− mice (data not shown). Taken together, these results suggest that, at least individually, endogenous IL-12, IL-18, and IFN-γ do not appear to have a major, nonredundant effect on the pathogenesis of infection-stimulated bone resorption in vivo.
There are several possible explanations for these findings. First, it is possible that the levels of these cytokines that are expressed within lesions (Fig. 3) are too low to have a regulatory effect on resorption. However, in a previous study (10) our investigators found that IFN-γ and IL-12 were induced at similar levels by infection, as observed in the present study (≤1 pg/mg of tissue), as was the Th2 cytokine IL-10 (1 to 1.5 pg/mg). Despite this modest induction, and in contrast to IFN-γ and IL-12, endogenous IL-10 has a profound effect in inhibiting bone resorption in this model, i.e., IL-10 knockout mice have a three to five times greater lesion size than wild-type controls (15). Thus, low levels of T-cell regulatory cytokines clearly can have dramatic effects on inflammatory resorption in vivo.
Alternatively, in addition to their proinflammatory effects, IL-12, IL-18, and IFN-γ also suppress the receptor-activator of NF-κB ligand (RANKL)-induced osteoclast differentiation and mature osteoclast function in vitro. The effect of IFN-γ is exerted at picomolar concentrations (6) and involves the accelerated degradation of the RANK adaptor TNF receptor-associated kinase 6 (18). Although IL-12 does not act directly on osteoclast precursors, it indirectly reduces RANKL-induced osteoclast differentiation alone or in synergy with IL-18 (7). IL-18, which may also be produced by osteoblasts, enhances granulocyte-macrophage colony-stimulating factor production by T cells (21). Granulocyte-macrophage colony-stimulating factor signaling in osteoclast precursors inhibits RANKL-induced osteoclast differentiation in an IFN-γ-independent manner (8, 21). It is therefore possible that the relative lack of effect seen in deficiencies of IL-12, IL-18, or IFN-γ on inflammatory resorption may reflect the balance of two opposing processes: the loss of osteoclast inhibitory activities versus the reduction in proinflammatory signals.
A third possibility is that the functional redundancy of these mediators may obscure their individual roles in inflammatory bone loss. Although the differences were not statistically significant, a small decrease (5 to 10%) in the extent of resorption was in fact seen in all three cytokine-deficient strains (Fig. 1). This consistent trend suggests that a real biological effect might be exerted in vivo but that very large numbers of animals and/or a deficiency of two or even all three mediators might be required to observe a significant reduction in bone resorption. A direct test of this possibility will require the generation of double or triple cytokine knockout animals. In addition, there are direct pathways, particularly those mediated by Toll-like receptors, which activate macrophage proinflammatory cytokine expression independent of Th1 cytokines. In this regard, our investigators have found that mice deficient in Toll-like receptor-4 have reduced expression of IL-1 and TNF-α, and they also have less bone resorption than wild-type animals following anaerobic infection (9).
Although the background strains were identical in the IL-12 and IL-18 experiments (C57BL/6) (Fig. 1), a larger absolute resorptive response was observed in the IL-18 mice. It should be noted that these represent independent experiments and that similar interexperimental variability in the size of lesions in infected teeth has been observed previously (H. Sasaki, unpublished data), emphasizing the need for an appropriate wild-type control assessed concurrently. A likely explanation is that such differences in lesion size are related to the viability and/or activity of the bacterial inoculum used, perhaps being in log phase of growth in studies in which larger lesions are induced.
These results with IFN-γ and its inducing cytokines are in sharp contrast to our previous findings with Th2 cytokines, several of which are potent suppressors of inflammatory resorption as noted above. Both IL-10-deficient and IL-6-deficient mice exhibited up-regulated local IL-1 production and increased infection-stimulated bone resorption in vivo compared to the wild type (2, 15). In the case of IL-10 deficiency, the increase in locally produced IL-1α production was dramatic (>10-fold) and correlated with severe bone resorption. These Th2 mediators thus play significant and nonredundant roles in suppressing bone inflammation and resorption in vivo. Interestingly, another Th2 cytokine, IL-4, had no effect on resorption in this model, indicating functional heterogeneity among this cytokine group (15). Taken together, these findings suggest that Th2 cytokines may represent more attractive targets for immunomodulation than Th1 and Th1-cytokine-inducing cytokines in preventing inflammatory bone loss.
Acknowledgments
We thank Ralph Kent, Jr. for his assistance in statistical analyses and Ralph Müller and Thomas Kohler for their assistance in micro-CT scanning.
This work was supported by grants DE-09018 and DE-13747 from the National Institutes of Health.
REFERENCES
- 1.Balto, K., R. Müller, D. C. Carrington, J. Dobeck, and P. Stashenko. 2000. Quantification of periapical bone destruction in mice by micro-computed tomography. J. Dent. Res. 79:35-40. [DOI] [PubMed] [Google Scholar]
- 2.Balto, K., H. Sasaki, and P. Stashenko. 2001. Interleukin-6 deficiency increases inflammatory bone destruction. Infect. Immun. 69:744-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boraschi, D., S. Censini, and A. Tagliabue. 1984. Interferon-gamma reduces macrophage-suppressive activity by inhibiting prostaglandin E2 release and inducing interleukin 1 production. J. Immunol. 133:764-768. [PubMed] [Google Scholar]
- 4.Chan, S. H., B. Perussia, J. W. Gupta, M. Kobayashi, M. Pospisil, H. A. Young, S. F. Wolf, D. Young, S. C. Clark, and G. Trinchieri. 1991. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J. Exp. Med. 173:869-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalton, D. K., S. Pitts-Meek, S. Keshav, I. S. Figari, A. Bradley, and T. A. Stewart. 1993. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 259:1739-1742. [DOI] [PubMed] [Google Scholar]
- 6.Gowen, M., G. E. Nedwin, and G. R. Mundy. 1986. Preferential inhibition of cytokine-stimulated bone resorption by recombinant interferon gamma. J. Bone Miner. Res. 1:469-474. [DOI] [PubMed] [Google Scholar]
- 7.Horwood, N. J., J. Elliott, T. J. Martin, and M. T. Gillespie. 2001. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J. Immunol. 166:4915-4921. [DOI] [PubMed] [Google Scholar]
- 8.Horwood, N. J., N. Udagawa, J. Elliott, D. Grail, H. Okamura, M. Kurimoto, A. R. Dunn, T. Martin, and M. T. Gillespie. 1998. Interleukin 18 inhibits osteoclast formation via T cell production of granulocyte macrophage colony-stimulating factor. J. Clin. Investig. 101:595-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou, L., H. Sasaki, and P. Stashenko. 2000. Toll-like receptor 4-deficient mice have reduced bone destruction following mixed anaerobic infection. Infect. Immun. 68:4681-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawashima, N., and P. Stashenko. 1999. Expression of bone-resorptive and regulatory cytokines in murine periapical inflammation. Arch. Oral Biol. 44:55-66. [DOI] [PubMed] [Google Scholar]
- 11.Kohno, K., J. Kataoka, T. Ohtsuki, Y. Suemoto, I. Okamoto, M. Usui, M. Ikeda, and M. Kurimoto. 1997. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J. Immunol. 158:1541-1550. [PubMed] [Google Scholar]
- 12.Nathan, C. F., H. W. Murray, M. E. Wiebe, and B. Y. Rubin. 1983. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 158:670-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamura, H., S. Kashiwamura, H. Tsutsui, T. Yoshimoto, and K. Nakanishi. 1998. Regulation of interferon-gamma production by IL-12 and IL-18. Curr. Opin. Immunol. 10:259-264. [DOI] [PubMed] [Google Scholar]
- 14.Pellat, C., Y. Henry, and J. C. Drapier. 1990. IFN-gamma-activated macrophages: detection by electron paramagnetic resonance of complexes between L-arginine-derived nitric oxide and non-heme iron proteins. Biochem. Biophys. Res. Commun. 166:119-125. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki, H., L. Hou, A. Belani, C. Y. Wang, T. Uchiyama, R. Müller, and P. Stashenko. 2000. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J. Immunol. 165:3626-3630. [DOI] [PubMed] [Google Scholar]
- 16.Stashenko, P., C. Y. Wang, N. Tani-Ishii, and S. M. Yu. 1994. Pathogenesis of induced rat periapical lesions. Oral Surg. Oral Med. Oral Pathol. 78:494-502. [DOI] [PubMed] [Google Scholar]
- 17.Takatsuki, F., A. Okano, C. Suzuki, Y. Miyasaka, T. Hirano, T. Kishimoto, D. Ejima, and Y. Akiyama. 1990. Interleukin 6 perfusion stimulates reconstitution of the immune and hematopoietic systems after 5-fluorouracil treatment. Cancer Res. 50:2885-2890. [PubMed] [Google Scholar]
- 18.Takayanagi, H., K. Ogasawara, S. Hida, T. Chiba, S. Murata, K. Sato, A. Takaoka, T. Yokochi, H. Oda, K. Tanaka, K. Nakamura, and T. Taniguchi. 2000. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408:600-605. [DOI] [PubMed] [Google Scholar]
- 19.Takeda, K., H. Tsutsui, T. Yoshimoto, O. Adachi, N. Yoshida, T. Kishimoto, H. Okamura, K. Nakanishi, and S. Akira. 1998. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8:383-390. [DOI] [PubMed] [Google Scholar]
- 20.Tani-Ishii, N., C. Y. Wang, and P. Stashenko. 1995. Immunolocalization of bone-resorptive cytokines in rat pulp and periapical lesions following surgical pulp exposure. Oral Microbiol. Immunol. 10:213-219. [DOI] [PubMed] [Google Scholar]
- 21.Udagawa, N., N. J. Horwood, J. Elliott, A. Mackay, J. Owens, H. Okamura, M. Kurimoto, T. J. Chambers, T. J. Martin, and M. T. Gillespie. 1997. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. J. Exp. Med. 185:1005-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang, C. Y., and P. Stashenko. 1993. The role of interleukin-1 alpha in the pathogenesis of periapical bone destruction in a rat model system. Oral Microbiol. Immunol. 8:50-56. [DOI] [PubMed] [Google Scholar]
- 23.Wang, C. Y., N. Tani-Ishii, and P. Stashenko. 1997. Bone-resorptive cytokine gene expression in periapical lesions in the rat. Oral Microbiol. Immunol. 12:65-71. [DOI] [PubMed] [Google Scholar]
- 24.Wu, C. Y., C. Demeure, M. Kiniwa, M. Gately, and G. Delespesse. 1993. IL-12 induces the production of IFN-gamma by neonatal human CD4 T cells. J. Immunol. 151:1938-1949. [PubMed] [Google Scholar]
- 25.Yamada, N., S. Niwa, T. Tsujimura, T. Iwasaki, A. Sugihara, H. Futani, S. Hayashi, H. Okamura, H. Akedo, and N. Terada. 2002. Interleukin-18 and interleukin-12 synergistically inhibit osteoclastic bone-resorbing activity. Bone 30:901-908. [DOI] [PubMed] [Google Scholar]