

Abstract
In the molecule of the title compound, C14H14ClNO2S, the N—H bond is in a syn position with respect to the meta-Cl atom in the aniline ring. The molecule is twisted about the N—S bond with a C—SO2—NH—C torsion angle of 44.55 (17)°. The two aromatic rings are inclined relative to each other by 66.2 (1)°. In the crystal, N—H⋯O hydrogen bonds link the molecules into infinite chains parallel to the b axis.
Related literature
For hydrogen bonding modes of sulfonamides, see: Adsmond & Grant (2001 ▶). For studies on the effects of substituents on the structures and other aspects of N-(aryl)-amides, see: Arjunan et al. (2004 ▶); Gowda et al. (1999 ▶), on N-(aryl)methanesulfonamides, see: Gowda et al. (2007 ▶) and on N-(aryl)arylsulfonamides, see: Gelbrich et al. (2007 ▶); Gowda et al. (2010 ▶); Perlovich et al. (2006 ▶). For the preparation of the title compound, See: Savitha & Gowda (2006 ▶)
Experimental
Crystal data
C14H14ClNO2S
M r = 295.77
Monoclinic,

a = 9.0385 (7) Å
b = 9.7830 (7) Å
c = 15.897 (1) Å
β = 97.284 (7)°
V = 1394.33 (17) Å3
Z = 4
Mo Kα radiation
μ = 0.42 mm−1
T = 293 K
0.44 × 0.40 × 0.32 mm
Data collection
Oxford Diffraction Xcalibur diffractometer with a Sapphire CCD detector
Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009 ▶) T min = 0.837, T max = 0.877
5245 measured reflections
2841 independent reflections
2438 reflections with I > 2σ(I)
R int = 0.009
Refinement
R[F 2 > 2σ(F 2)] = 0.036
wR(F 2) = 0.103
S = 1.06
2841 reflections
177 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.28 e Å−3
Δρmin = −0.36 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2009 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2009 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2009 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811029400/bt5582sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811029400/bt5582Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811029400/bt5582Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N⋯O1i | 0.84 (2) | 2.32 (2) | 3.117 (2) | 158 (2) |
Symmetry code: (i) 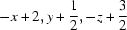 .
.
Acknowledgments
KS thanks the University Grants Commission, Government of India, New Delhi, for the award of a research fellowship under its faculty improvement program.
supplementary crystallographic information
Comment
The amide and sulfonamide moieties are the constituents of many biologically important compounds. The hydrogen bonding preferences of sulfonamides have been investigated (Adsmond & Grant, 2001). As part of our work on the substituent effects on the structures and other aspects of N-(aryl)-amides (Arjunan et al., 2004; Gowda et al., 1999), N-(aryl)-methanesulfonamides (Gowda et al., 2007) and N-(aryl)-arylsulfonamides (Gowda et al., 2010), in the present work, the crystal structure of N-(3-chlorophenyl)-2,4-dimethylbenzene- sulfonamide (I) has been determined (Fig. 1). The conformation of the N—C bond in the C—SO2—NH—C segment has gauche torsions with respect to the S═O bonds. The molecule is bent at the S atom with the C1—SO2—NH—C7 torsion angle of 44.6 (2)°, compared to the values of -54.9 (2)° in N-(2-chlorophenyl)-2,4-dimethylbenzenesulfonamide (II) (Gowda et al., 2010). Further, the conformation of the N—H bond in (I) is syn to the meta-chloro group in the anilino ring.
The two benzene rings in (I) are tilted relative to each other by 75.7 (1)°, compared to the value of 66.2 (1)° (II). The other bond parameters in (I) are similar to those observed in (II) and other aryl sulfonamides (Perlovich et al., 2006; Gelbrich et al., 2007).
The crystal packing of molecules in (I) via N—H···O(S) hydrogen bonds (Table 1) is shown in Fig.2.
Experimental
The solution of 1,3-xylene (1,3-dimethylbenzene) (10 ml) in chloroform (40 ml) was treated dropwise with chlorosulfonic acid (25 ml) at 0 ° C. After the initial evolution of hydrogen chloride subsided, the reaction mixture was brought to room temperature and poured into crushed ice in a beaker. The chloroform layer was separated, washed with cold water and allowed to evaporate slowly. The residual 2,4-dimethylbenzenesulfonylchloride was treated with 3-chloroaniline in the stoichiometric ratio and boiled for ten minutes. The reaction mixture was then cooled to room temperature and added to ice cold water (100 cc). The resultant solid N-(3-chlorophenyl)-2,4-dimethylbenzenesulfonamide was filtered under suction and washed thoroughly with cold water. It was then recrystallized to constant melting point from dilute ethanol. The purity of the compound was checked and characterized by recording its infrared and NMR spectra (Savitha & Gowda, 2006).
The prism like colourless single crystals used in X-ray diffraction studies were grown in ethanolic solution by a slow evaporation at room temperature.
Refinement
The H atom of the NH group was located in a difference map and later restrained to the distance N—H = 0.86 (2) Å. The other H atoms were positioned with idealized geometry using a riding model with the aromatic C—H = 0.93Å and methyl C—H = 0.96 Å, All H atoms were refined with isotropic displacement parameters set to 1.2 times of the Ueq of the parent atom.
Figures
Fig. 1.

Molecular structure of (I), showing the atom labelling scheme and displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

Molecular packing of (I) with hydrogen bonding shown as dashed lines.
Crystal data
| C14H14ClNO2S | F(000) = 616 |
| Mr = 295.77 | Dx = 1.409 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 2954 reflections |
| a = 9.0385 (7) Å | θ = 2.6–27.8° |
| b = 9.7830 (7) Å | µ = 0.42 mm−1 |
| c = 15.897 (1) Å | T = 293 K |
| β = 97.284 (7)° | Prism, colourless |
| V = 1394.33 (17) Å3 | 0.44 × 0.40 × 0.32 mm |
| Z = 4 |
Data collection
| Oxford Diffraction Xcalibur diffractometer with a Sapphire CCD detector | 2841 independent reflections |
| Radiation source: fine-focus sealed tube | 2438 reflections with I > 2σ(I) |
| graphite | Rint = 0.009 |
| Rotation method data acquisition using ω and φ scans | θmax = 26.4°, θmin = 2.6° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009) | h = −11→5 |
| Tmin = 0.837, Tmax = 0.877 | k = −12→5 |
| 5245 measured reflections | l = −19→19 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.036 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.103 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0568P)2 + 0.5258P] where P = (Fo2 + 2Fc2)/3 |
| 2841 reflections | (Δ/σ)max = 0.001 |
| 177 parameters | Δρmax = 0.28 e Å−3 |
| 1 restraint | Δρmin = −0.36 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.89077 (18) | 0.14785 (17) | 0.62875 (11) | 0.0313 (4) | |
| C2 | 0.90726 (19) | 0.19319 (18) | 0.54672 (11) | 0.0359 (4) | |
| C3 | 0.8365 (2) | 0.1167 (2) | 0.47948 (12) | 0.0407 (4) | |
| H3 | 0.8471 | 0.1439 | 0.4245 | 0.049* | |
| C4 | 0.7512 (2) | 0.0022 (2) | 0.49045 (12) | 0.0413 (4) | |
| C5 | 0.7383 (2) | −0.0396 (2) | 0.57271 (13) | 0.0453 (5) | |
| H5 | 0.6825 | −0.1170 | 0.5816 | 0.054* | |
| C6 | 0.8074 (2) | 0.03244 (19) | 0.64125 (12) | 0.0385 (4) | |
| H6 | 0.7980 | 0.0035 | 0.6960 | 0.046* | |
| C7 | 0.73064 (18) | 0.40051 (18) | 0.70252 (11) | 0.0328 (4) | |
| C8 | 0.67615 (19) | 0.50629 (19) | 0.64977 (12) | 0.0371 (4) | |
| H8 | 0.7410 | 0.5643 | 0.6259 | 0.045* | |
| C9 | 0.5229 (2) | 0.5243 (2) | 0.63306 (12) | 0.0402 (4) | |
| C10 | 0.4248 (2) | 0.4403 (2) | 0.66791 (13) | 0.0459 (5) | |
| H10 | 0.3224 | 0.4542 | 0.6563 | 0.055* | |
| C11 | 0.4810 (2) | 0.3354 (2) | 0.72028 (14) | 0.0467 (5) | |
| H11 | 0.4157 | 0.2778 | 0.7440 | 0.056* | |
| C12 | 0.6335 (2) | 0.3141 (2) | 0.73829 (13) | 0.0410 (4) | |
| H12 | 0.6703 | 0.2430 | 0.7738 | 0.049* | |
| C13 | 0.9916 (3) | 0.3198 (2) | 0.52740 (15) | 0.0575 (6) | |
| H13A | 1.0910 | 0.3159 | 0.5569 | 0.069* | |
| H13B | 0.9415 | 0.3991 | 0.5455 | 0.069* | |
| H13C | 0.9960 | 0.3252 | 0.4675 | 0.069* | |
| C14 | 0.6723 (3) | −0.0736 (3) | 0.41520 (15) | 0.0614 (6) | |
| H14A | 0.7398 | −0.0875 | 0.3740 | 0.074* | |
| H14B | 0.5881 | −0.0212 | 0.3904 | 0.074* | |
| H14C | 0.6387 | −0.1606 | 0.4334 | 0.074* | |
| N1 | 0.88789 (16) | 0.38382 (16) | 0.72030 (11) | 0.0376 (4) | |
| H1N | 0.943 (2) | 0.4465 (19) | 0.7057 (13) | 0.045* | |
| O1 | 0.93966 (15) | 0.15746 (14) | 0.79211 (8) | 0.0437 (3) | |
| O2 | 1.12517 (14) | 0.26929 (15) | 0.71487 (9) | 0.0469 (4) | |
| Cl1 | 0.45335 (6) | 0.65483 (6) | 0.56450 (4) | 0.06029 (19) | |
| S1 | 0.97232 (4) | 0.23553 (4) | 0.72011 (3) | 0.03353 (14) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0310 (8) | 0.0294 (8) | 0.0333 (8) | 0.0027 (6) | 0.0027 (6) | 0.0007 (7) |
| C2 | 0.0360 (9) | 0.0325 (9) | 0.0395 (9) | 0.0025 (7) | 0.0061 (7) | 0.0050 (7) |
| C3 | 0.0440 (10) | 0.0439 (10) | 0.0339 (9) | 0.0081 (8) | 0.0037 (7) | 0.0030 (8) |
| C4 | 0.0393 (9) | 0.0408 (10) | 0.0427 (10) | 0.0060 (8) | 0.0005 (8) | −0.0079 (8) |
| C5 | 0.0475 (11) | 0.0349 (10) | 0.0534 (12) | −0.0084 (8) | 0.0064 (9) | −0.0023 (8) |
| C6 | 0.0422 (9) | 0.0347 (9) | 0.0386 (9) | −0.0038 (8) | 0.0055 (7) | 0.0041 (7) |
| C7 | 0.0273 (8) | 0.0326 (8) | 0.0384 (9) | −0.0005 (7) | 0.0042 (7) | −0.0097 (7) |
| C8 | 0.0346 (9) | 0.0342 (9) | 0.0432 (10) | −0.0012 (7) | 0.0074 (7) | −0.0047 (8) |
| C9 | 0.0373 (9) | 0.0402 (10) | 0.0419 (10) | 0.0053 (8) | 0.0006 (7) | −0.0080 (8) |
| C10 | 0.0284 (9) | 0.0547 (12) | 0.0540 (12) | 0.0006 (8) | 0.0032 (8) | −0.0118 (10) |
| C11 | 0.0338 (9) | 0.0515 (12) | 0.0563 (12) | −0.0088 (8) | 0.0110 (9) | −0.0037 (10) |
| C12 | 0.0367 (9) | 0.0386 (10) | 0.0483 (11) | −0.0019 (8) | 0.0077 (8) | 0.0010 (8) |
| C13 | 0.0710 (14) | 0.0507 (13) | 0.0517 (13) | −0.0151 (11) | 0.0110 (11) | 0.0133 (10) |
| C14 | 0.0632 (14) | 0.0641 (15) | 0.0537 (13) | −0.0016 (12) | −0.0052 (11) | −0.0189 (12) |
| N1 | 0.0270 (7) | 0.0298 (8) | 0.0558 (10) | −0.0027 (6) | 0.0037 (7) | −0.0041 (7) |
| O1 | 0.0500 (8) | 0.0452 (8) | 0.0348 (7) | 0.0053 (6) | 0.0014 (6) | 0.0012 (6) |
| O2 | 0.0261 (6) | 0.0531 (8) | 0.0605 (9) | 0.0008 (6) | 0.0013 (6) | −0.0101 (7) |
| Cl1 | 0.0576 (3) | 0.0566 (3) | 0.0637 (4) | 0.0163 (3) | −0.0036 (3) | 0.0070 (3) |
| S1 | 0.0284 (2) | 0.0337 (2) | 0.0376 (2) | 0.00265 (16) | 0.00062 (16) | −0.00264 (17) |
Geometric parameters (Å, °)
| C1—C6 | 1.386 (2) | C9—C10 | 1.376 (3) |
| C1—C2 | 1.403 (2) | C9—Cl1 | 1.744 (2) |
| C1—S1 | 1.7655 (17) | C10—C11 | 1.378 (3) |
| C2—C3 | 1.392 (3) | C10—H10 | 0.9300 |
| C2—C13 | 1.506 (3) | C11—C12 | 1.388 (3) |
| C3—C4 | 1.383 (3) | C11—H11 | 0.9300 |
| C3—H3 | 0.9300 | C12—H12 | 0.9300 |
| C4—C5 | 1.389 (3) | C13—H13A | 0.9600 |
| C4—C14 | 1.508 (3) | C13—H13B | 0.9600 |
| C5—C6 | 1.379 (3) | C13—H13C | 0.9600 |
| C5—H5 | 0.9300 | C14—H14A | 0.9600 |
| C6—H6 | 0.9300 | C14—H14B | 0.9600 |
| C7—C8 | 1.383 (3) | C14—H14C | 0.9600 |
| C7—C12 | 1.391 (3) | N1—S1 | 1.6394 (15) |
| C7—N1 | 1.423 (2) | N1—H1N | 0.844 (15) |
| C8—C9 | 1.388 (2) | O1—S1 | 1.4375 (14) |
| C8—H8 | 0.9300 | O2—S1 | 1.4331 (13) |
| C6—C1—C2 | 120.98 (16) | C11—C10—H10 | 120.6 |
| C6—C1—S1 | 117.11 (13) | C10—C11—C12 | 121.16 (18) |
| C2—C1—S1 | 121.91 (13) | C10—C11—H11 | 119.4 |
| C3—C2—C1 | 116.81 (16) | C12—C11—H11 | 119.4 |
| C3—C2—C13 | 118.72 (17) | C11—C12—C7 | 119.09 (18) |
| C1—C2—C13 | 124.44 (18) | C11—C12—H12 | 120.5 |
| C4—C3—C2 | 123.20 (17) | C7—C12—H12 | 120.5 |
| C4—C3—H3 | 118.4 | C2—C13—H13A | 109.5 |
| C2—C3—H3 | 118.4 | C2—C13—H13B | 109.5 |
| C3—C4—C5 | 118.18 (17) | H13A—C13—H13B | 109.5 |
| C3—C4—C14 | 120.90 (19) | C2—C13—H13C | 109.5 |
| C5—C4—C14 | 120.92 (19) | H13A—C13—H13C | 109.5 |
| C6—C5—C4 | 120.60 (18) | H13B—C13—H13C | 109.5 |
| C6—C5—H5 | 119.7 | C4—C14—H14A | 109.5 |
| C4—C5—H5 | 119.7 | C4—C14—H14B | 109.5 |
| C5—C6—C1 | 120.23 (17) | H14A—C14—H14B | 109.5 |
| C5—C6—H6 | 119.9 | C4—C14—H14C | 109.5 |
| C1—C6—H6 | 119.9 | H14A—C14—H14C | 109.5 |
| C8—C7—C12 | 120.54 (16) | H14B—C14—H14C | 109.5 |
| C8—C7—N1 | 118.44 (16) | C7—N1—S1 | 123.69 (12) |
| C12—C7—N1 | 121.01 (17) | C7—N1—H1N | 118.4 (15) |
| C7—C8—C9 | 118.76 (17) | S1—N1—H1N | 110.5 (15) |
| C7—C8—H8 | 120.6 | O2—S1—O1 | 117.75 (9) |
| C9—C8—H8 | 120.6 | O2—S1—N1 | 104.39 (8) |
| C10—C9—C8 | 121.73 (18) | O1—S1—N1 | 109.03 (8) |
| C10—C9—Cl1 | 119.26 (15) | O2—S1—C1 | 111.66 (8) |
| C8—C9—Cl1 | 119.00 (15) | O1—S1—C1 | 106.89 (8) |
| C9—C10—C11 | 118.72 (17) | N1—S1—C1 | 106.59 (8) |
| C9—C10—H10 | 120.6 | ||
| C6—C1—C2—C3 | −0.1 (3) | C8—C9—C10—C11 | −0.4 (3) |
| S1—C1—C2—C3 | −179.26 (13) | Cl1—C9—C10—C11 | 178.36 (15) |
| C6—C1—C2—C13 | 177.94 (19) | C9—C10—C11—C12 | 0.3 (3) |
| S1—C1—C2—C13 | −1.2 (3) | C10—C11—C12—C7 | −0.1 (3) |
| C1—C2—C3—C4 | 1.0 (3) | C8—C7—C12—C11 | 0.0 (3) |
| C13—C2—C3—C4 | −177.20 (19) | N1—C7—C12—C11 | 179.00 (17) |
| C2—C3—C4—C5 | −1.3 (3) | C8—C7—N1—S1 | −136.09 (15) |
| C2—C3—C4—C14 | 177.68 (18) | C12—C7—N1—S1 | 44.9 (2) |
| C3—C4—C5—C6 | 0.8 (3) | C7—N1—S1—O2 | 162.85 (15) |
| C14—C4—C5—C6 | −178.21 (19) | C7—N1—S1—O1 | −70.50 (17) |
| C4—C5—C6—C1 | 0.0 (3) | C7—N1—S1—C1 | 44.55 (17) |
| C2—C1—C6—C5 | −0.3 (3) | C6—C1—S1—O2 | 133.91 (14) |
| S1—C1—C6—C5 | 178.83 (15) | C2—C1—S1—O2 | −46.93 (17) |
| C12—C7—C8—C9 | −0.2 (3) | C6—C1—S1—O1 | 3.82 (16) |
| N1—C7—C8—C9 | −179.16 (16) | C2—C1—S1—O1 | −177.03 (14) |
| C7—C8—C9—C10 | 0.4 (3) | C6—C1—S1—N1 | −112.67 (14) |
| C7—C8—C9—Cl1 | −178.42 (13) | C2—C1—S1—N1 | 66.48 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···O1i | 0.84 (2) | 2.32 (2) | 3.117 (2) | 158 (2) |
Symmetry codes: (i) −x+2, y+1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BT5582).
References
- Adsmond, D. A. & Grant, D. J. W. (2001). J. Pharm. Sci. 90, 2058–2077. [DOI] [PubMed]
- Arjunan, V., Mohan, S., Subramanian, S. & Gowda, B. T. (2004). Spectrochim. Acta Part A, 60, 1141–1159. [DOI] [PubMed]
- Gelbrich, T., Hursthouse, M. B. & Threlfall, T. L. (2007). Acta Cryst. B63, 621–632. [DOI] [PubMed]
- Gowda, B. T., Bhat, D. K., Fuess, H. & Weiss, A. (1999). Z. Naturforsch. Teil A, 54, 261–267.
- Gowda, B. T., Foro, S. & Fuess, H. (2007). Acta Cryst. E63, o2337.
- Gowda, B. T., Foro, S., Nirmala, P. G. & Fuess, H. (2010). Acta Cryst. E66, o1282. [DOI] [PMC free article] [PubMed]
- Oxford Diffraction (2009). CrysAlis CCD and CrysAlis RED Oxford Diffraction Ltd, Yarnton, England.
- Perlovich, G. L., Tkachev, V. V., Schaper, K.-J. & Raevsky, O. A. (2006). Acta Cryst. E62, o780–o782.
- Savitha, M. B. & Gowda, B. T. (2006). Z. Naturforsch. Teil A, 60, 600–606.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811029400/bt5582sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811029400/bt5582Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811029400/bt5582Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report