

Abstract
Melatonin mediates neuroprotection in several experimental models of neurodegeneration. It is not yet known, however, whether melatonin provides neuroprotection in genetic models of Huntington's disease (HD). We report that melatonin delays disease onset and mortality in a transgenic mouse model of HD. Moreover, mutant huntingtin (htt)-mediated toxicity in cells, mice, and humans is associated with loss of the type 1 melatonin receptor (MT1). We observe high levels of MT1 receptor in mitochondria from the brains of wild-type mice but much less in brains from HD mice. Moreover, we demonstrate that melatonin inhibits mutant htt-induced caspase activation and preserves MT1 receptor expression. This observation is critical, because melatonin-mediated protection is dependent on the presence and activation of the MT1 receptor. In summary, we delineate a pathologic process whereby mutant htt-induced loss of the mitochondrial MT1 receptor enhances neuronal vulnerability and potentially accelerates the neurodegenerative process.
Introduction
Huntington's disease (HD) is an autosomal-dominant chronic neurodegenerative disease that is universally fatal and lacks effective treatment. HD is caused by expansion of the CAG repeat in exon 1 of the HD gene (The Huntington's Disease Collaborative Research Group, 1993). Mutant huntingtin (htt) protein first kills selectively vulnerable medium spiny neurons (MSNs) in the striatum and thereafter the cortical neurons. As reported previously, we screened 1040 Food and Drug Administration-approved drugs for their ability to inhibit mitochondrial cytochrome c release triggered by proapoptotic stimuli (Wang et al., 2008). Melatonin inhibited cytochrome c release, activation of the caspase cascade, and cell death. Melatonin was also neuroprotective in a cellular model of HD (Wang et al., 2008).
Melatonin is an endogenously produced hormone secreted by the pineal gland and the retina (Cogburn et al., 1987). Within the body, the highest concentration of melatonin is in the brain (Reiter and Tan, 2002); within the cell, the highest concentration is in the mitochondria (Martín et al., 2000). Melatonin can be administered at high doses (Weishaupt et al., 2006) and is used clinically for sleep disturbances, circadian rhythm adjustment in the blind, and cancer (Ravindra et al., 2006). Most importantly, the potential benefits of melatonin have been evaluated in neurological diseases.
In addition to being a broad-spectrum antioxidant (Tan et al., 2002), melatonin is a ligand of several G-protein-coupled receptors. There are two mammalian isoforms of the melatonin receptor, MT1 and MT2. MT1 and MT2 are found in brain and peripheral tissues (Drew et al., 2001) (Allen Brain Atlas available at http://mouse.brain-map.org). Altered MT1 and MT2 expression levels have been reported in models of Alzheimer's disease (Savaskan et al., 2002, 2007), Parkinson's disease (Das et al., 2008), and normal aging (Sánchez-Hidalgo et al., 2009), as well as in senescence-accelerated mice (Caballero et al., 2008). In addition, melatonin blood levels are progressively reduced in HD patients (Aziz et al., 2009). Thus, the melatonin receptor–ligand axis may play a pathogenic role in HD and neurodegeneration.
Melatonin was shown to be protective in two HD-associated neurotoxin models, i.e., in rat brain homogenate treated with quinolinic acid (Southgate and Daya, 1999) and in striatal and cortical synaptosomes, isolated from rats treated with 3-nitropropionic acid (Túnez et al., 2004). However, melatonin has not been evaluated previously in transgenic mouse models of HD. Our data demonstrate that melatonin delays disease onset and prolongs lifespan in the R6/2 transgenic mouse HD model. In addition, we report the following. (1) MT1 receptor levels decrease in cultured striatal cells, mouse brain, and human striatum associated with mutant htt-mediated toxicity, and receptor depletion becomes greater as the disease progresses. (2) MT1 receptor knockdown by RNAi makes cells more vulnerable to cell death. Conversely, MT1 receptor overexpression increases resistance to cell death. (3) Melatonin administration counters MT1 receptor depletion attributable to mutant htt in vitro and in vivo. Together, these findings suggest that functional MT1 receptor depletion contributes to cell death from mutant htt.
Materials and Methods
Drugs.
Melatonin and luzindole were purchased from Sigma. 2-Iodo-melatonin was purchased from Tocris Bioscience.
Cell lines and induction of cell death.
Mutant htt ST14A striatal cells (8 Plx line) and parental ST14A striatal cells were kindly provided by Dr. Elena Cattaneo (University of Milan, Milan, Italy). Cell death was induced by shifting cells from the permissive temperature of 33°C to the nonpermissive temperature of 37°C and transferring them to serum-deprived medium (SDM) (Wang et al., 2005). Alternatively, cells were challenged by treatment with 1 mm H2O2 or with the combination of 10 ng/ml tumor necrosis factor α (TNFα) and 10 μm cycloheximide (CHX). In a second system, primary cerebrocortical neurons (PCNs) and primary striatal neurons (PSNs) were harvested from E14–E16 mice. Cultures of these cells were subject to oxygen–glucose deprivation (OGD) as described previously (Zhang et al., 2003a; Wang et al., 2009). In brief, culture medium was replaced with glucose-free Earle's balanced salt solution, and cells were incubated for 2 h with a defined concentration of melatonin. The cells were then placed in an anaerobic chamber with a BBL GasPak Plus (BD Biosciences), lowering the oxygen concentration to <100 ppm within 90 min. After 3 h, OGD was terminated by a return to normal culture conditions. Control cells were incubated for the same length of time in Earle's balanced salt solution with glucose in a normoxic incubator. Alternatively, PCNs (or PSNs) were treated for 18 h with 1 mm H2O2. In all experiments, cells were preincubated for 2 h with the test drug. Cell death was measured by the MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] or the LDH release assay (Roche Applied Science).
Expression vectors and transfection procedures.
The human MT1 receptor (cDNA clone MGC:104051 IMAGE: 30915497) cloned in the pCR4–TOPO vector was purchased from Invitrogen. The MT1 sequence was cut from pCR4–TOPO, epitope tagged, and transferred to the expression vector pcDNA3.1–Flag–hMT1a–GFP. Proper construction was confirmed by DNA sequencing (Biomyx Technology). Transient transfection of pcDNA3.1–Flag–hMT1a–GFP into mutant htt ST14A cells was performed using LipofectAMINE (Invitrogen) for 3 d. Cells were then shifted to nonpermissive conditions for 18 h. Cells were cotransfected with a plasmids encoding GFP. Morphology of cells was observed. This quantitative observation of cell death was made by counting the cells with morphologic features indicative of apoptosis (i.e., chromatin condensation, nuclear fragmentation, and cell shrinkage).
Western blotting.
Cells were collected in ice-cold lysis buffer [20 mm Tris, pH 8.0, 137 mm NaCl, 10% glycerol, 1% Nonidet P-40, and 2 mm EDTA with 5 mm Na2VO4, protease inhibitor mixture (Roche Molecular Biochemicals) supplemented with 0.2 mm PMSF]. The lysate was cleared by centrifugation at 19,720 × g for 10 min at 4°C, and the supernatant was analyzed by Western blotting. Samples of mouse brain were lysed in ice-cold RIPA buffer with protease inhibitors (Zhu et al., 2002; Wang et al., 2003). Antibodies to caspase-3 and caspase-9 were purchased from Cell Signaling Technology, ones to MT1 and MT2 from Millipore Bioscience Research Reagents, and ones to Rip2 from Alexis Biochemicals or BD Biosciences. Antibodies to caspase-1 were from Santa Cruz Biotechnology or BioSource and those to β-actin were from Sigma. Secondary antibodies and ECL reagents were from GE Healthcare.
Detection of factors released from mitochondria.
Separation of cytoplasmic and mitochondrial fractions of cells and tissues was conducted as described previously (Zhu et al., 2002; Wang et al., 2003). Antibodies to cytochrome c were purchased from BD Biosciences Pharmingen, ones to Smac/Diablo from Novus Biologicals, and ones to AIF from Sigma.
Isolation of brain mitochondria.
Brain mitochondria were isolated as described previously with some modification (Zhu et al., 2002; Krasnikov et al., 2005; Singh et al., 2006). Briefly, forebrains were quickly removed and homogenized in ice-cold buffer 1 (in mm: 320 sucrose, 10 HEPES, pH 7.4, and 1 EGTA, pH adjusted to 7.4 with KOH) using a tissue homogenizer (Kontes Glass). The homogenate was centrifuged twice at 3000 rpm for 3 min in a JA-20 rotor (Beckman Instruments), and the supernatant was further centrifuged at 11,000 rpm for 8 min. The pellet, containing crude mitochondria, was resuspended in buffer 1 and loaded atop a discontinuous Ficoll gradient of 7.5 and 10% in 320 mm sucrose, 10 mm HEPES-KOH, pH 7.4, and 0.02 mm EGTA, pH adjusted to 7.4 with KOH. The gradient was spun in a Beckman Instruments centrifuge with an SW-28 high-speed rotor at 23,400 rpm for 12 min. Pelleted non-synaptosomal mitochondria were rinsed twice, first with buffer 1 and then with buffer 2. Mitochondria were lysed in RIPA buffer containing protease inhibitors and were analyzed by immunoblotting.
Caspase activity assay.
Test cell cultures contain 5 μm melatonin, whereas controls are devoid of melatonin. Extracts were prepared and enzyme assays conducted as described previously (Wang et al., 2003) (Clontech). The ApoAlert caspase fluorescent assay kit was from Clontech, caspase-1-like substrate Ac-YVAD-AFC and caspase-9-like substrate Ac-LEHD-AFC were from Calbiochem, and caspase-3-like substrate Ac-DEVD-AFC was from BD Biosciences Pharmingen. Enzyme activity was determined with a Bio-Rad Versa Fluorometer. The evaluation of the effect of melatonin on enzymatic activities were performed in parallel with previously reported experiments evaluating methazolamide, but some of the controls are the same as in the previous report (Wang et al., 2008).
Human brain specimens.
Frozen postmortem samples of human brain were obtained from the Harvard Brain Tissue Resource Center (McLean Hospital, Belmont, MA). They were taken from the caudate and putamen of seven grade IV HD patients, four grade II HD patients, and six control samples from non-neurologic patients. There is no systematic difference between the average postmortem interval or age at death for persons in the three different groups (postmortem intervals, 21.9, 17.3, and 19.4 h; age, 58.9, 65.3, and 49.8 years for control, grade II, and grade IV group, respectively).
HD mice and treatment regimen.
R6/2 mice (The Jackson Laboratory) were used to establish a breeding colony. Male R6/2 mice were bred with congenic (i.e., B6SLJ) wild-types females (The Jackson Laboratory). In this study, R6/2 mice used were known to have a CAG repeat size of 110–115 (Wang et al., 2008). Starting at 6 weeks of age, test R6/2 mice were given daily intraperitoneal injections of 30 mg/kg melatonin, whereas control animals received the saline vehicle. Each group contained an equal number of male and female mice. Mice were evaluated weekly by placing them on a rotarod turning at 5 or 15 rpm and measuring the time until they fell (up to 7 min). Mortality was defined as the age of death or the age when the mouse was unable to right itself within 30 s or the age of death. In this study, we used nine mice in the melatonin group and compared them to six littermate mates in the saline control group. For molecular analysis of the effect of melatonin, matched littermates were killed at 25 weeks of age, and the brain tissue was analyzed. Experiments were conducted in accordance with protocols approved by the Harvard Medical School Animal Care Committee.
Stereology.
Serial-cut coronal tissue sections from the rostral segment of the neostriatum to the level of the anterior commissure (interaural 5.34 mm, bregma 1.54 mm to interaural 3.7 mm, bregma 0.10 mm; Franklin and Paxinos, 2000) were used for volumetric measurement of ventricular size. Ventricular areas were measured using Neurolucida Stereo Investigator software (MicroBrightField), with researchers (R.J.F. and K.S.) blinded to the experimental condition. All specimens were stained at the same time, photographed under the same conditions, and are age matched.
Measurement of huntingtin aggregates.
The extent of huntingtin protein aggregates was determined in serially cut coronal tissue sections from the most rostral segment of the neostriatum to the level of the anterior commissure (interaural 5.34 mm, bregma 1.54 mm to interaural 3.7 mm, bregma −0.10 mm). Neurolucida Stereo Investigator software was used to conduct unbiased stereological counts of huntingtin-positive aggregates (≥1.0 μm) in neostriatum from melatonin- and saline-treated R6/2 mice (25 weeks of age). The respective areas of the rostral brain, ventricles, and neostriatum were measured by imaging serial sections. To obtain unbiased measurements of aggregate density, counting frames were randomly sampled within each preparation from neostriatum. The number of huntingtin-positive aggregates was determined by the optical-dissector method reference. The areas of striatal neurons were determined by microscopic video capture with a Windows-based image analysis system (Optimas; Bioscan). Although this software identifies and measures profiles automatically, its reliability was verified by visual inspection of tissue preparations.
Immunohistochemistry.
Brains were cryoprotected in 30% sucrose and frozen in cold isopentane. Frozen sagittal sections (10 μm thick) were washed with 0.05% Tween 20 in PBS and permeabilized with 70% methanol and with 0.05% Triton X-100. The sections were blocked and incubated with MT1 antibody. After a second wash with 0.05% Tween 20 in PBS, the preparations were stained with FITC-conjugated secondary antibodies (a control procedure that omits staining with the primary antibodies determined the extent of background staining by the secondary antibody). After additional washes with 0.05% Tween 20 in PBS and DAPI counterstaining, sections were viewed with a Nikon Eclipse TE 200 fluorescence microscope.
RNAi.
Four siRNAs targeting MT1 expression were purchased from Qiagen. They were synthesized as pairs of oligoribonucleotides as follows. MT1 siRNA 1 targets sequence AACGCAATCATATACGGACTA and consists of 5′-CGCAAUCAUAUACGGACUAtt-3′ and 3′-ttGCGUUAGUAUAUGCCUGAU-5′ as sense and antisense strands, respectively. MT1 siRNA 2 targets sequence CGGGATCGCTATGAACCGCTA and consists of GGAUCGCUAUGAACCGCUAtt-3′, and 3′-GCCCUAGCGAUACUUGGCGAU-5′ as sense and antisense strands, respectively. MT1 siRNA 3 targets sequence CTCAGGAACGCAGGGAATATA and consists of 5′-CAGGAACGCAGGGAAUAUAtt-3′ and 3′-GAGUCCUUGCGUCCCUUAUAU-5′. MT1 siRNA 4 targets sequence 5′-GGCGCTGACGTCTATACTTAA-3′ and consists of 5′-CGCUGACGUCUAUACUUAAtt-3′ and 3′-ccGCGACUGCAGAUAUGAAUU-5′ as sense and antisense strands, respectively. Cells were transiently transfected with these siRNAs using HiPerFect Transfection Reagent (Qiagen). Forty hours after transfection, cells were analyzed using quantitative RT (qRT)-PCR and Western blotting. Apoptosis of mutant htt ST14A cells was induced in three different ways: (1) shifting to the nonpermissive temperature of 37°C with concomitant transfer to SDM; (2) exposure to H2O2 for 18 h; and (3) treatment with TNFα/CHX for 18 h.
Quantitative real-time RT-PCR.
Total RNA was isolated from mutant htt ST14A cells, mouse brains, or human striatum. The following pairs of primers were used: mouse MT1, 5′-TGAGTGTCATCGGCTCGATAT-3′ and 5′-TAGTAACTAGCCACGAACAGC-3′ (Niles et al., 2004); mouse MT2, 5′-CTCACTCTGGTGGCTTTGGTG-3′ and 5′-CTGCGCAAATCACTCGGTCTC-3′ (Kobayashi et al., 2005); human MT1, 5′-TGCTACATCTGCCACAGTCTC-3′ and 5′-CAGTAGCCCGTATATAATGGC-3′ (Niles et al., 1999); human MT2, 5′-TCATCGGCTCTGTCTTCAATA-3′ and 5′-ACTGGGTGCTGGCGGTCTGGA-3′ (Brydon et al., 2001); GAPDH, 5′-ATGGTGAAGGTCGGTGTCAACGGA-3′ and 5′-TTACTCCTTGGAGGCCATGTAGGC-3′ (Clontech). Real-time qRT-PCR was performed as described previously (Wang et al., 2005). The RT-PCR began by holding the temperature at 50°C for 30 min and then at 95°C for 10 min. Samples were then subject to 45 cycles of 95°C for 30 s, 55°C for 1 min, and 72°C for 30 s. Relative quantities of mRNAs were calculated from their Ct values (Wang et al., 2005). GAPDH was not variable in these experiments and was thus used as the control gene.
Statistical analysis.
Statistical significance was evaluated by the t test. Data on viability or the extent of cell death were compared by ANOVA or repeated-measures ANOVA and by nonpaired Student's t test. Statistical analyses were done using StatView 4.0 software (SAS Institute). Error bars represent SEM.
Results
Melatonin inhibits the mitochondrial and the Rip2/caspase-1 cell death pathways
Release of cytochrome c from mitochondria triggers a set of biochemical changes that lead to neuronal cell death. These molecular pathways are activated in a number of neurodegenerative disorders, including HD (Friedlander, 2003; Wang et al., 2003). We therefore tested whether melatonin inhibited the release of cytochrome c from the mitochondria of mutant htt ST14A cells. Mutant htt ST14A cells were constructed from the parental ST14A striatal cell line by transforming it with a temperature-sensitive large T antigen. Mutant htt ST14A cells undergo cell death during shift from 33°C to 37°C with concomitant transfer to SDM (i.e., change from permissive to nonpermissive conditions) (Rigamonti et al., 2001). Cytochrome c localization was determined by immunocytochemistry. Cells were stained with antibodies to cytochrome c and the mitochondrial marker MitoTracker (Fig. 1A). Merging the images revealed that the highest concentration of cytochrome c immunoreactivity colocalized with mitochondria. Temperature shift with transfer to SDM induced the release of cytochrome c from the mitochondria into the cytoplasm. As indicated by the arrows, we observed a more diffuse staining pattern in cells kept under nonpermissive versus permissive conditions. Indeed, melatonin inhibited cytochrome c release as indicated by the preservation of punctate staining after temperature shift. Cells treated with melatonin revealed a punctate pattern of staining even under nonpermissive conditions. Melatonin inhibits the release of cytochrome c from the mitochondria to the cytoplasm (Fig. 1A).
Figure 1.

Melatonin inhibits the mitochondrial cell-death pathway in cultured cells. A, Cell death is induced in mutant htt striatal cells by shifting cells to the nonpermissive temperature of 37°C in SDM. Test cell cultures contain 5 μm melatonin, whereas controls are devoid of melatonin. Five hours after being placed in nonpermissive conditions, cells are stained with MitoTracker, fixed, and stained with fluorescently tagged antibodies to cytochrome c. A punctate pattern colocalizing with MitoTracker indicates that cytochrome c is retained within mitochondria; a dim and diffuse one indicates that it has been released into the cytoplasm. The latter pattern is observed in stressed cells, confirming the release of cytochrome c. Even under nonpermissive conditions, however, melatonin-treated cells retain a bright and punctate appearance, suggesting the localization of cytochrome c in mitochondria (scale bar, 5 μm). B–E, Mutant htt striatal cells are shifted to nonpermissive conditions for 5 or 18 h (B–D) or 2 h (E) and treated as indicated. Cell lysates are centrifuged, and the cytosolic fractions (i.e., supernatant) are analyzed on Western blots probed with antibodies to cytochrome c, Smac, and AIF (B, top 3 blots). Alternatively, whole-cell lysates are resolved on Western blots and probed with antibodies to caspase-9, caspase-3, or caspase-1 or to Rip2 (2 bottom blots in B, D, E). In addition, caspase activities in whole-cell extract are measured by fluorogenic assays (C). Caspase-1 activation is evaluated by both Western blotting with an antibody to activated caspase-1 and fluorogenic assay (D). All Western blots have 50 μg/lane protein with β-actin as the loading control. A representative blot is shown. In all graphs, gray bars correspond to measurements on cells treated with melatonin, both with or without temperature shift/SDM. These are compared with white bars for unstressed cells and black bars for stressed cells without melatonin (null and negative controls, respectively). Values in bar graphs are the average of at least three independent measurements (*p < 0.05, **p < 0.001). F, Mutant htt ST14A cells are treated as indicated before being transferred to nonpermissive conditions. Living cells are stained with 2 μm Rh 123 to determine the mitochondrial membrane potential (Ψm). G, Mutant htt striatal cells are kept under nonpermissive conditions for 18 h in the presence or absence of 5 μm melatonin. Finally, chymotrypsin-like, trypsin-like, and caspase-like activity of proteasomes in cell lysate are measured using respective fluorogenic substrates, Suc-LLVY-MCA, Boc-LRR-AMC, and Z-LLE-AMC (Biomol). Results are the mean ± SEM for three independent experiments. MW, Molecular weight.
Smac and AIF are two other proteins whose release from mitochondria results in activation of cell death pathways. In a variety of neurodegenerative diseases, the appearance of these factors in the cytoplasm has been associated with mitochondrial dysfunction (Friedlander, 2003; Wang et al., 2003; Zhang et al., 2003a; Adamczyk et al., 2005). During disease progression in HD mice, release of cytochrome c/Smac/AIF triggers a set of events that activate mitochondrial cell-death pathways (Kiechle et al., 2002; Friedlander, 2003; Wang et al., 2003). Cytoplasmic cytochrome c, apaf-1, and pro-caspase-9 form a macromolecular assembly called the “apoptosome,” resulting in caspase-9 activation. In its turn, caspase-9 activates caspase-3, thereby triggering the terminal events in the cell-death pathway (Li et al., 1997). Melatonin partially inhibited mutant htt-induced release of mitochondrial apoptogenic factors and caspase activation (Fig. 1A--C). Caspase activation was evaluated by immunoblotting (Fig. 1B) and complementary fluorogenic activity assays (Fig. 1C).
In addition to its activation of caspase-9 and caspase-3 and cleavage at the caspase-6 site (Graham et al., 2006), mutant htt stimulates the Rip2/caspase-1 pathway in R6/2 mice and in mutant htt ST14A cultures (Ona et al., 1999; Chen et al., 2000; Zhang et al., 2003b; Wang et al., 2005). We demonstrated previously that increased abundance of Rip2 protein (a caspase-1 activator) (Thome et al., 1998) correlates with greater toxicity from mutant htt. We determined whether melatonin inhibits Rip2 accumulation and counters caspase-1 activation. Indeed, we observed that procaspase-1 is activated in mutant htt ST14A cells during transfer to nonpermissive conditions (Wang et al., 2003, 2005). As demonstrated by immunoblotting and complementary fluorogenic assay, melatonin inhibits mutant htt-induced caspase-1 activation (Fig. 1D).
Rip2 binds to caspase-1 via a caspase activation and recruitment domain (CARD)–CARD (for caspase recruitment domain domain) homotypic interaction resulting in caspase-1 activation. Rip2 protein levels increase in neurologic diseases, including HD (Wang et al., 2005), Alzheimer's disease (Engidawork et al., 2001), and stroke (Zhang et al., 2003a). Moreover, reducing the level of Rip2 by RNA interference inhibits mutant htt-induced cell death of ST14A cells (Wang et al., 2005). Given that melatonin inhibits caspase-1 activation, we determined whether this effect involves blocking Rip2 upregulation. Indeed, melatonin inhibited mutant htt-induced Rip2 upregulation in mutant htt ST14A cells (Fig. 1E).
Melatonin has been reported to modulate mitochondrial pathways through inhibition of the mitochondrial permeability transition (Andrabi et al., 2004). The collapse of the mitochondrial transmembrane potential (ΔΨm) is a central event in cell death (Petronilli et al., 1994; Friedlander, 2003; Stavrovskaya and Kristal, 2005). Consequently, preservation of ΔΨm by melatonin is a possible mechanism by which it inhibits cell death. We tested this hypothesis using rhodamine 123 (Rh 123), a fluorescent, hydrophobic, and positively charged molecule that accumulates in electrostatically charged mitochondria. We observed punctate staining by Rh 123, a pattern indicative of a negative mitochondrial membrane potential. Moreover, transferring cells to nonpermissive conditions resulted in dissipation of ΔΨm and ensuing cell death (Fig. 1F). Furthermore, melatonin preserved the punctate distribution of Rh 123 fluorescence within the cell. We conclude that the ability of melatonin to maintain ΔΨm likely contributes to the ability of the compound to rescue mutant htt ST14A from temperature shift-induced cell death.
Proteosomal dysfunction has been reported in HD (Bennett et al., 2007). We determined whether in our cellular system mutant htt causes malfunction of the ubiquitin–proteosome system. We found that, under nonpermissive conditions, mutant htt ST14A-expressing cells demonstrate a significant decline in proteosomal activity. Moreover, melatonin partially inhibits such stress-induced proteosomal dysfunction (Fig. 1G).
Melatonin-mediated protection requires the MT1 receptor
Neuroprotection by melatonin may be mediated by its ability to scavenge free radicals, by a traditional ligand–receptor interaction, or by a combination of the two. We addressed this question using two molecular analogs of melatonin: the melatonin-receptor antagonist luzindole and the agonist 2-iodomelatonin (Fig. 2A). As shown in Figure 2B, melatonin inhibited cell death as a result of temperature shift. To directly evaluate whether the effects of melatonin require receptor binding, we determined whether the receptor antagonist luzindole counters melatonin-mediated protection. In this in vitro system, luzindole almost completely eliminated neuroprotection by melatonin, indicative of a receptor-mediated effect as opposed to a free radical-scavenger mechanism (Fig. 2B). We repeated this experiment in other cell-death models, e.g., mutant htt ST14A cells exposed to H2O2 or treated with TNFα plus CHX (Fig. 2C,D). In both cases, luzindole blocked melatonin-mediated protection, confirming that receptor binding is central to the mechanism of protection.
Figure 2.

Melatonin inhibits cell death of both cell lines and primary neurons in culture; luzindole eliminates this neuroprotection. A, Chemical structures of melatonin, luzindole, and 2-iodomelatonin. B–G, Except in the presence of luzindole, the MT1 agonists melatonin and 2-iodomelatonin counter cell death in all of the five cellular systems: B, G, mutant htt ST14A placed in nonpermissive conditions; C, mutant htt ST14A exposed to 1 mm H2O2 for 18 h; D, mutant htt ST14A challenged with 10 ng/ml TNF-α and 10 μm CHX for 18 h; E, primary striatal neurons exposed to 1 mm H2O2 for 18 h; and F, primary cerebrocortical neurons exposed to 500 μm NMDA for 18 h. The data in B–D and G are from MTS assays of cell viability; those in E and F are from LDH assays of cell death. The bars are colored according to the presence of melatonin and luzindole: white, no agonist or antagonist of MT1/MT2; blue, just melatonin or (in G) 2-iodomelatonin; red, luzindole administered alone or with melatonin/2-iodomelatonin. In all systems, luzindole alone does not change the extent of cell death. In contrast, treatment with melatonin reduces cell death in a dose-dependent manner. When both compounds are present, however, luzindole (an antagonist of MT1 and MT2) eliminates rescue by melatonin. Data for each system are from at least three independent experiments. *p < 0.05; **p < 0.001, n = 3–6. H, Immunoblot of cytosolic fractions of mutant htt ST14A cells treated as indicated for 18 h. Luzindole blocks melatonin inhibition of cytochrome c release.
The most vulnerable cells in HD are the MSNs of the striatum followed by cortical neurons (Li, 1999). Melatonin prevents cell death induced by OGD (Cazevieille et al., 1997; Wang et al., 2009). We tested whether melatonin inhibits cell death of PSNs induced by H2O2. Indeed, treatment with melatonin provided dose-dependent protection of PSNs challenged by this oxidative stress (Fig. 2E). In addition, melatonin diminished the extent of NMDA-induced death of primary cerebrocortical neurons (PCNs) (Fig. 2F). Again, luzindole eliminated this neuroprotection, demonstrating that the action of melatonin is mediated by its G-protein-coupled receptor (Fig. 2E,F).
We next evaluated whether the MT1/MT2 agonist 2-iodomelatonin shares the protective properties of melatonin. As expected from their common affinity for the MT1 and MT2 receptors, 2-iodomelatonin inhibited cell death of mutant htt ST14A. Moreover, luzindole essentially eliminated neuroprotection by 2-iodomelatonin (Fig. 2G). As observed with melatonin, 2-iodomelatonin inhibited the increase in Rip2 expression in stressed mutant htt ST14A cells. This effect was observed both in cells challenged by temperature shift and in ones exposed to H2O2 (data not shown). A clue to the mechanism of these phenomena is the ability of melatonin to inhibit mutant htt-induced release of mitochondrial cytochrome c in ST14A cells. Moreover, luzindole eliminates this molecular activity of the MT1 receptor agonist. The correlation between inhibition of cytochrome c release and protection of neurons from cell death implicates the effect of melatonin on mitochondria in neuroprotection (Fig. 2H).
Mutant htt-mediated toxicity selectively causes MT1 receptor loss
We investigated whether the toxicity of mutant htt correlates with changes in the expression of melatonin receptor MT1, receptor MT2, or both. We found that levels of MT1 protein dropped significantly when mutant htt ST14A cells were shifted to a nonpermissive temperature. Decreased levels of MT1 receptor were also observed when these cells were exposed to H2O2 (Fig. 3A). In both conditions, statistical analysis demonstrate that melatonin treatment significantly ameliorated MT1 loss (Fig. 3A). In the same cells, however, neither stress altered the level of MT2 protein (Fig. 3A). These data, as well as the ability of luzindole to inhibit melatonin-mediated protection, points to the MT1 receptor as a key modulator of this effect.
Figure 3.
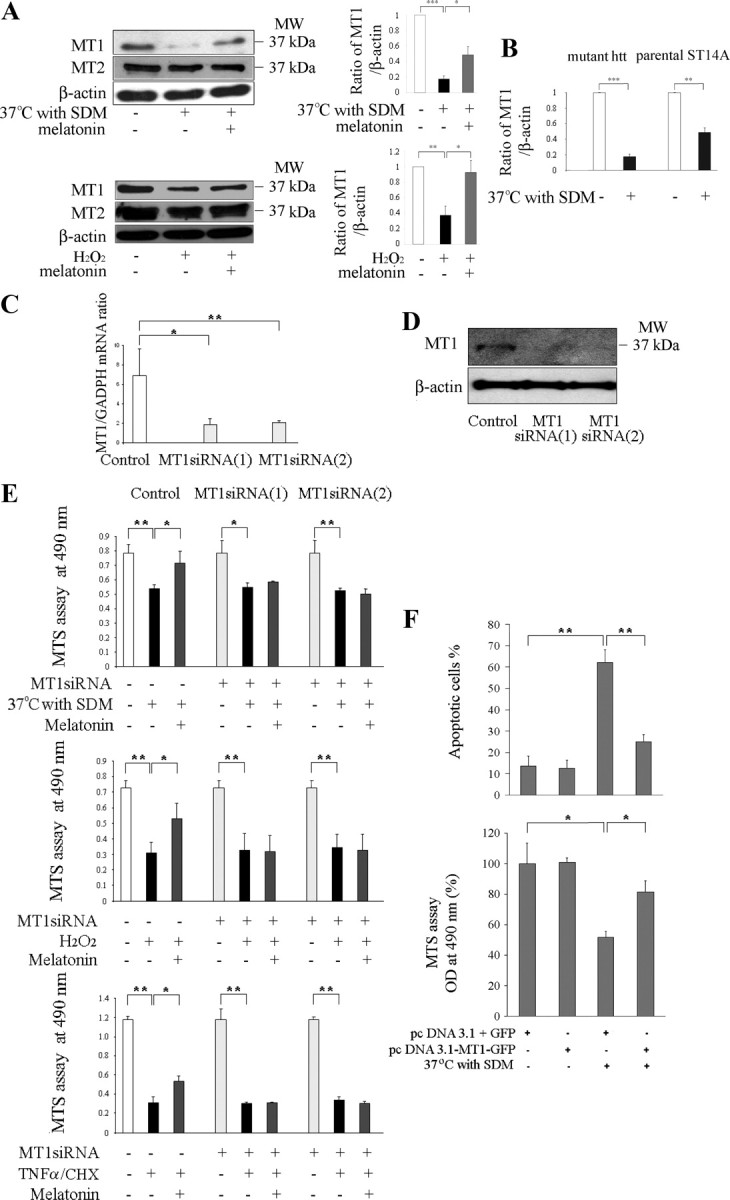
Knockdown of MT1 sensitizes cultured neurons to cell death; overexpression rescues them. Protein levels of MT1 and MT2 receptors were determined in mutant htt ST14A cells. During challenge by temperature shift in SDM or incubation for 18 h with 1 mm H2O2 (A), there is significant loss of MT1 receptor. In contrast, levels of MT2 receptor do not change. Pretreatment for 2 h with 5 μm melatonin significantly preserves levels of MT1 receptor (n = 3–4, *p < 0.05, **p < 0.01, ***p < 0.001). B, The comparison of protein levels of MT1 receptors were determined in mutant htt ST14A cells and parental ST14A cells during challenge by temperature shift in SDM (n = 4, **p < 0.01, ***p < 0.001). C–E, Neuroprotection by melatonin is eliminated by siRNAs, which target the MT1 receptor. Mutant htt ST14A cells were transiently transfected with MT1 siRNA 1 and siRNA 2 48 h before being challenged by the proapoptotic stimulus. Consequent depletion of MT1 mRNA was confirmed by qRT-PCR (C) and that of MT1 protein by immunoblotting (D). The mRNA for GAPDH and the protein for β-actin served as a loading controls for the PCR and the immunoblot, respectively (*p < 0.01, **p < 0.001). E, Neuroprotection afforded by melatonin is eliminated by knocking down levels of MT1 receptor. This phenomenon, apparent from MTS assays for cell viability, was observed in three different in vitro systems (i.e., mutant htt ST14A cells challenged by temperature shift in SDM, exposure to H2O2, or treatment with TNFα/CHX). F, Mutant htt ST14A cells challenged by temperature shift in SDM are rescued by an MT1 protein–GFP fusion but not by GFP alone. Cells were transfected with pcDNA3.1–MT1–GFP or pcDNA3.1/GFP, test and control plasmids, respectively. Forty-eight hours later, they were shifted to nonpermissive conditions for 18 h and then viewed under the fluorescence microscope. In the test group, stress caused significant cell death. In contrast, those cells expressing the MT1–GFP fusion protein were much more resistant to cell death. Parallel MTS assays of cell viability corroborated these results. Data are the mean ± SEM of three independent experiments. In all graphs, statistical significance is indicated: *p < 0.01, **p < 0.001.
Next we compared the effect of stress on MT1 expression in parental and mutant htt ST14A cells. We found that temperature shift with SDM [which also induces cell death in the parental cell line but to a lesser degree than in the mutant htt line (Wang et al., 2005)] induced a moderate decrease in expression of MT1 (Fig. 3B). However, the change in MT1 expression was less dramatic in parental ST14A cells (51%) compared with mutant htt ST14A striatal cell lines (82%). This suggests that loss of MT1 receptor is not a specific mutant htt-mediated phenomenon but rather a more fundamental response to cellular stress.
Knockdown of MT1 sensitizes neurons to cell death, whereas its overexpression is neuroprotective
To confirm the importance of the MT1 receptor to the action of melatonin, we used the technique of RNA interference. Four DNA fragments were synthesized, each encoding an siRNA targeting the MT1 message. These molecules were transfected into mutant htt ST14A striatal cells with ∼90% efficiency. Of the four oligonucleotides, MT1 siRNA1 and MT1 siRNA2 effectively diminished levels of the MT1 mRNA and the encoded protein (Fig. 3C,D).
We investigated the effects of RNAi-mediated knockdown of MT1 in mutant htt ST14A cells. Test cells expressing MT1 siRNA 1 or siRNA 2 and an appropriate control were challenged by shift to nonpermissive conditions, exposure to H2O2, or treatment with TNFα/CHX. In all three systems, MT1 knockdown completely eliminated melatonin-mediated protection (Fig. 3E).
Increasing the abundance of MT1 is expected to make cells more resistant to cell death. To test this hypothesis, we constructed the plasmid pcDNA3.1–MT1–GFP, a DNA molecule that expresses the MT1 receptor fused to GFP. Mutant htt ST14A cells were transiently transfected with either this plasmid or a control GFP-expressing plasmid. When shifted to 37°C with SDM, control cells underwent extensive cell death, whereas ones expressing the MT1–GFP fusion protein were relatively resistant. This qualitative observation was made rigorous by counting the cells with morphologic features indicative of apoptosis (i.e., cell shrinkage and chromatin condensation). Mitochondrial function was determined by the MTS assay, again showing that pcDNA3.1–MT1–GFP rescues mutant htt ST14A from cell death caused by this stress (Fig. 3F). The overexpression of the MT1 receptor (in the absence of exogenous melatonin) is sufficient to significantly attenuate the temperature shift-mediated cell death demonstrates that MT1 receptor itself is neuroprotective. Increasing the abundance of MT1 by either the transient transfection of MT1 receptor or the stimulation of melatonin (agonist of MT1) make cells more resistant to cell death. The correlation of MT1 concentration and cell rescue supports our contention that neuroprotection by melatonin depends on its interaction with this receptor molecule.
Melatonin delays disease onset and mortality in R6/2 mice
Having established the ability of melatonin to inhibit cell death in vitro, we investigated its effects in a mouse model of HD. We used the R6/2 mouse, a transgenic animal expressing a human gene encoding the N-terminal portion of polyQ-expanded huntingtin (Mangiarini et al., 1996). Beginning at 6 weeks of age, R6/2 littermate mice received daily intraperitoneal melatonin injections of 30 mg/kg or saline vehicle (n = 9 and 6, respectively; Fig. 4A--D). Disease onset, defined as the age when mice can no longer remain for 7 min on a rotarod turning at 15 rpm (Fig. 4A), was delayed 19% by melatonin treatment. Declining performance on a rotarod turning at 5 rpm (a more sensitive indicator of symptomatic disease progression) was also slowed by melatonin (Fig. 4B). Finally, melatonin prolonged the lifespan of these HD mice by 18% (Fig. 4D). In addition, using a larger sample size that includes both littermate and nonlittermate R6/2 mice that received daily intraperitoneal melatonin injections of 30 mg/kg (n = 12) or saline vehicle (n = 24), we found that disease onset was delayed 23% by melatonin treatment, whereas melatonin prolonged the lifespan of these HD mice by 21% (data not shown). In the course of disease progression, R6/2 mice lose a significant amount of body weight. The time course of weight loss is not significantly altered by melatonin in littermate mice (Fig. 4C) or in the entire sample, including littermates and non-littermates (data not shown). This remains true regardless of whether male and female mice are analyzed together or separately. It is noteworthy that, in many other studies using animal models for neurologic disease, drug and gene therapies are found not to change the pattern of weight loss (Ona et al., 1999; Chen et al., 2000; Chopra et al., 2007; Masuda et al., 2008; Wang et al., 2008).
Figure 4.

Melatonin slows disease progression in R6/2 mice. A, B, Treatment from 6 weeks of age with 30 mg · kg−1 · d−1 melatonin improves rotarod performance of R6/2 mice. Both melatonin- and saline-injected R6/2 littermates are challenged to remain for up to 7 min on a rotarod turning at 15 rpm (A) or 5 rpm (B). Data for many of the weekly tests show a statistically significant difference of rotarod performance of saline- and melatonin-treated R6/2 mice. When rotation is 15 rpm, we observe p < 0.05 for weeks 21, 22, and 25 and p < 0.001 for weeks 26–29. At 5 rpm, we find p < 0.05 for weeks 22, 28, and 29 and p < 0.001 for weeks 23–27. C, Weekly values for body weight are statistically indistinguishable between melatonin-treated animals and saline controls. D, The table summarizes data for the melatonin- and saline-treated groups (A–D, n = 9 and 6, respectively). Both the age at disease onset (i.e., when first the mouse falls from the rotarod before 7 min have elapsed) and that at death are greater for melatonin-treated mice (*p < 0.01). E, Melatonin reduces ventricular enlargement in the brains of R6/2 mice. The figure shows coronal sections from 25-week-old R6/2 mice treated with melatonin or the saline vehicle, as well as ones from age-matched wild-type littermates (n ≥ 3). F, Brain sections were prepared from 25-week-old R6/2 mice that had been treated with 30 mg · ml−1 · d−1 melatonin or saline. Slices of both cortical and striatal tissue were stained with anti-huntingtin antibodies. In both cases, any reduction in the extent of huntingtin aggregation was not statistically significant.
Progressive ventriculomegaly is observed in R6/2 mice (Ferrante et al., 2002). Moreover, neuroprotective drugs counter this pathology (Ferrante et al., 2002; Stack et al., 2006; Wang et al., 2008). Consequently, we monitored ventricular hypertrophy in melatonin- and saline-treated R6/2 mice. We found that melatonin treatment significantly reduced ventriculomegaly in this animal model for HD (saline, 4.07 ± 1.02 mm3; melatonin, 0.91 ± 0.39 mm3; F(2,18) = 15.12, *p < 0.01) (Fig. 4E).
Progressive accumulation of huntingtin-protein aggregates in neurons is a pathological hallmark in R6/2 mice (Turmaine et al., 2000). This process was modestly slowed by melatonin treatment, although our results were not statistically significant (saline, 5.27 × 106 ± 1.18; melatonin, 5.06 × 106 ± 1.04; F(2,12) = 21.61, p = 0.27) (Fig. 4F). The failure of melatonin to prevent formation of huntingtin aggregates resembles observations using a filter-retardation assay in vitro (Heiser et al., 2000). Furthermore, several neuroprotective in vivo regimens failed to stop such protein aggregation (Ona et al., 1999; Chen et al., 2000; Wang et al., 2008).
The most important finding discussed above is that melatonin inhibits mutant htt-induced activation of cell-death pathways in cultured cells (Fig. 1). Analogous experiments in R6/2 mice demonstrated the ability of melatonin to effect equivalent changes in vivo. In particular, mitochondrial release of cytochrome c, AIF, and Smac was significantly diminished in R6/2 mice by administration of melatonin (Fig. 5A). As reported previously, we found that brain tissue from R6/2 mice contains activated caspase-9 and caspase-3 (Chen et al., 2000; Kiechle et al., 2002; Wang et al., 2003). Moreover, administration of melatonin diminished these proapoptotic changes (Fig. 5A,B).
Figure 5.

Melatonin blocks the mitochondrial cell-death pathway in R6/2 mice. Beginning at 6 weeks of age, R6/2 mice were treated with either saline or melatonin. Wild-type littermates were used as controls. At 25 weeks, brains were removed and homogenized for Western blotting or sectioned for immunostaining. A, Some brain homogenates were fractionated by centrifugation, and the cytosolic supernatant was analyzed by Western blotting (top 3 blots). Probing with antibodies to cytochrome c, Smac, or AIF revealed the release of these proteins from mitochondria. Other samples were run as whole lysates and blotted for pro- and mature caspase-9, caspase-3, and caspase-1 or for Rip2 (bottom 4 blots). All blots were stripped and reprobed with antibodies to β-actin to confirm uniform loading. Test and control groups had three to five mice each. Bar graphs were generated by densitometry, thereby revealing the magnitude of each signal once normalized to that for β-actin (*p < 0.05, **p < 0.001). B, Brain sections were immunostained with antibodies to cytochrome c or activated caspase-3. Data from tissue slices were equivalent to those from Western blots: in R6/2 mice, there was cytochrome c release from mitochondria and caspase-3 activation. Both molecular changes were countered by administering melatonin.
The Rip2/caspase-1-mediated cell-death pathway is another physiological process important to neuronal dysfunction in R6/2 mice (Wang et al., 2005). Consequently, we determined whether the benefit of melatonin to these mice is associated with inhibition of this pathway. As described previously (Chen et al., 2000; Wang et al., 2003, 2005; Zhang et al., 2003b), Western blots of brain tissue from R6/2 mice revealed upregulation of Rip2 expression and increased caspase-1 cleavage (Fig. 5A). Consistent with our observations in cellular models for HD (Fig. 1D,E), we found that melatonin inhibits Rip2 upregulation and caspase-1 activation in R6/2 mice (Fig. 5A).
Levels of MT1 receptor decline as HD progresses; melatonin halts its depletion
It is evident from Figure 3A that depletion of MT1 receptor is associated with cell death in mutant htt ST14A cells. Knockdown of MT1 expression eliminates melatonin-mediated neuroprotection (Fig. 3E). In addition, overexpression of MT1 results in increased resistance to cell death (Fig. 3F). To evaluate the potential role of the MT1 receptor in vivo, we determined the abundance of protein in the brains of R6/2 mice. We found the MT1 receptor to be less abundant in brain tissue from symptomatic R6/2 mice than in equivalent samples from age-matched wild-type littermates. In contrast, levels of MT2 receptors were similar in brains from R6/2 and control mice. As observed for the receptor protein, there was less MT1 mRNA in R6/2 than in wild-type brains (Fig. 6A). Remarkably, treating R6/2 mice with melatonin countered the loss of MT1 receptor mRNA (p < 0.001). This increase in MT1 mRNA was reflected in a nonstatistically significant increase on MT1 protein. Consistent with observations in cellular models for HD, levels of MT2 receptor protein and mRNA were similar in brains of wild-type, saline-injected R6/2, and melatonin-treated R6/2 mice (Fig. 6B). The most significant reductions in MT1 receptor expression occurred in the cortex (Fig. 6C), with more subtle effects in the striatum (data not shown).
Figure 6.
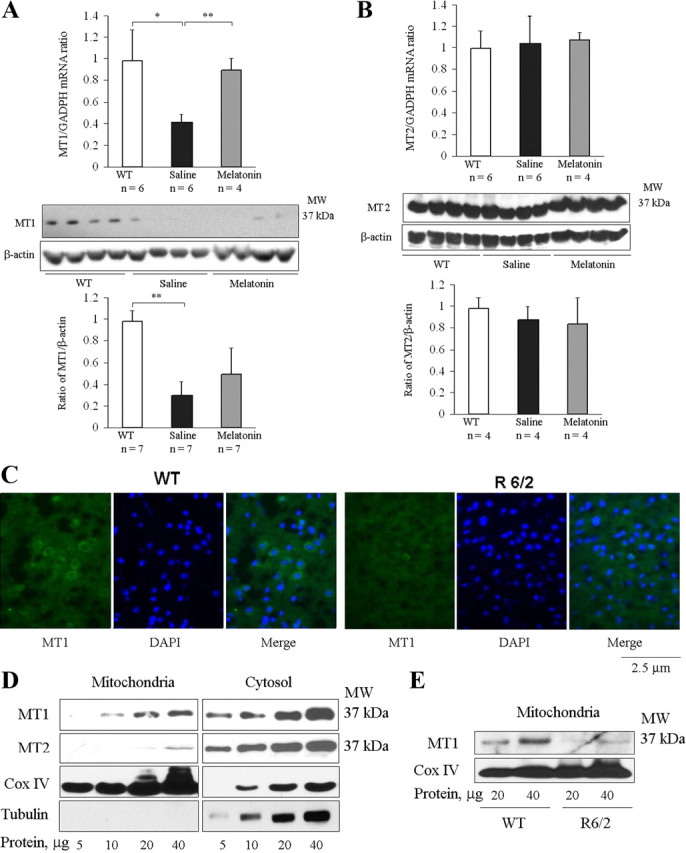
MT1 but not MT2 receptor is depleted in brains of R6/2 mice; mRNA MT1 levels are restored by melatonin. A, B, At 25 weeks, brains were removed from saline- and melatonin-treated R6/2 mice and from their wild-type littermates. Samples were homogenized and lysates were analyzed by qRT-PCR (top row) or Western blotting with antibodies to MT1 and MT2 (bottom 2 rows). Uniform loading was confirmed by reprobing with antibodies to β-actin. Images were analyzed by densitometry (bottom row) (test and control groups had n = 4–7. *p < 0.05, **p < 0.001). C, At 25 weeks, brains from R6/2 mice and their age-matched wild-type littermates were cryoprotected, sectioned, and stained with anti-MT1 antibodies. Preparations were blocked and stained with a secondary FITC-conjugated antibody. DAPI was used as a nuclear counterstain. Images (green from FITC-conjugated antibodies or blue from DAPI) are representative preparations from three R6/2 and three wild-type mice. D, E, Mitochondrial and cytosolic fractions of brain lysates were separated by centrifugation. Preparations in D were exclusively from wild-type mice; those in E were from both R6/2 and wild-type mice. D, MT1 protein was found in both cytoplasm and mitochondria, whereas MT2 was almost exclusively cytoplasmic. Blots were stripped and reprobed with antibodies to COX IV or tubulin, confirming the purity of the mitochondrial and cytoplasmic fractions. Each well was loaded with the indicated amount of protein. E, Levels of MT1 receptor in the mitochondria were lower in R6/2 brains than in wild-type controls. MW, Molecular weight.
The highest subcellular levels of melatonin are observed in mitochondria (Martín et al., 2000). Consequently, we measured levels of melatonin receptors in brain mitochondria from R6/2 and wild-type mice. We found that both MT1 and MT2 receptors are expressed in brain mitochondria (Fig. 6D), suggesting that the mitochondrial cell-death pathways may be mediated at least in part by the interactions of melatonin with its receptors, although it is possible that melatonin has additional, non-receptor-mediated targets. In whole-cell extract, the level of MT1 is lower than that of MT2 receptor (Fig. 6D, right). Consistent with its role in cell death pathways, MT1 receptor is enriched in the mitochondrial fraction (Fig. 6D, left). Furthermore, MT1 receptor is decreased fourfold in brain mitochondria from R6/2 mice compared with wild-type littermates (Fig. 6E).
Expression of MT1, MT2, and the encoding mRNAs in human striatum
To determine the relevance of our cellular and mouse models of HD to clinical cases, we investigated the expression of MT1 and MT2 in human striatum. Levels of MT1 and MT2 receptor proteins and their respective mRNAs were measured in postmortem samples of striatum from four persons who died at HD grade II and seven persons who survived until grade IV. Data from these samples were compared with that from controls, i.e., postmortem tissue from six persons deceased of non-neurologic causes. We observed progressive depletion of both the MT1 receptor protein and the encoding MT1 transcript as demonstrated by the visible MT1 lanes in the control samples, reduced intensity of MT1 bands in grade II samples, and the absence of MT1 bands in most grade IV samples (Fig. 7A). Positive bands were visible in all lanes using β-actin, demonstrating that sufficient protein was loaded in each lane. Minor differences in loading of protein were accounted for in the accompanying quantitative graph (Fig. 7A). In contrast, there was little correlation of the levels of MT2 mRNA or MT2 protein with the severity (or even presence) of HD (Fig. 7B). These analyses of melatonin receptors in human striatum confirm the relevance to clinical HD of our findings in cellular and mouse models.
Figure 7.

MT1 protein is progressively depleted in the brains of HD patients; MT2 protein is retained. A, B, Postmortem brain samples from human caudate and putamen were obtained from persons deceased at HD grades II and IV or from non-neurologic control patients. Tissue homogenate was analyzed by qRT-PCR using primers appropriate for the MT1 transcript and by Western blotting with antibodies to MT1 protein (A). Equivalent experiments were conducted using primers for MT2 mRNA and antibodies to MT2 protein (B). To confirm uniform loading, all gels were reprobed using antibodies to β-actin (n = 4 for group HD grade II, 7 for HD grade IV, and 6 for controls; *p < 0.05). MW, Molecular weight.
Discussion
Melatonin interferes with the mitochondrial and the Rip2/caspase-1 cell-death pathways
We have identified aberrant molecular events that favor activation of cell-death pathways in cellular and mouse models of HD. In particular, depletion of the MT1 receptor within mitochondria reduces the protective properties of melatonin, therefore increasing cellular vulnerability to proapoptotic challenge. An important result came from investigations on MT1 expression in conditionally immortalized mutant htt ST14A cells. In this system, we observed depletion of MT1 mRNA and protein during induction of cell death. In the mouse model of HD, melatonin treatment increases RNA expression of the MT1 receptor. However, the data do not exclude the possibility that melatonin treatment could also directly alter the expression of the huntingtin gene, affecting disease onset, progression, and survival. It is noteworthy that similar changes in the expression of melatonin receptor occur in other neurodegenerative diseases and during aging (Savaskan et al., 2002; Caballero et al., 2008; Sánchez-Hidalgo et al., 2009). Moreover, we observed low levels of MT1 (both protein and mRNA) in the brains of R6/2 mice and in postmortem specimens of HD human striatum. RNAi-mediated knockdown of MT1 receptor reduced the ability of melatonin to inhibit cell death in vitro, increasing cellular vulnerability. Conversely, overexpression of MT1 increased resistance to cellular stress.
The benefit of melatonin to stressed cells has been attributed to its ability to scavenge free radicals (Lena and Subramanian, 2003). More likely than not, melatonin counters cell death through both a receptor-mediated mechanism, as described above, and an antioxidant mechanism because the administration of a neuroprotective dose of melatonin does not completely restore MT1 expression. These actions become progressively attenuated as melatonin levels decline during aging (Mocchegiani et al., 1994; Carranza-Lira and Garcia Lopez, 2000). Changes in expression of the melatonin receptor may also contribute to this downward trend. There is a large variability in melatonin productivity depending on mouse genetic background (Kasahara et al., 2010), and inbred strains of laboratory mice such as C57BL/6J were found not to have detectable melatonin in their pineal glands.
Our investigations on melatonin were motivated by the ability of the compound to inhibit calcium-induced cytochrome c release from purified mitochondria (Wang et al., 2008). When administered to cells, melatonin blocked transfer to the cytoplasm of cytochrome c and blocked the release of mitochondrial factors Smac/Diablo and AIF. Because mitochondria are the organelles dedicated to generation of energy, they affect every aspect of cellular metabolism. Moreover, maintaining a rich store of ATP is particularly important at times of cellular stress, e.g., when cells are challenged by the expression of mutant htt. A molecular event characteristic of the cell-death process is dissipation of the mitochondrial membrane potential (ΔΨm). Not surprisingly, loss of ΔΨm is observed in models for many neurodegenerative disease, including ones for HD (Andrabi et al., 2004). We have now demonstrated that melatonin inhibits mutant htt-induced ΔΨm loss (Fig. 1F). This activity further inhibits the release of mitochondrial proapoptotic factors and makes melatonin all the more potent at countering cell death.
Rip2 dysregulation mediates aberrant caspase-1 activation in HD (Wang et al., 2003) and in cerebral ischemia (Zhang et al., 2003a). Other researchers have observed increased levels of Rip2 in Alzheimer's disease (Engidawork et al., 2001). We recently reported that melatonin inhibits caspase-1 activation in an experimental model of cerebral ischemia (Wang et al., 2009). We now show that melatonin counters the upregulation of Rip2 in both mutant htt ST14A cells transferred to nonpermissive conditions (Fig. 1) and R6/2 transgenic mice (Fig. 5). Moreover, Rip2 protein stimulates autocatalytic activation of procaspase-1 (Thome et al., 1998). The ability of melatonin to rescue stressed cells is apt to result, at least in part, from its targeting of this upstream event in the cell-death pathway. Alternatively, or in addition, the primary target of melatonin may be the mitochondria, because upstream modulation of cell death pathways may result from a feedback-loop response.
Melatonin and its analogs are candidate drugs for HD therapy
The potential benefits of melatonin have been evaluated in patients with and in animal models for Alzheimer's disease (Srinivasan et al., 2006), amyotrophic lateral sclerosis (Weishaupt et al., 2006), Parkinson's disease (Willis and Armstrong, 1999; Medeiros et al., 2007), stroke (Kilic et al., 2004; Dominguez-Rodriguez et al., 2007), epilepsy (Gupta et al., 2004; Savina et al., 2006), and traumatic brain injury (Ozdemir et al., 2005).
Melatonin is an endogenous hormone with great importance to health and disease. A gradual decrease in melatonin production has been observed in both a mouse model of aging (Mocchegiani et al., 1994) and investigations on human subjects (Carranza-Lira and Garcia Lopez, 2000). Recently, a significant delay was reported in melatonin peaks in humans with early-stage HD. Furthermore, there is a progressive reduction of melatonin blood levels associated with increasing disease severity (Aziz et al., 2009). Low levels of circulating melatonin is not specific to HD; it has also been observed in patients with Alzheimer's disease (Ozcankaya and Delibas, 2002) and Parkinson's disease (Blazejova et al., 2000).
2-Iodomelatonin, an agonist of MT1 and MT2, shares the neuroprotective properties of melatonin. Conversely, luzindole, an antagonist of these two receptors, eliminates neuroprotection by melatonin. The structures of these three molecules are shown in Figure 2, and their respective effects on cell death are noted. These observations are of practical importance: melatonin analogs and other agonists of the melatonin receptors are potential drugs for treating HD. In this manner, we hope to identify molecules that have a longer half-life than melatonin (20–50 min) (Waldhauser et al., 1984).
We and others have conducted drug trials in HD transgenic mice. Thirty-two potentially therapeutic agents are analyzed for their relative effectiveness in monotherapies (table not shown). Only five of these test compounds were observed to extend survival by >20%. Melatonin was one such drug, delaying mortality by 21%. The low toxicity of melatonin and its ability to cross the blood–brain barrier make it a potentially useful compound for the treatment of HD.
In conclusion, we have demonstrated that the interaction of melatonin with the mitochondrial MT1 receptor is critical for neuroprotection. Neither the localization of MT1 receptors to mitochondria nor their selective loss in HD has been reported previously. In HD, pathologically low levels of circulating melatonin and reduced expression of MT1 receptor increase the vulnerability of neurons to cell death. We have also demonstrated that therapies that restore high levels of melatonin–MT1 receptor complexes provide a potential avenue for neuroprotection in HD.
Footnotes
This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (R.M.F., X.W., R.J.F.), the Muscular Dystrophy Association (X.W.), the Huntington's Disease Society of America (R.M.F.,) and the Veterans Administration (R.J.F.). We thank Dr. Ethan M. Shimony and Dr. Diane Carlisle for editorial assistance, Dr. Irina Stavrovskaya, Jennifer Moody, and Diane Sheldon for experimental assistance, and Dr. Ahmad Aziz for insightful discussion.
References
- Adamczyk A, Czapski GA, Jesko H, Strosznajder RP. Non A beta component of Alzheimer's disease amyloid and amyloid beta peptides evoked poly(ADP-ribose) polymerase-dependent release of apoptosis-inducing factor from rat brain mitochondria. J Physiol Pharmacol. 2005;56(Suppl 2):5–13. [PubMed] [Google Scholar]
- Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004;18:869–871. doi: 10.1096/fj.03-1031fje. [DOI] [PubMed] [Google Scholar]
- Aziz NA, Pijl H, Frölich M, Schröder-van der Elst JP, van der Bent C, Roelfsema F, Roos RA. Delayed onset of the diurnal melatonin rise in patients with Huntington's disease. J Neurol. 2009;256:1961–1965. doi: 10.1007/s00415-009-5196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- Blazejová K, Nevsímalová S, Illnerová H, Hájek I, Sonka K. Sleep disorders and the 24-hour profile of melatonin and cortisol. Sb Lek. 2000;101:347–351. [PubMed] [Google Scholar]
- Brydon L, Petit L, Delagrange P, Strosberg AD, Jockers R. Functional expression of MT2 (Mel1b) melatonin receptors in human PAZ6 adipocytes. Endocrinology. 2001;142:4264–4271. doi: 10.1210/endo.142.10.8423. [DOI] [PubMed] [Google Scholar]
- Caballero B, Vega-Naredo I, Sierra V, Huidobro-Fernández C, Soria-Valles C, De Gonzalo-Calvo D, Tolivia D, Gutierrez-Cuesta J, Pallas M, Camins A, Rodríguez-Colunga MJ, Coto-Montes A. Favorable effects of a prolonged treatment with melatonin on the level of oxidative damage and neurodegeneration in senescence-accelerated mice. J Pineal Res. 2008;45:302–311. doi: 10.1111/j.1600-079X.2008.00591.x. [DOI] [PubMed] [Google Scholar]
- Carranza-Lira S, García López F. Melatonin and climactery. Med Sci Monit. 2000;6:1209–1212. [PubMed] [Google Scholar]
- Cazevieille C, Safa R, Osborne NN. Melatonin protects primary cultures of rat cortical neurones from NMDA excitotoxicity and hypoxia/reoxygenation. Brain Res. 1997;768:120–124. doi: 10.1016/s0006-8993(97)00611-2. [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P, Housman DE, Kazantsev A, Young AB, Hersch S. A small-molecule therapeutic lead for Huntington's disease: preclinical pharmacology and efficacy of C2–8 in the R6/2 transgenic mouse. Proc Natl Acad Sci U S A. 2007;104:16685–16689. doi: 10.1073/pnas.0707842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogburn LA, Wilson-Placentra S, Letcher LR. Influence of pinealectomy on plasma and extrapineal melatonin rhythms in young chickens (Gallus domesticus) Gen Comp Endocrinol. 1987;68:343–356. doi: 10.1016/0016-6480(87)90073-6. [DOI] [PubMed] [Google Scholar]
- Das A, Belagodu A, Reiter RJ, Ray SK, Banik NL. Cytoprotective effects of melatonin on C6 astroglial cells exposed to glutamate excitotoxicity and oxidative stress. J Pineal Res. 2008;45:117–124. doi: 10.1111/j.1600-079X.2008.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Rodriguez A, Abreu-Gonzalez P, Garcia-Gonzalez MJ, Kaski JC, Reiter RJ, Jimenez-Sosa A. A unicenter, randomized, double-blind, parallel-group, placebo-controlled study of Melatonin as an adjunct in patients with acute myocardial infarction undergoing primary angioplasty: the melatonin adjunct in the acute myocardial infarction treated with angioplasty (MARIA) trial: study design and rationale. Contemp Clin Trials. 2007;28:532–539. doi: 10.1016/j.cct.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Drew JE, Barrett P, Mercer JG, Moar KM, Canet E, Delagrange P, Morgan PJ. Localization of the melatonin-related receptor in the rodent brain and peripheral tissues. J Neuroendocrinol. 2001;13:453–458. doi: 10.1046/j.1365-2826.2001.00651.x. [DOI] [PubMed] [Google Scholar]
- Engidawork E, Gulesserian T, Yoo BC, Cairns N, Lubec G. Alteration of caspases and apoptosis-related proteins in brains of patients with Alzheimer's disease. Biochem Biophys Res Commun. 2001;281:84–93. doi: 10.1006/bbrc.2001.4306. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates. New York: Academic; 2000. [Google Scholar]
- Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Gupta M, Aneja S, Kohli K. Add-on melatonin improves quality of life in epileptic children on valproate monotherapy: a randomized, double-blind, placebo-controlled trial. Epilepsy Behav. 2004;5:316–321. doi: 10.1016/j.yebeh.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, Lehrach H, Wanker EE. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc Natl Acad Sci U S A. 2000;97:6739–6744. doi: 10.1073/pnas.110138997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Abe K, Mekada K, Yoshiki A, Kato T. Genetic variation of melatonin productivity in laboratory mice under domestication. Proc Natl Acad Sci U S A. 2010;107:6412–6417. doi: 10.1073/pnas.0914399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiechle T, Dedeoglu A, Kubilus J, Kowall NW, Beal MF, Friedlander RM, Hersch SM, Ferrante RJ. Cytochrome C and caspase-9 expression in Huntington's disease. Neuromolecular Med. 2002;1:183–195. doi: 10.1385/NMM:1:3:183. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Yulug B, Hermann DM, Reiter RJ. Melatonin reduces disseminate neuronal death after mild focal ischemia in mice via inhibition of caspase-3 and is suitable as an add-on treatment to tissue-plasminogen activator. J Pineal Res. 2004;36:171–176. doi: 10.1046/j.1600-079x.2003.00115.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Kromminga A, Dunlop TW, Tychsen B, Conrad F, Suzuki N, Memezawa A, Bettermann A, Aiba S, Carlberg C, Paus R. A role of melatonin in neuroectodermal-mesodermal interactions: the hair follicle synthesizes melatonin and expresses functional melatonin receptors. FASEB J. 2005;19:1710–1712. doi: 10.1096/fj.04-2293fje. [DOI] [PubMed] [Google Scholar]
- Krasnikov BF, Zorov DB, Antonenko YN, Zaspa AA, Kulikov IV, Kristal BS, Cooper AJ, Brown AM. Comparative kinetic analysis reveals that inducer-specific ion release precedes the mitochondrial permeability transition. Biochim Biophys Acta. 2005;1708:375–392. doi: 10.1016/j.bbabio.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Lena PJ, Subramanian P. Evaluation of the antiperoxidative effects of melatonin in ammonium acetate-treated Wistar rats. Pol J Pharmacol. 2003;55:1031–1036. [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li XJ. The early cellular pathology of Huntington's disease. Mol Neurobiol. 1999;20:111–124. doi: 10.1007/BF02742437. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Martín M, Macías M, Escames G, León J, Acuña-Castroviejo D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000;14:1677–1679. doi: 10.1096/fj.99-0865fje. [DOI] [PubMed] [Google Scholar]
- Masuda N, Peng Q, Li Q, Jiang M, Liang Y, Wang X, Zhao M, Wang W, Ross CA, Duan W. Tiagabine is neuroprotective in the N171-82Q and R6/2 mouse models of Huntington's disease. Neurobiol Dis. 2008;30:293–302. doi: 10.1016/j.nbd.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros CA, Carvalhedo de Bruin PF, Lopes LA, Magalhães MC, de Lourdes Seabra M, de Bruin VM. Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson's disease. A randomized, double blind, placebo-controlled study. J Neurol. 2007;254:459–464. doi: 10.1007/s00415-006-0390-x. [DOI] [PubMed] [Google Scholar]
- Mocchegiani E, Bulian D, Santarelli L, Tibaldi A, Muzzioli M, Pierpaoli W, Fabris N. The immuno-reconstituting effect of melatonin or pineal grafting and its relation to zinc pool in aging mice. J Neuroimmunol. 1994;53:189–201. doi: 10.1016/0165-5728(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Niles LP, Wang J, Shen L, Lobb DK, Younglai EV. Melatonin receptor mRNA expression in human granulosa cells. Mol Cell Endocrinol. 1999;156:107–110. doi: 10.1016/s0303-7207(99)00135-5. [DOI] [PubMed] [Google Scholar]
- Niles LP, Armstrong KJ, Rincón Castro LM, Dao CV, Sharma R, McMillan CR, Doering LC, Kirkham DL. Neural stem cells express melatonin receptors and neurotrophic factors: colocalization of the MT1 receptor with neuronal and glial markers. BMC Neurosci. 2004;5:41. doi: 10.1186/1471-2202-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- Ozcankaya R, Delibas N. Malondialdehyde, superoxide dismutase, melatonin, iron, copper, and zinc blood concentrations in patients with Alzheimer disease: cross-sectional study. Croat Med J. 2002;43:28–32. [PubMed] [Google Scholar]
- Ozdemir D, Uysal N, Gonenc S, Acikgoz O, Sonmez A, Topcu A, Ozdemir N, Duman M, Semin I, Ozkan H. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol Res. 2005;54:631–637. [PubMed] [Google Scholar]
- Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- Ravindra T, Lakshmi NK, Ahuja YR. Melatonin in pathogenesis and therapy of cancer. Indian J Med Sci. 2006;60:523–535. [PubMed] [Google Scholar]
- Reiter RJ, Tan DX. Role of CSF in the transport of melatonin. J Pineal Res. 2002;33:61. doi: 10.1034/j.1600-079x.2002.2e001.x. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Sipione S, Goffredo D, Zuccato C, Fossale E, Cattaneo E. Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J Biol Chem. 2001;276:14545–14548. doi: 10.1074/jbc.C100044200. [DOI] [PubMed] [Google Scholar]
- Sánchez-Hidalgo M, Guerrero Montávez JM, Carrascosa-Salmoral Mdel P, Naranjo Gutierrez Mdel D, Lardone PJ, de la Lastra Romero CA. Decreased MT1 and MT2 melatonin receptor expression in extrapineal tissues of the rat during physiological aging. J Pineal Res. 2009;46:29–35. doi: 10.1111/j.1600-079X.2008.00604.x. [DOI] [PubMed] [Google Scholar]
- Savaskan E, Wirz-Justice A, Olivieri G, Pache M, Kräuchi K, Brydon L, Jockers R, Müller-Spahn F, Meyer P. Distribution of melatonin MT1 receptor immunoreactivity in human retina. J Histochem Cytochem. 2002;50:519–526. doi: 10.1177/002215540205000408. [DOI] [PubMed] [Google Scholar]
- Savaskan E, Jockers R, Ayoub M, Angeloni D, Fraschini F, Flammer J, Eckert A, Müller-Spahn F, Meyer P. The MT2 melatonin receptor subtype is present in human retina and decreases in Alzheimer's disease. Curr Alzheimer Res. 2007;4:47–51. doi: 10.2174/156720507779939823. [DOI] [PubMed] [Google Scholar]
- Savina TA, Balashova OA, Shchipakina TG. Effect of chronic consumption of sodium valproate and melatonin on seizure activity in Krushinskii-Molodkina rats. Bull Exp Biol Med. 2006;142:601–604. doi: 10.1007/s10517-006-0429-0. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Southgate G, Daya S. Melatonin reduces quinolinic acid-induced lipid peroxidation in rat brain homogenate. Metab Brain Dis. 1999;14:165–171. doi: 10.1023/a:1020610708637. [DOI] [PubMed] [Google Scholar]
- Srinivasan V, Pandi-Perumal SR, Cardinali DP, Poeggeler B, Hardeland R. Melatonin in Alzheimer's disease and other neurodegenerative disorders. Behav Brain Funct. 2006;2:15. doi: 10.1186/1744-9081-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Smith KM, Ryu H, Cormier K, Chen M, Hagerty SW, Del Signore SJ, Cudkowicz ME, Friedlander RM, Ferrante RJ. Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic Huntington's disease mice. Biochim Biophys Acta. 2006;1762:373–380. doi: 10.1016/j.bbadis.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radic Biol Med. 2005;38:687–697. doi: 10.1016/j.freeradbiomed.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Tan DX, Reiter RJ, Manchester LC, Yan MT, El-Sawi M, Sainz RM, Mayo JC, Kohen R, Allegra M, Hardeland R. Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem. 2002;2:181–197. doi: 10.2174/1568026023394443. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–888. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- Túnez I, Montilla P, Del Carmen Muñoz M, Feijóo M, Salcedo M. Protective effect of melatonin on 3-nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of Huntington's disease. J Pineal Res. 2004;37:252–256. doi: 10.1111/j.1600-079X.2004.00163.x. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhauser F, Waldhauser M, Lieberman HR, Deng MH, Lynch HJ, Wurtman RJ. Bioavailability of oral melatonin in humans. Neuroendocrinology. 1984;39:307–313. doi: 10.1159/000123997. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang H, Figueroa BE, Zhang WH, Huo C, Guan Y, Zhang Y, Bruey JM, Reed JC, Friedlander RM. Dysregulation of receptor interacting protein-2 and caspase recruitment domain only protein mediates aberrant caspase-1 activation in Huntington's disease. J Neurosci. 2005;25:11645–11654. doi: 10.1523/JNEUROSCI.4181-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, Kristal BS, Friedlander RM. Inhibitors of cytochrome c release with therapeutic potential for Huntington's disease. J Neurosci. 2008;28:9473–9485. doi: 10.1523/JNEUROSCI.1867-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Sirianni AC, Zhu S, Day AL, Kristal BS, Friedlander RM. Methazolamide and melatonin inhibit mitochondrial cytochrome c release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40:1877–1885. doi: 10.1161/STROKEAHA.108.540765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weishaupt JH, Bartels C, Pölking E, Dietrich J, Rohde G, Poeggeler B, Mertens N, Sperling S, Bohn M, Hüther G, Schneider A, Bach A, Sirén AL, Hardeland R, Bähr M, Nave KA, Ehrenreich H. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J Pineal Res. 2006;41:313–323. doi: 10.1111/j.1600-079X.2006.00377.x. [DOI] [PubMed] [Google Scholar]
- Willis GL, Armstrong SM. A therapeutic role for melatonin antagonism in experimental models of Parkinson's disease. Physiol Behav. 1999;66:785–795. doi: 10.1016/s0031-9384(99)00023-2. [DOI] [PubMed] [Google Scholar]
- Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC, Friedlander RM. Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci U S A. 2003a;100:16012–16017. doi: 10.1073/pnas.2534856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ona VO, Li M, Drozda M, Dubois-Dauphin M, Przedborski S, Ferrante RJ, Friedlander RM. Sequential activation of individual caspases, and of alterations in Bcl-2 proapoptotic signals in a mouse model of Huntington's disease. J Neurochem. 2003b;87:1184–1192. doi: 10.1046/j.1471-4159.2003.02105.x. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]