

Abstract
Insulin resistance in skeletal muscle is a prominent feature of obesity and type 2 diabetes. The association between mitochondrial changes and insulin resistance is well known. More recently, there is growing evidence of a relationship between inflammation, extracellular remodeling, and insulin resistance. The intent of this review is to propose a potentially novel mechanism for the development of insulin resistance, focusing on the underappreciated connections among inflammation, extracellular remodeling, cytoskeletal interactions, mitochondrial function, and insulin resistance in human skeletal muscle. Several sources of inflammation, including expansion of adipose tissue resulting in increased lipolysis and alterations in pro- and anti-inflammatory cytokines, contribute to the insulin resistance observed in obesity and type 2 diabetes. In the experimental model of lipid oversupply, an inflammatory response in skeletal muscle leads to altered expression extracellular matrix-related genes as well as nuclear encoded mitochondrial genes. A similar pattern also is observed in “naturally” occurring insulin resistance in muscle of obese nondiabetic individuals and patients with type 2 diabetes mellitus. More recently, alterations in proteins (including α-actinin-2, desmin, proteasomes, and chaperones) involved in muscle structure and function have been observed in insulin-resistant muscle. Some of these cytoskeletal proteins are mechanosignal transducers that allow muscle fibers to sense contractile activity and respond appropriately. The ensuing alterations in expression of genes coding for mitochondrial proteins and cytoskeletal proteins may contribute to the mitochondrial changes observed in insulin-resistant muscle. These changes in turn may lead to a reduction in fat oxidation and an increase in intramyocellular lipid, which contributes to the defects in insulin signaling in insulin resistance.
Keywords: skeletal muscle, inflammation
insulin resistance in skeletal muscle is considered to be a prominent feature of obesity, type 2 diabetes, and other diseases associated with cardiometabolic risk. In point of fact, there is a broad range of insulin action in skeletal muscle, with many apparently healthy individuals having insulin action on the lower end of the distribution (Fig. 1). The position of an individual on the standard curve of insulin action, that is, their insulin action phenotype, is likely governed by familial, presumably genetic factors and the individual's environment, including variables such as physical activity and diet. Lower insulin action has been associated with lower insulin-stimulated activities of enzymes such as glycogen synthase and hexokinase (35, 45) and reduced ability of insulin to activate a variety of elements of the insulin signaling system, including tyrosine phosphorylation of the insulin receptor and insulin receptor substrate (IRS-1) (13, 30). More recently, lower insulin action has been associated with reduced content, and perhaps elements of function, of mitochondria in skeletal muscle that has more characteristics of fast-twitch, glycolytic, type II muscle fibers (11, 26, 46). As such, muscle exhibiting lower insulin action displays many characteristics of muscle subjected to “insufficient” physical activity, although the interplay between insulin action and exercise is complex (40).
Fig. 1.

The distribution of the M values (mg·kg−1·min−1), measured by insulin-stimulated glucose disposal during the euglycemic clamp in 45 normal, glucose-tolerant, healthy subjects. Data were obtained during an 80 mU·m−2·min−1 insulin infusion and were taken from unpublished (Mandarino LJ) and published (14, 23, 32) sources.
Related to these changes have been a number of gene expression differences between muscle from individuals with higher and lower levels of insulin action. For example, peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), a transcriptional activator with an important role in mitochondrial biogenesis and formation of type I muscle fibers (33), exhibits reduced expression in individuals with a family history of type 2 diabetes, obesity, or type 2 diabetes mellitus itself (37, 42). This lower expression of PGC-1α is accompanied by lower expression of mRNAs (23, 32) and protein abundance of a large number of nuclear-encoded mitochondrial genes (23, 32).
Recent evidence suggests that inflammation is associated with lower insulin action as well (19). It is not clear at this point whether inflammation precedes or merely coincides with lower insulin action, whether individuals who reside on the lower end of the insulin action curve have greater propensity for inflammatory processes, or whether inflammation itself moves an individual leftward along the insulin action curve to a state of reduced insulin action that eventually leads to disease propensity. At this point, the mechanistic or causal relationship between lower insulin action in skeletal muscle and disease is far from clear. Here, we review data from our laboratory that we believe point to a novel potential mechanism for the development of insulin resistance in skeletal muscle. This is not intended to be an exhaustive review of this subject, and the reader is referred to several excellent reviews in this area (1, 7, 17, 24, 38, 50, 51).
Sources of Inflammation
Several sources of inflammation have been associated with insulin resistance in muscle (Fig. 2). The recognition that adipose tissue can be a major source of proinflammatory cytokines has been a major advance in understanding the sources of inflammation associated with lower insulin action. Expansion of adipose tissue has been associated with increases in proinflammatory cytokines, including tumor necrosis factor-α (TNFα), interleukin 6 (IL-6), resistin, and retinol-binding protein, and insulin resistance is associated with reductions in plasma levels of the anti-inflammatory cytokine adiponectin (34, 57). It is conceivable that expansion of adipose tissue, accompanied by increased release of inflammatory cytokines, especially in individuals already on the lower end of the insulin action distribution, could tip the balance toward disease.
Fig. 2.
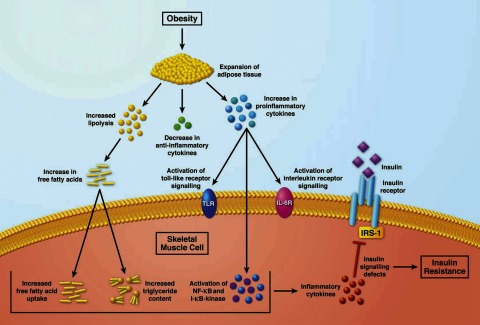
Sources of inflammation associated with insulin resistance in skeletal muscle. IRS-1, insulin receptor substrate-1.
These inflammatory cytokines can act on a variety of receptors in skeletal muscle to activate Toll-like receptor signaling, interleukin receptor signaling, NF-κB, and IκB kinase signaling, as well as a number of other inflammatory kinases (39, 47, 52, 54). It is believed, albeit far from demonstrated in humans, that many of these inflammatory kinases can reduce insulin signaling by phosphorylating IRS-1 on several serine residues that might lower the ability of IRS-1 to transduce the insulin signal (39, 52). It is not clear whether this is the only or primary mechanism responsible for lower insulin action in states of chronic inflammation, and much work remains to establish the validity of this model.
Expansion of adipose tissue is an inevitable outcome of a positive energy balance. In Western societies, a common route to a positive energy balance is a combination of low physical activity and a high-fat diet. A diet high in fat content not only increases adipose tissue mass but also increases plasma free fatty acid and triglyceride concentrations. In vitro, elevated free fatty acids can inhibit insulin action in a variety of ways (20, 55, 58). In vivo, higher free fatty acids and triglycerides induced by an infusion of lipid emulsions, with or without heparin to activate lipase, have been shown to reduce insulin action and insulin signaling, as measured by a decreased ability of insulin to stimulate glucose disposal (8, 9, 12, 15, 27, 28, 48). Infusion of lipids itself increases inflammatory cytokines and activates inflammatory signaling in muscle (4, 49).
Toll-like receptors (TLRs) play an important role in the innate immune system by activating inflammatory pathways. TLR4, the best-characterized TLR, functions as the receptor for lipopolysaccharide (LPS) of gram-negative bacterial cell walls (36). In addition, there is evidence that saturated and polyunsaturated fatty acids signal through TLR, particularly TLR4 (22). Activation of the TLR4 pathway induces the expression of a large number of proinflammatory genes (47). It does so by regulating the activities of signal-dependent transcription factors that include NF-κB, activator protein-1, and interferon regulatory factor family members (39). Obese and type 2 diabetic subjects had significantly elevated TLR4 gene expression and protein content in muscle, which correlated with the severity of insulin resistance. Obese and type 2 diabetic subjects also had lower IκBα content, an indication of elevated IκB/NF-κB signaling. The increase in TLR4 and NF-κB signaling was accompanied by elevated expression of the NF-κB-regulated proinflammatory genes IL-6 and superoxide dismutase 2 (47).
Changes in Plasma Lipid Levels Alter Gene Expression in Skeletal Muscle
The decrease in expression of PGC-1α referred to above was related to plasma free fatty acid concentrations in vivo in humans (42). This finding led to a test of the hypothesis that raising plasma lipid levels in healthy individuals itself was sufficient to lower PGC-1α expression (49). In this experiment, a lipid emulsion was infused for 48 h, followed by a euglycemic clamp experiment to assess insulin action along with percutaneous needle muscle biopsies (49). A 48-h saline infusion served as the control experiment. Using a combination of quantitative PCR and oligonucleotide microarrays, the lipid infusion was found to lower PGC-1α expression as well as suppress the mRNA levels for a number of genes coding for proteins involved in mitochondrial function (49). However, the use of global gene expression analysis revealed that unexpected and more profound changes were occurring simultaneously. The mRNA levels for a number of extracellular matrix-related genes were increased profoundly, including various collagens, fibronectin, proteoglycans, matrix metalloproteinases, and others (Fig. 3). These changes were confirmed at the protein level using immunoblot analysis. The pattern of gene and protein changes was strikingly similar to typical fibrotic responses seen in response to inflammation in a number of other circumstances, including (perhaps not coincidentally) nonalcoholic steatohepatitis (4). In some diseases, such as scleroderma or pulmonary fibrosis, connective tissue growth factor (CTGF) expression is increased in response to inflammatory stimuli such as TGFβ (49). After lipid infusion, CTGF expression levels were increased, although there was no evidence of increases in expression of TGFβ or other inflammatory cytokines directly in muscle (49), suggesting that circulating cytokines could be responsible for these effects.
Fig. 3.

mRNA expression levels from 7 subjects for collagens (COL), lumican (LUM), fibronectin (FN1), and connective tissue growth factor (CTGF) following a 48-h lipid infusion in normal, glucose-tolerant, healthy subjects normalized to 18S ribosomal RNA and shown as means ± SE of fold increase compared with a 48-h saline infusion in the same participants. *P < 0.05, lipid infusion vs. saline. Data are redrawn from Richardson et al. (49).
Collagen Content Is Increased in Naturally Occurring Insulin Resistance
On the basis of our findings that experimental insulin resistance induced by a lipid infusion also profoundly increases expression of a wide array of genes encoding extracellular matrix proteins, we set out to determine whether similar changes could be found in the setting of “naturally” occurring insulin resistance in muscle of obese nondiabetic individuals and patients with type 2 diabetes mellitus. To answer this, we used percutaneous biopsies of the vastus lateralis muscle taken from lean, healthy volunteers and insulin-resistant obese nondiabetic and diabetic participants (4). As expected, the obese and diabetic individuals were insulin resistant compared with lean controls. Collagen content of muscle was assessed by two means. First, hydroxyproline content of acid hydrolysates of muscle was used as a marker for collagen (4), and second, immunofluorescence histochemistry was employed to localize the site or sites of collagen deposition. Hydroxyproline content of tissue from both obese and diabetic volunteers showed a profound increase, providing evidence for increased collagen content. Immunofluorescence staining also showed increased collagen types I and III deposition and revealed that, similar to data from the lipid infusion experiments (Fig. 4A), the major increases in collagen deposition occurred in the endomysium, the matrix layer surrounding individual muscle fibers. Excess deposition of collagen appeared to be similar in obese nondiabetic and type 2 diabetic individuals (Fig. 4B), suggesting that this was associated with insulin resistance rather than hyperglycemia.
Fig. 4.
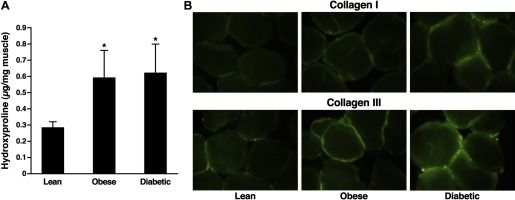
A: hydroxyproline content of acid hydrolysates of biopsies of vastus lateralis muscle from lean (n = 10), obese (n = 10), and diabetic (n = 10) subjects. Data are shown as means ± SE. *P < 0.05 vs. lean control values. Data are redrawn from Berria et al. (4). B: immunofluorescence staining of 5-μm sections of biopsies of vastus lateralis muscle from lean, obese, and diabetic subjects for types I (top) and III (bottom) collagen. Data are redrawn from Berria et al. (4).
A study in human adipose tissue demonstrated that collagen VI-α3 (COL6A3)-subunit mRNA expression was increased significantly in obese and type 2 diabetic patients (41). The obese subjects with high COL6A3 mRNA had greater visceral adipose tissue mass, lower size of small and medium adipocytes, more CD68+ and CD163/MAC2+ macrophages, and increased macrophage inflammatory protein-1α and macrophage chemoattractant protein-1α mRNA (41). These results suggest that adipose tissue fibrosis is present in human obesity.
Similar to the human studies described above, high-fat-fed mice had insulin resistance and increased muscle collagen III and collagen IV protein (25). Rescue of muscle insulin resistance by genetic, muscle-specific, mitochondria-targeted catalase overexpression or by the phosphodiesterase 5a inhibitor sildenafil reversed high-fat feeding effects on extracellular matrix remodeling and increased muscle vascularity (25). These results in mice further demonstrate that extracellular matrix collagen expansion is tightly associated with muscle insulin resistance.
Taking all these results together, we predicted that decreasing plasma free fatty acids would decrease the expression of genes encoding for these extracellular proteins. Although a reduction in lipid supply by acipimox treatment reduced free fatty acids and improved insulin sensitivity (2), which was concomitant with a decrease in intramuscular fatty acyl-CoA, it did not reverse the molecular changes associated with lipid oversupply in muscle. Paradoxically, the expression of collagens and other extracellular matrix genes such as connective tissue growth factor and TGFβ1 was increased following treatment with acipimox (2). This suggests that changes in insulin sensitivity in either direction, induced by changes in plasma free fatty acids, are accompanied by a remodeling of the extracellular matrix.
Global Proteomics Analysis Leads to Potentially Novel Mechanisms of Insulin Resistance
On the basis of these surprising and sizeable changes in extracellular matrix protein abundance found in insulin-resistant muscle, a global mass spectrometry-based proteomics discovery experiment was undertaken to find other unexpected changes in insulin-resistant muscle. As before, we used lean, healthy controls, obese nondiabetics, and patients with type 2 diabetes and subjected muscle biopsies to mass spectrometry analysis using a normalized spectrum abundance factor method (44, 60, 61) for quantification (23).
On the basis of earlier studies (37, 42, 53), we expected that we would find decreases in mitochondrial proteins. Mitochondrial proteins make up more than 20% of the human muscle proteome (18). In all, 1,218 proteins were assigned in the study, and 400 could be quantified in at least one-half of all subjects. Muscle from patients with type 2 diabetes demonstrated the largest decreases in abundance of a variety of mitochondrial proteins in a wide range of functional categories, including electron transport proteins (23). Other unexpected proteins and groups of proteins also emerged from this analysis as being potentially dysregulated in insulin-resistant muscle. For example, chaperone proteins, including the TCP1 family of cytosolic protein chaperones, were increased dramatically in insulin-resistant muscle (23). The TCP1 proteins comprise the major cytosolic chaperone complex that is responsible for folding proteins such as actin (31, 59). In addition, a large number of proteasome subunits were increased in abundance, suggesting increased protein degradation in insulin-resistant muscle. The combination of increased chaperone and proteasome proteins in insulin resistance suggests a generalized increase in protein turnover. This, for example, could reflect inflammation or endoplasmic reticulum stress (5, 6, 21), although specific proteins involved in the unfolded protein response to endoplasmic reticulum stress were not represented in the proteins assigned or quantified in this experiment, perhaps due to low abundance.
Perhaps most dramatic of all were decreases in insulin-resistant muscle in proteins involved in muscle structure and function. Both α-actinin-2 and desmin were decreased profoundly in insulin-resistant muscle (23). Desmin is the major component of intermediate filaments in skeletal muscle and heart. Intermediate filaments tie together the sarcomere, the fundamental contractile unit, with the extracellular matrix through dystroglycan and sarcoglycan complexes in the sarcolemma. These intermediate filaments can be thought of as “cables” that transmit force from the sarcomere to the extracellular matrix and then eventually to tendons to apply force for movement. In addition, intermediate filaments are mechanosignal transducers that allow muscle fibers to sense contractile activity and respond appropriately with changes in gene expression that can accommodate increases or decreases in activity (10). Of potential import in this regard are findings that insulin-resistant muscle is also “exercise resistant” in its response to a bout of exercise or muscle contraction (14). Lack of desmin produces a variety of “desminopathies” that affect both heart and skeletal muscle (43).
The other major structural protein that is altered in abundance in insulin resistance is α-actinin-2. Actinin-2 is the major isoform of actinin in skeletal muscle and heart, with actinin 3 being a minor isoform that is not expressed in about 20% of humans without adverse consequences (3). Actinin-2 binds to and anchors α-actin at the Z-disk of the sarcomere and is the major component of Z-disks (10). Additionally, actinin-2 participates in protein-protein interactions of a variety of cellular proteins with the actin cytoskeleton and participates in a variety of other functions in addition to skeletal muscle contraction via α-actin (10, 29). We found actinin-2 to be decreased by nearly 50% by both mass spectrometry analysis and immunoblot analysis in insulin-resistant muscle (23). At this point, the decrease has not been localized to Z-disk or cytoskeletal actinin-2, but the magnitude of the decrease makes it likely that the Z-disk actinin is decreased because it comprises the majority of cellular actinin in skeletal muscle. The extent of a decrease in cytoskeletal-associated actinin-2 could not be determined from these studies.
With respect to actinin in insulin resistance, it may be significant that, in L6 myotubes, α-actinin-4, which binds filamentous cytosolic actin, associates with glucose transporter 4 (GLUT4) either directly or indirectly (16), and even more significantly, knockdown of actinin-4 abrogates insulin-stimulated GLUT4 translocation in these cells (56). Actinin-4 abundance is much lower than actinin-2 in human skeletal muscle. In fact, in our proteomics analysis, only a single spectrum from a peptide unique to actinin-4 was assigned in only one of 24 total biopsies analyzed (23). In contrast, hundreds of actinin-2 spectra were assigned in human skeletal muscle; in all, there were 24 biopsies. It is tempting to speculate that, although the direct applicability of the actinin-4 results in L6 myotubes to human muscle can be questioned, it is possible that actinin-2 serves the same function in human muscle. This could imply that a possible mechanism of defects in GLUT4 translocation in human muscle involves the deficit in actinin-2 abundance observed (23). Changes in mitochondrial, extracellular matrix, and cytoskeletal/structural genes and proteins in human skeletal muscle insulin sensitivity are summarized in Table 1.
Table 1.
Changes in mitochondrial, extracellular matrix, and cytoskeletal/structural genes and proteins in human skeletal muscle insulin sensitivity
| Naturally Occurring Insulin Resistance | Lipid-Induced Insulin Resistance | Reduction in Lipid Supply by Acipimox Treatment | |
|---|---|---|---|
| Decreased PGC-1α and oxidative phosphorylation | Hwang et al. (23), Lefort et al. (23), Patti et al. (42), Mootha et al. (37) | Richardson et al. (49) | Bajaj et al. (2) |
| Increased collagens and other extracellular matrix genes, such as CTGF | Berria et al. (4) | Richardson et al. (49) | Bajaj et al. (2) |
| Increased proteasomes and chaperones | Hwang et al. (23) | No change | No change |
| Decreased structural proteins | Hwang et al. (23) | No change | No change |
PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; CTGF, connective tissue growth factor.
Proposed Model of the Relationships Among Changes in Expression of Extracellular Matrix and Cytoskeletal Proteins and Mitochondrial Changes in Insulin-Resistant Muscle
Our proposed model of the relationship between inflammation and insulin resistance in skeletal muscle is shown in Fig. 5. In this model, an inflammatory response leads to changes in the extracellular matrix that are reminiscent of fibrosis. The extracellular matrix remodeling that is observed in insulin resistance may then alter mechanosignal transduction mediated by cytoskeletal elements such as intermediate (desmin) filaments or the actin cytoskeleton, resulting in altered sensing of contractile activity and ensuing gene expression changes that lead to decreased muscle fiber type I remodeling and regeneration and reduced mitochondrial number and function (perhaps mediated by changes in PGC-1α expression). Changes in sensing of contractile activity could then result in alterations in expression of cytoskeletal and structural proteins by a feedback mechanism, reinforcing this vicious cycle. Changes in these genes and proteins contribute to the mitochondrial abnormalities observed in insulin-resistant muscle and may in the end lead to decreased fat oxidation, accumulation of ectopic lipid, insulin-signaling abnormalities, and ultimately insulin resistance. We point out that this mechanism is compatible with and complementary to other current hypotheses regarding the vicious cycle connecting inflammation, mitochondrial changes, lipid accumulation, and insulin-signaling defects. The novel aspect of this mechanism is that it connects inflammatory processes with changes in insulin sensitivity by means of altered mechanosignal transduction due to fibrotic changes.
Fig. 5.

Proposed model of the relationships among inflammation, changes in the extracellular matrix, cytoskeletal elements and mechanosignal transduction, and mitochondrial function in insulin-resistant muscle.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-47936 and R01-DK-66483 (both to L. J. Mandarino) and by grants from Arizona State University and the Mayo Clinic in Arizona.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We thank Irene Beauvais for administrative assistance and Marv Ruona for graphic design assistance.
REFERENCES
- 1. Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol 2010: 476279, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bajaj M, Medina-Navarro R, Suraamornkul S, Meyer C, DeFronzo RA, Mandarino LJ. Paradoxical changes in muscle gene expression in insulin-resistant subjects after sustained reduction in plasma free fatty acid concentration. Diabetes 56: 743–752, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Berman Y, North KN. A gene for speed: the emerging role of alpha-actinin-3 in muscle metabolism. Physiology (Bethesda) 25: 250–259, 2010 [DOI] [PubMed] [Google Scholar]
- 4. Berria R, Wang L, Richardson DK, Finlayson J, Belfort R, Pratipanawatr T, De Filippis EA, Kashyap S, Mandarino LJ. Increased collagen content in insulin-resistant skeletal muscle. Am J Physiol Endocrinol Metab 290: E560–E565, 2006 [DOI] [PubMed] [Google Scholar]
- 5. Boden G. Endoplasmic reticulum stress: another link between obesity and insulin resistance/inflammation? Diabetes 58: 518–519, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 57: 2438–2444, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonen A. PGC-1alpha-induced improvements in skeletal muscle metabolism and insulin sensitivity. Appl Physiol Nutr Metab 34: 307–314, 2009 [DOI] [PubMed] [Google Scholar]
- 8. Bonora E, Bonadonna RC, Del Prato S, Gulli G, Solini A, Matsuda M, DeFronzo RA. In vivo glucose metabolism in obese and type II diabetic subjects with or without hypertension. Diabetes 42: 764–772, 1993 [DOI] [PubMed] [Google Scholar]
- 9. Campbell PJ, Carlson MG, Nurjhan N. Fat metabolism in human obesity. Am J Physiol Endocrinol Metab 266: E600–E605, 1994 [DOI] [PubMed] [Google Scholar]
- 10. Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res 313: 2063–2076, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Chomentowski P, Coen PM, Radikova Z, Goodpaster BH, Toledo FG. Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab 96: 494–503, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Colberg SR, Simoneau JA, Thaete FL, Kelley DE. Skeletal muscle utilization of free fatty acids in women with visceral obesity. J Clin Invest 95: 1846–1853, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest 105: 311–320, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Filippis E, Alvarez G, Berria R, Cusi K, Everman S, Meyer C, Mandarino LJ. Insulin-resistant muscle is exercise resistant: evidence for reduced response of nuclear-encoded mitochondrial genes to exercise. Am J Physiol Endocrinol Metab 294: E607–E614, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 103: 253–259, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foster LJ, Rudich A, Talior I, Patel N, Huang X, Furtado LM, Bilan PJ, Mann M, Klip A. Insulin-dependent interactions of proteins with GLUT4 revealed through stable isotope labeling by amino acids in cell culture (SILAC). J Proteome Res 5: 64–75, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Højlund K, Bowen BP, Hwang H, Flynn CR, Madireddy L, Geetha T, Langlais P, Meyer C, Mandarino LJ, Yi Z. In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. J Proteome Res 8: 4954–4965, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Højlund K, Yi Z, Hwang H, Bowen B, Lefort N, Flynn CR, Langlais P, Weintraub ST, Mandarino LJ. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol Cell Proteomics 7: 257–267, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord 27, Suppl 3: S53–S55, 2003 [DOI] [PubMed] [Google Scholar]
- 20. Hubert P, Bruneau-Wack C, Cremel G, Le Marchand-Brustel Y, Staedel C. Lipid-induced insulin resistance in cultured hepatoma cells is associated with a decreased insulin receptor tyrosine kinase activity. Cell Regul 2: 65–72, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res 107: 579–591, 2010 [DOI] [PubMed] [Google Scholar]
- 22. Hwang D. Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways. FASEB J 15: 2556–2564, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Højlund K, Yi Z, Mandarino LJ. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 59: 33–42, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johannsen DL, Ravussin E. The role of mitochondria in health and disease. Curr Opin Pharmacol 9: 780–786, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kang L, Ayala JE, Lee-Young RS, Zhang Z, James FD, Neufer PD, Pozzi A, Zutter MM, Wasserman DH. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 60: 416–426, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Kelley DE, Mandarino LJ. Hyperglycemia normalizes insulin-stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J Clin Invest 86: 1999–2007, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kelley DE, Mokan M, Simoneau JA, Mandarino LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest 92: 91–98, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khurana T, Khurana B, Noegel AA. LIM proteins: association with the actin cytoskeleton. Protoplasma 219: 1–12, 2002 [DOI] [PubMed] [Google Scholar]
- 30. Krook A, Björnholm M, Galuska D, Jiang XJ, Fahlman R, Myers MG, Jr, Wallberg-Henriksson H, Zierath JR. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 49: 284–292, 2000 [DOI] [PubMed] [Google Scholar]
- 31. Kubota H, Yokota S, Yanagi H, Yura T. Structures and co-regulated expression of the genes encoding mouse cytosolic chaperonin CCT subunits. Eur J Biochem 262: 492–500, 1999 [DOI] [PubMed] [Google Scholar]
- 32. Lefort N, Glancy B, Bowen B, Willis WT, Bailowitz Z, De Filippis EA, Brophy C, Meyer C, Højlund K, Yi Z, Mandarino LJ. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 59: 2444–2452, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418: 797–801, 2002 [DOI] [PubMed] [Google Scholar]
- 34. Maachi M, Pieroni L, Bruckert E, Jardel C, Fellahi S, Hainque B, Capeau J, Bastard JP. Systemic low-grade inflammation is related to both circulating and adipose tissue TNFalpha, leptin and IL-6 levels in obese women. Int J Obes Relat Metab Disord 28: 993–997, 2004 [DOI] [PubMed] [Google Scholar]
- 35. Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O'Doherty RM, Osawa H, Sewell C, Consoli A, Granner DK, DeFronzo RA. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Am J Physiol Endocrinol Metab 269: E701–E708, 1995 [DOI] [PubMed] [Google Scholar]
- 36. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol 1: 135–145, 2001 [DOI] [PubMed] [Google Scholar]
- 37. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273, 2003 [DOI] [PubMed] [Google Scholar]
- 38. Muoio DM. Intramuscular triacylglycerol and insulin resistance: guilty as charged or wrongly accused? Biochim Biophys Acta 1801: 281–288, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246, 2010 [DOI] [PubMed] [Google Scholar]
- 40. Ostergard T, Jessen N, Schmitz O, Mandarino LJ. The effect of exercise, training, and inactivity on insulin sensitivity in diabetics and their relatives: what is new? Appl Physiol Nutr Metab 32: 541–548, 2007 [DOI] [PubMed] [Google Scholar]
- 41. Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, Ravussin E, Bray GA, Smith SR. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab 94: 5155–5162, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100: 8466–8471, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paulin D, Huet A, Khanamyrian L, Xue Z. Desminopathies in muscle disease. J Pathol 204: 418–427, 2004 [DOI] [PubMed] [Google Scholar]
- 44. Pavelka N, Fournier ML, Swanson SK, Pelizzola M, Ricciardi-Castagnoli P, Florens L, Washburn MP. Statistical similarities between transcriptomics and quantitative shotgun proteomics data. Mol Cell Proteomics 7: 631–644, 2008 [DOI] [PubMed] [Google Scholar]
- 45. Pendergrass M, Koval J, Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, Ardehali H, Printz R, Granner D, DeFronzo RA, Mandarino LJ. Insulin-induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes 47: 387–394, 1998 [DOI] [PubMed] [Google Scholar]
- 46. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664–671, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A, Musi N. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57: 2595–2602, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reynisdottir S, Ellerfeldt K, Wahrenberg H, Lithell H, Arner P. Multiple lipolysis defects in the insulin resistance (metabolic) syndrome. J Clin Invest 93: 2590–2599, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, DeFronzo RA, Jenkinson CP, Mandarino LJ. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem 280: 10290–10297, 2005 [DOI] [PubMed] [Google Scholar]
- 50. Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375: 2267–2277, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 118: 2992–3002, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sreekumar R, Halvatsiotis P, Schimke JC, Nair KS. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 51: 1913–1920, 2002 [DOI] [PubMed] [Google Scholar]
- 54. Sriwijitkamol A, Ivy JL, Christ-Roberts C, DeFronzo RA, Mandarino LJ, Musi N. LKB1-AMPK signaling in muscle from obese insulin-resistant Zucker rats and effects of training. Am J Physiol Endocrinol Metab 290: E925–E932, 2006 [DOI] [PubMed] [Google Scholar]
- 55. Svedberg J, Björntorp P, Smith U, Lönnroth P. Effect of free fatty acids on insulin receptor binding and tyrosine kinase activity in hepatocytes isolated from lean and obese rats. Diabetes 41: 294–298, 1992 [DOI] [PubMed] [Google Scholar]
- 56. Talior-Volodarsky I, Randhawa VK, Zaid H, Klip A. Alpha-actinin-4 is selectively required for insulin-induced GLUT4 translocation. J Biol Chem 283: 25115–25123, 2008 [DOI] [PubMed] [Google Scholar]
- 57. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6: 772–783, 2006 [DOI] [PubMed] [Google Scholar]
- 58. Usui I, Takata Y, Imamura T, Morioka H, Sasaoka T, Sawa T, Ishihara H, Ishiki M, Kobayashi M. Fatty acid induced insulin resistance in rat-1 fibroblasts overexpressing human insulin receptors: impaired insulin-stimulated mitogen-activated protein kinase activity. Diabetologia 40: 894–901, 1997 [DOI] [PubMed] [Google Scholar]
- 59. Yamazaki Y, Kubota H, Nozaki M, Nagata K. Transcriptional regulation of the cytosolic chaperonin theta subunit gene, Cctq, by Ets domain transcription factors Elk-1, Sap-1a, and Net in the absence of serum response factor. J Biol Chem 278: 30642–30651, 2003 [DOI] [PubMed] [Google Scholar]
- 60. Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 5: 2339–2347, 2006 [DOI] [PubMed] [Google Scholar]
- 61. Zybailov BL, Florens L, Washburn MP. Quantitative shotgun proteomics using a protease with broad specificity and normalized spectral abundance factors. Mol Biosyst 3: 354–360, 2007 [DOI] [PubMed] [Google Scholar]