

Abstract
Renal cancers are highly aggressive and clinically challenging, but a transgenic mouse model to promote pathologic studies and therapeutic advances has yet to be established. Here we report the generation of a transgenic mouse model of von Hippel-Lindau (VHL) renal cancer termed the TRACK model (transgenic cancer of the kidney). TRACK mice specifically express a mutated, constitutively active HIF1α in kidney proximal tubule (PT) cells. Kidney histologies displayed by TRACK mice are highly similar to histologies seen in patients with VHL disease, including areas of distorted tubular structure, cells with clear cytoplasm and increased glycogen and lipid deposition, multiple renal cysts, and early onset of clear cell renal cell carcinoma (ccRCC). Distorted tubules in TRACK mice exhibit higher levels of CA-IX, Glut1, and VEGF than tubules in non-transgenic control mice. Further, these tubules exhibit increased numbers of endothelial cells, increased cell proliferation, and increased expression of the human ccRCC marker CD70 (TNFSF7). Moreover, PT cells in kidney tubules from TRACK mice exhibit increased genomic instability, as monitored by elevated levels of γH2AX. Our findings establish that activated HIF1α in murine kidney PT cells is sufficient to promote cell proliferation, angiogenesis, genomic instability, and other phenotypic alterations characteristic of human VHL kidney disease, establishing the TRACK mouse as a valid preclinical model of human renal cell carcinoma.
Keywords: HIF1α, DNA repair, genomic instability, mouse model, clear cell renal cell carcinoma (ccRCC)
Introduction
Von Hippel-Lindau (VHL) disease is a hereditary cancer syndrome caused by germline mutations of the VHL tumor suppressor gene (1). Patients with VHL disease have a greatly increased risk of developing various types of tumors, including hemangioblastomas, clear cell renal cell carcinomas (ccRCC), and pheochromocytomas (1). The lifetime risk of developing ccRCC in VHL disease patients is >70% by the age of 60 years (2). Loss of expression or mutation of the VHL tumor suppressor gene plays an etiological role in this cancer syndrome (1). The associated, increased expression of two transcription factors, the alpha subunits of hypoxia inducible factor 1 (HIF1α) and HIF2α, may be critical for carcinogenesis (3-5).
VHL disease patient kidneys display cystic changes or distortion of the tubular structure adjacent to cells, which can be differentiated from normal renal parenchyma by histological evaluation (6). In addition to expressing HIF1α, cells in these early lesions overexpress HIF1α target genes, such as Glucose Transporter 1(Glut-1), VEGF, and carbonic anhydrase IX(CA-IX) (6). The majority of these early abnormalities are of the “clear” cell type, and a small percentage show changes in nuclear morphology, nuclear to cytoplasm ratio, and tubular architecture. Surrounding areas show increased vascularization. Importantly, HIF1α, but not HIF2α activation occurs in the earliest lesions (6), suggesting a role for HIF1α activation in early renal carcinogenesis. However, this hypothesis remains untested, as many genes are aberrantly expressed in the absence of VHL (1).
The HIF1α protein is regulated post-translationally under normoxic conditions (7) by interaction with and poly-ubiquitination by an E3 ubiquitin ligase complex containing VHL protein(pVHL) (8). The poly-ubiquitinated HIF1α is then targeted to the 26S proteasome for degradation (8). The interaction of HIF1α with pVHL is mediated by hydroxylation of two proline residues (P402, 564) in the oxygen-dependent degradation domain (9, 10). Under hypoxic conditions, these proline residues are not hydroxylated. HIF1α, therefore, can't be recognized by pVHL and becomes stabilized. Dimerization of the stabilized HIF1α with the constitutively active HIF1β results in binding of the HIF heterodimer complex to hypoxia responsive elements (HRE, consensus sequence: 5′-RCGTG-3′) (11). Under normoxic conditions HIF1α is also hydroxylated at a conserved asparagine residue (N803) by factor inhibiting HIF1α (FIH-1), whose activity is also oxygen-dependent (12). The asparaginyl hydroxylation prevents recruitment of p300 and the transcriptional co-activator protein CBP, resulting in attenuated transcription of HIF1α target genes by HIF1α (13). Since the asparaginyl hydroxylation is also oxygen dependent, under hypoxic conditions HIF1α is not inhibited by FIH-1 and can activate transcription. Mutation of these three key amino acids should mimic hypoxic conditions by preventing HIF1α from being degraded by pVHL and by allowing HIF1α to recruit p300 and CBP, resulting in constitutive expression of HIF1α target genes.
Few mouse models that exhibit the pertinent features of human VHL disease in the kidney exist. Recapitulation of human kidney carcinogenesis by inactivation of the human tumor suppressor gene VHL has not been achieved (14-16). We created a triple mutant (P402A, P564A, N803A) human HIF1α construct using the kidney proximal tubule specific type 1 γ-glutamyl transpeptidase(GGT or γGT) promoter (17, 18) to drive expression of this triple mutant, constitutively active HIF1α in the proximal tubule cells (PTCs). Mice expressing this triple mutant, constitutively active HIF1α construct in the kidney exhibit “clear” cells, renal cysts, disorganized PTs, and cystic clear cell carcinoma, consistent with ccRCC.
Materials and Methods
Plasmid construction
Mutated, constitutively active HIF1α cDNA was created by site-directed mutagenesis (Invitrogen, Carlsbad, CA) of conserved proline residues(402, 564) and a conserved asparagine(803) into alanine residues. GGT is specifically expressed in the PTs of the kidney starting at about 3 weeks (17, 18). The rat GGT promoter (-1930–+246) was amplified by PCR from a plasmid from Dr. Terzi (18). The GGT promoter, mutated HIF1α, and beta-globin poly-A were cloned into pBlueScript and named γGT-HIF1α triple mutant(γ-HIF1α-M3).
Generation of Transgenic Mice
DNA was prepared as a linearized ClaI-XbaI fragment (vector sequence removed) and was injected into pronuclei of one-cell embryos (C57BL/6 × C57BL/6) at the WCMC Mouse Genetics Core. Southern Blot analysis was then performed (19).
Tissue Dissection, Processing, and Pathological review
Tissues were fixed, processed, sectioned, and H&E stained following standard protocols (20). Slides were reviewed blindly by Dr. Shevchuk, an experienced clinical pathologist specializing in kidney cancer, and independently by a veterinary pathologist, Dr. Sébastien Monette, from the Laboratory of Comparative Pathology, WCMC.
Reverse Transcriptase PCR (RT-PCR)
Total RNA was extracted using mini-RNAeasy columns (Qiagen, Valencia, CA). RT-PCR was then performed (21).
Immunostaining
Immunohistochemistry and immunofluorescence were performed (22). Antibodies: HIF1α (610958, BD-Transduction); CA-IX (sc-25600, Santa Cruz); Glut-1 (ab14683, Abcam, Cambridge, MA); VEGF (sc-507, Santa Cruz); Cd-31 (550274, BD Pharmingen, San Diego, CA); PCNA (M0879, Dako, Carpinteria, CA) and γH2AX (9718S, Cell Signaling, Danvers, MA). Cd-31 was stained on cryo-preserved, frozen sections. Periodic Acid/Schiff (PAS) stain was performed on paraffin-embedded (23) and cryo-preserved sections. Oil red O (ORO) staining was also performed (24).
Statistical Analysis
Results are expressed as mean ± SEM. Student's t test was used to determine the statistical significance of the γH2AX+ and Ki67+ cell number differences between TG+ and TG- mice.
Results
Generation of transgenic mice expressing mutated, constitutively active HIF1α
A high level of HIF1α protein is a prominent feature of ccRCC (8). Furthermore, increased expression of HIF1α and/or HIF2α, which has 48% amino acid homology with HIF1α, is thought to be a key event in ccRCC carcinogenesis (3). To examine the role of HIF1α in ccRCC carcinogenesis we constructed a GGT-HIF1α triple mutant plasmid (γ-HIF1α-M3, Fig. 1A). After confirmation of activity in cultured normal kidney proximal tubule cells, γ-HIF1α-M3 transgenic mice were generated from the γHIF1α-M3 plasmid. Five of 51 founder mice harbored the integrated target gene (Fig. 1B, founders #8 to #14). Four lines were evaluated by RT-PCR, using a transgene specific primer pair (primers 1 and 2, Fig. 1A) for kidney, spleen, liver, heart, lung, intestine, skeletal muscle, and testis/ovary mutant HIF1α mRNA. The triple mutant HIF1α was expressed only in the kidneys of transgenic positive (TG+) lines #8, #25, #32 and #43 (Fig. 1C, panel 1, #43 as an example). The transgene was not expressed in other organs analyzed (Fig. 1D). The specific expression of HIF1α in kidney proximal tubules (PTs) was also confirmed by immunohistochemistry (see below). VHL, endogenous HIF1α, and endogenous HIF2α mRNA levels were not changed in the kidneys of TG+ compared to the kidneys of transgenic negative (TG-) mice (Fig. 1C, panels 2, 3, 4). All four TG+ founder lines, γ-HIF1α-M3-8, γ-HIF1α-M3-25, γ-HIF1α-M3-32, and γ-HIF1α-M3-43, developed normally and passed the transgene to offspring following a Mendelian inheritance pattern.
Figure 1. Generation of γ-HIF1α-M3 transgenic lines.
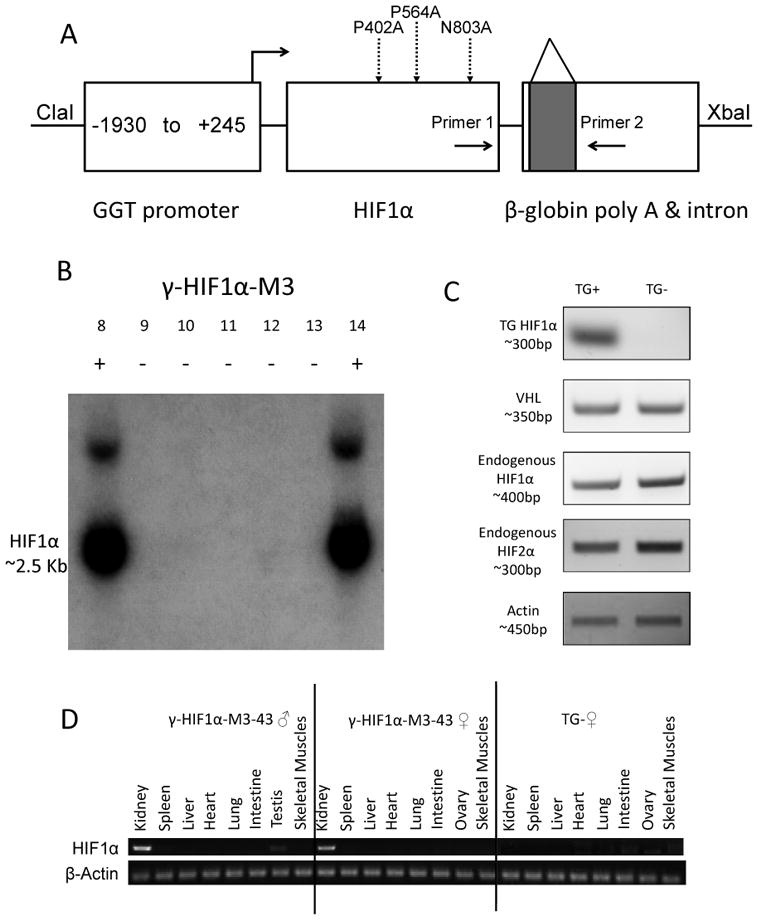
A. Construction of γ-HIF1α-M3 plasmid. Three mutations (P402A, P564A, N803A), dashed arrows. Intron (shaded square) in β-globin poly-A. Primers 1, 2 amplify the transgene by RT-PCR. B. Southern Blot of some TG+ and TG- Founders. Founders #8 and #14, TG+; others are TG-. C. HIF1α transgene, endogenous VHL, HIF1α, and HIF2α RT-PCR γ-HIF1α-M3-43 kidneys. HIF1α transgene, detected only in TG+. Endogenous VHL, HIF1α, and HIF2α mRNAs are expressed at similar levels in TG+ and TG-. β-Actin, loading control. D. HIF1α transgene RT-PCR in multiple organs of γ-HIF1α-M3-43 mice. HIF1α transgene, detected specifically in γ-HIF1α-M3-43 TG+ kidneys. No transgene expression in any organ of TG- mice. β-Actin, loading control.
γ-HIF1α-M3-43 TG+ mice exhibit kidney lesions that histologically resemble human VHL disease
Kidney, spleen, liver, heart, lung, intestine, skeletal muscle, and testis/ovary were removed from TG+ and TG- littermates, aged 6-7 months. Areas of distorted tubular structure were identified with clusters of “clear” cells in the outer cortex of TG+ kidneys from all four TG+ lines. These “clear” cells have characteristic proximal tubule cell features, like abundant, acidophilic cytoplasm (Fig. 2A; Fig. 2B, TG-). The γ-HIF1α-M3-43 line exhibited the strongest phenotype (Fig. 2A); the other TG+ lines had similar phenotypes, but they possessed fewer PTs containing “clear” cells (not shown). The distorted tubule cells showed moderate to marked cellular swelling, cytoplasmic vacuolation (Fig. 2A, black arrows), and prominent cell membranes, a feature of human ccRCC (Fig. 2A). We identified two types of vacuolation: large round, discrete vacuoles displacing the nucleus (Fig. 2A, solid black arrow), and vacuoles with pale, eosinophilic to clear feathery cytoplasm without displacement of the nucleus (Fig. 2A, dashed black arrow). The morphology of the large round, discrete vacuoles is consistent with lipid accumulation, while the morphology of the feathery cytoplasm is consistent with glycogen accumulation and hydropic degeneration. These “clear” cell clusters were morphologically strikingly similar to early lesions reported to occur in the “normal” kidneys of patients with VHL disease (6). Since the GGT promoter only drives the expression of the transgene in the PTs, we did not expect abnormalities in the renal medulla or papilla and did not observe any morphological abnormalities in these regions (not shown). Not all PTs contained these “clear” cell clusters; areas of “normal” PTs were present in the cortex, as assessed by histological morphology (Fig. 2A, yellow arrow). “Clear” cell PTs covered about 30% of the cortex in the 6 month γ-HIF1α-M3-43 transgenic mice (not shown). The majority of histologically abnormal PT cells in TG+ mice were large, simple cuboidal epithelial cells (Fig. 2A, black arrows) and were surrounded by tubular basement membrane, suggesting that these cells are still under proper growth control. Variations in the sizes of the nuclei (anisokaryosis) were identified in these “clear” cells. HIF1α protein was overexpressed in “clear” cells of TG+ (Fig. 2C, arrow), but not TG- kidneys (Fig. 2D) by immunohistochemistry.
Figure 2. “Clear” cells in γ-HIF1α-M3-43 mice.

Representative images of histological morphology (A,B), HIF1α immunohistochemistry (C,D), PAS stain (E,F), and ORO stain (G,H) “clear” cells in 6 month γ-HIF1α-M3-43 male TG+ kidneys (A, C, E, G) and TG- kidneys (B, D, F, H). “Clear” cells, TG+ tubule cells (A, solid, dashed arrows). Large round vacuole displacing the nucleus, solid arrow; feathery cytoplasm without nucleus displacement, dashed arrow. “Normal” PTCs in TG+ kidney (yellow arrow). Increased HIF1α protein in TG+ nuclei (C, arrow). Strong PAS (E, arrow), ORO (G, arrow) staining in cytoplasm of TG+ cells (E, G, arrows). Abnormal PTs with “clear” cells, panels A, C, E, G. All PTs stained by PAS or ORO were abnormal PTs with “clear” cells (panels E, G). Scale, 100 μm.
In ccRCC, the clear cytoplasm is caused by deposition of glycogen, phospholipids, and neutral lipids, particularly cholesterol esters (25). Similarly, the “clear” cells observed in our TG+ mice contained large amounts of cytoplasmic glycogen and lipid, as demonstrated by Periodic Acid/Schiff (PAS) (Fig. 2E, arrow; Fig. 2F, TG-) and Oil Red O (ORO) staining (Fig. 2G, arrow; Fig. 2H, TG-), respectively. In normal PTs, we detected strong PAS stain only in the basement membrane and the brush border/luminal side of the PT cells, and not in the cytoplasm. In contrast, in PT cells from TG+ mice strong PAS stain was identified in the cytoplasm, consistent with glycogen accumulation. Similarly, we detected ORO staining in the cortex of TG+ (Fig. 2G) but not TG- mice (Fig. 2H).
The multiple renal cysts observed (26) in VHL disease patients are believed to be precursors of ccRCC (1, 27). We identified cysts composed of a single layer of epithelial cells in 12 month TG+ mice (Fig. 3A). Dilated blood vessels filled with red blood cells were typically found near cysts. The majority of the cysts had clear spaces within, while some cysts contained amorphous, pale, eosinophilic material (Fig. 3A). While both tubular and glomerular cysts were found in VHL conditional knockout mice (16), we observed tubular cysts, but not glomerular cysts, in the γ-HIF1α-M3-43 TG+ mice.
Figure 3. The γ-HIF1α-M3-43 mice show morphological changes consistent with in situ ccRCC.

Representative images of renal cysts (A), a “clear” cell with enlarged nuclei and conspicuous nucleoli (B,C), disorganized PTs (B,D), and cystic ccRCC (E) in γ-HIF1α-M3-43 mice. A. Two adjacent renal tubular cysts, one filled with amorphous, pale, eosinophilic material, one empty. These structures are not observed in TG- mice (Fig. 2B and 3F). B. Low magnification of cortex showing a “clear” cell with enlarged nuclei and one conspicuous nucleolus (C. high magnification) and several TG+ PTs with disorganized, multilayer “clear” cells (D. high magnification). E. Cystic ccRCC. F. TG- control. Scale bars, 100 μm.
The PT cells from 7 month γ-HIF1α-M3-43 TG+ mice show simple, cuboidal epithelial cells with a “clear” morphology. The nuclei of some “clear” cells were large and hyperchromatic, with conspicuous nucleoli, suggestive of a neoplastic change (Fig. 3B and 3C, arrow). In 14-20 month TG+ mice, we observed abnormal PTs with multiple layers of epithelial cells (Fig. 3B and 3D, arrow). The normal proximal tubule structure is completely disrupted by these disorganized “clear” cells. These abnormal, disorganized “clear” cells form intratubular nests, consisting of multiple layers of cells, and are surrounded by cells with spindle-like nuclei, some of which are endothelial cells (see below), and red blood cells. This has also been observed in foci of VHL disease patient kidneys (6). These abnormal tubules display an intratubular proliferation of atypical clear cells, indicating carcinoma in situ. We examined 15 male γ-HIF1α-M3-43 TG+ mice from 3 to 20 months old. Kidneys of all 15 mice show the expected phenotype. The severity of these histologic changes increases with age (Table 1). Twenty-two month-old TG+ mice had cystic ccRCC (Fig. 3E). VHL kidney disease features such as “clear” cells, renal cysts, hyperchromatic nuclei, disorganized PTs, and cystic ccRCC were not observed in TG- mice (Fig. 3F).
Table 1. Summary of male TG+ mice showing clear cell PTs, renal cysts or carcinoma in situ.
Results from 15 TG+ and 15 TG- mice. A summary of TG+ mice that exhibit clear cell PTs, hyperchromatic nuclei, renal cysts, or carcinoma in situ. These abnormalities are not seen in male TG- littermates. Results from TG- littermates are not included.
| Animal ID | Age at sacrifice (months) | Clear cell PT area (%) | Number of hyperchromatic nuclei/300× field | Renal cysts (/section) | Carcinoma in situ (/section) |
|---|---|---|---|---|---|
| 43-102 | 3 | 40.4 | 24.4 | 0 | 0 |
| 43×17-2 | 3 | 31.6 | 8.6 | 0 | 0 |
| 43-143 | 4 | 31.6 | 7.4 | 0 | 0 |
| 43-9 | 6 | 36.4 | 11.4 | 0 | 0 |
| 43-93 | 7 | 30.9 | 16 | 0 | 0 |
| 43-12 | 12 | 35.0 | 1.8 | 1 | 0 |
| 43-24 | 13 | 38.2 | 4.8 | 0 | 0 |
| 43-13 | 14 | 43.0 | 6.6 | 1 | 6 |
| 43 Founder | 14 | 44.4 | 4.4 | 26 | 5 |
| 43-72 | 14 | 46.1 | 7.2 | 1 | 8 |
| 43-27 | 18 | 27.9 | 1.8 | 2 | 7 |
| 43-10 | 19 | 39.1 | 5.6 | 3 | 13 |
| 43-21 | 20 | 46.5 | 6 | 6 | 17 |
| 43-20 | 22 | 45.5 | 19.8 | 8 | 19 |
| 43-22 | 22 | 52.4 | 22.2 | 5 | 18 |
γ-HIF1α-M3-43 TG+ murine kidneys molecularly resemble human VHL kidney disease
In ccRCC there is increased vascularization surrounding tumor cells (28). We detected increased CD31 staining surrounding the morphologically abnormal PTs in TG+ (Fig. 4A, arrow) as compared to TG- mice. In TG- mice we detected many CD31+ endothelial cells (29) in glomeruli (Fig. 4B), but not in blood vessels surrounding the PTs (Fig. 4B).
Figure 4. Expression of ccRCC markers in γ-HIF1α-M3-43 kidneys.

Representative images of immunostaining using CD31 (A,B), CA-IX (C,D), Glut-1 (E,F), and VEGF antibodies (G,H) in 6 month γ-HIF1α-M3-43 TG+ PTs (A, C, E, G) and TG- PTs (B, D, F, H). Increased staining of CD31 (A), CA-IX (C), Glut-1 (E), and VEGF (G) is only observed in the abnormal PTs of TG+ mice. Arrow (panel A), strong CD31 staining (red) around PT. Nuclei stained with DAPI (blue). Arrow (panel C), cup-like CA-IX staining of basal and lateral membranes of “clear” cells. Arrow (panel E), strong Glut-1 staining around empty vacuole in “clear” cells. Arrow (panel G), strong VEGF staining around empty vacuole in “clear” cells. Representative Western Blot (I) of CA-IX protein and RT-PCR (J) of CA-IX and CD70 mRNA and β-Actin (I,J), control. Scale bars, 100 μm.
CA-IX(NP_647466), Glut-1(NP_035530), and VEGF(NP_001020421) are HIF1α target genes that show increased protein expression in human ccRCC (30). Using immunohistochemistry (Fig. 4C-H), we observed strong CA-IX, Glut-1, and VEGF signals in the abnormal, but not in the morphologically normal, PTs of the same TG+ mice (Fig. 4C, E, G, TG+), however, we saw weak or no CA-IX, Glut-1, and VEGF signals in the PTs of TG- mice (Fig. 4D, F, H, TG-). The “clear” cells in the abnormal PTs were identified by the clear cytoplasm with vacuoles (Fig. 4C, E, G).
CA-IX is the major marker used to diagnose ccRCC. CA-IX protein expression increases in ccRCC carcinogenesis (6) and CA-IX protein is highly expressed in ccRCC (31). High CA-IX protein expression is a diagnostic and prognostic marker for human ccRCC (32, 33). We detected greatly increased CA-IX expression in the basal and lateral membranes of the abnormal PTCs in the γ-HIF1α-M3-43 TG+ kidneys, but CA-IX was not easily identified in the luminal membranes/brush borders (Fig. 4A, arrow). The staining pattern was membranous and cup-like. The cup-like Glut-1 and CA-IX staining patterns suggest that the brush borders of the abnormal PTCs are aberrant. By Western analysis, the CA-IX protein is elevated 30-40 fold in the TG+ kidneys relative to TG- littermates (Fig. 4I). CA-IX is a direct target of HIF1α, and CA-IX mRNA is highly expressed in the kidneys of TG+ relative to TG- littermates (Fig. 4J) but not in other tissues in TG+ mice (not shown). We also detected a much higher CD70 mRNA level in kidneys of TG+ vs. TG- mice (Fig. 4J). CD70 (TNFSF7) is a marker of human ccRCC (34).
We detected increased Glut-1 in the basal and lateral membranes of the “clear” cells of abnormal PTs (Fig. 4C), but not in the luminal membranes/brush borders in the γ-HIF1α-M3-43 TG+ mice. We also observed a stronger Glut-1 signal in some membranes surrounding the clear vacuoles (Fig. 4C, arrow). The “normal” PTs in TG+ kidneys, which are similar in morphology to normal PTs in TG- kidneys, show much weaker Glut-1 staining (Fig. 4D).
We detected increased VEGF staining, assessed by immunohistochemistry, in the cytoplasm of the “clear” cells in the abnormal PTs (Fig. 4E) in the γ-HIF1α-M3-43 TG+ mice. In some “clear” cells we detect intense VEGF staining close to empty vacuoles (Fig. 4E, arrow). The morphologically “normal” PT cells in TG+ mice exhibit lower VEGF staining, similar to the normal PTs in TG- mice (Fig. 4F).
γ-HIF1α-M3-43 TG+ mice express Ki67 and γH2AX, molecular markers of carcinogenesis
Proliferating Cell Nuclear Antigen (PCNA) is an essential component of DNA polymerase δ (35) and is expressed during S phase of the cell cycle; Ki67 is a marker for proliferation (36). We detected increased numbers of PCNA+ or Ki67+ cells in the morphologically abnormal PTs of TG+mice (Fig. 5A, arrows), but not in the “normal” PTs. In TG- mice, we saw few PCNA+ (not shown) or Ki67+ cells in the PTs (Fig. 5B, TG-).
Figure 5. Increased γH2AX and Ki67 γ-HIF1α-M3-43 kidneys.

Representative images of immunohistochemistry staining using Ki67 antibody (A,B), and γ-H2AX antibody (C,D) in 14 month γ-HIF1α-M3-43 TG+ PTs (A,C) and TG- PTs (B,D). Increased Ki67 and γH2AX staining in TG+ (A, C, arrows). No Ki67 or γH2AX stain in TG- (B,D), but Ki67 stain in glomeruli of both TG+ and TG- kidneys (A,B). Arrows, highly stained Ki67+ (A) and γH2AX+ (C) “clear” cells. The Ki67+ and γH2AX+ cells in 10 random fields of sections of 4 TG+ and 4 TG- were counted, panel E. All TG+ mice show a statistically significant increase in Ki67+ and γH2AX+ cells (P<0.001). Scale bars, 100 μm.
Genomic instability is another universal feature of tumor cells (37). Premalignant and malignant cells accumulate more mutations than normal cells (37). An increased DNA mutation rate/genomic instability may facilitate neoplastic transformation (38). Since the DNA mutation rate is difficult to measure, we measured the numbers of DNA double strand breaks (DSBs) in these “clear” cells. The serine(139) phosphorylated form of H2A histone family, member X (γH2AX) is a widely used marker for DSBs (39). Using a γH2AX antibody, we demonstrated that TG+ mice show a greater number of γH2AX+ cells in the kidneys, especially in the regions of “clear” cells (Fig. 5C, arrow) than TG- mice (Fig. 5D). Almost all of the γH2AX+ cells in the TG+ mice were in clusters of “clear” cells in morphologically abnormal PTs (Fig. 5C). We detected 6 Ki67+ cells and 8 γH2AX+ cells in the TG+ mice compared with 1.5 Ki67+ cells and 2 γH2AX+ cells per high-power field in the TG- mice (P<0.001) (Fig. 5E).
Additional TG+ mouse lines show a similar phenotype
We have four independently derived HIF1α TG+ lines, γ-HIF1α-M3-8, γ-HIF1α-M3-25, γ-HIF1α-M3-32 and γ-HIF1α-M3-43. We analyzed the γ-HIF1α-M3-43 line in greatest detail because it expresses the highest level of the HIF1α transgene. The other three TG+ lines express a lower level of the HIF1α transgene (Fig. 6). We observe “clear” cell morphological abnormalities and increased expression of CA-IX, and γH2AX in PT cells in these additional 3 TG+ lines (Fig. 6A, B, C). We detect increased Ki67 in the γ-HIF1α-M3-32 line, the line that expresses the second highest level of HIF1α protein (Fig. 6D). We also observe renal cysts in older TG+ mice (Fig. 6E). Thus, the phenotype we describe here is the result of mutant, constitutive HIF1α overexpression.
Figure 6. Staining in four independently derived TG+ lines.

“Clear” cell morphological abnormalities are identified in all four TG+ lines (panel A). Increased CA-IX and γH2AX in all four TG+ lines (panels B,C). Increased Ki67 only in γ-HIF1α-M3-43 and γ-HIF1α-M3-32 (panel D). Renal cysts in all four TG+ lines (panel E). Scale bars, 100 μm.
Discussion
The kidneys of γ-HIF1α-M3 transgenic mice exhibit features of human VHL disease
Loss of the VHL gene plays an important role in the development of ccRCC in some VHL disease patients (3). VHL knockout mouse models have been established (14-16) but they don't recapitulate early human ccRCC well. For example, the “clear” cell phenotype has not been reported (14-16). Although some genes (VEGF, CA-IX) upregulated in human ccRCC showed increased expression and renal cysts were observed in old mice (16), other obvious ccRCC characteristics, such as disorganized, “clear” cell proliferation in PTs, were not observed in the kidneys of such mice.
In contrast, all TG+ mice that express the constitutively active HIF1α construct show the characteristic human VHL disease phenotype in kidney, including “clear” cells, abnormal vascularization, and extremely high CA-IX expression. We also observe some molecular abnormalities usually seen in tumor cells, such as increased cell proliferation and DNA DSBs. Furthermore, we observe abnormalities characteristic of ccRCC, such as renal cysts, disorganized “clear” cell PTs, and CD70 overexpression (34). We observed no abnormalities in other organs of the γ-HIF1α-M3 TG+ lines, most likely because the GGT promoter drives transgene expression only in kidney PT cells, thought to be the cells of origin of human ccRCC (40). We observed that only a subgroup of all PTs exhibited “clear” cells; this may occur because the truncated GGT promoter drives expression primarily in the S3 segment of PTs (41).
HIF1α activation is responsible for the phenotype of VHL disease in the kidney
This characteristic human ccRCC phenotype is most likely the result of the increased expression of well-known HIF1α target genes such as Glut-1, VEGF, and CA-IX (42, 43). The upregulation of these genes is only seen in HIF1α expressing, “clear” cell PTs, indicating that these changes are directly related to HIF1α overexpression in these “clear” cell PTs. Increased transcription of HIF1α and Glut-1 changes the metabolism and glucose uptake in HIF1α expressing kidney PTs (11), which may account for the large increase in the accumulation of glycogen and lipids in the cytoplasm of PTCs in TG+ mice. Up-regulation of VEGF induces the proliferation of endothelial cells (44), which then likely generates the enhanced vascularization around the abnormal PTs with “clear” cells. CA-IX is up-regulated early in carcinogenesis, and CA-IX is now used as the major diagnostic and prognostic marker of human ccRCC (32, 33). Increased expression of CA-IX, observed in TG+ mice and in early VHL disease patients, may play a role in ccRCC carcinogenesis (45).
HIF1α activation can induce genomic instability and cell proliferation
A most interesting observation in the γ-HIF1α-M3 TG+ lines is the induction of DNA DSBs in a small percentage of “clear” cells, as indicated by γH2AX staining (Fig. 5C, E). DNA DSBs can induce genomic instability and carcinogenesis (46). Almost every DNA DSB forms a γH2AX+ focus, and therefore γH2AX is used to detect DNA DSBs (39). The increased γH2AX staining in some abnormal, “clear” cells indicates increased numbers of DNA DSBs in these TG+ cells. Why over-expression of a mutated, constitutively active HIF1α is associated with increased numbers of DNA DSBs is not known. RAD51, a key mediator of homologous recombination, shows reduced expression under hypoxic conditions (47). In preliminary experiments, we found that RAD51 mRNA was lower in TG+ than in WT kidneys. Whether this is associated with increased numbers of DSBs and/or genomic instability is under investigation.
Increased numbers of PCNA or Ki67+ cells indicate that mutated, constitutively active HIF1α is driving some cells to re-enter the cell cycle and proliferate. This is very similar to what has been observed in early kidney lesions of VHL disease patients (6). In these patients, 1–2% of the cells in the HIF1α activated, early lesions show a 4-8-fold higher Ki67 level as compared to normal cells.
HIF1α activation can induce VHL disease in the kidney
Renal cysts are observed in more than 60% of VHL disease patients and are thought to be precursors of ccRCC (1). The “clear” cells in the disorganized PTs of TG+ mice (Fig. 3D) are between the stages of carcinoma in situ and frank carcinoma. However, we did not detect late stage ccRCC with invasion and metastasis in TG+ mice. CD70 protein is upregulated in the majority of human ccRCC and is a marker of early human ccRCC (34). CD70 mRNA was higher in older TG+ mice relative to TG- controls (Fig. 4I). This result suggests that there may be some neoplastically transformed, “clear” cells present. Cigarette smoking, obesity, and hypertension are three well-established risk factors for ccRCC (48). We are testing these TG+ mice to determine if exposure to these conditions results in more rapid neoplastic transformation of PT cells in our TG+ mice.
Our results suggest that activated HIF1α functions as an oncogene in clear cell renal carcinogenesis. Although increased HIF1α protein expression is an early event in many solid tumors (4, 30), HIF1α has only been implicated as an etiological factor of ccRCC (3). The role of HIF1α in VHL disease, especially in ccRCC carcinogenesis, is of interest because HIF1α is directly regulated by pVHL, which is usually lost or silenced in ccRCC (3). In an earlier publication, when pVHL expression was restored in VHL-/- human ccRCC cells tumorigenesis in a xenograft tumor model was blocked, and stabilization of HIF1α alone didn't reproduce the tumor phenotype in these ccRCC cells expressing ectopic pVHL (49). However, HIF1α is not fully activated in this model (49) because hydroxylation of the asparagine(N803) can still occur. Moreover, fully tumorigenic ccRCC cells were used in a xenograft model so the carcinogenesis process was not analyzed (49). In contrast, our TG+ mice allow us to follow the carcinogenesis process over time and assess the early molecular changes that occur.
In summary, our results indicate that this constitutively active HIF1α functions as an oncogene in renal carcinogenesis. Expression of a mutated, constitutively active HIF1α protein in kidney PTs results in a phenotype similar to that observed in patients with VHL disease, including premalignant lesions, multiple renal cysts, and ccRCC.
Acknowledgments
We thank Dr. Semenza for the HIF1α and Dr. Terzi for the GGT promoter construct.
Grant Support: Research support by WCMC, the Turobiner Kidney Cancer Research Fund, and the Robert H. McCooey Genitourinary Oncology Research Fund. Dr. Fu holds the Robert H. McCooey Genitourinary Oncology Research Fellowship. Dr. Wang was supported by NCI-R25105012.
Footnotes
Notes: All authors declare no conflicts of interests.
References
- 1.Kaelin WG. Von hippel-lindau disease. Annual review of pathology. 2007;2:145–73. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 2.Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151–63. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 3.Kaelin WG., Jr The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007;13:680s–4s. doi: 10.1158/1078-0432.CCR-06-1865. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–34. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Current opinion in genetics & development. 2007;17:71–7. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer cell. 2002;1:459–68. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 7.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 9.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. Science. Vol. 292. New York, NY: 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing; pp. 464–8. [DOI] [PubMed] [Google Scholar]
- 10.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Science. Vol. 292. New York, NY: 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation; pp. 468–72. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Hypoxia-Inducible Factor 1 (HIF-1) Pathway. Sci STKE. 2007;2007:cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 12.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes & development. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci U S A. 2002;99:5271–6. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–8. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleymenova E, Everitt JI, Pluta L, Portis M, Gnarra JR, Walker CL. Susceptibility to vascular neoplasms but no increased susceptibility to renal carcinogenesis in Vhl knockout mice. Carcinogenesis. 2004;25:309–15. doi: 10.1093/carcin/bgh017. [DOI] [PubMed] [Google Scholar]
- 16.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer research. 2006;66:2576–83. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquemin E, Bulle F, Bernaudin JF, Wellman M, Hugon RN, Guellaen G, et al. Pattern of expression of gamma-glutamyl transpeptidase in rat liver and kidney during development: study by immunochemistry and in situ hybridization. Journal of pediatric gastroenterology and nutrition. 1990;11:89–95. doi: 10.1097/00005176-199007000-00018. [DOI] [PubMed] [Google Scholar]
- 18.Terzi F, Burtin M, Hekmati M, Federici P, Grimber G, Briand P, et al. Targeted expression of a dominant-negative EGF-R in the kidney reduces tubulo-interstitial lesions after renal injury. The Journal of clinical investigation. 2000;106:225–34. doi: 10.1172/JCI8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. The Journal of biological chemistry. 2005;280:40226–34. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- 20.Sheren-Manoff M, Shin SJ, Su D, Bok D, Rando RR, Gudas LJ. Reduced lecithin:retinol acyltransferase expression in human breast cancer. Int J Oncol. 2006;29:1193–9. [PubMed] [Google Scholar]
- 21.Gillespie RF, Gudas LJ. Retinoic acid receptor isotype specificity in F9 teratocarcinoma stem cells results from the differential recruitment of coregulators to retinoic response elements. The Journal of biological chemistry. 2007;282:33421–34. doi: 10.1074/jbc.M704845200. [DOI] [PubMed] [Google Scholar]
- 22.Tang XH, Vivero M, Gudas LJ. Overexpression of CRABPI in suprabasal keratinocytes enhances the proliferation of epidermal basal keratinocytes in mouse skin topically treated with all-trans retinoic acid. Experimental cell research. 2008;314:38–51. doi: 10.1016/j.yexcr.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 23.Sjolund J, Johansson M, Manna S, Norin C, Pietras A, Beckman S, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. The Journal of clinical investigation. 2008;118:217–28. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–56. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 25.Gebhard RL, Clayman RV, Prigge WF, Figenshau R, Staley NA, Reesey C, et al. Abnormal cholesterol metabolism in renal clear cell carcinoma. Journal of lipid research. 1987;28:1177–84. [PubMed] [Google Scholar]
- 26.Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. The New England journal of medicine. 1996;335:865–75. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- 27.Montani M, Heinimann K, von Teichman A, Rudolph T, Perren A, Moch H. VHL-gene deletion in single renal tubular epithelial cells and renal tubular cysts: further evidence for a cyst-dependent progression pathway of clear cell renal carcinoma in von Hippel-Lindau disease. Am J Surg Pathol. 2010;34:806–15. doi: 10.1097/PAS.0b013e3181ddf54d. [DOI] [PubMed] [Google Scholar]
- 28.Haase VH. The VHL tumor suppressor in development and disease: functional studies in mice by conditional gene targeting. Seminars in cell & developmental biology. 2005;16:564–74. doi: 10.1016/j.semcdb.2005.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman PJ, Berndt MC, Gorski J, White GC, 2nd, Lyman S, Paddock C, et al. Science. Vol. 247. New York, NY: 1990. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily; pp. 1219–22. [DOI] [PubMed] [Google Scholar]
- 30.Semenza GL. Targeting HIF-1 for cancer therapy. Nature reviews. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 31.Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer research. 2000;60:7075–83. [PubMed] [Google Scholar]
- 32.Bui MH, Seligson D, Han KR, Pantuck AJ, Dorey FJ, Huang Y, et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: implications for prognosis and therapy. Clin Cancer Res. 2003;9:802–11. [PubMed] [Google Scholar]
- 33.Sandlund J, Oosterwijk E, Grankvist K, Oosterwijk-Wakka J, Ljungberg B, Rasmuson T. Prognostic impact of carbonic anhydrase IX expression in human renal cell carcinoma. BJU Int. 2007;100:556–60. doi: 10.1111/j.1464-410X.2007.07006.x. [DOI] [PubMed] [Google Scholar]
- 34.Adam PJ, Terrett JA, Steers G, Stockwin L, Loader JA, Fletcher GC, et al. CD70 (TNFSF7) is expressed at high prevalence in renal cell carcinomas and is rapidly internalised on antibody binding. British journal of cancer. 2006;95:298–306. doi: 10.1038/sj.bjc.6603222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prelich G, Tan CK, Kostura M, Mathews MB, So AG, Downey KM, et al. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-delta auxiliary protein. Nature. 1987;326:517–20. doi: 10.1038/326517a0. [DOI] [PubMed] [Google Scholar]
- 36.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. Journal of cellular physiology. 2000;182:311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Charames GS, Bapat B. Genomic instability and cancer. Current molecular medicine. 2003;3:589–96. doi: 10.2174/1566524033479456. [DOI] [PubMed] [Google Scholar]
- 38.Loeb KR, Loeb LA. Genetic instability and the mutator phenotype. Studies in ulcerative colitis. The American journal of pathology. 1999;154:1621–6. doi: 10.1016/S0002-9440(10)65415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nature reviews. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace AC, Nairn RC. Renal tubular antigens in kidney tumors. Cancer. 1972;29:977–81. doi: 10.1002/1097-0142(197204)29:4<977::aid-cncr2820290444>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 41.Dworniczak B, Skryabin B, Tchinda J, Heuck S, Seesing FJ, Metzger D, et al. Inducible Cre/loxP recombination in the mouse proximal tubule. Nephron Exp Nephrol. 2007;106:e11–20. doi: 10.1159/000100554. [DOI] [PubMed] [Google Scholar]
- 42.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 43.Grabmaier K, AdW MC, Verhaegh GW, Schalken JA, Oosterwijk E. Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene. 2004;23:5624–31. doi: 10.1038/sj.onc.1207764. [DOI] [PubMed] [Google Scholar]
- 44.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Science. Vol. 246. New York, NY: 1989. Vascular endothelial growth factor is a secreted angiogenic mitogen; pp. 1306–9. [DOI] [PubMed] [Google Scholar]
- 45.Parkkila S. Significance of pH regulation and carbonic anhydrases in tumour progression and implications for diagnostic and therapeutic approaches. BJU Int. 2008;101 4:16–21. doi: 10.1111/j.1464-410X.2008.07643.x. [DOI] [PubMed] [Google Scholar]
- 46.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 47.Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, Roth ME, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Molecular and cellular biology. 2004;24:8504–18. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pascual D, Borque A. Epidemiology of kidney cancer. Adv Urol. 2008:782381. doi: 10.1155/2008/782381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer cell. 2002;1:247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]