

Abstract
Franceschetti syndrome is an autosomal dominant disorder of craniofacial development with variable expressivity. It is commonly known as Treacher Collins syndrome (TCS). It is named after E. Treacher Collins who described the essential components of the condition. It affects both genders equally. This article reports a case of TCS in an 18-year-old female.
Keywords: Hypoplasia, mandible, palpebral fissures, Treacher-Collins syndrome, zygoma
Introduction
Treacher Collins syndrome (TCS) is a severe congenital disorder of craniofacial development characterized by numerous developmental anomalies that are restricted to the head and face.[1] Hypoplasia of the facial bones, particularly the mandible and zygomatic complex, is an extremely common feature of TCS. Although the condition was probably first described by Thomson (1846)[2] followed by Toynbee (1847)[3] and Berry (1889),[4] it is named after E. Treacher Collins[5] who described the essential components of the syndrome in 1900. The first extensive review of the condition was done by Franceschetti and Klein[6] in 1949, who coined the term mandibulofacial dysostosis to describe the clinical features. In England and in the American continent this abnormality is described as the “Treacher Collins syndrome” and in the European Continent as “Mandibulo-facial dysostosis” or “Franceschetti syndrome.”[5] This article reports a case of TCS in an 18-year-old female.
Case Report
An 18-year-old female reported to the Department of Oral Medicine and Radiology with the chief complaint of forwardly placed teeth in the upper anterior region of jaw. Family history revealed that her father and grandmother had typical signs of the Treacher Collins syndrome. On extra oral examination, she presented with antimongloid slanting of the palpebral fissures with sparse eyelashes on the lower eyelid. There was hypoplasia of the malar prominence [Figure 1]. Micrognathia was also evident [Figure 2]. The maxilla appeared to be prognathic. Thus face had “bird like” appearance. Intraoral examination revealed narrow high arched palate. The maxillary incisors were proclined and there were spacing in the maxillary and mandibular anteriors. There were root stumps in place of 26, 36, and 46. Generalized gingival recession and grade I mobility was present. Radiographs revealed prominent antigonial notch, short ramus [Figure 3], hypoplasia of the mandible, proclined upper anteriors [Figure 4], hypoplasia of zygomatic bone, and maxillary sinus [Figure 5].
Figure 1.
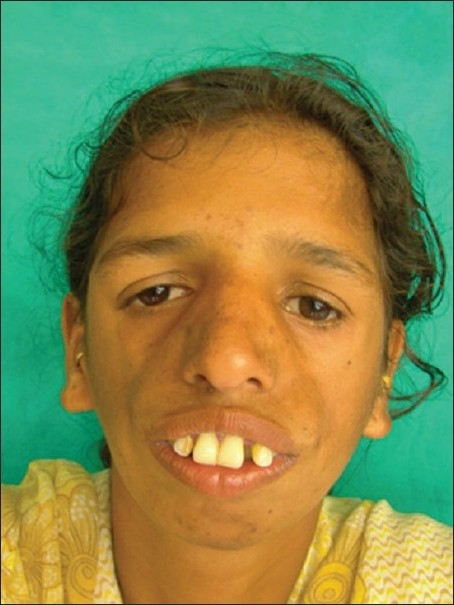
Front view of the patient showing hypoplasia of facial bones, downsloping palpabral fissures, with scanty lower eye lashes
Figure 2.
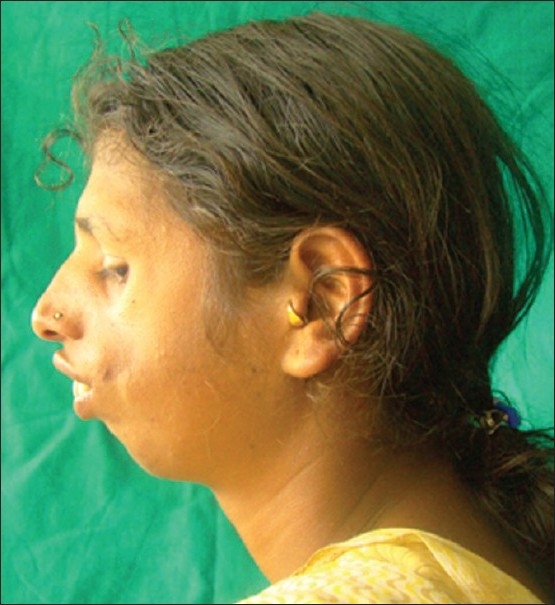
Lateral view showing retruded mandible and prominent nose
Figure 3.

Orthopantomogram showing prominent antigonial notch, short ramus
Figure 4.
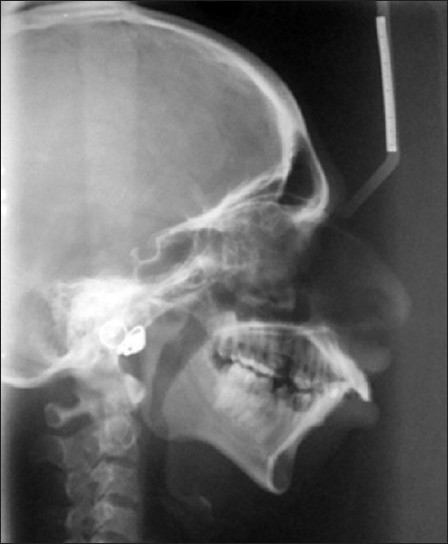
Lateral cephalogram showing hypoplastic mandible, prominent nose, and proclined upper anteriors
Figure 5.
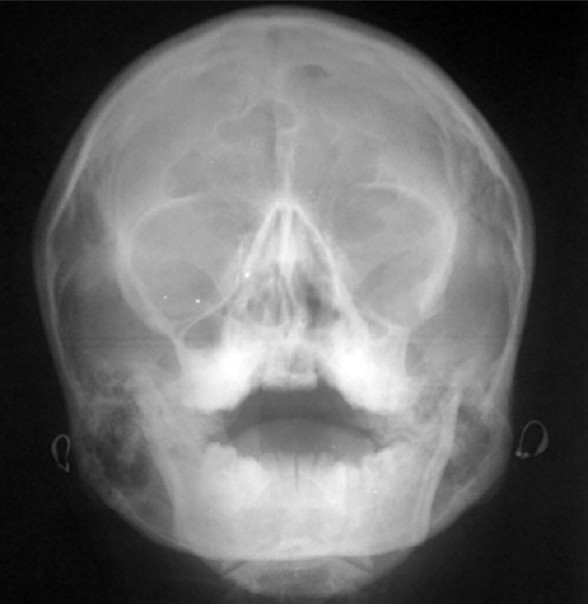
PA water showing hypoplasia of zygomatic bone and maxillary sinus
Discussion
TCS or Franceschetti syndrome is an autosomal dominant disorder of craniofacial development which has an incidence of approximately one in 50,000 live births.[7] It affects both genders equally. Nonpenetrance is rare.[1] While 40% of cases have a previous family history, the remaining 60% appear to arise as a result of a de novo mutation.[8] In our case, patient had family history of the syndrome.
Several hypotheses were proposed to explain the pathogenesis of TCS including abnormal patterns of neural crest cell migration,[9] abnormal domains of cell death,[10] improper cellular differentiation during development,[11] or an abnormality of the extracellular matrix;[12] however, there was little experimental evidence to support any of these hypotheses. Recently genetic, physical, and transcript mapping techniques have identified the gene mutated in TCS which is designated as TCOF1 and mapped to human chromosome 5q32--33.2 locus. It was found to encode a low complexity, serine/alanine-rich, nucleolar phosphoprotein termed Treacle.-[13] Valdez et al.[14] suggested that haploinsufficiency of treacle in TCS patients might cause insufficient rRNA production in the prefusion neural folds, resulting in abnormal craniofacial development. (The cephalic neural crest cells probably require a higher threshold concentration of rRNA for their survival and proper differentiation during early embryogenesis.)
The structures affected in TCS are derived from the first and second pharyngeal arch, groove, and pouch.[15] Franceschetti and Klein[6] (1949) reviewed the literature and described the typical characteristics of the syndrome as follows: (1) Antimongoloid palpebral fissures with either a notch or coloboma of the outer third of the lower lid, and occasional absence or paucity of the lashes of the lower lid. (2) Hypoplasia of the facial bones, especially the malar bones and mandible. (3) Malformation of the external ear, and occasionally of the middle and inner ear, with low implantation of the auricle. (4) Macrostomia, high palate, malocclusion, and abnormal position of the teeth. (5) Atypical hair growth in the form of tongue-shaped processes of the hairline extending toward the cheeks in the preauricular region. (6) Association at times with other anomalies, such as obliteration of the nasofrontal angle, pits, or clefts between the mouth and ear, and skeletal deformities.
After this description was published, some of these features were regarded as being of lesser importance and some were emphasized in the diagnosis. Thus Axelsson et al.[16] (1963) named the following features as “obligatory”: (1) antimongoloid palpebral fissures, (2) Anomaly of the lower lid: coloboma of the outer third, or deficient lashes, or both, (3) hypoplasia of the malar bones, (4) hypoplasia of the mandible. In our case, all obligatory features were present.
While the clinical features are generally bilaterally symmetrical, expression of the mutated gene is highly variable. At one extreme, the features can be so mild that it may be difficult to reach a diagnosis.[17] At the other extreme, the facial complex may be so severely hypoplastic that prenatal death ensues as a result of compromisation of the airway.
Prenatal diagnosis of TCS has only been performed in families with a history of TCOF1 using either fetoscopy[18] or ultrasound imaging[19] in the second trimester of pregnancy (approximately 18 weeks) when termination of pregnancy is a particularly traumatic procedure psychologically as it involves the induction of labor. However, the onset of TCS abnormalities occurs very early during human embryonic development, typically within the first 4--8 weeks. Hence first trimester prenatal diagnosis would seem to be preferable, particularly if the family feel that termination of pregnancy is desirable in the event that the fetus is affected. Phenotypic diagnosis at this stage (first trimester) even with the most sophisticated ultrasonography available today is not possible. Although linkage analysis (molecular analysis) has been used to make first trimester diagnostic predictions in a pregnancy at high risk of producing an affected child, it is not possible to predict how severely affected a fetus might be using this approach alone; consequently, ultrasonography is an invaluable aid to prenatal diagnosis, as this technique may provide information about the severity of affected pregnancies and can be used to evaluate fetal progression.[20]
Nager and Miller syndrome should be included in the differential diagnosis of TCS. Nager syndrome has similar facial features to TCS, particularly in the region of the eyes (downward slanting with a deficiency of eyelashes). However, the mandible is usually more hypoplastic, the lower lid colobomas are rare, and preaxial limb abnormalities (hypoplastic, or aplastic thumbs , fused radius and ulna) are a consistent feature of Nager syndrome, unlike TCS.[21] Miller syndrome also has some similarity in the facial features to TCS; in addition it has postaxial limb defects (absence or incomplete development of the fifth digital ray of all four limbs) and ectropion or outturning of the lower lids.[15] Also cleft lip, with or without cleft palate, is more common than in TCS and some patients may exhibit congenital heart defects.
The development of speech and language skills depends on the child's ability to hear during the first 3 years of life. As the great majority of these patients are of normal intelligence, early recognition of deafness and its correction with hearing aids or surgery, when possible, is of great importance for development. An affected parent of either sex will transmit the defect to 50% of his or her offspring in accordance with mendelian laws of genetics. This emphasizes the importance of genetic counseling to affected individuals.
In animal models it is shown that chemical and genetic inhibition of p53 function can repress the wave of neuroepithelial apoptosis associated with TCS and in doing so prevents the pathogenesis of craniofacial anomalies. However, p53 performs many critically important cellular functions and suppressing p53 function completely is a very risky approach given that loss-of-function mutations in p53 are the most common mutations associated with cancer and tumorigenesis. Therefore appropriate titration of p53 function is key in preventing the pathogenesis of TCS without risking the onset of tumorigenesis.[22]
Conclusions
Individuals diagnosed with severe TCS typically undergo, over several years, multiple major reconstructive surgeries that are rarely fully corrective. Although stem cell therapy holds promise for treating degenerative diseases, it is unlikely to benefit the reconstructive repair of severe craniofacial malformations. Consequently, prevention provides the best hope for improving the prognosis of at-risk or affected individuals.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Marres HA, Cremers CW, Dixon MJ, Huygen PL, Joosten FB. The Treacher Collins Syndrome.A clinical, radiological, and genetic linkage study on two pedigrees. Arch Otolaryngol Head Neck Surg. 1995;121:509–14. doi: 10.1001/archotol.1995.01890050009002. [DOI] [PubMed] [Google Scholar]
- 2.Thompson A. Notice of several cases of malformation of the external ear, together with experiments on the state of hearing in such persons. Monthly J Med Sci. 1846;7:420. [Google Scholar]
- 3.Toynbee J. Description of a congenital malformation in the ears of a child. Monthly J Med Sci. 1847;10:738–42. [Google Scholar]
- 4.Berry G. Note on congenital defect (coloboma?) of the lower lid. R Lond Ophthalmic Rep. 1889;12:255–57. [Google Scholar]
- 5.Treacher Collins E. Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones. Trans Ophthalmol Soc UK. 1900;20:190–92. [Google Scholar]
- 6.Franceschetti A, Klein D. The mandibulofacial dysostosis; a new hereditary syndrome. Acta Ophthalmol (Copenh) 1949;27:143–224. [PubMed] [Google Scholar]
- 7.Koppel D, Moos K. Mandibulofacial Dysostosis. In: Booth Peter Ward, Schendel Stephen A., editors. Maxillofacial surgery. 2nd ed. Vol. 2. St. Louis, Mo: Churchill Livingstone/Elsevier; 2007. pp. 799–805. [Google Scholar]
- 8.Jones KL, Smith DW, Harvey MA, Hall BD, Quan L. Older paternal age and fresh gene mutation: data on additional disorders. J Pediatr. 1975;86:84–8. doi: 10.1016/s0022-3476(75)80709-8. [DOI] [PubMed] [Google Scholar]
- 9.Poswillo D. The pathogenesis of the Treacher Collins syndrome (mandibulofacial dysostosis) Br J Oral Surg. 1975;13:1–26. doi: 10.1016/0007-117x(75)90019-0. [DOI] [PubMed] [Google Scholar]
- 10.Sulik KK, Johnston MC, Smiley SJ, Speight HS, Jarvis BE. Mandibulofacial dysostosis (Treacher Collins syndrome): a new proposal for its pathogenesis. Am J Med Genet. 1987;27:359–72. doi: 10.1002/ajmg.1320270214. [DOI] [PubMed] [Google Scholar]
- 11.Wiley MJ, Cauwenbergs P, Taylor IM. Effects of retinoic acid on the development of the facial skeleton in hamsters: early changes involving cranial neural crest cells. Acta Anat (Basel) 1983;116:180–92. doi: 10.1159/000145741. [DOI] [PubMed] [Google Scholar]
- 12.Herring SW, Rowlatt UF, Pruzansky S. Anatomical abnormalities in mandibulofacial dysostosis. Am J Med Genet. 1979;3:225–9. doi: 10.1002/ajmg.1320030303. [DOI] [PubMed] [Google Scholar]
- 13.Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 1996;12:130–6. doi: 10.1038/ng0296-130. [DOI] [PubMed] [Google Scholar]
- 14.Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci U S A. 2004;101:10709–14. doi: 10.1073/pnas.0402492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorlin R, Cohen M, Levin L. Oxford, UK: Oxford University Press; 1990. Syndromes of the Head and Neck. [Google Scholar]
- 16.Axelsson A, Brolin I, Engstrom H, Liden G. J Laryngol Otol. 1963;77:575–92. doi: 10.1017/s002221510006103x. [DOI] [PubMed] [Google Scholar]
- 17.Dixon MJ, Marres HA, Edwards SJ, Dixon J, Cremers CW. Treacher Collins syndrome: correlation between clinical and genetic linkage studies. Clin Dysmorphol. 1994;3:96–103. [PubMed] [Google Scholar]
- 18.Nicolaides KH, Johansson D, Donnai D, Rodeck CH. Prenatal diagnosis of mandibulofacial dysostosis. Prenat Diagn. 1984;4:201–05. doi: 10.1002/pd.1970040307. [DOI] [PubMed] [Google Scholar]
- 19.Meizner I, Carmi R, Katz M. Prenatal ultrasonic diagnosis of mandibulofacial dysostosis (Treacher Collins syndrome) J Clin Ultrasound. 1991;19:124–27. doi: 10.1002/jcu.1870190213. [DOI] [PubMed] [Google Scholar]
- 20.Edwards SJ, Fowlie A, Cust MP, Liu DT, Young ID, Dixon MJ. Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging. J Med Genet. 1996;33:603–06. doi: 10.1136/jmg.33.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chemke J, Mogilner B, Ben-Itzhak I, Zurkowski L, Ophir D. Autosomal recessive inheritance of Nager acrofacial dysostosis. J Med Genet. 1988;25:230–2. doi: 10.1136/jmg.25.4.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trainor PA, Dixon J, Dixon MJ. Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur J Hum Genet. 2009;17:275–83. doi: 10.1038/ejhg.2008.221. [DOI] [PMC free article] [PubMed] [Google Scholar]