

Abstract
RNase mitochondrial RNA processing (RNase MRP) mutants have been shown to have an exit-from-mitosis defect that is caused by an increase in CLB2 mRNA levels, leading to increased Clb2p (B-cyclin) levels and a resulting late anaphase delay. Here we describe the molecular defect behind this delay. CLB2 mRNA normally disappears rapidly as cells complete mitosis, but the level remains high in RNase MRP mutants. This is in direct contrast to other exit-from-mitosis mutants and is the result of an increase in CLB2 mRNA stability. We found that highly purified RNase MRP cleaved the 5′ untranslated region (UTR) of the CLB2 mRNA in several places in an in vitro assay. In vivo, we identified RNase MRP-dependent cleavage products on the CLB2 mRNA that closely matched in vitro products. Disposal of these products was dependent on the 5′→3′ exoribonuclease Xrn1 and not the exosome. Our results demonstrate that the endoribonuclease RNase MRP specifically cleaves the CLB2 mRNA in its 5′-UTR to allow rapid 5′ to 3′ degradation by the Xrn1 nuclease. Degradation of the CLB2 mRNA by the RNase MRP endonuclease provides a novel way to regulate the cell cycle that complements the protein degradation machinery. In addition, these results denote a new mechanism of mRNA degradation not seen before in the yeast Saccharomyces cerevisiae.
RNase mitochondrial RNA processing (RNase MRP) is a site-specific endoribonuclease ribonucleoprotein (8). RNase MRP was initially isolated from mammalian mitochondria, where its activity is consistent with its playing a role in primer formation in the initiation of mitochondrial DNA replication. Cellular fractionation and immunolocalization experiments revealed that the majority of the complex is localized to the nucleolus (31), where a role in processing of rRNAs has been identified (9, 16, 36). Mutations in the RNA component of the human RNase MRP have been shown to be the cause of the genetic disease cartilage hair hypoplasia (32), which is characterized by short stature, brittle and sparse hair, and immunodeficiency (11, 26).
The protein and RNA components of RNase MRP are highly conserved in both structure and sequence in all eukaryotes (1, 17, 23, 42, 44). Biochemically, the enzymes from yeasts to humans have been found to be similar in both substrate specificity and activity (41). In addition, the RNA component of RNase MRP is structurally related the RNA component of RNase P, both enzymes contain identical protein components, and both can cleave common substrates (6, 7).
The gene for the Saccharomyces cerevisiae MRP RNA is called NME1 for nuclear mitochondrial endonuclease 1 (35). In addition, at least nine yeast proteins associated with the MRP RNA in vivo have been identified. Eight of these proteins are shared with the ribonucleoprotein endoribonuclease RNase P (6, 10, 12, 23, 42). One protein encoded by the SNM1 gene encodes an RNA binding protein that is associated only with the RNase MRP RNA and not the RNase P RNA (37).
All of the components of RNase MRP are essential for the viability of S. cerevisiae. Mutations in the yeast RNase MRP components leads to a defect in 5.8S rRNA processing, specifically at the A3 site in the pre-rRNA. In the yeasts S. cerevisiae and Schizosaccharomyces pombe, a mutation in the MRP RNA was found to lead to a cell cycle delay (4, 27). Extensive genetic interaction between RNase MRP mutants and cell cycle mutations in S. cerevisiae that are involved in the exit from mitosis was also found.
RNase MRP mutants accumulate late in the mitotic cycle with large budded cells, dumbbell-shaped nuclei, and extended spindles, identical to what is seen in previously described exit-from-mitosis mutants (43). Western and Northern analyses of the mitotic cyclin Clb2 showed that Clb2 protein and mRNA accumulate in late mitosis (4). It is the increased Clb2p/ cyclin-dependent kinase (CDK) activity that is believed to cause the cell cycle delay (18). This cell cycle delay may lend insights to the cause of the pleotropic phenotypes seen in cartilage hair hypoplasia (26).
Here we demonstrate that the CLB2 mRNA is a direct target for the RNase MRP endoribonuclease. RNase MRP cleaves this mRNA in its 5′ untranslated region (UTR) at the end of mitosis to remove the 5′ cap. In addition, Xrn1, a 5′→3′ exoribonuclease was found to be necessary to degrade the CLB2 mRNA following RNase MRP cleavage.
MATERIALS AND METHODS
Strains and media.
Yeast media and genetic manipulations have been described (5). The Escherichia coli strain used for cloning, DH5α, has the genotype φ80dlacZΔM15 endA1 recA1 hsdR17 (rk− mk+) supE44 thi-1 λ− gyrA96 relA1 Δ(lacIZYA-argF)U169 F−. Basic molecular biology techniques were performed as described (34). The relevant genotypes of the yeast strains used are shown in Table 1. All strains are congenic with MES111 unless noted.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| MES111 | MATα his3-Δ200 leu2-3,112 ura3-52 trp1-Δ1 ade2 nme1-Δ2::TRP1 pMES127[CEN URA3 NME1] | 37 |
| MES111-140 | MATα his3-Δ200 leu2-3,112 ura3-52 trp1-Δ1 ade2 nme1-Δ2::TRP1 pMES140[CEN LEU2 NME1] | 37 |
| MES111-P6 | MATα his3-Δ200 leu2-3,112 ura3-52 trp1-Δ1 ade2 nme1-Δ2::TRP1 pMES140-P6[CEN LEU2 nme1-P6] | 37 |
| TLG105-140 | MATα his3-Δ200 leu2-3,112 ura3-52 trp1-Δ1 ade2 nme1-Δ2::TRP1 xrn1-Δ1::KanMX4 pMES140[CEN LEU2 NME1] | This study |
| TLG105-P6 | MATα his3-Δ200, leu2-3,112 ura3-52 trp1-Δ1 ade2 nme1-Δ2::TRP1 xrn1-Δ1::KanMX4 pMES140-P6[CEN LEU2 nme1-P6] | This study |
| TLG136 | MATaade2-1 his3-Δ200 leu2-3,112 trp1-Δ1 ura3-52 cdc15-1 | This study |
| YJA103 | MATα ade2-1 his3-Δ200 leu2-3,112 met15 trp1-Δ1 ura3-52 nme1-Δ2::TRP1 clb2Δ::KanMX4 pMES140 [LEU2, CEN, NME1] | 4 |
| YSW1 | MATaPOP4::TAPTAG::TRP1ks pep4::LEU2 nuc1::LEU2 sep1::URA3 trp1-1 his3-11,15 can-100 ura3-1 leu2-3,112 | This study |
Cell synchronization and arrest experiments.
For cell arrest experiments, yeast strains were grown to 106 cells/ml at 25°C in SCD (0.67% yeast nitrogen base, 0.5% Casamino Acids, 2% dextrose, 20 mg each of adenine, tryptophan, and uracil per liter), and then shifted to 37°C for 3 h. Cells were harvested and RNA was purified and examined by Northern analysis (5, 38).
For monitoring mRNA levels after hydroxyurea arrest, yeast strains were grown to 106 cells/ml at 25°C in SCD, arrested in hydroxyurea (15 mg/ml) for 2 h at 25°C, and then shifted to 37°C for 1 h. Cells were washed, resuspended in fresh medium, and kept growing at 37°C. Total RNA was made at 0, 15, 30, 45, 60, 75, 90, 105, and 120 min after release from hydroxyurea arrest. Northern analysis was performed.
To determine mRNA decay rates, yeast strains were grown to 106 cells/ml at 25°C in SCD, arrested in hydroxyurea (15 mg/ml) for 2 h at 25°C, and then shifted to 37°C for 1 h. Cells were washed, resuspended in fresh medium, and kept growing at 37°C for 60 min. To halt transcription, 1,10-phenanthroline (28) was added at 100 μg/ml, and total RNA was isolated at 0, 3, 6, 10, 15, 20, and 30 min after adding 1,10-phenanthroline. Northern analysis was performed. In the above experiments, cell synchrony and release from arrest were monitored by examination of cell budding and morphology.
Analysis of yeast RNA.
RNA was extracted as previously described (38). Approximately 10 μg of whole-cell RNA was separated on 1% agarose gels (34). Gels were stained with ethidium bromide to ensure equal loading by visualizing the rRNA, and Northern blot analysis was performed (34, 36). The probes used were CLB2 (653-bp BglII-HindIII fragment of the CLB2 gene) (14) and ACT1 (1,141-bp XhoI-KpnI fragment of the ACT1 gene) (39). Probes were radiolabeled for hybridization with α-[32P]dCTP with the Prime-It kit (Stratagene, Inc., La Jolla, Calif.). Radioactive blots were analyzed on a Molecular Dynamics PhosphoImager.
In vitro cleavage assay.
The RNase MRP cleavage assays and the generation of 3′ end-labeled substrates were performed as previously described (3). Internally labeled substrates were generated with SP6 RNA polymerase and α-[32P]UTP. The template for the rRNA substrate (A3 cleavage, positive control) was generated by amplifying the ITS1 region from yeast genomic DNA with the oligonucleotides SP6-A2 (AGA TTT AGG TGA CAC TAT AGA ATA CAA CAC ACT GTG GAG) and OMS-13 (ATA TTT TAA AAT TTC CAG TTA CGA). This fragment was then cloned with the Qiagen PCR cloning kit into the pDrive vector (Qiagen Corporation, Valencia, Calif.) to generate plasmid pJA110. For RNA synthesis with SP6 RNA polymerase, the plasmid was first digested with DraI.
The template for the CLB2 substrate was generated by amplifying the 5′-UTR with the oligonucleotides CLB2-SP6 (GAA GCT TAT TTA GGT GAC ACT ATA GAA TAT AAG AAG TAA AGT AT) and CLB2-EcoR1 (CGA ATT CAT TGG GTT GGA CAT CTA TAA GAT C). This generated four substrates of different lengths due to nonspecific termination of the SP6 RNA polymerase. RNase MRP efficiently cleaved all four substrates (data not shown), but the shortest gave the best results.
The negative control was a portion of the 5.8S rRNA that is not processed by RNase MRP. Subsequently, for the generation of 3′ end-labeled substrate and mapping of RNase MRP cleavage sites, a template for the CLB2 substrate was generated by amplifying the 5′-UTR with the oligonucleotides CLB2-SP6 and O-CLB-12 (TAC AAT GAT TAA AAT TTC TCC AAT GTC G). This fragment was cloned into the pDrive vector and digested with BamHI for the synthesis of RNA with SP6 RNA polymerase.
Purification of RNase MRP.
RNase MRP was purified from S. cerevisiae with the tandem affinity purification (TAP) tag protocol developed by Rigout et al. (33), with the following modifications. The YSW1 strain was used for all purifications; this strain contains an integration of the TAP tag cassette from pBS1479 (33) into the carboxy-terminal codon of the chromosomal POP4 gene (Table 1).
Strain YSW1 was grown in 16 liters of YPD [1% (wt/vol) yeast extract, 2% (wt/vol) peptone, 2% (wt/vol) dextrose] at 30°C to 2 × 108 cells/ml with vigorous aeration. Cells were harvested by centrifugation at 4,000 × g for 5 min and yielded approximately 3 g of cell paste per liter of culture. Cells could be frozen at −80°C or used directly. All subsequent procedures were carried out at 4°C unless noted otherwise. Cells were thawed or resuspended in 3 volumes of water and recentrifuged at 4,000 × g for 5 min. Cells were washed one time in 3 volumes of buffer Z [20 mM Tris-HCl (pH 8.0), 150 mM KCl, 1 mM EDTA, 10% (vol/vol) glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride]. Following the wash, the cells were resuspended in 1 volume of buffer Z and broken by being passed through a French pressure cell two times. The broken cells were centrifuged at 10,000 × g for 5 min, and the partially clarified extract was collected and recentrifuged to remove any remaining cells. The extract was then centrifuged at 100,000 × g for 100 min, and the clarified extract was collected carefully, avoiding the loose lipid layer on the top of the tube and the loose pellet on the bottom. This low-salt (150 mM KCl) extraction removed most of the RNase MRP but does not extract RNase P.
The extract was made 0.1% Tween 20 by the slow addition of a 10% (vol/vol) solution of Tween 20. The extract was then mixed with 1 ml of rabbit IgG-agarose beads (Sigma, St. Louis, Mo.) that had been equilibrated in buffer Z+T [buffer Z with 0.1% (vol/vol) Tween 20] and incubated at 4°C for 4 h with moderate agitation. The sample was then centrifuged at 500 × g for 5 min to collect the beads, and the extract was removed. The beads were washed six times with 10 volumes of buffer Z+T. Phenylmethylsulfonyl fluoride and dithiothreitol were excluded from the last two washes to avoid inhibition of the tobacco etch virus protease. After the last wash, the beads were resuspended in 5 ml of buffer Z+T without phenylmethylsulfonyl fluoride and dithiothreitol, and 200 units of tobacco etch virus protease (Invitrogen, Carlsbad, Calif.) were added. The sample was incubated overnight at 4°C with moderate agitation.
The beads were then centrifuged at 500 × g for 10 min, and the supernatant was removed and saved. An additional 5 ml of buffer Z+T was added to resuspend the beads and they were recentrifuged as before. The second supernatant was pooled with the first. The sample was prepared for calmodulin binding with the addition of 100 μl of 100X CBS [calmodulin-binding supplement: 100 mM sodium imidizole (pH 7.4), 150 mM magnesium acetate, 500 mM CaCl2]. Calmodulin affinity resin (Stratagene, La Jolla, Calif.) (500 μl) that had been equilibrated in buffer Z+T with 1× CBS was added to the pooled sample. This was incubated for 3 h at 4°C with moderate agitation. The resin was then washed six times in 10 ml of buffer Z+T with 1× CBS. After the final wash, as much supernatant as possible was removed, the resin was resuspended in 10 ml of buffer Z+T with 1× CEB [1× calmodulin elution buffer: 1 mM sodium imidizole (pH 7.4), 1.5 mM magnesium acetate, 10 mM EGTA], and incubated at 4°C for 2 h with moderate agitation. The sample was then centrifuged at 500 × g for 10 min, and the supernatant was collected and concentrated in a Centricon-YM100 (Amicon, Bedford, Mass.). Purified preparations were stored at −80°C and diluted in buffer Z+T before use in enzymatic assays. This preparation was at least 98% pure as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The preparation contained a single RNA, the MRP RNA, and all nine known yeast RNase MRP proteins, each of which was identified by mass spectrometry.
Primer extension.
Cells were grown at 24°C to 106 cells/ml, and RNA was purified as described above. Equal amounts of RNA were subjected to Northern analysis for the CLB2 mRNA. RNA from the same preparation was then used for primer extension. Primer extension was carried out as previously described (34) with an oligonucleotide (labeled on its 5′ end with γ-[32P]ATP and polynucleotide kinase) that hybridizes from nucleotides −218 to −240 (from the CLB2 translational start; O-CLB2-10) or from nucleotides −107 to −134 (O-CLB2-12). The same primer was used in a DNA sequencing reaction and run in parallel to identify 5′ ends. For mapping of RNase MRP in vitro cleavage sites, RNA cleavage products generated as described above were extended with the oligonucleotide O-CLB2-12.
RESULTS
Increased CLB2 mRNA levels in an RNase MRP mutant.
RNase MRP is a well-characterized site-specific endoribonuclease with only two known substrates (8). We would predict that the function of RNase MRP in the cell cycle involves the processing of some cellular RNA. Previously we showed that CLB2 mRNA levels are increased in nme1-P6 mutants (MES111-P6; nme1-P6 is a strong temperature-sensitive point mutation, G122A, in the gene for the RNase MRP RNA component) (4). The increase in CLB2 mRNA was very similar to the changes that were seen at the protein level, suggesting that the increased mRNA level is the direct cause of the increased Clb2p levels leading to the delay in cell cycle progression.
In order to ensure that the increase in CLB2 mRNA levels was not the result of an accumulation of cells at a point in the cell cycle where CLB2 mRNA levels are normally high, we also examined a cdc15 mutant. Cdc15 is a protein kinase that is necessary for exit from mitosis. cdc15 mutants arrest with a phenotype identical to that of RNase MRP mutants (large budded cells with well-divided nuclei, and an extended contiguous spindle). As can be seen in Fig. 1A, CLB2 mRNA levels were elevated in the nme1-P6 mutant, but were actually lower than the wild-type levels in the cdc15 mutant. This is consistent with what has been seen before in cdc15 mutants (14, 40). This result indicates that RNase MRP is playing a direct role in regulating CLB2 mRNA levels, as opposed to an indirect effect of the cells accumulating at a particular stage of the cell cycle.
FIG. 1.

Analysis of CLB2 mRNA levels through the cell cycle. (A) Comparison of CLB2 mRNA levels between wild-type and mutant strains carrying the nme1-P6 or cdc15-1 mutation. Cells were grown at 24°C, shifted to 37°C for 2 h, and RNA was isolated as described in Materials and Methods. An equal amount of RNA from each strain was subjected to Northern analysis. All blots were first probed for CLB2 mRNA and subsequently for ACT1 mRNA. The locations of the relevant transcripts are shown (wild-type, MES111-140; nme1-P6, MES111-P6; cdc15-1, TLG136). (B) Analysis of CLB2 mRNA levels in cell cycle-synchronized S. cerevisiae. Wild-type (MES111-140) and nme1-P6 mutant (MES111-P6) strains were grown to 106 cells/ml at 25°C in SCD, arrested in hydroxyurea for 2 h at 25°C, and then shifted to 37°C for 1 h. Cells were then washed to remove the hydroxyurea, resuspended in fresh medium, and maintained at 37°C. Total RNA was made at 0, 15, 30, 45, 60, 75, 90, 105, and 120 min after release from hydroxyurea arrest (see Materials and Methods). RNA was harvested at the indicated times, and CLB2 and ACT1 mRNA levels were measured. Cell synchrony and release from arrest were monitored by examination of cell budding and morphology. The signals corresponding to the CLB2 and ACT1 mRNAs are shown.
CLB2 mRNA levels remain high after M phase in an RNase MRP RNA mutant.
CLB2 mRNA is differentially transcribed as a function of the stage of the cell cycle (25). To further characterize CLB2 mRNA throughout the cell cycle, S. cerevisiae cells were synchronized with hydroxyurea, and CLB2 mRNA levels were followed (Fig. 1B). In wild-type cells, CLB2 mRNA increased shortly after removal of hydroxyurea, peaking 45 to 60 min after release. Levels then fell rapidly as the cells exited mitosis and entered G1 of the next cell cycle. At the 120-min time point, CLB2 levels started to rise again as the cells approached the next mitotic division. In nme1 mutant cells, CLB2 mRNA accumulated to a peak in 45 to 60 min, but it remained at a high level throughout the experiment. This result confirms that CLB2 mRNA accumulates for an extended period in RNase MRP mutants.
Degradation of CLB2 mRNA in an RNase MRP RNA mutant.
Based on our observation that the CLB2 mRNA levels failed to fall at the appropriate time in the RNase MRP RNA mutant, we examined the stability of CLB2 mRNA in the nme1-P6 mutant. Cells were synchronized with hydroxyurea, shifted to the nonpermissive temperature, released from the arrest, and allowed to progress for 60 min. This is the point at which CLB2 mRNA levels started to fall. Transcription was then arrested with the RNA polymerase II inhibitor 1,10-phenanthroline (28), and CLB2 mRNA levels were followed by Northern analysis (see Fig. 2). In wild-type cells, most of the CLB2 mRNA was rapidly degraded, with a half-life of less than 6 min. But in the nme1-P6 mutant, nearly 70% of the mRNA was present after 30 min, giving it a calculated half-life of over 58 min. This result indicates that normally RNase MRP affects the accumulation of CLB2 mRNA by promoting its instability at the end of mitosis.
FIG. 2.

Degradation of CLB2 mRNA in the nme1-P6 mutant. Wild-type (MES111-140) and nme1-P6 mutant (MES111-P6) strains were arrested in hydroxyurea for 2 h at 25°C and then shifted to 37°C for 1 h. Cells were then washed to remove the hydroxyurea, resuspended in fresh medium, and maintained at 37°C for 60 min. The transcriptional inhibitor 1,10-phenanthroline was added, and RNA was harvested at the indicated times (see Materials and Methods). The CLB2 and ACT1 mRNAs are shown.
Cleavage of the CLB2 5′-UTR by purified RNase MRP.
RNase MRP has been demonstrated to act as an endoribonuclease on in vitro-transcribed RNA substrates corresponding to both the mitochondrial origin region and the rRNA ITS1 (3, 22, 41). However, these are the only two substrates that have ever been identified, indicating the enzyme's high degree of specificity. The CLB2 mRNA is unusual for a yeast transcript in that the predominant transcript contains a long 364-nucleotide 5′-UTR (25). If RNase MRP is acting directly on the CLB2 transcript, this is a likely location.
In order to test for direct cleavage by RNase MRP, we examined a series of in vitro-generated RNA transcripts across the CLB2 5′-UTR for cleavage by highly purified RNase MRP.
Though specific cleavage was seen with several of these transcripts (data not shown), the most efficient cleavage was seen with a short 95-nucleotide RNA that started at the transcription initiation site (Fig. 3A and B). Levels of cleavage by RNase MRP of this substrate were comparable to that seen with the rRNA substrate (Fig. 3A). The purified RNase MRP used in the assay was at least 98% pure as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis protein analysis, and contained a single RNA, the MRP RNA, and all nine known protein components. The cleavage pattern of this enzyme on the rRNA substrate was identical to that reported previously (Fig. 3A) (22).
FIG. 3.

In vitro cleavage of the CLB2 5′-UTR by RNase MRP. (A) Internally labeled RNA substrates containing the rRNA A3 cleavage site (positive control), the CLB2 5′-UTR or the 5.8S rRNA (noncleaved control RNA) were generated. Highly pure yeast RNase MRP was purified with the TAP tag protocol and used in a standard RNase MRP in vitro assay (3, 22). (B) The schematic indicates the source of the RNA substrates and the sizes of the RNAs.
To investigate this cleavage further, we cloned the first 252 nucleotides of the CLB2 mRNA into an expression vector and used this as a substrate to map the cleavage sites. As can be seen in Fig. 4A, when this substrate was labeled at its 3′ end and subjected to a MRP cleavage assay, it demonstrated at least five distinct cleavage sites. The occurrence of multiple cleavages is similar to what has been reported for the mouse nuclear RNase MRP enzyme on the mitochondrial substrate (19). This is only the third in vitro substrate for RNase MRP that has been identified, exemplifying the high degree of specificity of RNase MRP. Our results indicate that the effect of RNase MRP on CLB2 mRNA is a direct result of site-specific cleavage of the CLB2 mRNA by RNase MRP. The exact cleavage sites for RNase MRP on the CLB2 mRNA in this in vitro reaction were mapped by primer extension (Fig. 4B) and are summarized in Fig. 5.
FIG. 4.

In vitro cleavage of the CLB2 5′-UTR and mapping of cleavage sites. (A) 3′-end-labeled RNA substrates containing the rRNA A3 cleavage site (positive control) and a 268-nucleotide CLB2 5′-UTR were generated. Highly pure yeast RNase MRP was purified with the TAP tag protocol and used in a standard RNase MRP in vitro assay (3, 22). Increasing amounts of enzyme were used in the assay: from left to right, 0 ng, 20 ng, 40 ng, 80 ng, 200 ng, and 800 ng of protein for both substrates. (B) A second RNase MRP assay was performed exactly as in panel A but then subjected to primer extension with the primer O-CLB2-12 (34). The positions of the cleavage sites are shown in Fig. 5.
FIG. 5.

CLB2 5′-UTR. Both DNA strands are shown, with the transcribed RNA above. The major transcriptional initiation sites are at positions 6 and 39. The translational initiation site is at position 400 in this figure. Sites of both in vitro cleavage by RNase MRP and mapped in vivo ends are indicated. Numbering of cleavage sites corresponds to the positions in Fig. 4. O-CLB2-10 and O-CLB2-12 are the two major oligonucleotides used in primer extension reactions.
Identification of RNase MRP products in vivo.
It has previously been reported that nme1 mutations have increased temperature sensitivity when present in combination with a deletion in the gene for the xrn1 nuclease (13). Xrn1 is a 5′→3′ exoribonuclease that is involved in mRNA decay. We examined levels of the CLB2 mRNA in xrn1Δ deletion and xrn1Δ nme1-P6 double mutants (see Fig. 6); our version of this strain was very slow growing but viable at 24°C. As shown earlier, levels of the CLB2 mRNA increased in MRP RNA mutants. Interestingly, levels also increased in the xrn1Δ mutant and were much higher in the double mutant (17.9-fold higher than the wild-type level as quantitated on a phosphoimager).
FIG. 6.

Both RNase MRP and Xrn1 participate in CLB2 mRNA degradation. The relevant genotypes of the yeast strains used are indicated; complete genotypes can be found in Table 1 (wild-type, MES111-140; nme1-P6, MES11-P6; xrn1Δ, TLG105; xrn1Δ nme1-P6, TLG105-P6; clb2Δ, YJA103). Cells were grown at 24°C, and equal amounts of RNA (based on both rRNA staining [bottom panel] and subsequent ACT1 mRNA analysis) were subjected to Northern analysis for the CLB2 mRNA. The CLB2 mRNA is indicated.
The very large increase in CLB2 mRNA may explain the increased temperature sensitivity of the double mutant strain. We also noted that there was a slight increase in mobility of the CLB2 mRNA in the xrn1Δ strains. This was consistently reproduced in four separate experiments. Based on our in vitro cleavage assay, the approximate change in size of the CLB2 mRNA (50 to 100 nt) is compatible with this species being an RNase MRP cleavage product. Indeed, as demonstrated in Fig. 7, one of the CLB2 mRNAs found in the xrn1 strains has a 5′ end that mapped extremely close to the cleavage site produced in vitro with purified RNase MRP. These 5′ ends were absent from the wild-type and nme1-P6 mutant strains. The double mutant strain contained increased amounts of full-length transcript compared to the xrn1Δ mutant, indicating a dependence on RNase MRP to produce the cleavage product. With different oligonucleotides, we were also able to map in vivo 5′ ends near other in vitro sites (data not shown), and these results are summarized in Fig. 5. We were unable to detect the presence of a long-lived 5′ end in either a cytoplasmic exosome mutant (ski7) or a nuclear exosome mutant (rrp6) (data not shown) (45, 46).
FIG. 7.
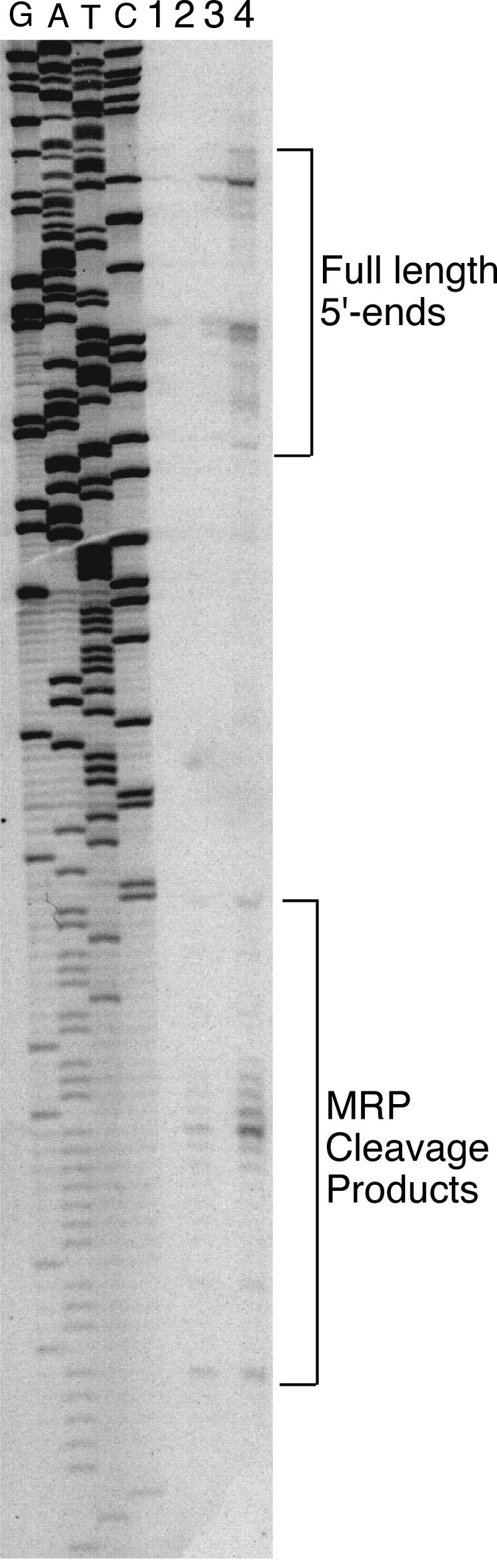
Identification of in vivo-generated RNase MRP products. Primer extension was performed on the same RNA used in Fig. 6 (34), with an oligonucleotide that hybridizes from positions −240 to −218 (from the CLB2 translational start). Previously identified full-length and novel xrn1Δ-specific ends are indicated (14). Lanes 1 to 4, wild-type, xrn1Δ, nme1-P6, and xrn1Δ nme1-P6, respectively. A sequencing reaction generated with the same primer on a plasmid version of the CLB2 gene is shown.
DISCUSSION
Role of RNase MRP in cell cycle control.
In a previous study we found that mutations in RNase MRP lead to cell cycle delay by accumulating the mitotic cyclin Clb2p (4). Furthermore, we showed that the accumulation of Clb2p is a result of an increase in steady-state levels of CLB2 mRNA. Here we continued this work by identifying the molecular mechanism of the effect of RNase MRP on the CLB2 mRNA. We show that this is a direct result of a failure to degrade the CLB2 message at a normal rate. In Saccharomyces cerevisiae, Clb2p is the most important B-type cyclin required for initiation and completion of mitosis. CLB2 transcripts begin to accumulate late in S phase, remain elevated until late in mitosis, and are degraded rapidly as cells complete mitosis (14). Increased levels of CLB2 transcripts at this time correlate with Clb2p-associated CDK activity, which peaks just before and disappears immediately following anaphase. This suggests that CLB2 mRNA levels play an important role in control of Clb2p-CDK activity.
Transcription plays a major part in the control of CLB2 mRNA levels in the cell cycle, but little has been reported about how CLB2 mRNA is degraded, even though degradation of the transcript is essential for tight control of mRNA levels. It is noteworthy that a cdc15-1 mutant arrested in telophase has very low levels of the CLB2 mRNA (14) (Fig. 1A). This indicates that the CLB2 mRNA is degraded in anaphase immediately before or concomitant with the first wave of Clb2 protein degradation (43). This is in contrast to RNase MRP mutants, which show increased and sustained levels of CLB2 mRNA at the telophase arrest. This result indicates that either the cdc15 block is downstream of CLB2 mRNA degradation step or the CLB2 mRNA is constitutively unstable.
Eukaryotic mRNAs are degraded by several mechanisms (2). Since RNase MRP is also an endoribonuclease, we propose that RNase MRP is able to directly cleave CLB2 mRNA specifically and cause its destabilization. The CLB2 transcript has an unusually long 5′-UTR (362 nucleotides) (25), and we hypothesized that it may play a role influencing transcript stability (30). Indeed, the results indicate that this is the case. With an in vitro-generated transcript identical to the CLB2 5′-UTR, we were able to demonstrate cleavage by highly purified RNase MRP. Many different RNA substrates have been tested for cleavage by purified RNase MRP without success (M. Schmitt, unpublished results). This is the first substrate outside of the mitochondrial substrate or the rRNA substrate that we have detected cleavage on, indicating the high degree of specificity of RNase MRP.
There may be a number of mRNAs that are degraded by RNase MRP at this late stage of the cell cycle, and they may share some common recognition element for RNase MRP-directed cleavage. However, previous analysis of other RNase MRP substrates indicated that RNase MRP, like its close relative RNase P, recognizes a structural motif and that the primary RNA sequence plays little role in enzyme recognition (15, 19). Indeed, the mRNAs for several genes that are expressed in parallel with CLB2 are made with long 5′-UTRs (40). Many of these may also contain RNase MRP processing sites, and we are testing this possibility. It should be noted that there is no apparent sequence or structural homology between the rRNA ITS1 substrate, the mitochondrial ORI5 substrate, or the CLB2 mRNA substrate.
One interesting facet of the cleavage assay is the appearance of multiple cleavage sites, as has been reported previously for the mouse nuclear RNase MRP enzyme on the mitochondrial substrate (19). This type of cleavage may be a distinct feature of all nuclear RNase MRP enzymes and may represent a distinct difference between RNase MRPs from the two different compartments. Our RNase MRP preparation is of nuclear origin, consistent with this hypothesis. Careful examination of substrate differences between RNase MRPs purified from mitochondria and nuclei could prove quite interesting.
Further support for RNase MRP playing a role in mRNA degradation comes from results that have shown that a mutation in the gene for the RNase MRP RNA is synthetically lethal with a deletion in the gene for the Xrn1 5′-3′ exonuclease (13). Xrn1 is involved in the general degradation of many mRNAs. We found elevated levels of the CLB2 mRNA in the xrn1Δ mutant (2.5-fold) and highly elevated levels in the xrn1Δ nme1 strain (more than 17-fold). Much of this accumulation appears to be RNase MRP cleaved products in the xrn1Δ strains. Our results indicate that the Xrn1 nuclease is responsible for degrading the CLB2 mRNA following RNase MRP cleavage. This result explains the increase in temperature sensitivity of the double nme1 xrn1Δ mutant strains (13). Rapidly degrading the CLB2 mRNA following RNase MRP cleavage should promote an increase in the rate of cleavage by eliminating product inhibition of the enzyme. These results suggest a model in which RNase MRP performs the initial endonuclease cleavage of the CLB2 mRNA, opening up a free 5′ end for Xrn1p-mediated exonuclease activity to destroy the message (Fig. 8).
FIG. 8.

Model of the role of RNase MRP in cell cycle control. Based on our results, RNase MRP is responsible for degradation of the CLB2 mRNA. This is accomplished by processing the CLB2 mRNA in its 5′-UTR, resulting in an uncapped transcript. This uncapped transcript is then efficiently degraded by the Xrn1 5′-3′ exoribonuclease. Defects in RNase MRP increase levels of the CLB2 mRNA, producing more Clb2 protein. Sustained levels of Clb2 protein keep the cyclin-dependent kinase Cdc28 active and inhibit the end of mitosis. Genetic interactions between RNase MRP and the exit from the mitosis pathway may indicate potential regulation points of RNase MRP or a bypass involving activation of the Clb2 protein degradation pathway (4).
Our model for RNase MRP function is shown in Fig. 8. RNase MRP is required for destabilization of the CLB2 mRNA during telophase. A mutation in RNase MRP leads to continued presence of the CLB2 mRNA and concomitant increases in the Clb2 protein level. The continuously high Clb2p keeps the CDK active and delays exit from mitosis, resulting in the accumulation of cells in telophase with an elongated mitotic spindle and separated nuclei. Genetic interactions previously identified between RNase MRP mutants and genes involved in the exit from mitosis are consistent with this model (4).
In recent years a tremendous amount of excitement has surrounded the finding that regulated proteolysis is intricately involved in controlling the cell cycle in S. cerevisiae and mammals (20, 21). We report that RNase digestion can also play a crucial role in controlling the cell cycle in S. cerevisiae. This may also be true in mammals, where RNase MRP is highly conserved (17, 24, 32). Conservation of a similar function in humans could explain the molecular cause of the genetic disease cartilage hair hypoplasia (11, 32). Cell lines generated from these patients have been shown to have both a cell cycle delay and changes in expression of several cell cycle-regulated transcripts (29). This provides us with a simple model system for studying a complex human disease.
It is not clear whether the activity of RNase MRP on the CLB2 mRNA is regulated. Preliminary results indicate that this may be the case. Clearly, regulation of mRNA degradation is essential for rapid removal of a gene product from a cell at a critical time in the cell cycle. Many mRNA transcripts may be degraded in a regulated fashion by RNase MRP and a variety of other ribonucleases at various times in the cell cycle, providing for the efficient elimination of different cell cycle components. Future studies aimed at revealing how CLB2 mRNA stability might be regulated should be extremely interesting.
Acknowledgments
We thank Roy Parker and David Amberg for comments and helpful discussions during the preparation of the manuscript. We are grateful to D. Amberg, SUNY Upstate Medical University, D. Hallberg, Syracuse University, and R. Parker, University of Arizona, for strains and plasmids.
This work was supported by grants RPG-96-109 from the American Cancer Society and GM063798 from the National Institute of General Medical Sciences.
REFERENCES
- 1.Aulds, J., T. Cai, and M. E. Schmitt. 2002. RNase MRP from yeast to humans, cell cycle control and cartilage hair hypoplasia. Recent Res. Dev. Mol. Cell. Biol. 3:371-378. [Google Scholar]
- 2.Beelman, C. A., and R. Parker. 1995. Degradation of mRNA in eukaryotes. Cell 81:179-183. [DOI] [PubMed] [Google Scholar]
- 3.Cai, T., and M. E. Schmitt. 2001. Characterization of ribonuclease MRP function. Methods Enzymol. 342:135-142. [DOI] [PubMed] [Google Scholar]
- 4.Cai, T., J. Auld, T. Gill, M. Cerio, and M. E. Schmitt. 2002. The Saccharomyces cerevisiae RNase Mitochondrial RNA Processing is critical for cell cycle progression at the end of mitosis. Genetics 161:1092-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai, T., T. R. Reilly, M. Cerio, and M. E. Schmitt. 1999. Mutagenesis of SNM1, which encodes a protein component of the yeast RNase MRP, reveals a role for this ribonucleoprotein endoribonuclease in plasmid segregation. Mol. Cell. Biol. 19:7857-7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamberlain, J. R., E. Pagan-Ramos, D. W. Kindelberger, and D. R. Engelke. 1996. An RNase P RNA subunit mutation affects ribosomal RNA processing. Nucleic Acids Res. 24:3158-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain, J. R., Y. Lee, W. S. Lane, and D. R. Engelke. 1998. Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes Dev. 12:1678-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, D. D., and D. A. Clayton. 1987. A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J. 6:409-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu, S., J. M. Zengel, and L. Lindahl. 1994. The RNA of RNase MRP is required for normal processing of ribosomal RNA. Proc. Natl. Acad. Sci. 18:659-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu, S., J. M. Zengel, and L. Lindahl. 1997. A novel protein shared by RNase MRP and RNase P. RNA 3:382-391. [PMC free article] [PubMed] [Google Scholar]
- 11.Clayton, D. A. 2001. A big development for a small RNA. Nature 410:29-31. [DOI] [PubMed] [Google Scholar]
- 12.Dichtl, B., and D. Tollervey. 1997. Pop3p is essential for the activity of the RNase MRP and RNase P. EMBO J. 16:417-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dichtl, B., A. Stevens, and D. Tollervey. 1997. Lithium toxicity in yeast is due to the inhibition of RNA processing enzymes. EMBO J. 16:7184-7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitch, I., C. Dahmann, U. Surana, A. Amon, K. Nasmyth, L. Goetsch, B. Byers, and B. Futcher. 1992. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell 3:805-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forster, A. C., and S. Altman. 1990. External guide sequences for an RNA enzyme. Science 249:783-786. [DOI] [PubMed] [Google Scholar]
- 16.Henry, Y., H. Wood, J. P. Morrissey, E. Petfalski, S. Kearsey, and D. Tollervey. 1994. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 13:2452-2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarrous, N., P. S. Eder, D. Wesolowski, and S. Altman. 1999. Rpp14 and Rpp29, two protein subunits of human ribonuclease P. RNA 5:153-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaspersen, S. L., J. F. Charles, R. L. Tinker-Kulberg, and D. O. Morgan. 1998. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol. Biol. Cell 9:2803-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karwan, R., J. L. Bennett, and D. A. Clayton. 1991. Nuclear RNase MRP processes RNA at multiple discrete sites: interaction with an upstream G box is required for subsequent downstream cleavages. Genes Dev. 5:1264-1276. [DOI] [PubMed] [Google Scholar]
- 20.Koepp, D. M., J. W. Harper, and S. J. Elledge. 1999. How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell 97:431-434. [DOI] [PubMed] [Google Scholar]
- 21.Lew, D. J., T. Weinert, and J. R. Pringle. 1997. Cell cycle control in Saccharomyces cerevisiae, p. 607-695. Cold Spring Harbor Press, Cold Spring Harbor, N.Y.
- 22.Lygerou, Z., C. Allmang, D. Tollervey, and B. Seraphin. 1996. Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science 272:268-270. [DOI] [PubMed] [Google Scholar]
- 23.Lygerou, Z., P. Mitchell, E. Petfalski, B. Seraphin, and D. Tollervey. 1994. The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev. 8:1423-1433. [DOI] [PubMed] [Google Scholar]
- 24.Lygerou, Z., H. Pluk, W. J. van Venrooij, and B. Seraphin. 1996. hPop1: an autoantigenic protein subunit shared by the human RNase P and RNase MRP ribonucleoproteins. EMBO J. 15:5936-5948. [PMC free article] [PubMed] [Google Scholar]
- 25.Maher, M., F. Cong, D. Kindelberger, K. Nasmyth, and S. Dalton. 1995. Cell cycle-regulated transcription of the CLB2 gene is dependent on Mcm1 and a ternary complex factor. Mol. Cell. Biol. 15:3129-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mäkitie, O., T. Sulisalo, A. de la Chapelle, and I. Kaitila. 1995. Cartilage-hair hypoplasia. J. Med. Genet. 32:39-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paluh, J. L., and D. A. Clayton. 1996. A functional dominant mutation in Schizosaccharomyces pombe RNase MRP RNA affects nuclear RNA processing and requires the mitochondrial-associated a nuclear mutation ptp1-1 for viability. EMBO J. 15:4723-4733. [PMC free article] [PubMed] [Google Scholar]
- 28.Parker, R., D. Herrick, S. W. Peltz, and A. Jacobson. 1991. Measurement of mRNA decay rates in Saccharomyces cerevisiae. Methods Enzymol. 194:415-423. [DOI] [PubMed] [Google Scholar]
- 29.Pierce, G. F., and S. H. Polomar. 1982. Lymphocyte dysfunction in cartilage hair hypoplasia. II. Evidence for a cell cycle specific defect in T cell growth. Clin. Exp. Immunol. 50:621-628. [PMC free article] [PubMed] [Google Scholar]
- 30.Pierrat, B., F. Lacroute, and R. Losson. 1993. The 5′ untranslated region of the PPR1 regulatory gene dictates rapid mRNA decay in yeast. Gene 131:43-51. [DOI] [PubMed] [Google Scholar]
- 31.Reimer, G., I. Raska, V. Scheer, and E. M. Tan. 1988. Immunolocalization of 7-2 ribonucleoprotein in the granular component of the nucleolus. Exp. Cell Res. 176:117-128. [DOI] [PubMed] [Google Scholar]
- 32.Ridanpää, M., H. van Eenennaam, K. Pelin, R. Chadwick, C. Johnson, B. Yuan, W. vanVenrooij, G. Pruijn, R. Salmela, S. Rockas, O. Mäkitie, I. Kaitila, and A. de la Chapelle. 2001. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell 104:195-203. [DOI] [PubMed] [Google Scholar]
- 33.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Séraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030-1032. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Press, Cold Spring Harbor, N.Y.
- 35.Schmitt, M. E., and D. A. Clayton. 1992. Yeast site-specific ribonucleoprotein endoribonuclease MRP contains an RNA component homologous to mammalian RNase MRP RNA and essential for cell viability. Genes Dev. 6:1975-1985. [DOI] [PubMed] [Google Scholar]
- 36.Schmitt, M. E., and D. A. Clayton. 1993. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 13:7935-7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt, M. E., and D. A. Clayton. 1994. Isolation of a unique protein component of the yeast RNase MRP: an RNA-binding protein with a zinc-cluster domain. Genes Dev. 8:2617-2628. [DOI] [PubMed] [Google Scholar]
- 38.Schmitt, M. E., T. A. Brown, and B. L. Trumpower. 1990. A rapid, improved method for isolation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18:3091-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shortle, D., J. E. Haber, and D. Botstein. 1982. Lethal disruption of the yeast actin gene by integrative DNA transformation. Science 217:371-373. [DOI] [PubMed] [Google Scholar]
- 40.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9: 3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stohl, L. L., and D. A. Clayton. 1992. Saccharomyces cerevisiae contains an RNase MRP that cleaves at a conserved mitochondrial RNA sequence implicated in replication priming. Mol. Cell. Biol. 12:2561-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stolc, V., and S. Altman. 1997. Rpp1, an essential protein subunit of nuclear RNase P required for processing of precursor tRNA and 35S precursor rRNA in Saccharomyces cerevisiae. Genes Dev. 11:2414-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Surana, U., A. Amon, C. Dowzer, J. McGrew, B. Byers, and K. Nasmyth. 1993. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 12:1969-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Eenennaam, H., N. Jarrous, W. J. van Venrooij, and G. J. Pruijn. 2000. Architecture and function of the human endonucleases RNase P and RNase MRP. IUBMB Life 49:265-272. [DOI] [PubMed] [Google Scholar]
- 45.van Hoof, A., P. Lennertz, and R. Parker. 2000. Yeast exosome mutants accumulate 3′-extended polyadenylated forms of U4 small nuclear RNA and small nucleolar RNAs. Mol. Cell. Biol. 20:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Hoof, A., P. A. Frischmeyer, H. C. Dietz, and R. Parker. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262-2264. [DOI] [PubMed] [Google Scholar]