

Abstract
Single crystals of a new iron containing oxide, Ba4KFe3O9, were grown from a hydroxide melt and the crystal structure was determined by single crystal x-ray diffraction. This ferrite represents the first complex oxide containing isolated 6-member rings of corner sharing FeO4 tetrahedra. Mössbauer measurements are indicative of two tetrahedral high-spin Fe3+ coordination environments. The observed magnetic moment (~3.9 BM) at 400 K is significantly lower than the calculated spin-only (~5.2 BM) value indicating the presence of strong antiferromagnetic interactions in the oxide. Our density functional calculations confirm the strong antiferromagnetic coupling between adjacent Fe3+ sites within each 6-member ring and estimate the nearest neighbor spin exchange integral as ~200 K; next nearest neighbor interactions are shown to be negligible. The lower than expected effective moment for Ba4KFe3O9 calculated from χT data is explained as resulting from the occupation of lower lying magnetic states in which more spins are paired. X-band (9.5 GHz) electron paramagnetic resonance (EPR) spectra of powder sample consist of a single line at g~2.01 that is characteristic of Fe3+ ions in a tetrahedral environment, thus, confirming the Mössbauer results. Further analysis of the EPR line shape reveals the presence of two types of Fe6 magnetic species with an intensity ratio of ~1:9. Both species have Lorentzian line shapes and indistinguishable g-factors but differ in the peak-to-peak line widths (δBpp). The line width ratio δBpp(major)/δBpp(minor) ~ 3.6 correlates well with the ratio of the Weiss constants, θminor/θmajor ~ 4.
Introduction
Among extended oxide compounds, structures containing rings of transition metal-oxygen polyhedra are uncommon, with the exception of silicates and germanates, where they are ubiquitous. Natural silicate minerals like beryl and tourmaline have 6-member rings of corner-sharing SiO4 tetrahedra,1 while some germinates, such as Ge9O19(OH)2(N2C2H10)2(N2C2H8)0.5•H2O, contain both 8- and 11-membered rings of corner-sharing GeO4 tetrahedra.2 In contrast, only a small number of iron-containing oxides adopt ring structures. The room temperature AFeO2 (A = K, Rb and Cs) structures are of the KGaO2 type and contain six member rings of corner-sharing FeO4 tetrahedra that are connected to adjacent rings by additional corner-sharing.3 Na14Fe6O16 contains linked 4- and 6-member rings of corner sharing FeO4 tetrahedra,4 while in Ba3NbFe3Si2O14, FeO4 tetrahedra form trimers that are interconnected via NbO6 octahedra.5 Other iron containing ring structures include Na10Fe4O9,6 and Ba3Fe2O67 In most iron containing oxides the rings are interconnected but exceptions exist, including the FeO4 tetrahedra trimers in Ba3NbFe3Si2O14,5 and the 12-member rings in Ba3Fe2O6.7
A number of molecular inorganic ring structures are known8 and are of interest both for the aesthetics of the structures9 as well as for potential applications in catalysis and gas adsorption.10 Furthermore, when such rings contain magnetic ions, such as manganese, molybdenum or iron, they typically exhibit complex magnetic interactions.11 Over the years, a number of iron containing molecular ring structures, referred to as ferric wheels,12,13 have been crystallized via solution routes, including [Fe(OMe)2(O2CCH2Cl)]10,13 [Fe14O10(OH)4(Piv)18 (HPiv = pivalic acid)14 and [Fe(OH)(C30H38N2O8)Fe2(OCH304(O2CCH3)2]6.15
We have been exploring the crystallization of iron containing complex oxides using high temperature solutions, including carbonate and hydroxide melts, and have synthesized complex oxides containing di-, tri- and tetravalent iron centers.16,17 Of the fluxes used, the molten hydroxides work especially well for the crystal growth of complex oxides,18 as noted by other groups that have also investigated the crystal growth of iron containing oxides out of hydroxide fluxes.19
Recently, we succeeded in using hydroxide melts to crystallize a new iron containing oxide, Ba4KFe3O9. To our knowledge, this is the only reported complex iron oxide containing isolated 6-member rings. We have structurally characterized this new oxide and have collected Mössbauer spectroscopy, EPR and magnetic data to investigate the magnetic interactions between the iron centers. To supplement these data, we have carried out density functional calculations to study the magnetic coupling between adjacent iron centers. Herein we report the crystal growth of Ba4KFe3O9, its structure determination and the investigation of its magnetic properties.
Experimental Section
Crystal Growth
Single crystals of Ba4KFe3O9 were grown from molten hydroxides. In a typical synthesis 1.9 g of dehydrated Ba(OH)2 (prepared by heating Ba(OH)2·8H2O (Alfa Aesar, 98%) at 280°C for 3 h), 2.5 g of KOH (Fisher, reagent grade), and 0.4 g of Fe2O3 (Alfa Aesar, 99.998%) were loaded into an alumina crucible, which was then covered with a lid. The crucible was placed into a box furnace, heated to 750 °C in 5 h, held at that temperature for 5 h, cooled to 600 °C in 32 h, and finally cooled to room temperature by turning off the furnace. Upon completion of the reaction, removal of the crucible lid revealed a bed of loose crystals. The crystals were twice washed with 36 ml iso-propanol under N2 to remove any remaining flux particles and then, since the crystals are very moisture sensitive, stored in a glovebox.
Structure Determination
X-ray data from an irregular shape black-looking single crystal were obtained at 150(2) K using a Bruker SMART APEX (Billerica, MA) diffractometer (Mo Kα radiation, λ = 0.71073 Å).20 The crystal was separated from a rough chunk covered with 1 mL of decane right after the reaction. When thin enough to transmit light, the crystals appear dark brown. The data collection covered 100% of the reciprocal space to 2θmax = 66.3°, with an average redundancy of 23.2 and Rint = 0.0995 after absorption correction. Raw area detector data frame integration was performed with SAINT+.20 Final unit cell parameters were determined by least-squares refinement of 5628 reflections from the data set in the range 6.8°<2θ<64.9°. Direct methods structure solution, difference Fourier calculations and full-matrix least-squares refinement against F2 were performed with SHELXTL.21
The compound crystallizes in the cubic space group Pa 3̄ (No. 205), as determined uniquely by the pattern of systematic absences in the intensity data. There are three barium, two iron, one potassium, three mixed K+/Ba2+ positions and six oxygen atom positions in the asymmetric unit. All atoms are in general positions (Wyckoff symbol 24d) except Ba3, K1, and K2/Ba2A (8c, site symmetry 3), K3/Ba3A (4a, site symmetry 3̄) and Ba4/K4A (4b, site symmetry 3̄). Unreasonably large or small displacement parameters were observed for sites K2/Ba2A (8c), K3/Ba3A (4a) and Ba4/K4A (4b) if refined as fully occupied by Ba2+ or K+, respectively. Trial refinement of the affected site occupancies always showed significant deviations from the unit occupancy by a single atom type. Each site was therefore refined as a mixture of Ba2+/K+, with total occupancy of each site constrained to sum of unity. The refined occupancy values are 0.569(5)/0.431(5) for K2/Ba2A, 0.934(5)/0.066(5) for K3/Ba3A, and 0.962(5)/0.038(5) for Ba4/K4A Occupancies of the other metal atom sites deviated from full occupancy by less than 3%, and were not refined. The site mixing model results in a formula of Ba3.97K1.03Fe3O9. This formula implies a small excess negative charge of 0.03 e− per formula unit (assuming all Fe3+). This is very close to electro-neutrality and supports the site mixing model over a vacancy model. All atoms were refined with anisotropic displacement parameters. The largest residual electron density extrema are +1.88 and −2.07 e−/Å located 0.61 and 0.01 Å from Ba2 and Ba4/K4A, respectively. The reported atomic coordinates were standardized with the StructureTidy program implemented in PLATON.22
Magnetic Susceptibility
The magnetic susceptibility χ of powder samples of Ba4KFe3O9 was measured as a function of temperature T using a Quantum Design PPMS (San Diego, CA) equipped with a vibrating sample magnetometer (VSM) attachment. The sample was contained in a gelatin capsule and held in place using small pieces of tissue paper. The capsule was fastened inside a plastic straw that was connected to the PPMS sample holder. χ(T) were collected in an applied magnetic field of 1 kG under both field-cooled (FC) and zero-field cooled (ZFC) conditions. ZFC data were collected in the temperature range 2 – 400 K, while FC data were collected in the temperature range of 2 – 300 K. An empty capsule was prepared in the identical fashion and its χ(T) was collected in order to correct the collected data for the contribution of the sample holder.
Heat Capacity
A representative group of crystals were weighed and placed in an aluminum Tzero sample pan that was coated on the inside with a thin layer of Apiezon N grease to increase thermal contact. The pans were hermetically sealed under helium, which provides further thermal contact with the small crystals. Heat capacity measurements were conducted between 5 and 300 K in zero magnetic field using a Quantum Design PPMS. The lattice contribution was not subtracted from the heat capacity.
Mössbauer
The 57Fe transmission Mössbauer spectrum was measured at room temperature using a γ-ray source from 57Co embedded in a rhodium matrix. The sample was sealed within 2 mylar tapes in a glove box before the measurement. The spectrum was fitted with Lorenzian lines by the MOSFIT program. The isomer shift was referenced to metallic α-Fe at 293 K.
Electron paramagnetic resonance (EPR) spectroscopy
Ba4KFe3O9 powder was placed in a 3×4 mm (i.d. × o.d.) quartz EPR tube (Wilmad, Vineyard, NJ), filled with nitrogen gas and flame-sealed. EPR spectra were collected at ~9.5 GHz (X-band) in the 190 K – 360 K temperature range using a Bruker Elexsys 580 spectrometer equipped with an ER4131 variable temperature unit (all from Bruker Biospin, Billerica MA). Electronics g-factors, peak-to-peak line widths (δBpp) and double-integrated intensities (I) were obtained by performing a Levenberg-Marquardt least-squares fitting of the EPR spectra to a two-component model using EWVOIGTN program.23,24
Results and Discussion
Crystal structure
Single crystals of Ba4KFe3O9 were grown out of molten KOH and carefully isolated to prevent exposure to atmospheric moisture to avoid rapid decomposition. Any exposure to moisture causes the structure to visibly degrade, as evidenced by a color change to light brown and the appearance of additional diffraction lines in the powder X-ray diffraction pattern. This extreme moisture sensitivity is likely directly related to this structure type that contains isolated anionic Fe6O18 rings held in a salt like environment made up of barium and potassium cations.
The crystallographic data and atomic coordinates are summarized in Table 1 and Table 2. The structure consists of a complex cubic barium/potassium framework in which Fe6O18 rings are located. One Fe6O18 ring consisting of corner shared FeO4 tetrahedra that encapsulates a potassium ion is shown in Figure 1a. There are two different tetrahedral iron sites in the ring structure that are arranged such that each Fe(1)O4 tetrahedron shares one corner with each of two Fe(2)O4 tetrahedra and vice-versa. The two FeO4 tetrahedra are slightly distorted and each has four different Fe-O bond lengths, ranging from 1.83 – 1.89 Å with O-Fe-O angles from 102.7 to 106.9° (Table 3). The 6-membered ring has a shape similar to the chair-conformation of cyclohexane and holds one potassium cation, K(1), in the middle (Figure 1b). Unlike other FeO4 tetrahedra-based 6-membered ring structures, the rings in this compound are isolated from each other (Figure 2). Each Fe6O18 ring has a formal charge of 18-, which is balanced by the Ba2+ and K+ counterions. The ionic radii for 9-coordinate Ba and K are quite similar, 1.47 and 1.55 Å, respectively. Hence, it is not surprising that some of the counterion sites have mixed Ba2+ and K+ occupancy. The cation site in the middle of the Fe6O18 ring is, however, occupied by potassium only. In this structure, the coordination numbers for Ba2+, K+, and mixed occupied metal sites range from 9 to 10.
Table 1.
Crystal and refinement data
| Empirical formula | Ba3.97Fe3K1.03O9 |
| Formula weight | 897 |
| Temperature | 150(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Cubic |
| Space group | Pa-3 |
| a = 16.7644(2) Å α = 90° | |
| b = 16.7644(2) Å β = 90° | |
| Unit cell dimensions | c = 16.7644(2) Å γ = 90° |
| Volume | 4711.55(10) Å3 |
| Z | 16 |
| Density (calculated) | 5.058 Mg/m3 |
| Absorption coefficient | 17.030 mm−1 |
| F(000) | 6270 |
| Crystal size | 0.12 × 0.08 × 0.06 mm3 |
| Theta range for data collection | 2.10 to 33.14°. |
| Index ranges | −25<=h<=25, −25<=k<=25, −25<=l<=25 |
| Reflections collected | 104498 |
| Independent reflections | 3008 [R(int) = 0.0995] |
| Completeness to theta = 33.14° | 100.00% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 1.0000 and 0.3407 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3008/0/108 |
| Goodness-of-fit on F2 | 1.078 |
| Final R indices [I>2sigma(I)] | R1 = 0.0314, wR2= 0.0632 |
| R indices (all data) | R1 = 0.0451, wR2= 0.0679 |
| Extinction coefficient | 0.000018(3) |
| Largest diff. peak and hole | 1.878 and −2.073 e.Å−3 |
Table 2.
Atomic coordinates (× 104) and equivalent isotropic displacement parameters (Å2 × 103). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
| x | y | z | U(eq) | Occupancy | |
|---|---|---|---|---|---|
| Ba(1) | 1227(1) | 3811(1) | 3881(1) | 9(1) | |
| Ba(2) | 1359(1) | 3712(1) | 1374(1) | 15(1) | |
| Ba(3) | 3754(1) | 3754(1) | 3754(1) | 12(1) | |
| Ba(4) | 5000 | 5000 | 5000 | 12(1) | 0.962(5) |
| K(4A) | 5000 | 5000 | 5000 | 12(1) | 0.038(5) |
| Fe(1) | 145(1) | 180(1) | 2542(1) | 8(1) | |
| Fe(2) | 42(1) | 2407(1) | 2374(1) | 8(1) | |
| K(1) | 1236(1) | 1236(1) | 1236(1) | 21(1) | |
| K(2) | 2559(1) | 2559(1) | 2559(1) | 10(1) | 0.574(3) |
| Ba(2A) | 2559(1) | 2559(1) | 2559(1) | 10(1) | 0.426(3) |
| K(3) | 0 | 0 | 0 | 12(1) | 0.937(5) |
| Ba(3A) | 0 | 0 | 0 | 12(1) | 0.063(3) |
| O(1) | 2332(2) | 4074(2) | 2821(2) | 14(1) | |
| O(2) | 56(2) | 2575(2) | 1262(2) | 12(1) | |
| O(3) | 1025(2) | 2710(2) | 2757(2) | 13(1) | |
| O(4) | 166(2) | 4861(2) | 3453(2) | 12(1) | |
| O(5) | 1297(2) | 2424(2) | 4964(2) | 12(1) | |
| O(6) | 235(2) | 4745(2) | 1521(2) | 20(1) |
Figure 1.
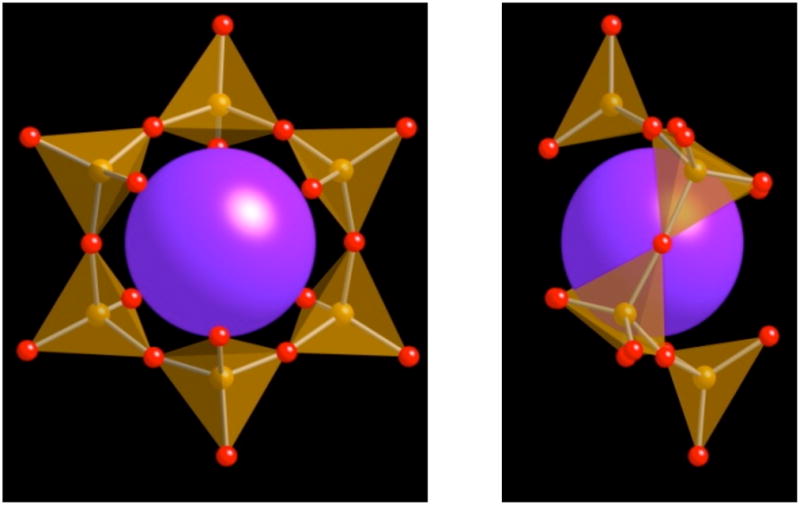
(a) One 6-member Fe6O18 ring with K atom in center viewed straight on (left) and (b) sideways to emphasize chair-configuration (right). FeO4 tetrahedra are shown in orange, oxygen atoms in red and potassium atoms in purple.
Table 3.
Selected bond lengths (Å) and bond angles (°)
| Fe(1)-O(4)#15 | 1.828(3) |
| Fe(1)-O(6)#15 | 1.846(3) |
| Fe(1)-O(2)#9 | 1.885(3) |
| Fe(1)-O(5)#16 | 1.898(3) |
| Fe(2)-O(1)#2 | 1.831(3) |
| Fe(2)-O(3) | 1.840(3) |
| Fe(2)-O(2) | 1.885(3) |
| Fe(2)-O(5)#16 | 1.896(3) |
| O(4)#15-Fe(1)-O(6)#15 | 124.20(15) |
| O(4)#15-Fe(1)-O(2)#9 | 106.19(13) |
| O(6)#15-Fe(1)-O(2)#9 | 105.92(15) |
| O(4)#15-Fe(1)-O(5)#16 | 105.73(14) |
| O(6)#15-Fe(1)-O(5)#16 | 107.95(15) |
| O(2)#9-Fe(1)-O(5)#16 | 105.53(13) |
| O(1)#2-Fe(2)-O(3) | 126.48(14) |
| O(1)#2-Fe(2)-O(2) | 102.77(14) |
| O(3)-Fe(2)-O(2) | 106.97(13) |
| O(1)#2-Fe(2)-O(5)#16 | 105.23(13) |
| O(3)-Fe(2)-O(5)#16 | 105.67(13) |
| O(2)-Fe(2)-O(5)#16 | 108.94(14) |
Symmetry transformations used to generate equivalent atoms:
#1 −z+1/2,x+1/2,y
#2 y−1/2,z,−x+1/2
#3 y,−z+1/2,x+1/2
#4 −x,y+1/2,−z+1/2
#5 z,−x+1/2,y+1/2
#6 y,z,x
#7 x,−y+1/2,z−1/2
#8 z,−x+1/2,y−1/2
#9 z,x,y
#10 x+1/2,y,−z+1/2
#11 −y+1,z+1/2,−x+1/2
#12 z+1/2,−x+1/2,−y+1
#13 −x+1/2,−y+1,z+1/2
#14 −x+1,−y+1,−z+1
#15 −x,y−1/2,−z+1/2
#16 z−1/2,x,−y+1/2
Figure 2.
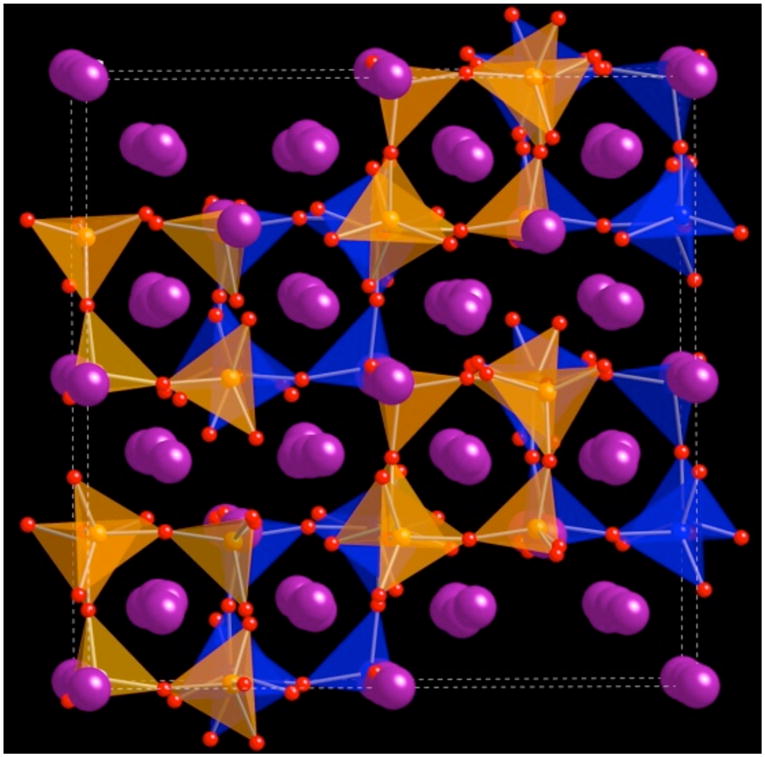
Crystal structure of Ba4KFe3O9 depicting the isolated nature of the 6-member rings. FeO4 tetrahedra belonging to different rings are shown in orange and blue to illustrate the isolated nature of the rings. Ba and K atoms are both shown in purple.
The counterions (Ba2+, K+, and Ba2+/K+ mixed sites) form the cubic framework shown in Figure 3, within which the Fe6O18 rings are located. The cation arrangement is best related to a defect body centered cubic structure, where the atoms at the corners of the cubes (K(1)) are located in the middle of the ring, while the atoms at the body center of the cubes lie adjacent to the rings. By comparison, in RbFeO2, an oxide with connected 6-membered FeO4 tetrahedra rings, the Rb+ counterions arrange in a diamond structure 25
Figure 3.
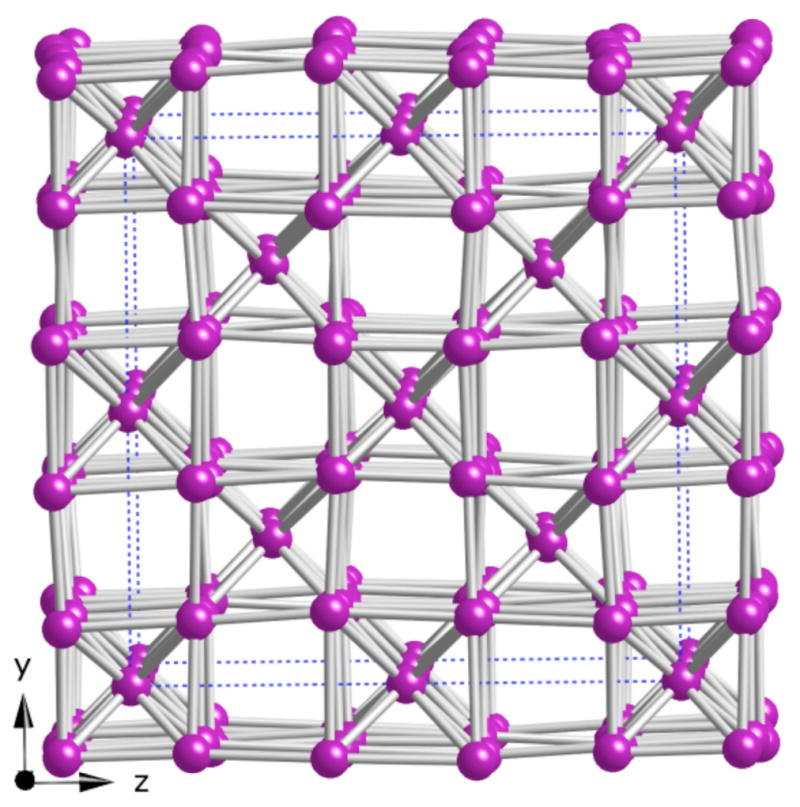
The arrangement of Ba and K atoms. Ba and K atoms are both shown in purple.
Mössbauer, Magnetic Susceptibility and Heat Capacity
The room temperature Mössbauer spectrum of Ba4KFe3O9 (Figure 4) is composed of two quadrupole doublets with isomer shifts and quadrupole splittings (δ1 = 0.41 mm/s, ΔEQ1 = 0.64 mm/s) and (δ2 = 0.25 mm/s, ΔEQ2 = 0.55 mm/s) consistent with Fe(III) in a high-spin state and tetrahedral coordination environment.17 The ratio between these two sites is 0.52/0.48 and is in agreement with the crystal structure. No magnetic hyperfine interactions are observed, consistent with reports for other cyclic iron structures.12
Figure 4.

Mössbauer spectrum of Ba4KFe3O9.
To investigate the possible existence of long-range magnetic order in this oxide, magnetic susceptibility data, χ(T), were collected between 2 K and 400 K. The χ vs. T and χ−1 vs. T plots are shown in Figure 5, and do not contain any feature indicative of a long-range magnetic order. The inverse susceptibility plot is non-linear over the entire temperature range measured and cannot be readily fit to the Curie law. Furthermore, the χT vs. T plot shows a complex change in the moment as a function of temperature. The observed effective magnetic moment of ~3.933 μB(Bohr magneton) at 400 K is lower than the expected spin-only value of 5.92 μB (for g = 2) for a tetrahedral high spin Fe3+ ion. This behavior indicates the presence of antiferromagnetic interactions between Fe3+ ions within the Fe6O18 ring. A similar effect was observed for [Fe14O10(OH)4(Piv)18] where the room temperature moment was only about 20% of the spin-only value.14
Figure 5.
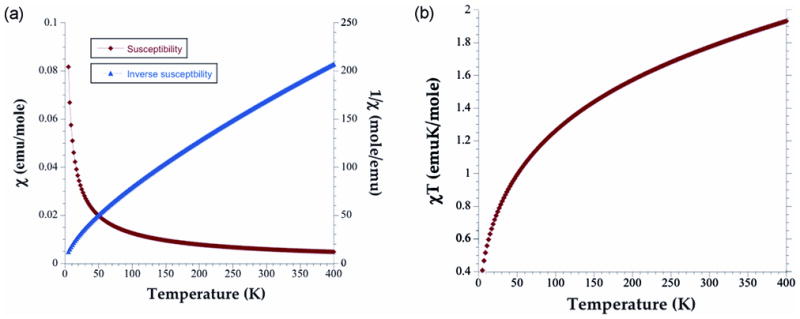
(a) Magnetic Susceptibility and inverse susceptibility vs. Temperature plot per mole of Fe3+ for Ba4KFe3O9 (left), (b) χT vs. Temperature plot for per mole of Fe3+ for Ba4KFe3O9 (right).
It has previously been observed that the measured effective magnetic moment is much smaller than the theoretical magnetic moment not only in molecular ring structures but also in related iron and cobalt oxides such as, for example, BaKFeO319 and Ba3Fe2O67 as well as Co(IV) compounds Cs2CoO3, Rb2CoO3 and K2CoO3.26 In some of these cases, it was suggested that the large difference between the observed and theoretical magnetic moments is due to a long-range antiferromagnetic ordering with Néel temperatures above the upper limit of the magnetic measurement temperature.7,19 It is unlikely that this scenario applies to our system, because a long-range antiferromagnetic order was not detected in the Mössbauer spectrum. Nonetheless, in order to investigate a possibility of a magnetic order we collected the heat capacity data shown in Figure 6. The data give no indication of an identifiable magnetic ordering transition between 5 and 300 K.
Figure 6.

Zero –field heat capacity data for Ba4KFe3O9.
EPR spectroscopy
Figure 7a shows a typical room temperature X-band EPR spectrum from Ba4KFe3O9 powder. The spectrum consists of a single line at g~2.01 and this g-factor is characteristic of Fe3+ ion in a tetrahedral environment.27,28 The line is sharp at the center but has some broad wings and cannot be fitted to a single Lorentzian line shape (not shown). EPR spectra with broad wings were previously observed for other types of strongly-coupled spin systems29,30 and were explained by either a dipolar broadening29 or a two-component Lorentzian model.30 It was found that Ba4KFe3O9 EPR spectra over the entire temperature range studied could be modeled as a superposition of two Lorentzian shapes that have the same field position but differ in widths with one component by a factor of ~3.6 broader than the other one (see Fig. 7a). Note that the number of electronic spins is proportional to the double integral of the corresponding EPR spectra. Further, for the first-derivative components appear to be of comparable peak-to-peak amplitudes (Fig. 7a), the spectra of the same (Lorentzian) shape, such as shown in Fig. 7, the double integral will be proportional to the square of the peak-to-peak width. Then, even though the two components appear to be of comparable peak-to-peak amplitudes (Fig. 7a), the double integrated intensity of the broad component represents ~90% of all the electronic spins because the peak-to-peak width is broader by a factor of ~3.6. Therefore this component is further referred as ‘major’ whereas the narrower component, which accounts for the remaining ~10% of the double integrated intensity, is referred to as ‘minor’. Both components are attributed to Fe3+ species based on g-factor gmajor = gminor ≈ 2.01. Based on the x-ray structure, we assign the major component in the EPR spectra to Fe6 clusters with an encapsulated K+ ion in the chair conformation. One may consider a possibility that the two crystallographically distinct tetrahedral Fe3+ ions could give rise to the two distinct EPR spectral peaks observed experimentally (Fig. 7). However, we believe that this is not the case. X-ray studies show only single type of Fe6 ring with two crystallographically distinct Fe3+ ions in equal amounts i.e., each ring is made-up of three ions of each type of Fe3+ ion. This does not match the 9:1 ratio of the EPR double-integrated intensities. Furthermore, even though the two crystallographically distinct tetrahedral Fe3+ ions could yield distinguishable anisotropic EPR spectra for isolated ions, we show that the adjacent Fe3+ sites within the 6-member rings are strongly spin exchange coupled. Then in the limit of strong spin exchange all the anisotropic features of Fe3+ EPR spectra and, hence, the differences between the EPR spectra corresponding to two crystallographically distinct Fe3+ sites would average out yielding an isotropic Lorentzian shape observed in our experiment. Thus, the presence of the two crystallographically distinct Fe3+ sites was ruled out as the origin of the two Lorentzian EPR components. It is more likely that the minor component would originate from a small fraction of a polymorph in the sample or even a partially decomposed sample due to its extreme moisture sensitivity. It is worthwhile to note here that the ability to resolve two components in EPR spectra reinforces the statue of EPR spectroscopy as one of the best analytical tools for detecting and differentiating the electronic spin environments in various systems.
Figure 7.

(a) Room temperature X-band EPR spectrum (black solid line) of Ba4KFe3O9 powder along with the major (dotted line) and minor (dashed line) components. The fit residual (red solid line), a difference between the experimental and simulated spectra, demonstrates an exceptional quality of the two-component model. The two components and the residual are displaced along the ordinate for clarity, (b) X-band EPR spectra of Ba4KFe3O9 powder at three representative temperatures.
To further understand the spin environment in Fe6 clusters and estimate the nearest neighbor spin exchange integral, Jnn, we have performed variable temperature EPR studies from 190 to 360 K (see Figure 7b for representative EPR spectra). Figure 8a shows that the peak-to-peak line width (δBpp) of both the major and the minor components decreases monotonously as the temperature increases while maintaining the δBpp(major)/δBpp(minor) ratio of ~3.6. The observed line narrowing with temperatures is attributed to further averaging of the local magnetic fields caused by an increased rate of spin fluctuations.
Figure 8.

(a) Peak-to-peak EPR line width (δBpp) as a function of temperature for the major and minor components. The solid lines are guides to the eye. (b) Inverse EPR double-integral signal intensity of the major and minor components as a function of temperature. Solid red lines are the best linear fit to the Curie-Weiss law. See the text for further details.
The isotropic exchange integral J can be estimated from δBpp(T) according to the Anderson-Weiss (AW) EPR line width theory.31 In a system with strong spin exchange interactions and containing a uniform distribution of magnetic ions in a simple cubic lattice, the δBpp parameter is related to the dipolar and spin exchange fields as:31
| (1) |
| (2) |
| (3) |
where Bd is the dipolar field, Bex is the spin exchange field, g is the g-factor, μB is the electronic Bohr magneton, r is the distance between the magnetic ions, S is the spin of the magnetic ion, and k the Boltzmann constant. Finally, the 10/3 factor in Eq. 1 accounts for additional broadening contributions to the line width from other magnetic interactions such as zero-field splitting from crystal field interactions, hyperfine interactions, etc.31
Let us now assume that the minor component in the EPR spectra originates from a very similar Fe6 cluster but with different Jnn and that these clusters are non-interacting with each other. Since the EPR spectral line shapes of both components of Ba4KFe3O9 are Lorentzian, the spin exchange field in Fe6 clusters must be sufficiently strong to average out the magnetic anisotropy and local dipolar fields. Therefore, it is reasonable to apply the AW theory to estimate, at least qualitatively, the Jnn from Eqs. 1 – 3. Since our sample contains discrete Fe6 wheels as opposed to the uniform distribution of Fe3+ ions expected in the AW model, we assume that (i) the effective dipolar field Bd at each Fe3+ site of the Fe6 cluster is the sum of dipolar fields due to the remaining Fe3+ ions in the Fe6 cluster (see Figure 9). (ii) The Fe6 clusters corresponding to the minor and major components have essentially identical bond lengths and angles resulting in very similar effective dipolar fields Bd. (iii) Since the density functional calculations, vide infra, show that the intra-cluster next nearest neighbor interaction Jnnn ≪ Jnn, only the two intra-cluster nearest neighbors are considered in calculating Jnn. (iv) Note that Eqs. 1 – 3 are only valid for the regime of the extreme narrowing of the line width due to large spin exchange interactions. Thus, only the lowest experimental δBpp values were used for estimating numerical values of Jnn. Table 4 lists the Jnn values corresponding to the major and minor components calculated using gmajor = gminor ≈ 2.01 and Smajor = Sminor = 5/2.
Figure 9.
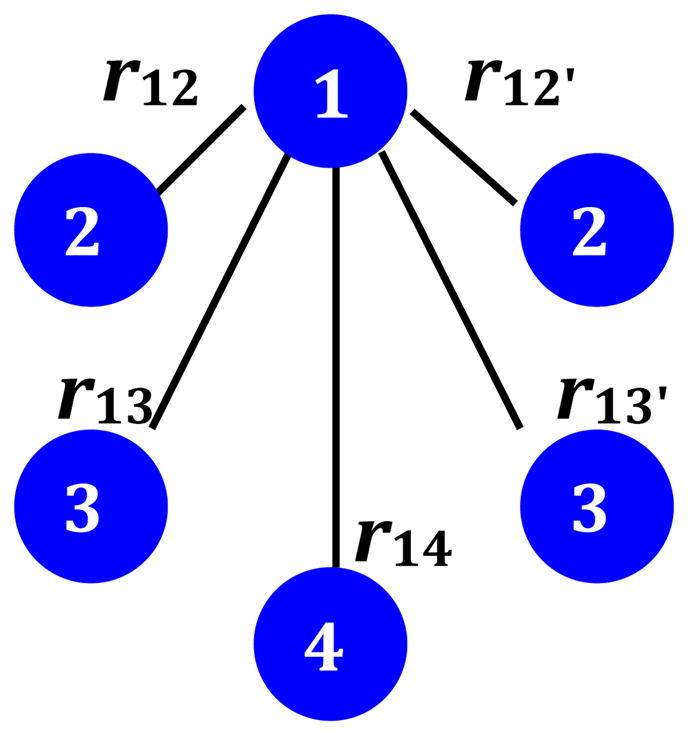
A diagram depicting dipolar interactions considered in estimating the total dipolar field Bd. rij is the distance between ith and jth ions and is assumed to be same for both minor and major components of Ba4KFe3O9. The distances are: r12 = r12′ = 3.750 Å, r13 = r13′= 5.642 Å, and r14 = 6.745 Å.
Table 4.
The Curie-Weiss temperature θ obtained from the Curie-Weiss fits (Eq. 4) of the EPR intensities as a function of temperature (Fig. 8b) and the nearest neighbor spin exchange integrals Jnn calculated from Eq. 5. The last column lists the |Jnn| values calculated from the Anderson-Weiss theory of EPR line width (Eqs. 1 – 3). The error in θ and Jnn is ±5 K.
| I(T) | δBpp (T) | ||
|---|---|---|---|
| Θ (K) | Jnn (K) | |Jnn| (K) | |
| Major component | −125 | −21 | 12 |
| Minor component | −470 | −81 | 38 |
Figure 8b shows a plot of inverse of the double-integrated EPR intensity (I) as a function of the temperature for both the major and the minor components. Notably, Imajor/Iminor remains at ~10 over the entire temperature range, thus, reinforcing the validity of the two-component model. As long as the thermal energy kT ~ J,J can be estimated from the I(T) measurements.28 Since the double-integrated EPR intensity is proportional to the paramagnetic component of magnetic susceptibility χ, Imajor(T) and Iminor(T) of Ba4KFe3O9 could be fitted to the Curie-Weiss law27 (cf. Eq. 4) to extract the Jnn according to the molecular field theory (MFT) (cf. Eq. 5):27
| (4) |
| (5) |
where, θ is the Weiss constant, C is the Curie constant and z (=2 for our Fe6 clusters) is the number of the nearest neighbors. The solid lines in Figure 8b are the best fits to the Eq. 4 with parameters shown in Table 4 along with the Jnn values obtained from the MFT relation shown in Eq. 5.
It is worth noting that the spin exchange integral ratio Jnn(minor)/Jnn(major) ~ 3 – 3.5, determined from 1/I vs T plot as well as the AW model, correlate well with the observed line width ratio δBpp(major)/δBpp(minor) ~ 3.6. Indeed, if all the parameters of the AW theory are the same with exception of Jnn, then δBpp ~ 1/Jnn ~ 1/θ. Thus, the major difference in the two magnetic environments corresponding to the narrow and broad EPR components is likely to be in the spin exchange Jnn integral with the rest of parameters being about the same. We speculate that the observed discrepancy in Jnn obtained from the AW theory and the Curie-Weiss fits is either due to the assumptions (i) – (iv) adapted for our Fe6 clusters or the inherent simplifications of the AW model. In fact, (δBpp)observed/(δBpp)calculated ~ 2 was expected by the authors of the AW model31 and noticed in other experiments32. Authors in references [32] attributed this discrepancy to the truncated Lorentzian line shape employed in the AW theory. We also note that the AW theory neglects the partial averaging of the dipolar fields due to spin-flips on the time scale of EPR experiment.
Spin exchange interactions
To better understand the magnetic properties of the Ba4KFe3O9, we examined the spin-exchange interactions within the iron hexamer ring by performing energy-mapping analysis based on density functional electronic band structure calculations.33–35 Since the cation disorder was observed for some positions of Ba/K, we assumed an ordered stoichiometric arrangement of Ba/K for our calculations by arranging them with largest distance from each other, unless the occupancies of their positions are higher than 0.9. Only the intra-ring spin-exchange interactions were considered, namely, the nearest-neighbors (Jnn) and next-nearest-neighbors (Jnnn) exchanges within each iron hexamer, while the inter-ring spin exchange interactions are expected to be negligible. To further simplify our discussion, it was assumed that all the nearest-neighbor interactions Jnn are identical, and so are the next-nearest-neighbor interactions Jnnn.
To determine the values of Jnn and Jnnn, we first consider the relative energies of the three ordered spin states (AF1, AF2 and AF3) of each iron hexamer ring shown in Figure 10 on the basis of density functional calculations. The calculations employed the projector-augmented wave (PAW) method encoded in the VASP code36 with the generalized-gradient approximation (GGA)37 for the exchange–correlation functional, the plane-wave cutoff energy of 485 eV, and either a set of 2×2×2 k-points or one k-point (i.e., Γ point). To consider the electron correlation associated with Fe 3d states, GGA plus on-site repulsion (GGA+U) calculations38 with U = 3 eV were also carried out. In evaluations of Jnn and Jnnn, the optimized crystal structure of Ba4KFe3O9 was employed. With the lattice parameters kept constant, the atomic positions of Ba4KFe3O9 were relaxed by GGA+U calculations (with U = 3 eV) using the AF1 state until the force acting on each ion is reduced below 0.01 eV/Å.
Figure 10.

Three ordered spin arrangements of each iron hexamer ring used for the energy-mapping analysis, where the filled and empty circles represent down-spin and up-spin Fe3+ sites, respectively. For each state, the expression of the total spin exchange energy per hexamer ring is given, where N is the number of unpaired spins at each Fe3+ site (i.e., N = 5), and the four numbers in the square bracket from left to right are the relative energies in meV per unit cell in the order of the entry given in Table 5.
The relative energies of the AF1, AF2 and AF3 states obtained from our GGA+U calculations are summarized in Figure 10. The total spin exchange energies of these states, per hexamer ring, can be expressed in terms of the spin Hamiltonian Ĥ = −Σi<j JijŜi · Ŝj, where Jij = Jnn, Jnnn. By applying the energy expressions obtained for spin dimers with N unpaired spins per spin site (in the present case, N = 5),39 the total spin exchange energies of the three ordered spin states per six-membered ring are written as summarized in Figure 10. Therefore, we determine Jnn and Jnnn by mapping the relative energies of the three ordered states obtained from the GGA+U calculations onto those expected from the total spin exchange energies (Table 5).
Table 5.
Values in K of the spin exchange parameters Jnn and Jnnn obtained from GGA+U calculations for the experimental (Exp) and optizmized (Opt) crystal structures of Ba4KFe3O9.
| U (eV) | Structure | k-points | Jnn | Jnnn |
|---|---|---|---|---|
| 0 | Exp | Γ | −347 | −5.3 |
| 3 | Exp | Γ | −206 | −0.9 |
| 3 | Exp | 2×2×2 | −207 | −1.3 |
| 3 | Opt | 2×2×2 | −199 | −1.0 |
In all cases, Jnn is strongly antiferromagnetic (of the order of −200 K), and Jnnn is negligible. It should be noted that since S = 5/2 for the high-spin Fe3+ ions, the effective nearest-neighbor spin exchange is given by J′nn = S2Jnn = 25Jnn/4. The width of the magnetic energy spectrum of Ba4KFe3O9 scales as J′nn, and hence should spread over a wide range of energy. Then, even at room temperature the Boltzmann distribution favors the occupation of lower-lying magnetic states in which more spins are paired resulting in lower moments. Consequently, the effective moment calculated from χT will be much smaller than expected from the paramagnetic state in which all the allowed magnetic states are equally occupied. As a result, the effective moment calculated from χT decreases continuously with lowering the temperature. In general, this kind of observation should be expected not only for magnetic oxides made up of corner-sharing FeO4 tetrahedra with Fe3+ (S = 5/2) ions but also for those made up of corner-sharing CoO4 tetrahedra with Co4+ (S = 5/2) ions as found for Cs2CoO3, Rb2CoO3, and K2CoO3.25
The value of Jnn ≈ −200 K obtained from the present GGA+U calculations is stronger than Jnn ≈ −20 K for the major component estimated from the EPR measurements by an order of magnitude. In general, GGA+U electronic structure calculations overestimate the magnitude of spin exchange interactions by a factor approximately up to four to five.34, 35, 40 Thus, we cannot exclude the possibility that our EPR analysis underestimated the value of Jnn by a factor of two or greater.
Conclusion
In summary, a new ferrite containing discrete 6-member rings of FeO4 tetrahedra has been synthesized and characterized experimentally and theoretically. In spite of the relatively short Fe-O-Fe contacts, no evidence of long-range magnetic order has been found in the magnetic susceptibility, Mössbauer, or the heat capacity data. X-band EPR indicated presence of two different magnetic species, with the Fe6 clusters in Ba4KFe3O9 accounting for ~90% of the total spin count. The temperature dependence of the EPR signal intensity of the Fe6 clusters is analyzed in terms of the Curie-Weiss law yielding the Weiss constant of ~ −125 K. This corresponds to the spin exchange integral Jnn of ~ −20 K that agrees well with |Jnn|~12 K estimated from the Anderson-Weiss EPR line shape theory. Our density functional calculations reveal that all nearest neighbor magnetic interactions are strongly antiferromagnetic, while next nearest neighbor interactions are negligible. The lower than expected effective moment calculated from χT is explained as resulting from the occupation of lower lying magnetic states in which more spins are paired, resulting in a lower moments.
Supplementary Material
Acknowledgments
Financial support from the National Science Foundation through Grants DMR:0804209 and DMR:0706593 is gratefully acknowledged. W.-J. S and M.-H. W acknowledge the support from the Office of Basic Energy Sciences, Division of Materials Sciences, U. S. Department of Energy, under Grant DE-FG02-86ER45259, and also from the computing resources of the NERSC center and the HPC center of NCSU. EPR instrumentation is supported by grants NIH S10RR023614, NSF CHE-0840501, and NCBC 2009-IDG-1015. S. N. and A. I. S. are grateful to U.S. Department of Energy’s Office of Basic Energy Sciences for financial support (Grant DE-FG02-02ER15354). T. M. P. is grateful for support by UNF’s Terry Presidential Professorship.
References
- 1.Deer WA, Howie RA, Zussman J. Rock Forming Minerals. John Wiley & Sons; New York: 1986. [Google Scholar]
- 2.Medina ME, Iglesias M, Snejko N, Gutierrez-Puebla E, Monge MA. Chem Mater. 2004;16:594. [Google Scholar]
- 3.Ali NZ, Nuss J, Sheptyakov D, Jansen M. J Solid State Chem. 2010:752. [Google Scholar]
- 4.Brachtel G, Hoppe R. Angew Chem Int Ed. 1977;16:43. [Google Scholar]
- 5.Zhou HD, Lumata LL, Kuhns PL, Reyes AP, Choi ES, Dalal NS, Lu J, Jo YJ, Baicas L, Brooks JS, Wiebe CR. Chem Mater. 2009;21:156. [Google Scholar]
- 6.Amann P, Sobotka BM, Fastje O, Moller A. Z Anor Allg Chem. 2007;633:2579. [Google Scholar]
- 7.Delattre JL. Chemistry. 2002;PhD:143. [Google Scholar]
- 8.Hoshino N, Ako AM, Powell AK, Oshio H. Inorg Chem. 2009;48:3396. doi: 10.1021/ic801776w. [DOI] [PubMed] [Google Scholar]; Müller A, Peters F, Pope MT, Gatteschi D. Chem Rev. 1998;98:239. doi: 10.1021/cr9603946. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman R. Sci Am. 1993;268:66. [Google Scholar]; Kortz U, Müller A, van Slageren J, Schnack J, Dalaf NS, Dressel M. Coord Chem Rev. 2009;253:2315. [Google Scholar]
- 10.Beck JS, Vartuli JC. Curr Opin Solid State Mater Sci. 1996;1:76. [Google Scholar]; Breck DW. Zeolite Molecular Sieves. John Wiley; New York: 1974. [Google Scholar]
- 11.Benelli C, Parsons S, Solan GA, Winpenny REP. Angew Chem. 1996;108:1967. [Google Scholar]; Caneschi A, Cornia A, Fabretti AC, Foner S, Gatteschi D, Grandi R, Schenetti L. Chem Eur J. 1996;11:1379. [Google Scholar]
- 12.Taft KL, Delfs CD, Papaefthymiou GC, Foner S, Gatteschi D, Lippard SJ. J Am Chem Soc. 1994;116:823. [Google Scholar]
- 13.Taft KL, Lippard SJ. J Am Chem Soc. 1990;112:9629. [Google Scholar]
- 14.Baca SG, Speldrich M, Ellern A, Kögerler P. Materials. 2011;4:300–310. doi: 10.3390/ma4010300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watton SP, Fuhrmann P, Pence LE, Caneschi A, Corina A, Abbati GL, Lippard SJ. Angew Chem Int Ed. 1997;36:2774. [Google Scholar]
- 16.Giaquinta DM, Davis WM, zur Loye H-C. Acta Cryst. 1994;C50:5–7. [Google Scholar]; Mwamuka JN, Gemmill WR, Stitzer KE, Smith MD, zur Loye H-C. J Alloys Comp. 2004;377:91–97. [Google Scholar]; Gemmill WR, Smith MD, zur Loye H-C. Solid State Sci. 2007;9:380–384. [Google Scholar]; Roof IP, Smith MD, zur Loye H-C. Solid State Sci. 2010;12:1211–1214. [Google Scholar]; Zhao Q, Smith MD, zur Loye H-C. J Chem Cryst. 2011 in press. [Google Scholar]
- 17.Giaquinta DM, Papaefthymiou GC, zur Loye H-C. J Solid State Chem. 1995;114:199. [Google Scholar]
- 18.Mugavero SJ, III, Gemmill WR, Roof IP, zur Loye H-C. J Solid State Chem. 2009;182:1950–1963. [Google Scholar]; Bugaris DE, zur Loye H-C. Angew Chem Int Ed. 2011 in press. [Google Scholar]
- 19.Delattre JL, Stacy AM. J Solid State Chem. 2003;172:261. [Google Scholar]
- 20.SMART Version 5.625, SAINT+ Version 6.45 and SADABS Version 2.05. Bruker Analytical X-ray Systems, Inc; Madison, Wisconsin, USA: 2001. [Google Scholar]
- 21.Sheldrick GM. Acta Cryst A. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 22.PLATON, A Multipurpose Crystallographic Tool. Utrecht University; Utrecht, The Netherlands, Spek, A. L: 1998. [Google Scholar]
- 23.Smirnov AI, Belford RL. J Magn Reson A. 1995;113:65. [Google Scholar]
- 24.Smirnov AI, Smirnova TI. Bio Magn Reson. 2004;21:277. [Google Scholar]
- 25.Nuss J, Ali NZ, Jansen M. Acta Cryst. 2007;B63:719. doi: 10.1107/S0108768107037147. [DOI] [PubMed] [Google Scholar]
- 26.Jansen M, Hoppe R. Z Anor Allg Chem. 1974;408:75. [Google Scholar]
- 27.Carlin RL. Magnetochemistry. Springer-Verlag; New York: 1986. [Google Scholar]
- 28.Abragam A, Bleaney B. Electron Paramgnetic Resonance of Transition Ions. Dover publications; New York: 1986. [Google Scholar]
- 29.Smirnova TI, Smirnov AI, Belford RL, Clarkson RB. J Am Chem Soc. 1998;120:5060. [Google Scholar]
- 30.Smirnov AI, Sen S. J Chem Phys. 2001;115:7650. [Google Scholar]
- 31.Anderson PW, Weiss PR. Rev Mod Phys. 1953;25:269. [Google Scholar]
- 32.Dalal NS, Millar JM, Jagadeesh MS, Seehra MS. J Chem Phys. 1981;74:1916. [Google Scholar]; Gulley JE, Hone D, Scapino DJ, Silbernagel BG. Phys Rev B. 1970;1:1020. [Google Scholar]
- 33.Whangbo M-H, Koo H-J, Dai D. J Solid State Chem. 2003;176:417. [Google Scholar]
- 34.Koo HJ, Whangbo M-H. Inorg Chem. 2008;47:4779. doi: 10.1021/ic800216j. [DOI] [PubMed] [Google Scholar]
- 35.Koo HJ, Whangbo M-H. Inorg Chem. 2008;47:128. doi: 10.1021/ic701153z. [DOI] [PubMed] [Google Scholar]
- 36.Kresse G, Hafner J. Phys Rev B. 1993;47:558. doi: 10.1103/physrevb.47.558. [DOI] [PubMed] [Google Scholar]; Kresse G, Furthmüller J. Compt Mater Sci. 1996;6:15. [Google Scholar]; Kresse G, Furthmüller J. Phys Rev B. 1996;54:11169. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 37.Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 38.Dudarev SL, Botton GA, Savrasov SY, Humhreys CJ, Shutton AP. Phys Rev B. 1998;57:1505. [Google Scholar]
- 39.Dai D, Whangbo M-H. J Chem Phys. 2001;114:2887. [Google Scholar]; Dai D, Whangbo M-H. J Chem Phys. 2003;118:29. doi: 10.1063/1.1760749. [DOI] [PubMed] [Google Scholar]
- 40.Xiang HJ, Lee C, Whangbo MH. Phys Rev B. 2007;76:220411. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.