

Abstract
Although flaviviruses cause significant human diseases, no antiviral therapy is currently available for clinical treatment of these pathogens. To identify flavivirus inhibitors, we performed a high-throughput screening of compound libraries using cells containing luciferase-reporting replicon of West Nile viruses (WNV). Five novel small molecular inhibitors of WNV were identified from libraries containing 96,958 compounds. The inhibitors suppress epidemic strain of WNV in cell culture, with EC50 (50% effective concentration) values of <10 µM and TI (therapeutic index) values of >10. Viral titer reduction assays, using various flaviviruses and nonflaviviruses, showed that the compounds have distinct antiviral spectra. Mode-of-action analysis showed that the inhibitors block distinct steps of WNV replication: four compounds inhibit viral RNA syntheses, while the other compound suppresses both viral translation and RNA syntheses. Biochemical enzyme assays showed that two compounds selectively inhibit viral RNA-dependent RNA polymerase (RdRp), while another compound specifically inhibits both RdRp and methyltransferase. The identified compounds could potentially be developed for treatment of flavivirus infections.
Keywords: West Nile virus, antiviral drug discovery, high-throughput screening, flavivirus replication, RNA-dependent RNA polymerase, RNA cap methyltransferase
1. Introduction
The family Flaviviridae includes three genera, the flaviviruses, the pestiviruses, and the hepatitis C viruses. West Nile virus (WNV), a member from the Flavivirus genus, is an arthropod-borne pathogen that cycles between Culex species of mosquitoes and birds; the virus can also infect humans and other vertebrate species. Together with WNV, the four serotypes of dengue virus (DENV), yellow fever virus (YFV), Japanese encephalitis virus (JEV), and tick-borne encephalitis virus (TBEV) are global emerging and reemerging pathogens. More than 50 million, 200,000, and 50,000 humans are infected by DENV, YFV and JEV every year, respectively (Gubler, Kuno, and Markoff, 2007). Besides parts of Africa, Asia, and Australia, WNV has also been found in Canada, Mexico, and South America (Kramer, Li, and Shi, 2007). Since 1999, WNV has caused thousands of human infections in the United States, representing the largest meningoencephalitis outbreak in the Western Hemisphere and the biggest WNV outbreak ever reported (Kramer, Li, and Shi, 2007). Although human vaccines are currently available for YFV, JEV, and TBEV, no effective antiviral therapy has been approved for the treatment of flavivirus infections. Therefore, it is important to develop antiviral agents for the treatment of flavivirus infections.
Flavivirus virions are about 50 nm in diameter, and harbor a plus-sense, single-stranded RNA genome of about 11 kb (Chambers et al., 1990). The genomic RNA contains a 5’-untranslated regions (UTR) and a 3’-UTR, and a single open reading frame (ORF) encoding a long polyprotein that is co- and post-translationally cleaved into 10 mature proteins by viral and cellular proteases. The N- and C-terminal portions of the ORF encode 3 structural proteins (capsid [C], premembrane [prM] or membrane [M], and envelope [E]) and 7 nonstructural proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5) (Chambers et al., 1990). The structural proteins are components of virions. The NS proteins are involved in the viral RNA replication; additionally, some NS proteins play roles in virion assembly (Kummerer and Rice, 2002; Liu, Chen, and Khromykh, 2003) and in anti-immune responses (Best et al., 2005; Guo, Hayashi, and Seeger, 2005; Lin et al., 2004; Liu et al., 2005; Munoz-Jordan et al., 2005; Munoz-Jordan et al., 2003). Enzymatic activities have been reported only for NS3 and NS5 proteins. NS3 is a multifunctional protein with activities of a serine protease (with NS2b as a cofactor), RNA helicase, 5’-RNA triphosphatase (RTPase), and nucleoside triphosphatase (NTPase) (Wengler and Wengler, 1991). NS5 functions as an RNA-dependent RNA polymerase (RdRp) (Tan et al., 1996) and an RNA cap methyltransferase (MTase) (Egloff et al., 2002; Koonin, 1993; Ray et al., 2006). The NS proteins form replication complexes on ER membrane and transcribe genomic plus-sense RNA into a complementary minus-sense RNA, which in turn, serves as the template for the synthesis of more plus-sense RNA. The plus-sense RNA is then packaged by viral C protein to form nucleocapsid, which is enclosed in an envelope consisting of host-derived lipid bilayer and the viral prM/M and E proteins (Chambers et al., 1990).
Different classes of inhibitors have been reported for flaviviruses (Shi, 2008). (1) Antibody-mediated treatment has generated promising therapeutic results. Passive administration of monoclonal antibodies prevented and alleviated encephalitis caused by JEV (Kimura-Kuroda and Yasui, 1988), YFV (Schlesinger, Brandriss, and Walsh, 1985), and St. Louis encephalitis virus (SLEV) (Mathews and Roehrig, 1984). Humanized monoclonal antibodies against WNV E protein were shown to be efficacious in mice, even administered as a single dose at day 5 post-infection (p.i.), when WNV has already infected the CNS (Ben-Nathan et al., 2003; Engle and Diamond, 2003; Julander et al., 2005). (2) A number of inhibitors have been reported to antagonize viral enzymes of NS3 and NS5 (see review [Shi, 2008]). (3) Interferon-α-2b is under clinical trial for treatment of WNV-mediated meningoencephalitis. Because several flaviviral nonstructural proteins can block interferon signaling, the antiviral effect of interferon is dramatically reduced once viral replication has initiated (Anderson and Rahal, 2002). (4) Inhibitors of other mechanisms have also been reported. Inhibitors of host α-glucosidase, such as castanospermine and N-nonyl-deoxynojirimycin, suppress JEV, DENV, and WNV (Courageot et al., 2000; Whitby et al., 2005; Wu et al., 2002). Minocycline inhibits WNV replication and apoptosis in human neuronal cells (Michaelis et al., 2007). Triaryl pyrazoline (Puig-Basagoiti et al., 2006), pyrozolopyrimidine (Gu et al., 2006), sulfonamide (Noueiry et al., 2007), and lycorine (Zou et al., 2008) compounds can also suppress WNV replication; however, the targets of these inhibitors remain to be identified. (5) RNAi and antisense oligomers were reported to inhibit flaviviruses. Synthetic siRNA or vectors expressing shRNA potently inhibit JEV and WNV in cell culture, and protect mice from viral disease (Bai et al., 2005; Kumar et al., 2006; Murakami et al., 2005). Antisense phosphorodiamidate morpholino oligomers (PPMO) showed effective antiviral activities against DENV and WNV (Deas et al., 2007; Deas et al., 2005; Holden et al., 2006; Kinney et al., 2005).
The goal of this study was to identify small molecule inhibitors of flaviviruses. Five novel inhibitors have been identified through high-throughput (HTS) screening of nearly 100,000 compounds in a WNV replicon assay. The compounds inhibit WNV in cell culture, with single-digit micromolar EC50 (50% effective concentration) values and >10 therapeutic indices (TI = CC50/EC50; CC50 represents 50% cytotoxic concentration). Mode-of-action study indicated that distinct steps of viral replication were suppressed by these compounds, some of which specifically target viral MTase and RdRp. Furthermore, the identified compounds were shown to have distinct antiviral spectra.
2. Materials and methods
2.1. Cells and viruses
Vero cells were maintained in DMEM with 10% FBS in 5% CO2 at 37°C. A Vero cell line containing persistently replicating WNV replicon (Rluc-Neo-Rep) with a Renilla luciferase (Rluc) reporter and a neomycin phosphotransferase (Neo) gene was maintained with 1 mg/ml G418 in DMEM with 10% FBS (Lo, Tilgner, and Shi, 2003). WNV was derived from a full-length infectious cDNA clone of the New York strain 3356 (Shi et al., 2002). YFV (17D vaccine strain), DENV-2 (New Guinea strain), and Western Equine encephalitis virus (WEEV; Cova 746 strain) were generously provided by Laura Kramer at Arbovirus laboratory at Wadsworth Center, Albany, New York. Vesicular stomatitis virus (VSV; New Jersey serotype; a gift from Paul Masters at the Wadsworth Center) was also included in the antiviral spectrum assay.
2.2. HTS of small molecular compound libraries
The WNV Rluc-Neo-Rep Vero cells were previously established and validated for screening inhibitors of WNV (Puig-Basagoiti et al., 2005). To increase the throughput, we developed the assay into a 384-well format. Briefly, Rluc-Neo-Rep Vero cells (5,000 cells in 30 µl of DMEM plus 10% FBS without G418 were seeded to each well of the 384-well plate. After incubating the plates at 37°C for 12 h, 0.1 µl of each compound stock (5 mg/ml in dimethyl sulfoxide [DMSO]) were added to each well. The plates were incubated for 48 h, after which 10 µl of luciferase substrate EndurRen (Promega) was added to each well to a final concentration of 10 µM. After incubating the plates at 37°C for another 2 h, the luciferase activities were measured in an Envision 2102 multilabel reader (Perkin Elmer). Since the assay does not involve infectious particles, the screening was performed in a biosafety level-2 laboratory.
Using the above assay method, we screened a total of 96,958 small molecular compounds from the commercial libraries, collected at the National Screening Laboratory for the Regional Centers of Excellence for Biodefense and Emerging Infectious Disease (NSRB; Harvard Medical School). Each compound was screened in duplicate. Mycophenolic acid (1 µM), a known WNV inhibitor (Lo, Tilgner, and Shi, 2003), was included in each plate as a positive control.
2.3. MTT cell proliferation assay
Compound cytotoxicity was determined in Vero cells using an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] cell proliferation assay (ATCC). Approximately 1×104 Vero cells in 100 µl DMEM medium were seeded per well in a 96-well plate. After 6 h incubation, 1 µl of compound, dissolved in 100% DMSO, was added to cells at indicated concentrations. After incubating the plates for 48 h, 10 µl of MTT reagent was added. The cells were then incubated for another 3.5 h, after which 100 µl of detergent reagent was added. The plates were incubated in the dark at room temperature for 4 h. Absorbance was recorded in a microtiter plate reader with a 550 nm filter.
2.4. Viral titer reduction assay
Viral titer reduction assay was performed to examine the antiviral spectra of the “hit” compounds in WNV, YFV, DENV-2, WEEV, and VSV. Approximately 2.5×105 Vero cells were seeded to each well of a 24-well plate. After incubation for 12 h, the cells were infected with individual virus (0.1 MOI) and treated immediately with compounds at indicated concentrations. For WNV, YFV, DENV-2 and WEEV, culture medium was collected at 42 h p.i., stored at −80°C, and subjected to plaque assays. For VSV, culture medium was collected at 16 h post-infection. A double overlayer protocol was followed for plaque assays on Vero cells. For WNV, 6×105 Vero cells per well were seeded in a 6-well plate. After incubating for 3 days to reach confluence, the cells were infected with 100 µl of 10-fold serial dilutions of the virus for 1 h at 37°C. Afterwards, 3 ml of a first layer containing 0.6% Oxoid agar, basal medium Eagle with 1% FBS, 0.02% DEAE Dextran, and 0.13% NaHCO3 was added to the infected cells. Two days later, 3 ml of a second layer containing 1% Noble agar, basal medium Eagle medium with 1% FBS, 0.02% DEAE Dextran, 0.13% NaHCO3, and 0.004% neutral red was added over the first layer. The plates were further incubated for 12 h before counting the plaques. The protocol for WEEV was identical to that for the WNV. For other viruses, the time between the addition of the first and the second layer of agar was 1, 4, and 5 days for VSV, YFV, and DENV-2, respectively.
2.5. Transient replicon assay
A transient replicon assay of WNV was used to quantify compound-mediated inhibition of viral translation and suppression of RNA replication. The replicon contained a Renilla luciferase reporter and the foot-and-mouth disease virus 2A gene in place of the deleted viral structural genes, and the hepatitis delta virus ribozyme (HDVR) was engineered at the 3’ end of the WNV genome, resulting in Rluc2A-Rep (Tilgner, Deas, and Shi, 2005). For the transient assay, 10 µg of Rluc2A-Rep RNA was electroporated into Vero cells (8×106) as previously described (Lo et al., 2003b). The transfected cells were suspended in 25 ml of DMEM with 10% FBS; 1.5 ml of the cell suspension was seeded to each well of a 12-well plates, and immediately treated with inhibitors at indicated concentrations. The cells were assayed for luciferase activities at 4 h post-transfection (p.t.; representing peak translation), and at 36 h and 48 h p.t. (representing RNA replication), as previously reported (Tilgner, Deas, and Shi, 2005).
2.6. RdRp assay
The RdRp reaction (25 µl) contained 50 mM Tris, 20 mM NaCl, 5 mM MgCl2, 0.5 mM ATP, 0.5 mM GTP, 0.5 mM CTP, 5 µM UTP, 15 µCi α-32P-UTP (10 µCi/µl, 3,000 Ci/mmol; Perkin Elmer Life Sciences), 3 µg NS5, 1 µg RNA template, and indicated concentrations of compounds. The RNA template consisted of the 5’-terminal 269 nt that are directly connected to the 3’-terminal 622 nt of the WNV genome (Zhang et al., 2008). The reactions were incubated at 33°C for 2 h; the mixtures were passed through a MicroSpin G-25 column (GE Healthcare) to remove unincorporated NTPs; and the elute was mixed with an equal volume of denaturing Gel Loading Buffer II (Ambion) and loaded into a 6% denaturing polyacrylamide gel (PAGE) with 7M urea. A PhosphorImager was used to quantify the 32P-labeled RNA products.
2.7. Protease assay
A recombinant WNV protease, containing amino acids 50–90 of NS2b, a (Gly)4Ser(Gly)3-linker, and amino acids 1–183 of NS3, was cloned into plasmid pET-28a at the BamH I and EcoR I sites. E.coli Rosetta (DE3), harboring the pET-28a-WNV protease plasmid, was grown in LB medium with 30 µg/ml kanamycin at 37°C to absorbance of 0.6 at 600 nm, after which the culture was induced with 0.3 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 16°C for 16 h. The recombinant protease, with an N-terminal (His)6-tag, was purified through a nickel-nitrilotriacetic acid column (Qiagen) using an elution buffer containing 200 mM imidazole.
We used a fluorogenic peptide substrate pERTKR-AMC (purchased from R&D systems) to measure the protease activity. One microliter of the purified protease (1µg/µl) was mixed with 97 µl of buffer (50 mM Tris, pH 9.5, and 30% glycerol) plus 1µl of compound at indicated concentrations in a 96-well plate. The reaction was initiated by the addition of 1 µl of the peptide substrate to a final concentration of 20 µM. Fluorescence (excitation 380 nm and emission 460 nm) was monitored every minute using a Modulus™ Microplate Multimode Reader (Turner Biosystems) at room temperature.
2.8. Methylation assay
An N-7 methylation assay was used to examine compound-mediated inhibition of WNV MTase. The methylation assay has been previously described (Dong et al., 2007). Briefly, the 5’-end labeled substrates, G*pppA-RNA (* indicates that the following phosphate is 32P-labeled), representing the first 190 nt of the WNV genome were prepared using a vaccinia virus capping enzyme, following the manufacturer’s protocol (Epicentre). The N7 methylation activity was quantified by measuring the G*pppA-RNA→ m7G*pppA-RNA conversion, catalyzed by the WNV MTase domain in a pH 7.0 buffer. At pH 7.0, no 2’-O methylation can occur, whereas N7 methylation is fully active (Zhou et al., 2007). The methylation reactions were then digested with nuclease P1 to release cap structures (GpppA and m7GpppA), and were analyzed on polyethyleneimine cellulose thin layer chromatography (TLC) plates. The spots on TLC plates, representing the different cap structures, were quantified using a PhosphorImager.
2.9. NTPase assay
The standard NTPase reaction (10 µl) contained 20 mM Tris (pH 7.5), 2.5 mM MgCl2, 2 mM dithiothreitol (DTT), 1 mM cold GTP spiked with 1 µCi corresponding [α-P32] GTP (3000 Ci/mmol; Perkin Elmer), and 0.5 µg of NS3 protein. The reaction was incubated at 37°C for 30 minutes, and terminated by the addition of 1 µl of 0.5 M ethylenediaminetetraacetic acid disodium salt (EDTA). One microliter of the reaction was then spotted onto a plastic-backed polyethyleneimine cellulose F sheet (J.T. Baker), and analyzed by ascending TLC using 0.4 M ammonium sulfate as a running buffer. The TLC plate was dried, and quantified using a PhosphorImager.
3. Results
3.1. Primary HTS
We previously established a WNV replicon Vero cell line that can be used for antiviral screening (Lo, Tilgner, and Shi, 2003; Puig-Basagoiti et al., 2005). The replicon contained dual reporters: a Renilla luciferase and a neomycin phosphotransferase (Rluc-Neo-Rep, Fig. 1A). Here we adapted this cell line into a 384-well plate HTS assay. Fig. 1B summarizes the optimal condition of the assay. The assay procedure is homogeneous, without any washing steps. In the absence of compound, the assay showed an average luciferase signal of 1.2×105 ± 6.0×103 light units, an assay window (a ratio of luciferase signal from the replicon-containing Vero cells to a background luciferase signal from the naïve Vero cells without replicon) of >103, and a Z-factor value of >0.9. These results demonstrate the reliability of the assay.
Fig. 1.

WNV replicon-based HTS. (A) A schematic diagram of the WNV replicon (Rluc-Neo-Rep) containing a Renilla luciferase reporter and a neomycin phosphotransferase gene. The Rluc reporter is in-frame fused with the WNV genome to replace the viral structural C-prM-E genes. The Neo gene is located at the upstream end of the 3’ UTR, and is driven by the encephalomyocarditis virus internal ribosomal entry site (IRES). (B) Flowchart of the replicon-based HTS. Vero cells harboring the WNV Rluc-Neo-Rep were seeded into 384-well plates, incubated with compounds, and assayed for luciferase activities.
A total of 96,958 compounds were screened in duplicate on separate plates. Since the concentration of compound stock was at 5 mg/ml (molecular weights ranged from 300 to 700), the compounds were screened at a final concentration between 24 µM and 55 µM. The primary screening revealed 2,260 “hits”, each of which suppressed the luciferase activity by >90%. Because limited number of compound reordering (i.e., “cherry-picking”) was allowed from the screening facility, we were only able to obtain the most active 291 compounds for “hit” reconfirmation.
3.2.“Hit” reconfirmation
“Hit” reconfirmation was performed in the WNV Rluc-Neo-Rep Vero cells through determining the antiviral efficacy (EC50). Cytotoxicity (CC50) of each compound was measured using a cell proliferation-based MTT assay. From the 291 “hits”, four compounds (number 11, 17, 35, and 42) exhibited EC50 values of 0.2–12 µM (Fig. 2, third column) and TI values of >10 (Table 1). Another compound (number 39) showed an EC50 value of 14 µM (Fig. 2) and a TI of <10 in the replicon assay; however, the TI value of this compound was >10 when tested in the viral titer reduction assay (Table 1). The other 286 “hits” either did not reproduce the antiviral activities using the reordered compounds, or showed TI values of >10 (indicating that the observed antiviral activities were due to cytotoxicity). Therefore, only five compounds (11, 17, 35, 39, and 42) were selected for further analysis.
Fig. 2.

Anti-WNV activities of compounds 11 (A), 17 (B), 35 (C), 39 (D), and 42 (E). For each compound, chemical structure, MTT-based cytotoxicity, replicon-based efficacy, and viral titer reduction-based efficacy are presented, with EC50 and CC50 values indicated. Vero cells were used in all presented experiments. For the results of WNV infection, the percentage of viral titer after compound treatment is indicated on the top of each bar (with the viral titer from the mock-treated sample set as 100%). Average results from three to four independent experiments are presented; error bars represent the standard deviations.
Table 1.
Summary of antiviral activity
| Compound | Structure | CC50 (µM)a |
WNV replicon | WNV infection | Inhibition step |
Potential target |
Antiviral spectrum |
||
|---|---|---|---|---|---|---|---|---|---|
| EC50 (µM)b | TIc | EC50 (µM)b | TIc | ||||||
| 11 |  |
125 | 12 | 10 | 9 | 14 | Translation and RNA synthesis | RdRp | WNV, DENV-2 YFV, WEEV, and VSV |
| 17 | 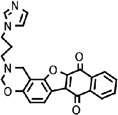 |
15 | 0.2 | 150 | 0.7 | 21 | RNA synthesis | MTase and RdRp | WNV, DENV-2 and YFV |
| 35 |  |
80 | 8 | 10 | 7 | 11 | RNA synthesis | N.D.d | WNV and DENV-2 |
| 39 |  |
50 | 14 | 4 | 4 | 13 | RNA synthesis | RdRp | WNV |
| 42 |  |
45 | 0.7 | 450 | 2 | 22 | RNA synthesis | N.D.d | WNV, DENV-2 YFV, WEEV, and VSV |
CC50 values were derived from Vero cells using an MTT assay.
EC50 values were derived from assays using the WNV replicon Vero cells or from assays using WNV infection on Vero cells.
TI (therapeutic index) values were calculated by dividing the CC50 values by the EC50 values.
N.D.: not detectable.
3.2. Validation of “hits” in WNV infection assay
To validate the results derived from the replicon cells, we assayed the “hits” in a viral titer reduction assay. Vero cells were infected with an epidemic strain of WNV (0.1 MOI). The infected cells were immediately treated with different concentrations of inhibitors. The tested concentration range of each compound was selected according to the cytotoxicity results. At 42 h p.i., viral titers in supernatants were determined by plaque assays. As shown in Fig. 2, the efficacies derived from the viral titer reduction assay in general correlated with those obtained from the replicon cell-based assay (compare the third and fourth columns). The difference in the EC50 values between the two assays is ≤3.5-fold. Overall, the viral titer reduction results confirmed the antiviral activities of the identified compounds.
3.3. Antiviral spectrum
To examine the antiviral spectrum of the inhibitors, we performed viral titer reduction assays, using flaviviruses other than WNV (YFV and DENV-2), WEEV (a plus-sense RNA alphavirus), and VSV (a negative-sense RNA rhabdovirus). Vero cells were infected with individual virus (0.1 MOI), treated immediately with various concentrations of compounds, and assayed for viral yields in culture medium (see details in Section 2.4). As shown in Table 2, compounds 11 and 42 inhibited all tested viruses; compound 17 specifically suppressed flaviviruses (WNV, DENV-2, and YFV); compound 35 selectively inhibited WNV and DENV-2; and compound 39 suppressed only WNV. The results demonstrated the antiviral selectivity of the compounds.
Table 2.
Antiviral spectruma
| WNV | DENV-2 | YFV | WEEV | VSV | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | EC50 (µM) | TI | EC50 (µM) | TI | EC50 (µM) | TI | EC50 (µM) | TI | EC50 (µM) | TI |
| 11 | 9 | 14 | 12 | 10 | 5 | 25 | 12 | 10 | 12 | 10 |
| 17 | 0.7 | 21 | 1.5 | 10 | 1.5 | 10 | 5 | 3 | 12 | 1 |
| 35 | 7 | 11 | 8 | 10 | 15 | 5 | 15 | 5 | 15 | 5 |
| 39 | 4 | 13 | 15 | 3 | 15 | 3 | 15 | 3 | 12 | 4 |
| 42 | 2 | 22 | 2 | 23 | 3 | 15 | 3.5 | 13 | 5 | 9 |
EC50 values were determined from viral titer reduction assays using Vero cells; CC50 values were obtained from Vero cells using an MTT assay (Fig. 2); TI = CC50 / EC50.
3.4. Selection of resistance to WNV
We attempted to select WNVs that are resistant to the identified inhibitors. Wild-type virus was cultured on Vero cells for eight rounds (MOI 0.1; 2–3 days per incubation round) in the presence of compounds. Based on the antiviral efficacy and cytotoxicity (Fig. 2), we chose to select resistant virus at concentrations of 33 µM, 3.7 µM, 33 µM, 33 µM, and 11 µM for compounds 11, 17, 35, 39, and 42, respectively. At the chosen concentrations, ≥80% cell viability was maintained after incubating the cells with individual compounds. No resistant viruses were recovered after eight rounds of selection (data not shown). As a positive control, resistant viruses were readily obtained after six rounds of selection in the presence of 0.8 µM of lycorine (data not shown). Lycorine was recently reported to have antiviral activities against flaviviruses (Zou et al., 2008).
3.5. Differentiation of inhibition between viral translation and RNA synthesis
We analyzed the inhibitors in a transient replicon assay to dissect the steps at which the compounds suppress WNV replication. The transient replicon assay is able to differentiate between viral translation and RNA synthesis (Deas et al., 2005; Lo et al., 2003a). Vero cells were transfected with a luciferase-reporting replicon of WNV (Rluc2A-Rep, Fig. 3A), immediately treated with inhibitors; and measured for luciferase activities at 4 h p.t. (indicative of translation), and 36 h and 485 h p.t. (indicative of RNA synthesis). It should be noted that, according to the MTT results (Fig. 2), each compound was tested at a subtoxic concentration (with ≥90% cell viability). As shown in Fig. 3, at 4 h p.t., compound 11 suppressed the luciferase signal by 55%, indicating that the compound inhibits RNA translation; in contrast, other four compounds had minor effects on the luciferase activities (<20%). At 36 h and 48 h p.t., all compounds inhibited luciferase activities by >70%, indicating that each compound suppresses viral RNA synthesis. Overall, the results showed that the identified compounds block distinct steps of WNV replication. Four compounds (number 17, 35, 39, and 42) inhibit viral RNA syntheses. For compound 11, the suppression of RNA synthesis may result from the suppression of RNA translation; however, biochemistry assays showed that this compound weakly inhibited polymerase activity (see below), indicating that the compound blocks both translation and RNA synthesis.
Fig. 3.

Inhibition of WNV translation and RNA synthesis. (A) A WNV reporting-replicon (Rluc2A-Rep) that can be used to differentiate between viral translation and RNA replication. The replicon contains a Renilla luciferase and the foot-and-mouth disease virus (FMDV) 2A at a position where WNV structural genes were deleted; the hepatitis delta virus ribozyme (HDVR) was engineered at the 3’ end of the replicon. (B) Compound-mediated inhibition of viral translation and RNA synthesis. Immediately after electroporation with Rluc2A-Rep RNA, Vero cells were treated with compounds at indicated concentrations, and assayed for luciferase activities at 4 h, 36 h, and 48 h post transfection. Inhibition of viral translation was quantified by luciferase signals at 4 h p.t., while inhibition of RNA synthesis was monitored by luciferase activities at 36 h and 48 h post transfection. Each bar indicates the percentage of luciferase activity derived from the compound-treated cells versus the luciferase signal derived from the mock-treated cells. Average results from three experiments are presented.
3.6. Inhibition of WNV enzymes
We directly tested the compounds in four WNV enzyme assays: protease, NTPase, RdRp, and MTase. None of the five compounds inhibited the protease and NTPase activities, up to 100 µM (the highest tested concentration). In contrast, control compounds, nonradioactive GTP (1 µM) and aprotinin (25 µM) respectively suppressed the NTPase and the protease activities by >85% (data not shown). For the MTase, we examined the compounds in an N7-methylation (the conversion of GpppA-RNA to m7GpppA-RNA) assay. Only compound 17 suppressed the N7-methylation (IC50 ≈ 54 µM) (Fig. 4). For the RdRp, compounds 11, 17, and 39, but not compounds 35 and 42, showed weak inhibition (Fig. 5). These results suggest that compounds 11, 17, and 39 inhibit WNV infection, possibly through suppression of the NS5-encoded enzymatic activities.
Fig. 4.

Inhibition of the WNV MTase. (A) TLC analysis of N7-methylation. WNV 5’ G*pppA-190-nt RNA (the asterisk indicates that the following phosphate is 32P-labeled) was methylated to m7G*pppA-190-nt RNA by recombinant MTase domain of the WNV NS5. The methylation reaction mixtures were digested with nuclease P1, to release G*pppA (residual substrate) and m7G*pppA (product), and then analyzed on TLC plates. The G*pppA→m7G*pppA conversion was quantified using a PhosphorImager. The compound-mediated inhibition was indicated by the remaining methylation activity, as compared to the mock-treated reaction (set as 100%). 32P-labeled markers, G*pppA and m7G*pppA, are shown at the top. The positions of the origin and the migration positions of G*pppA and m7G*pppA are shown to the left of the TLC image. (B) Summary of methylation results. Average results of three experiments are presented.
Fig. 5.

Inhibition of the WNV RdRp activity. An RNA containing the 5’ 269 nt directly fused to the 3’ 622 nt of the WNV genome was used as a template for the RdRp assay. The 32P-UMP-labeled products were resolved on a denaturing PAGE gel, and quantified using a PhosphorImager. Two forms of RNA products (input length [1X] and double the length of the input RNA [2X]) are indicated. The compound-mediated inhibitions were indicated by the remaining RdRp activities, as compared to the mock-treated reaction (set as 100%). A representative result from three experiments is shown.
4. Discussion
As an initial step towards development of small molecule-based therapeutics of flaviviruses, we established a 384-well formatted screening assay using a WNV replicon cell line (Fig. 1). The current study has demonstrated the reliability and reproducibility of the assay, as evidenced by an assay window of >103 and a Z-factor of >0.9. This assay can be used to screen compound libraries for inhibitors of viral translation and RNA synthesis. Theoretically, inhibitors of both viral enzymes and viral targets without enzymatic activities (e.g., NS1, NS2a, NS2b, NS4a, and NS4b) could be identified through the replicon screening; additionally, inhibitors of host factors that are critical for viral replication can also be identified. Compared with enzyme-based assays, such cell-based assay has the advantage of being able to filter compound libraries for molecules that can penetrate cellular membrane as well as have antiviral activities. Since WNV and many other flaviviruses are biosafety level-3 or level-4 pathogens, the other major advantage of the replicon assay is that the antiviral screening can be performed in a biosafety level-2 laboratory, due to the lack of viral structural genes in the replicon.
Five novel inhibitors of WNV were identified after screening libraries of 96,958 compounds in the replicon assay (Table 1). When tested in the WNV infection assay, all five compounds showed EC50 values of <10 µM and TI values of >10. The antiviral efficacy derived from the replicon assay was in general agreement with that derived from the authentic viral infection assay, with the difference in EC50 value of ≤3.5 folds between the two assay systems. The difference in EC50 value is expected, given the different nature of the two assays. Specifically, the viral RNA and host cell in the replicon assay are different from those in the viral infection assay. During the selection of replicon-cell line, both the replicon RNA and host cells had accumulated adaptations to reach a balance between viral replication level and cell viability (Puig-Basagoiti et al., 2007); these adaptations may change the antiviral sensitivity of the replicon assay. Alternatively, if a compound inhibits multiple steps of viral life cycle (e.g., viral translation or RNA synthesis plus virion entry or assembly), the compound may exhibit a better potency in the viral infection assay than that in the replicon assay.
Target deconvolution for inhibitors identified from the replicon assay can be challenging. Three approaches were taken to determine the antiviral target. First, we examined the antiviral spectra of the inhibitors by performing viral titer reduction assays, using various flaviviruses and nonflaviviruses. The antiviral spectrum results would illuminate whether the compounds inhibit host or viral targets. It is reasonable to speculate that a compound capable of inhibiting viruses from different families is likely to suppress a host target. Along this line, compounds 11 and 42 inhibited flavivirus, alphavirus, and rhabdovirus (Table 2), suggesting that these two compounds may interfere with host target(s). In contrast, compound 17 inhibited all three tested flaviviruses (WNV, DENV-2, and YFV), but not the nonflaviviruses, indicating that its antiviral target is flavivirus-specific. Interestingly, compound 39 only suppressed WNV, and compound 35 selectively inhibited WNV and DENV-2, suggesting that these compounds target factor(s) unique to the sensitive virus(es).
Second, we attempted to select resistance WNV to illuminate the targets of the inhibitors. Unfortunately, no resistant virus was obtained for any of the identified compounds. The failure of resistance virus selection was most likely due to the low TI values (TI = 11–22) of the compounds in the viral infection assay. Because of the low TI values, we were unable to select resistant viruses at compound concentrations high enough to impose sufficient pressure for virus to evolve (Note that high concentrations of these compound would cause cytotoxicity). This notion is supported by the fact that resistant WNV was consistently recovered after selection with lycorine, a flavivirus inhibitor with a higher TI value (EC50 = 0.23 µM; CC50 = 24 µM; TI = 104) (Zou et al., 2008).
Third, we directly analyzed the inhibitors in four enzyme assays of WNV, including protease, NTPase, MTase, or RdRp (See summary in Table 1). Compounds 11 and 39 selectively inhibited RdRp (Fig. 5), while compound 17 suppressed both the MTase and RdRp (Figs. 4 and 5). The observed inhibitions of MTase and RdRp were not due to the promiscuous nature of the compounds, because the same inhibitors did not suppress the protease and NTPase. However, it should be noted that the observed inhibitions were weak, with IC50 values >50 µM. We are puzzled by the results that the IC50 values (derived from the enzyme assay) were much higher than the EC50 values (<10 µM; derived from the viral infection assay). If a compound suppresses only one target, it is expected to show an EC50 value higher than the IC50 value; this is because the compound could nonspecifically bind to cell culture components (e.g., serum and cellular proteins), resulting in a decrease in potency. These results prompted us to speculate that, besides suppressing the identified enzymes, certain compounds may also work on other unknown targets.
Considering the available results, we conclude the following for the identified compounds. (1) Compound 11 may inhibit a host target as well as viral RdRp because (i) it has a broad antiviral spectrum, (ii) it inhibits both viral translation and RNA synthesis in the replicon assay, and (iii) it weakly suppresses viral RdRp. (2) Compound 17 inhibits both MTase and RdRp. Since the two enzymes are physically linked within a single protein NS5, inhibition of both enzymes may result in a synergy, leading to a potent antiviral activity in cell culture. (3) Compound 35 may inhibit virus through suppression RNA synthesis through an unidentified viral target, as evidenced by (i) it specifically suppressed RNA synthesis in the replicon assay, and (ii) it selectively inhibited WNV and DENV-2. (4) Compound 39 selectively inhibits WNV, possibly through suppression of viral RdRp. However, since the in vitro activity against RdRp is weak, the compound may also inhibit another unidentified viral target. (5) Compound 42 inhibits a broad spectrum of viruses through suppression of RNA synthesis, possibly at a host target. Although the mode-of-action of these compounds remained to be further analyzed, the potent antiviral activities in cell culture (EC50 = 0.7–9 µM; TI = 11–22) warrant these “hits” for future development.
Acknowledgments
We are grateful to Su Chiang, Caroline Shamu, David Wrobel, Sean Johnson, and Dave Fletcher at the NSRB (Harvard Medical school) for their help and assistance with the compound library screening. We thank the Cell Culture Facility at the Wadsworth Center for the maintenance of Vero cells. The work was partially supported by federal funds from the National Institute of Allergy and Infectious Disease, National Institutes of Health (NIH), under contract N01-AI-25490, and by NIH grants 1U01AI061193 and U54-AI057158 (Northeast Biodefense Center).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson JF, Rahal JJ. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg. Infect. Dis. 2002;8(1):107–108. doi: 10.3201/eid0801.010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai F, Wang T, Pal U, Bao F, Gould LH, Fikrig E. Use of RNA interference to prevent lethal murine west nile virus infection. J Infect Dis. 2005;191(7):1148–1154. doi: 10.1086/428507. [DOI] [PubMed] [Google Scholar]
- Ben-Nathan D, Lustig S, Tam G, Robinzon S, Segal S, Rager-Zisman B. Prophylactic and therapeutic efficacy of human intravenous immunoglobulin in treating West Nile virus infection in mice. J. Infect. Dis. 2003;188(1):5–12. doi: 10.1086/376870. [see comment] [DOI] [PubMed] [Google Scholar]
- Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, Boer E, Wolfinbarger JB, Bloom ME. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005;79(20):12828–12839. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- Courageot MP, Frenkiel MP, Dos Santos CD, Deubel V, Despres P. Alpha-glucosidase inhibitors reduce dengue virus production by affecting the initial steps of virion morphogenesis in the endoplasmic reticulum. J. Virol. 2000;74(1):564–572. doi: 10.1128/jvi.74.1.564-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas TS, Bennett CJ, Jones SA, Tilgner M, Ren P, Behr MJ, Stein DA, Iversen PL, Kramer LD, Bernard KA, Shi P-Y. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob. Agents Chemother. 2007;51:2470–2482. doi: 10.1128/AAC.00069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas TS, Binduga-Gajewska I, Tilgner M, Ren P, Stein DA, Moulton HM, Iversen PL, Kauffman EB, Kramer LD, Shi P-Y. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J. Virol. 2005;79(8):4599–4609. doi: 10.1128/JVI.79.8.4599-4609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Ray D, Ren S, Zhang B, Puig-Basagoiti F, Takagi Y, Ho CK, Li H, Shi PY. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J Virol. 2007;81(9):4412–4421. doi: 10.1128/JVI.02455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21(11):2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle M, Diamond M. Antibody prophylaxis and therapy against West Nile virus infection in wild-type and immunodeficient mice. J. Virol. 2003;77(24):12941–12949. doi: 10.1128/JVI.77.24.12941-12949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Ouzunov S, Wang L, Mason P, Bourne N, Cuconati A, Block TM. Discovery of small molecule inhibitors of West Nile virus using a high-throughput sub-genomic replicon screen. Antiviral Res. 2006;70(2):39–50. doi: 10.1016/j.antiviral.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Gubler D, Kuno G, Markoff L. Flaviviruses. In: Knipe DM, Howley PM, editors. Fields virology. 5th. vol. 1. Philadelphia, Pa: Lippincott William & Wilkins; 2007. pp. 1153–1253. [Google Scholar]
- Guo J, Hayashi J, Seeger C. West nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 2005;79(3):1343–1350. doi: 10.1128/JVI.79.3.1343-1350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden K, Stein D, Pierson T, Ahmed A, Clyde K, Iversen P, Harris E. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3' stem-loop structure. Virology. 2006;344(2):439–452. doi: 10.1016/j.virol.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Julander JG, Winger QA, Olsen AL, Day CW, Sidwell RW, Morrey JD. Treatment of West Nile virus-infected mice with reactive immunoglobulin reduces fetal titers and increases dam survival. Antiviral Res. 2005;65(2):79–85. doi: 10.1016/j.antiviral.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Kimura-Kuroda J, Yasui K. Protection of mice against Japanese encephalitis virus by passive administration with monoclonal antibodies. J. Immunol. 1988;141(10):3606–3610. [PubMed] [Google Scholar]
- Kinney R, Huang C, Rose B, Kroeker A, Dreher T, Iversen P, Stein D. Inhibition of dengue virus serotypes 1 to 4 in vero cell cultures with morpholino oligomers. J. Virol. 2005;79(8):5116–5128. doi: 10.1128/JVI.79.8.5116-5128.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. Computer-assisted identification of a putative methyltransferase domain in NS5 protein of flaviviruses and lambda 2 protein of reovirus. J. Gen. Virol. 1993;74(Pt 4):733–740. doi: 10.1099/0022-1317-74-4-733. [DOI] [PubMed] [Google Scholar]
- Kramer L, Li J, Shi P-Y. West Nile virus. The Lancet Neurology. 2007;6:171–182. doi: 10.1016/S1474-4422(07)70030-3. [DOI] [PubMed] [Google Scholar]
- Kumar P, Lee SK, Shankar P, Manjunath N. A single siRNA suppresses fatal encephalitis induced by two different flaviviruses. PLoS Med. 2006;3(4):e96. doi: 10.1371/journal.pmed.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummerer BM, Rice CM. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002;76(10):4773–4784. doi: 10.1128/JVI.76.10.4773-4784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R-J, Liao C-L, Lin E, Lin Y-L. Blocking the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J. Virol. 2004;78(17):9285–9294. doi: 10.1128/JVI.78.17.9285-9294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wang X, Mokhonov V, Shi P-Y, Randall R, Khromykh A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 2005;79(3):1934–1942. doi: 10.1128/JVI.79.3.1934-1942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WJ, Chen HB, Khromykh AA. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J. Virol. 2003;77(14):7804–7813. doi: 10.1128/JVI.77.14.7804-7813.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo L, Tilgner M, Bernard K, Shi P-Y. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3' untranslated region of West Nile virus using a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 2003a;77(18):10004–10014. doi: 10.1128/JVI.77.18.10004-10014.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo L, Tilgner M, Shi P-Y. A potential high-throughput assay for screening inhibitors of West Nile virus replication. J. Virol. 2003;77(23):12901–12906. doi: 10.1128/JVI.77.23.12901-12906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo MK, Tilgner M, Bernard KA, Shi PY. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3' untranslated region of West Nile virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J Virol. 2003b;77(18):10004–10014. doi: 10.1128/JVI.77.18.10004-10014.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews JH, Roehrig JT. Elucidation of the topography and determination of the protective epitopes on the E glycoprotein of Saint Louis encephalitis virus by passive transfer with monoclonal antibodies. J. Immunol. 1984;132(3):1533–1537. [PubMed] [Google Scholar]
- Michaelis M, Kleinschmidt M, HW D, Cinatl JJ. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J. Antimicrob. Chemother. 2007;60(5):981–986. doi: 10.1093/jac/dkm307. [DOI] [PubMed] [Google Scholar]
- Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005;79(13):8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA. 2003;100(24):14333–14338. doi: 10.1073/pnas.2335168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Ota T, Nukuzuma S, Takegami T. Inhibitory effect of RNAi on Japanese encephalitis virus replication in vitro and in vivo. Microbiol Immunol. 2005;49(12):1047–1056. doi: 10.1111/j.1348-0421.2005.tb03701.x. [DOI] [PubMed] [Google Scholar]
- Noueiry A, Olivo P, Slomczynska U, Zhou Y, Buscher B, Geiss B, Engle M, Roth R, Chung K, Samuel M, Diamond M. Identification of novel small-molecule inhibitors of West Nile virus infection. J. Virol. 2007;81(21):11992–12004. doi: 10.1128/JVI.01358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Basagoiti F, Deas TS, Ren P, Tilgner M, Ferguson DM, Shi P-Y. High-throughput assays using luciferase-expressing replicon, virus-like particle, and full-length virus for West Nile virus drug discovery. Antimicrob. Agent. Chemother. 2005;49(12):4980–4988. doi: 10.1128/AAC.49.12.4980-4988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Basagoiti F, Tilgner M, Bennett C, Zhou Y, Munoz-Jordan J, Garcia-Sastre A, Bernard K, Shi P-Y. A mouse cell-adapted NS4B mutation attenuates West Nile virus RNA synthesis. Virology. 2007;361:229–241. doi: 10.1016/j.virol.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Basagoiti F, Tilgner M, Forshey B, Philpott S, Espina N, Wentworth, Goebel S, Masters PS, Falgout B, Ren P, Ferguson, Shi PY. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob. Agents Chemother. 2006;50(4):1320–1329. doi: 10.1128/AAC.50.4.1320-1329.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Shah A, Tilgner M, Guo Y, Zhao Y, Dong H, Deas T, Zhou Y, Li H, Shi P-Y. West nile virus 5'-cap structure is formed by sequential guanine N-7 and ribose 2'-O methylations by nonstructural protein 5. J. Virol. 2006;80(17):8362–8370. doi: 10.1128/JVI.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger JJ, Brandriss MW, Walsh EE. Protection against 17D yellow fever encephalitis in mice by passive transfer of monoclonal antibodies to the nonstructural glycoprotein gp48 and by active immunization with gp48. J. Immuno. 1985;135(4):2805–2809. [PubMed] [Google Scholar]
- Shi P-Y. Novel therapeutics against West Nile virus. In: Diamond MS, editor. West Nile Encephalitis Virus Infection: Viral pathogenesis and the Host Immune Response. Springer Publisher; 2008. (In press) [Google Scholar]
- Shi PY, Tilgner M, Lo MK, Kent KA, Bernard KA. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 2002;76(12):5847–5856. doi: 10.1128/JVI.76.12.5847-5856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BH, Fu J, Sugrue RJ, Yap EH, Chan YC, Tan YH. Recombinant dengue type 1 virus NS5 protein expressed in Escherichia coli exhibits RNA-dependent RNA polymerase activity. Virology. 1996;216(2):317–325. doi: 10.1006/viro.1996.0067. [DOI] [PubMed] [Google Scholar]
- Tilgner M, Deas TS, Shi P-Y. The flavivirus-conserved penta-nucleotide in the 3' stem-loop of the West Nile virus genome requires a specific sequence and structure for RNA synthesis, but not for viral translation. Virology. 2005;331:375–386. doi: 10.1016/j.virol.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Wengler G, Wengler G. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology. 1991;184(2):707–715. doi: 10.1016/0042-6822(91)90440-m. [DOI] [PubMed] [Google Scholar]
- Whitby K, Pierson T, Geiss B, Lane K, Engle M, Zhou Y, Doms R, Diamond M. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 2005;79(14):8698–8706. doi: 10.1128/JVI.79.14.8698-8706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-F, Lee C-J, Liao C-L, Dwek R, Zitzmann N, Lin Y-L. Antiviral effects oa an iminosugar derivative on flavivirus infections. J. Virol. 2002;76(8):3596–3604. doi: 10.1128/JVI.76.8.3596-3604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Dong H, Zhou Y, Shi PY. Genetic interactions among the West Nile virus methyltransferase, the RNA-dependent RNA polymerase, and the 5' stem-loop of genomic RNA. J. Virol. 2008;82(14):7047–7058. doi: 10.1128/JVI.00654-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Ray D, Zhao Y, Dong H, Ren S, Li Z, Guo Y, Bernard K, Shi P-Y, Li H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007;81(8):3891–3903. doi: 10.1128/JVI.02704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou G, Puig-Basagoiti F, Zhang B, Qing M, Chen L, Pankiewicz K, Felczak K, Yuan Z, Shi P. A single-amino acid substitution in West Nile virus 2K peptide between NS4A and NS4B confers resistance to lycorine, a flavivirus inhibitor. Virology. 2008 doi: 10.1016/j.virol.2008.11.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]