

Abstract
We report the α-arylation of ketone enolates with a range of aryl chlorides with enantioselectivities from 90% to 99% catalyzed by the combination of Ni(COD)2 and (R)-BINAP, and the coupling of ketone enolates with a range of heteroaryl chlorides with enantioselectivities up to 99% catalyzed by Ni(COD)2 and (R)-DIFLUORPHOS. In contrast to the typical relative reactivities of haloarenes, the analogous reactions of bromoarenes occur with much lower rates and enantioselectivities. Mechanistic studies showed that the stoichiometric reactions of both arylnickel bromide and chloride complexes [(R)-BINAP]Ni(X)(Ar) (X = Br and Cl) with the sodium enolate of 2-methyl-1-indanone afforded the α-aryl indanone product with >99% ee, even though the catalytic reactions of the two types of aryl halides occurred with much different enantioselectivity. Monitoring the enantioselectivity vs time showed that the reactions of aryl chlorides occurred with constant high ee, while the reactions of aryl bromides occurred with decreasing ee. The decomposition products of [(R)-BINAP]Ni(X)(Ar), {[(R)-BINAP]Ni(μ-X)}2, catalyze the α-arylation of ketones with slower rates and lower enantioselectivities than does Ni(COD)2 and (R)-BINAP. Therefore, the difference in rate of decomposition of the arylnickel halide complexes likely accounts for the difference in the enantioselectivities of the reactions of bromoarenes and chloroarenes. This catalyst decomposition can be overcome by conducting the reactions with the Ni(0) species, [(R)-BINAP]Ni(η2-NC-Ph) (4), which adds haloarenes at room temperature. α-Arylations catalyzed by this complex occur at room temperature with high yields and enantioselectivities for a wide range of both chloro- and bromoarenes.
The α-arylation of ketones has become a mainstream synthetic method, but asymmetric α-arylation of carbonyl compounds remains a challenge.1–2 Several catalysts for the coupling of specific classes of ketones with aryl halides have been reported,3 and improved selectivities have been achieved by using aryl triflates instead of aryl bromides.4 However, simple systems that catalyze a wide range of couplings of ketones with aryl halides have not been reported, and catalysts suitable for asymmetric α-heteroarylation with nitrogen heterocycles, arguably more important for applications in medicinal chemistry than asymmetric α-arylation, have not been identified.
Most asymmetric couplings of enolates with aryl halides have been conducted with palladium catalysts.4–10 Asymmetric couplings of enolates with aryl halides and pseudohalides catalyzed by nickel complexes have been limited to those of electron-poor aryl triflates and to reactions of aryl halides with γ-lactones.4,11–12 While investigating simple nickel catalysts for the asymmetric α-arylation with heteroaryl halides, we identified a dramatic effect of the halide on enantioselectivity: the coupling of 2-methyl indanone with 2-chloropyridine occurred in higher yield and with much higher enantioselectivity than the analogous reaction of the typically more reactive 2-bromopyridine. This observation led us to identify the most general system for the asymmetric α-arylation of ketones, a system for asymmetric α-heteroarylation of ketones, and an ability to distinguish between catalysis by Ni(0) and by Ni(I). We report the combination of the scope of nickel-catalyzed asymmetric α-arylation and heteroarylation of a series of cyclic ketones with a range of aryl and heteroaryl chlorides and mechanistic studies that reveal the origins of the difference in enantioselectivity. These mechanistic studies, in turn, led to a system for asymmetric α-arylations of both chloroarenes and bromoarenes at room temperature.
Reactions of 2-methyl-1-indanone with phenyl halides and phenyl triflate catalyzed by complexes generated from Ni(COD)2 and a series of bidentate phosphine ligands (L1–L6) are summarized in Table 1. We focused initially on reactions of 2-methyl-1-indanone because published enantioselective α-arylation of this ketone with aryl bromides catalyzed by the combination of BINAP and palladium occurred with variable selectivities,5 and the corresponding reactions with aryl triflates catalyzed by Pd(dba)2 and DIFLUORPHOS occurred with only modest ee’s (55–84%).4
Table 1.
Screening of Ligands and Electrophiles for Ni-catalyzed Asymmetric α-Arylation of 2-Methyl-1-indanone.a
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | PhX | Ligand | yield (%) | ee (%) | entry | PhX | Ligand | yield (%) | ee (%) |
| 1 | PhBr | L1 | 61 | 76 | 7 | PhI | L1 | 36 | 37 |
| 2 | PhBr | L2 | 40 | 47 | 8 | PhOTf | L1 | 33 | 70 |
| 3 | PhBr | L3 | 53 | 50 | 9 | PhCl | L1 | 78 | 93 |
| 4 | PhBr | L4 | 44 | 42 | 10 | PhCl | L3 | 67 | 94 |
| 5 | PhBr | L5 | 55 | 60 | 11 | PhCl | L5 | 77 | 96 |
| 6 | PhBr | L6 | 59 | 60 | 12 | PhCl | L6 | 80 | 94 |
| |||||||||
Conditions: 2-Methyl-1-indanone (0.200 mmol), PhX (0.400 mmol), NaOtBu (0.400 mmol), Ni(COD)2 (0.010 mmol), and Ligand (0.012 mmol); solvent: toluene (1.0 mL); temperature: 80 °C; ee was determined by chiral HPLC analysis.
Reactions of bromobenzene catalyzed by the combination of Ni(C)OD)2 and L1–L6 occurred in only modest yields (40–61%) and with low enantioselectivities (42–76% ee) (entries 1–6). The reaction of iodobenzene catalyzed Ni(COD)2/(R)-BINAP occurred with both low yield and ee (entry 7), and the analogous reaction of phenyl triflate occurred in low yield and modest ee (entry 8). In contrast, the reaction of chlorobenzene (entries 9–12) occurred in much higher yields (67– 80%) and with high enantioselectivities (93–96%). Moreover, reactions of aryl chlorides conducted with ligands L1, L3, L5, and L6 all occurred with similar enantioselectivity. These results indicate that the leaving group in the aryl electrophile significantly affects both yield and enantioselectivity of the reactions in Table 1 catalyzed by the combination of Ni(COD)2 and the bisphosphines.
Because of the accessibility and low cost of (R)-BINAP and chloroarenes, we studied the scope of the asymmetric α-arylation with a range of chloroarenes catalyzed by the combination of Ni(COD)2 and (R)-BINAP (Table 2). We studied reactions of 1-indanones and 1-tetralones bearing different substituents (Me, Et, and Bn) on the α-carbon. A range of electron-rich, electron-neutral, and electron-deficient chloroarenes reacted to give the coupled product in good isolated yields (58–83%) with high enantiomeric excess (90–99%). Reactions of ketones containing varied substituents at the α-position of the carbonyl group of 1-indanone and 1-tetralone (e.g., see entries 2, 11, and 14) occurred with a series of electronically varied chloroarenes (e.g., see entries 11–16) in good to excellent yield with enantioselectivity. When reactions occurred in moderate yield (e.g., entries 6, 20, and 25), the starting ketones were not fully converted. Side products of ketone self-condensation and β-hydrogen elimination were not observed by GC-MS. Finally, the reaction of 2-methyl-1-indanone with chlorobenzene run on a 2.0-mmol scale occurred in a yield (80%) and with an enantioselectivity (96%) as high as those run on smaller scale. Thus, these coupling reactions are suitable for synthetic applications.
Table 2.
Asymmetric α-Arylation of Indanones and Tetralones with Chloroarenes Catalyzed by Ni(COD)2/(R)-BINAPa
 | |||
|---|---|---|---|
| entry | ArX | yield (%) | ee (%) |
| n = 0, R = CH3 | |||
| 1b |
|
75 | 96 |
| 2b | 81 | 95 | |
| 3b |
|
58 | 92 |
| 4 |
|
72 | 94 |
| 5 | 75 | 95 | |
| 6b |
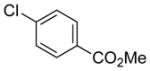
|
41 | 99 |
| 7b |
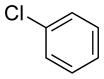
|
82 | 96 |
| 8 |
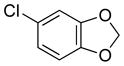
|
73 | 98 |
| 9b |
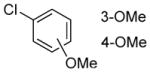
|
79 | 96 |
| 10b | 81 | 96 | |
|
| |||
| n = 0, R = CH2CH3 | |||
| 11 |
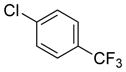
|
66 | 92 |
| 12 |
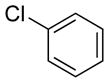
|
77 | 98 |
| 13 |
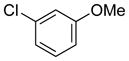
|
72 | 94 |
|
| |||
| n = 0, R = CH2Ph | |||
| 14 |
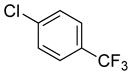
|
89 | 99 |
| 15 |
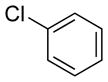
|
77 | 98 |
| 16 |
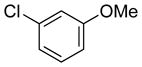
|
83 | 98 |
|
| |||
| n = 1, R = CH3 | |||
| 17 |
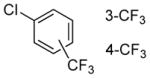
|
74 | 96 |
| 18 | 80 | 94 | |
| 19 |
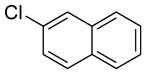
|
73 | 90 |
| 20 |
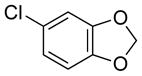
|
61 | 96 |
| 21 |
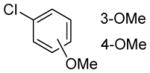
|
69 | 92 |
| 22 | 72 | 97 | |
|
| |||
| n = 1, R = CH2Ph | |||
| 23 |
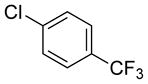
|
76 | 94 |
| 24 |
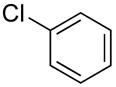
|
72 | 98 |
| 25 |
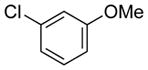
|
53 | 99 |
Conditions: Indanones or tetralones (0.200 mmol), chloroarenes (0.400 mmol), NaOtBu (0.400 mmol), Ni(COD)2 (0.010 mmol), and (R)-BINAP (0.012 mmol) for indanones, and Ni(COD)2 (0.020 mmol) and (R)-BINAP (0.024 mmol) for tetralones; solvent: toluene (1.0 mL); temperature: 80°C; reaction time: 36 h for indanones and 48 h for tetralones; ee was determined by chiral HPLC analysis.
60 °C.
To determine the absolute configuration of the arylation products, we compared the optical rotation of 2-methyl-2-phenyl-2,3-dihydro-1H-inden-1-one obtained by the Ni(COD)2/(R)-BINAP catalyst (entry 7) and the Pd(dba)2/(R)-DIFLUORPHOS catalyst.4 The values of the optical rotation of the products by these two catalysts are both negative. Thus, the α-arylation of ketones catalyzed by these Ni- and Pd-catalyst systems lead to the same enantiomers. These relative configurations contrast the absolute configurations of the products from the arylation of γ-lactones. Ni- and Pd-catalysts were reported to form products with opposite configurations.11
Asymmetric α-heteroarylation is as important as asymmetric α-arylation because heteroaromatic units are ubiquitous in medicinal chemistry and can be reduced to saturated heterocycles. However, asymmetric α-heteroarylation is more challenging than asymmetric α-arylation because ligation of heteroarenes can lead to poisoning of the catalyst or displacement of the chiral ligand to form an achiral catalyst.13,14 Because this nickel catalyst contains a bidentate ligand, rather than the monodentate ligands of some palladium systems for asymmetric α-arylation,7,9–10 we hypothesized that nickel-bisphosphine systems would be suitable for asymmetric α-heteroarylation.
Initial studies on asymmetric heteroarylation showed that the reaction of 2-methyl-1-indanone with 2-bromopyridine under the conditions for the arylation of 2-methyl-1-indanone in Table 2 did not yield the α-heteroaryl indanone. The same reaction conducted with (R)-DIFLUORPHOS (L6) in place of (R)-BINAP (L1) gave the desired product in 69% isolated yield, but only 57% ee (entry 1, Table 3). Most striking, the same reaction conducted with 2-chloropyridine in place of 2-bromopyridine afforded the heteroarylation product in 93% yield and 97% ee (entry 2, Table 3). The leaving group had a similar effect on the heteroarylation of 2-methyl-1-tetralone (entries 13 and 14, Table 3). Thus, further studies on the asymmetric coupling of ketones with heteroaryl electrophiles were conducted with heteroaryl chlorides.
Table 3.
Asymmetric Heteroarylation of Ketones with Heteroaryl Halides Catalyzed by Ni(COD)2/(R)-DIFLUORPHOSa
 | ||||
|---|---|---|---|---|
| entry | ArX | yield (%) | ee (%) | |
| n = 0 | ||||
|
| ||||
| 1 |
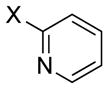
|
X = Br | 69 | 57 |
| 2 | X = Cl | 93 | 97 | |
| 3 |
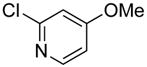
|
54 | 91 | |
| 4 |
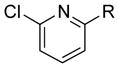
|
R = OMe | 75 | 41 |
| 5 | R = Me | 87 | 94 | |
| 6 |
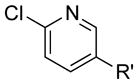
|
R′ = CF3 | 85 | 81 |
| 7 | R′ = F | 86 | 98 | |
| 8 | R′ = CN | 87 | 21 | |
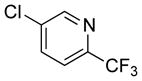
| 9 |
|
76 | 96 | |
| 10 |
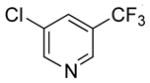
|
54 | 95 | |
| 11 |
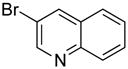
|
90 | 99 | |
| 12 |
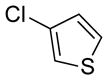
|
85 | 90 | |
|
| ||||
| n = 1 | ||||
|
| ||||
| 13 |
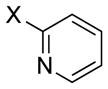
|
X = Br | 73 | 55 |
| 14 | X = Cl | 89 | 99 | |
| 15 |
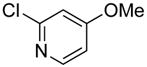
|
68 | 94 | |
| 16 |
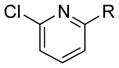
|
R = OMe | 72 | 35 |
| 17 | R = Me | 86 | 98 | |
| 18 |
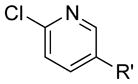
|
R′ = CF3 | 85 | 96 |
| 19 | R′ = F | 89 | 99 | |
| 20 | R′ = CF3 | 84 | 93 | |
| 21 |
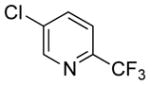
|
54 | 96 | |
| 22 |
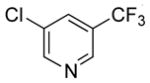
|
45 | 97 | |
| 23 |
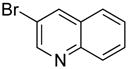
|
87 | 99 | |
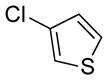
| 24 |
|
79 | 92 | |
Conditions: Indanones or tetralones (0.200 mmol), chloro- or bromoarenes (0.400 mmol), NaOtBu (0.400 mmol), Ni(COD)2 (0.020 mmol), and (R)-DIFLUORPHOS (0.024 mmol); solvent: toluene (1.0 mL); temperature: 70 °C; reaction time: 28 h; ee was determined by chiral HPLC analysis.
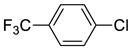
The reactions of a series of nitrogen- and sulfur-containing heteroaryl chlorides possessing electron-withdrawing groups (i.e., –CF3, –CN, and –F) and electron-donating groups (i.e., –OMe and –Me) with 2-methyl-1-indanone and 2-methyl-1-tetralone are summarized in Table 3. In general, these reactions occurred with high enantioselectivities (90–99%) and in modest to high yields (45–93%). The few exceptions included the reactions of 2-chloro-6-methoxypyridine (entries 4 and 16) and the reactions of two electron-poor 2-chloropyridines. 2-Chloro-6-methoxypyridine has the potential to chelate the nickel center – the reactions of isosteric 2-chloro-6-methylpyridine afforded products with high ee’s (entries 5 and 17). The low ee’s obtained in entries 6 and 8 appear to be caused by competing uncatalyzed nucleophilic aromatic substitution between the sodium enolate of 2-mthyl-1-indanone and activated 2-chloropyridine bearing a highly electron-withdrawing group on the 2-position (see supporting information for background reactions).
To understand the effect of the leaving group on yield and enantioselectivity, we monitored by 31P NMR spectroscopy the reaction of 2-methyl-1-indanone with 4-chlorobenzotrifluoride and with 4-bromobenzotrifluoride in the presence of NaOtBu catalyzed by Ni(COD)2/(R)-BINAP at 60 °C. Two nickel species, [(R)-BINAP]2Ni and [(R)-BINAP]Ni(COD), were detected in both reactions. Stoichiometric reactions of [(R)-BINAP]2Ni with 4-chloro- or 4-bromobenzotrifluoride at 60 °C gave no reaction. The same reaction of [(R)-BINAP]Ni(COD) led to complete consumption of the nickel complex within 1 h, but no new species was detected by 31P NMR spectroscopy. These data suggest that [(R)-BINAP]2Ni is not the active catalyst and that [(R)-BINAP]Ni(COD) reacts with bromo- or chloroarenes to give products that are thermally labile toward the generation of Ni(I) species in the absence of enolate at elevated temperatures.
To determine whether catalyst decomposition relates to the difference in reactivity of bromoarenes and chloroarenes, we monitored the enantiomeric excess as a function of time. As shown in Figure S1 in the supporting information, the ee of the product from reaction of the bromoarene decreased from 92% to 60% during the course of the reaction, while that of the product from reaction of the chloroarene remained above 94%. These data are consistent with the hypothesis that the catalyst decomposes in the reaction of the bromoarene to a second species that catalyzes the reaction with lower enantioselectivity.
To determine the species that accounts for the high enantioselectivity and to identify potential modes of decomposition, we studied the formation and reactivity of arylnickel halide complexes. The reactions of [(R)-BINAP]Ni(COD) with the series of chloroarenes in Table 2 at room temperature showed that only 4-chlorobenzonitrile reacted to give a stable arylnickel chloride complex. [(R)-BINAP]Ni(Cl)(C6H4-4-CN) (1) was isolated from this reaction in 77% yield.
We speculated that the cyano group in 4-chlorobenzonitrile plays a role in triggering the oxidative addition reaction. Indeed, the reaction of [(R)-BINAP)]Ni(COD) (generated in situ) with 4-chlorobenzotrifluoride in the presence of a catalytic amount of benzonitrile (15 mol%) (eq 1) afforded [(R)-BINAP]Ni(Cl)(C6H4-4-CF3) (2, its structure is shown in Figure S2 of the supporting information) in 75% isolated yield. The bromide congener, [(R)-BINAP]Ni(Br)(C6H4-4-CF3) (3), was isolated in 80% yield by an analogous procedure.
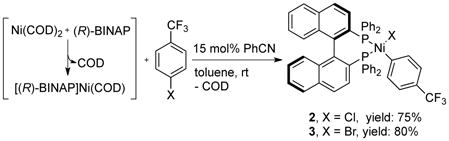 |
(eq 1) |
To determine the origin of the effect of benzonitrile on the oxidative additions of chloroarenes to [(R)-BINAP]Ni(COD), we studied the reaction of [(R)-BINAP]Ni(COD) with benzonitrile in toluene. The 31P NMR spectrum of the reaction containing 2 equiv of benzonitrile led to the conversion of about 45% of [(R)-BINAP]Ni(COD) to the new species [(R)-BINAP]Ni(η2-NC-Ph) (4). Addition of 20 equiv of benzonitrile to the same mixture led to 95% conversion of [(R)-BINAP]Ni(COD) and a 86% isolated yield of 4(eq 2). Single-crystal X-ray diffraction (Figure S3 of the supporting information) confirmed its structural assignment. This complex is stable on its own at room temperature, but reacts rapidly with chloroarenes. Isolated 4 reacted with 5 equiv of 4-chlorobenzotrifluoride in toluene at room temperature to form [(R)-BINAP]Ni(Cl)(C6H4-4-CF3) (2) in quantitative yield within 5 min, as determined by 31P NMR spectroscopy.
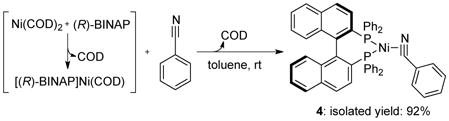 |
(eq 2) |
Benzonitrile also promoted the catalytic asymmetric α-arylation. The reaction of 2-methyl-1-indanone with 4-chlorobenzotrifluoride in the presence of NaOtBu catalyzed by the combination of 5 mol% Ni(COD)2, 6 mol% (R)-BINAP, and 20 mol% PhCN at room temperature led to full conversion of 2-methyl-1-indanone within 3 h and a 92% isolated yield of the 2-aryl indanone in over 99% ee. The same reaction at room temperature without added benzonitrile afforded less than 5% product after 3 h.
 |
(eq 3) |
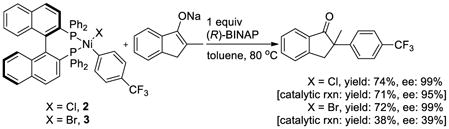
Because of the ambiguity of the oxidation state of the nickel species in cross-coupling reactions, we assessed the competence of the arylnickel(II) halide compounds to be intermediates in asymmetric α-arylation. To do so, we conducted the stoichiometric reaction of [(R)-BINAP]Ni(X)(C6H4-4-CF3) (X = Cl, 2; X = Br, 3) with the sodium enolate of 2-methyl-1-indanone (eq 3) at 80 °C in the presence of 1 equivalent of (R)-BINAP to trap a Ni(0) product. The reaction of [(R)-BINAP]Ni(Cl)(C6H4-4-CF3) (2) afforded the α-aryl indanone in 74% yield and 99% ee in 3 h. This yield and ee match those of the corresponding catalytic reaction (71% yield and 95% ee), indicating that [(R)-BINAP]Ni(Cl)(C6H4-4-CF3) (2) is kinetically and chemically competent to be an intermediate in the catalytic process. The same reaction of [(R)-BINAP]Ni(Br)(C6H4-4-CF3) (3) yielded the α-aryl indanone in 72% yield and 99% ee. These values are much higher than those of the corresponding catalytic reaction (38% yield and 39% ee). This result provides further evidence that [(R)-BINAP]Ni(Br)(C6H4-4-CF3) (3) decomposes in the catalytic system to form a less reactive and selective catalyst for the asymmetric coupling of aryl bromides.
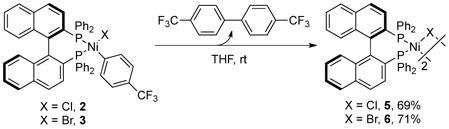 |
(eq 4) |
To identify this less reactive and selective species, we studied the thermal reactivity of the arylnickel chloride and bromide complexes, [(R)-BINAP]Ni(X)(C6H4-4-CF3) (X = Cl, 2; X = Br, 3). Both 2 and 3 fully decomposed in THF within two days at room temperature to form {[(R)-BINAP]Ni(μ-X)}2 (X = Cl, 5, 69%; X = Br, 6, 71%) with concomitant release of 4,4′-bis(trifluoromethyl)-1,1′-biphenyl (detected by GC-MS analysis) (eq 4). Both 5 and 6 were paramagnetic, and the structures of these complexes were determined by single-crystal X-ray diffraction (see Supporting Information for structural data).
| |||||
|---|---|---|---|---|---|
|
|
|
|||
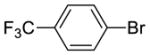
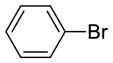
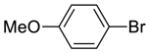
| {[(R)-BINAP]NiCl}2 (5), | yield: 33%, ee: 50% | yield: 34%, ee: 81% | yield: 13%, ee: 89% | ||
| Ni(COD)2/(R)-BINAP, | yield: 71%, ee: 95% | yield: 78%, ee: 93% | yield: 73%, ee: 94% | ||
|
|
|
|||
| {[(R)-BINAP]NiBr}2 (6), | yield: 32%, ee: 7% | yield: 49%, ee: 34% | yield: 28%, ee: 51% | ||
| Ni(COD)2/(R)-BINAP, | yield: 38%, ee: 39% | yield: 61%, ee: 76% | yield: 44%, ee: 77% | ||

The reactions of 2-methyl-1-indanone with electronically varied chloro- and bromoarenes catalyzed by Ni(I) halides 5 and 6 (eq 5) were slower than those catalyzed by nitrile complex 4. Neither the reactions catalyzed by 5 nor by 6 occurred at room temperature, and reactions at 80 °C formed the α-aryl indanone in low yields (13–49%). Moreover, the ee of the product was only 50–89% and 7–51% for reaction of the chlorides and bromides (eq 5) catalyzed by 5 and 6. These data imply that the loss of activity and enantioselectivity during the reactions of bromides results from competing reactions catalyzed by accumulating Ni(I) species.
The oxidative additions of chloro- and bromoarenes to [(R)-BINAP]Ni(η2-NC-Ph) (4) at room temperature suggest that the catalytic process could also occur at this mild temperature.15 As shown in Table 4, both chloro- and bromoarenes reacted with 2-methyl-1-indanone, in high yield and enantioselectivities (entries 1–5) at rom temperature. Under these mild conditions, the difference in enantioselectivities with chloro and bromoarenes for most reactions was small. The reaction of 2-methyl-1-tetralone with 4-chlorobenzotriflouride, however, did occur with significantly higher enantioselectivity than that of 4-bromobenzotriflouride (entry 6).
Table 4.
Room-Temperature Asymmetric α-Arylation of Ketones with Chloro- and Bromoarenes Catalyzed by [(R)-BINAP]Ni(η2-NC-Ph)a
 | |||||
|---|---|---|---|---|---|
| 1 |
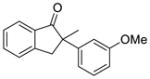 X = Cl (10%) yield: 91%; ee: >99% X = Br (5%) yield: 95%; ee: >99% |
2 |
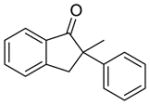 X = Cl (10%) yield: 64%; ee: 98% X = Br (5%) yield: 74%; ee: 99% |
3 |
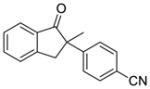 X = Cl (10%) yield: 93%; ee: >99% X = Br (5%) yield: 61%; ee: 97% |
| 4 |
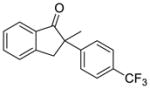 X = Cl (5%) yield: 92%; ee: >99% X= Br (5%) yield: 96%; ee: >99% |
5 |
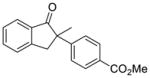 X = Cl (10%) yield: 72%; ee: >99% X = Br (5%) yield: 50%; ee: 98% |
6 |
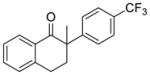 X = Cl (10%) yield: 59%; ee: 95% X = Br (10%) yield: 74%; ee: 83% |
Conditions: Ketones (0.200 mmol), ArX (0.400 mmol), NaOtBu (0.400 mmol), and [(R)-BINAP]Ni(η2-NC-Ph) (0.010 mmol, 5% or 0.020 mmol, 10%); solvent: toluene (1.0 mL); ee was determined by chiral HPLC analysis.
In summary, the α-arylations of ketones with aryl and heteroaryl chlorides catalyzed by the combination of Ni(COD)2 and (R)-BINAP (for α-arylation) or (R)-DIFLOURPHOS (for α-heteroarylation) occur with high enantioselectivity. In contrast to the usual reactivity of haloarenes, bromoarenes reacted with lower yield and enantioselectivity than the corresponding chloroarenes, most likely due to greater reactivity of the bromoarene through a less selective catalyst formed by decomposition of the Ni(0) or Ni(II) species. Like the dibenzylidene acetone in electron-rich L2Pd(dba) complexes,16–17 the COD in [(R)-BINAP]Ni(COD) and related complexes is slow to dissociate and makes the oxidative addition of haloarenes to the Ni(0) species slow. However, the discrete Ni(0) precursor, [(R)-BINAP]Ni(η2-NC-Ph) (4) adds chloro- and bromoarenes fast at room temperature, and this fast rate allows the α-arylation of ketones also to occur at room temperature and to attenuate the decomposition to form the less selective Ni(I) species. Future work will focus on asymmetric α-arylation reactions of other types of carbonyl compounds and on additional cross-coupling reactions initiated with the discrete Ni(0) complexes ligated by benzonitrile and bidentate phosphines.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-58108).
Footnotes
Supporting information. Detailed experimental procedures, characterizations of all compounds, and X-ray crystallographic data for compounds 2, 4, 5, and 6 are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Johansson CCC, Colacot TJ. Angew Chem, Int Ed. 2010;49:676. doi: 10.1002/anie.200903424. [DOI] [PubMed] [Google Scholar]
- 2.For an alternative approach to the asymmetric α-arylation of carbonyl compounds involving the coupling of α-halo carbonyl compounds with organometallic reagents, see: Dai X, Strotman NA, Fu GC. J Am Chem Soc. 2008;130:3302. doi: 10.1021/ja8009428.Lou S, Fu GC. J Am Chem Soc. 2010;132:1264. doi: 10.1021/ja909689t.Lundin PM, Fu GC. J Am Chem Soc. 2010;132:11027. doi: 10.1021/ja105148g.
- 3.Burtoloso ACB. Synlett. 2009:320. [Google Scholar]
- 4.Liao X, Weng Z, Hartwig JF. J Am Chem Soc. 2008;130:195. doi: 10.1021/ja074453g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahman J, Wolfe JP, Troutman MV, Palucki M, Buchwald SL. J Am Chem Soc. 1998;120:1918. [Google Scholar]
- 6.Lee S, Hartwig JF. J Org Chem. 2001;66:3402. doi: 10.1021/jo005761z. [DOI] [PubMed] [Google Scholar]
- 7.Hamada T, Chieffi A, Ahman J, Buchwald SL. J Am Chem Soc. 2002;124:1261. doi: 10.1021/ja011122+. [DOI] [PubMed] [Google Scholar]
- 8.Kündig EP, Seidel Thomas M, Jia Y-x, Bernardinelli G. Angew Chem, Int Ed. 2007;46:8484. doi: 10.1002/anie.200703408. [DOI] [PubMed] [Google Scholar]
- 9.García-Fortanet J, Buchwald SL. Angew Chem, Int Ed. 2008;47:8108. doi: 10.1002/anie.200803809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor AM, Altman RA, Buchwald SL. J Am Chem Soc. 2009;131:9900. doi: 10.1021/ja903880q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spielvogel DJ, Buchwald SL. J Am Chem Soc. 2002;124:3500. doi: 10.1021/ja017545t. [DOI] [PubMed] [Google Scholar]
- 12.As described in more detail previously (reference 4), we have not been able to reproduce the enantioselectivity and yields reported previously for a nickel-catalyzed asymmetric α-arylation (Chen GC, Kwong FY, Chan HO, Yub WY, Chan ASC. Chem Commun. 2006:1413. doi: 10.1039/b601691j.) The much lower optical rotations for the products reported by Kwong and Chan than for analogous products we reported in reference 4 are consistent with this finding. Moreover, we find that the products from α-arylation of indanone catalyzed by the combination of Ni(COD)2 and (R)-P-Phos have a levorotatory rotation, but they are reported to have a dextrorotatory rotation. The detailed studies in the current paper are consistent with our prior findings.
- 13.Döbler C, Kreuzfeld HJ, Michalik M, Krause HW. Tetrahedron: Asymmetry. 1996;7:117. [Google Scholar]
- 14.Jiang L, Weist S, Jansat S. Org Lett. 2009;11:1543. doi: 10.1021/ol900131q. [DOI] [PubMed] [Google Scholar]
- 15.For prior asymmetric α-arylation of ketones with a palladium-monophosphine system, see reference 7.
- 16.Amatore C, Jutand A, Khalil F, M’Barki MA, Mottier L. Organometallics. 1993;12:3168. [Google Scholar]
- 17.Amatore C, Broeker G, Jutand A, Khalil F. J Am Chem Soc. 1997;119:5176. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.