

Abstract
We have accomplished a parallel screen of cycloaddition partners for ortho-quinols utilizing a plate-based microwave system. Microwave irradiation improves the efficiency of retro-Diels-Alder/Diels-Alder cascades of ortho-quinol dimers which generally proceed in a diastereoselective fashion. Computational studies indicate that asynchronous transition states are favored in Diels-Alder cycloadditions of ortho-quinols. Subsequent biological evaluation of a collection of cycloadducts has identified an inhibitor of activator protein-1 (AP-1), an oncogenic transcription factor.
INTRODUCTION
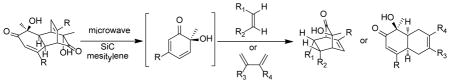
ortho-Quinols are highly reactive 2,4-cyclohexadienones and have been proposed as precursors for a number of natural products. ortho-Quinol dimers are common building blocks for preparation of bicyclo[2.2.2]octenones via retro-Diels-Alder reaction followed by a Diels-Alder cycloaddition with external dienophiles.1 We recently reported the synthesis of the natural product chamaecypanone C (1)2 involving a retro-Diels-Alder/Diels-Alder cascade of 2,4-cyclohexadienone (o-quinol) dimer 2 and diaryl enone 3 under thermal, oxidative conditions (Scheme 1). [4+2] Cycloaddition of the derived ortho-quinol monomer 4 and 2,4-diaryl cyclopentadienone 5 was found to proceed in a highly regio- and diastereoselective manner. This result, along with reactions with a few dienophiles, prompted us to further evaluate cycloadditions of 4 and related ortho-quinols with a range of reaction partners, in order to better understand the mode of reactivity for dienophiles as well as diastereoselectivity with respect to the o-quinol.
Scheme 1.

Synthesis of (+)-chamaecypanone C utilizing a retro-[4+2]/[4+2] cascade
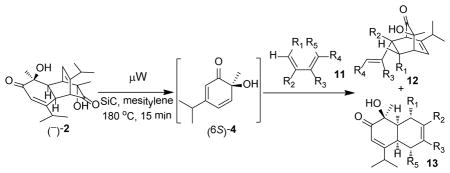
Retro-[4+2]/[4+2] reactions of dimers derived from o-quinols and masked o-benzoquinones (MOBs) have been utilized for preparation of reactive 2,4-cyclohexadienones enroute to bicyclo[2.2.2]octenones.1,3,4 However, most reported examples involve thermolysis at high temperatures for extended reaction times. In some cases, sealed tubes have been used at temperatures up to 220 °C,3b where safety issues may become a concern. In comparison to conventional heating, microwave heating has been proven to be efficient in many cases leading to a dramatic reduction in reaction time.5 In particular, use of thermal susceptors or “sensitizers” including graphite6 and silicon carbide (SiC),7 has been shown to further improve the efficiency of thermal transformations, as these materials can reach high temperature under microwave irradiation in spite of the nature of reaction solvents.8 Recently, Kappe and coworkers have reported the development of sintered SiC microtiter plates for performing parallel synthesis in a multimode microwave reactor.9 Considering the reactivity of ortho-quinol dimers and the available literature precedents, we wished to utilize SiC plates in a high-throughput reaction partner assessment.10 In the current study, a broad panel of reaction partners in the retro-[4+2]/[4+2] cascade of o-quinol dimers have been evaluated under microwave conditions in an effort to study the scope and limitations of this methodology, to obtain both bicyclo[2.2.2]octenone and cis-decalin frameworks, and to understand the factors determining regio- and diastereoselectivity. To explain the observed reactivities and diastereoselectivities, in particular the regioselectivity for cycloadditions, computational studies of Diels-Alder reactions between o-quinols and select dienophiles/dienes were also conducted.11 In this paper, we report the results of this study as well as biological screening of the resulting collection of complex cycloadducts.
RESULTS AND DISCUSSION
Reaction Optimization
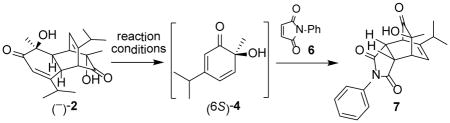
We first evaluated reactions between the readily accessible dimer (−)-212 and N-phenylmaleimide (6) under microwave conditions in various solvents (Table 1).13 Although reactions went to completion within 30 min in either DMF (entry 1) or DMA (entry 2) to afford cycloadduct 7, we noticed that use of mesitylene as solvent afforded cleaner reactions (entry 3). Further experiments (entry 4) indicated that reactions could be completed within 15 min (μW, 180 °C) in comparison to use of conventional heating (entry 5) which required approximately 90 minutes for completion.2
Table 1.
Optimization of the Microwave-mediated retro-[4+2]/[4+2] Cascade
 | ||||
|---|---|---|---|---|
| entry | solvent | conditionsa | time (min) | conversionb (%) |
| 1 | DMF | A | 30 | >99 (90)c,d |
| 2 | DMA | A | 30 | >99 (92)d |
| 3 | mesitylene | A | 30 | >99 (98) |
| 4e | mesitylene | A | 15 | >99 (98) |
| 5f | mesitylene | B | 90 | >99 (97) |
Reaction conditions: A, 3.0 equiv 6, SiC chip, μW, 188 °C (IR temperature); B, 3.0 equiv N-phenylmaleimide (6), 150 °C;
Conversion based on UPLC analysis.
Isolated yield of product 7 in parentheses.
Aqueous workup required.
Reaction conducted at 180 °C (IR temperature) under microwave irradiation.
Reaction reported in Reference 2.
Screening of Reaction Partners
Utilizing the optimized microwave conditions, 57 reaction partners were evaluated in a reaction screen.12 Representative structures were shown in Figure 1. A number of alkene partners were selected based on retro-[4+2]/[4+2] cycloadditions of o-quinol dimers and MOB dimers reported in the literature.1,3 Representative dipolarophiles and dienes were also included. Reactions were conducted on a 0.009 mmol scale using 3.0 mg of dimer (−)-2 as substrate, 10 equiv of the corresponding reaction partner, and 100 μL of mesitylene under microwave irradiation at 188 °C. After 60 min, reaction mixtures were filtered through a silica gel plug, concentrated, and evaluated using ultra performance liquid chromatography (UPLC)-MS which allows for short analytical run times (approx. three minutes).14 Based on UPLC-MS data, reactions showing major peaks in the HPLC trace were scaled up (0.06 mmol, 20 mg substrate) using a SiC chip as passive heating element in a single mode microwave reactor.12 The results of the reaction screening indicated that dimer 2 was completely consumed in all cases. Of the 57 reactions, 12 afforded products corresponding to [4+2] adducts between the o-quinol and alkene reaction partners, and other reactions afforded product 8 (Scheme 2), likely generated from a dienone-phenol rearrangement of intermediate 4.15 The formation of 8 is in accordance with literature reported thermal degradation results for dimer 2,16 and suggests that the corresponding reaction partners are unreactive in the cycloaddition event.
Figure 1.

Reaction Partners in Retro-[4+2]/[4+2] Cascade.
Scheme 2.

Dienone-phenol rearrangement of o-quinol 4
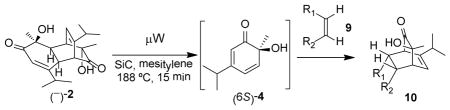
As shown in Table 2, o-quinol 4 may react with activated alkenes, including the normal-demand dienophiles MVK (9a), indene (9b), 4-methoxy styrene (9c), the inverse-demand dienophiles dihydrofuran (DHF 9d), vinylene carbonate (9e), and the alkynes dimethyl acetylenedicarboxylate (DMAD, 9f) and phenylacetylene (9g). In all cases, Diels-Alder cycloaddition between o-quinol 4 and dienophiles 9 proceeded in a highly diastereoselective fashion with [4+2] adducts 10a–g isolated in moderate to excellent yields. Structure elucidation of products using 2D NMR experiments or X-ray crystallography confirmed that endo-[4+2] cycloadducts were the predominate products isolated from the reactions,12 which is in accordance with both experimental results and theoretical calculations in literature that secondary orbital overlap is believed to govern endo-selectivity.17 Facial selectivity could be explained by either sterics or hyperconjugation effects according to our computational studies (vide infra).
Table 2.
Retro-[4+2]/[4+2] reactions to afford bicyclo[2.2.2]-octenonesa
 | |||
|---|---|---|---|
| entry | dienophile (equiv) | cycloadductb | yieldc (%) |
| 1 |
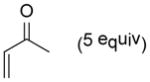 9a |
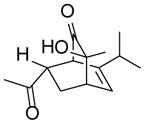 10a |
85 |
| 2 |
9b |
 10b |
99 |
| 3 |
9c |
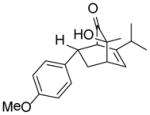 10cd |
98 |
| 4 |
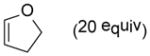 9d |
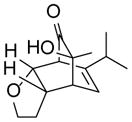 10d |
85 |
| 5 |
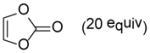 9e |
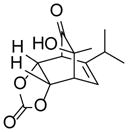 10ed |
76 |
| 6 |
9f |
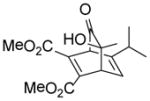 10f |
90 |
| 7 |
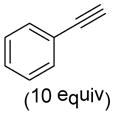 9g |
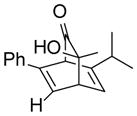 10gd |
80 |
Reaction conditions: dimer (−)-2, dienophile, SiC chip, mesitylene, μW, 180 °C, 15 min;
Single diastereomer isolated unless otherwise noted;
Isolated yield after column chromatography;
Approximately 9% of an inseparable minor product observed by 1H-NMR (ca. 10:1 isomeric ratio).
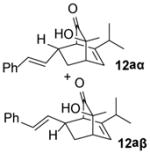
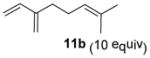
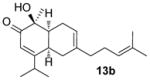
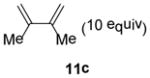
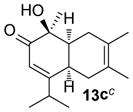
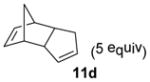
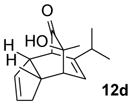
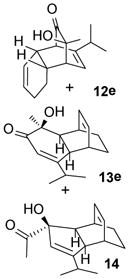
In the cases of reaction with dienes (11, Table 3), o-quinol 4 was found to react as either a 4π or 2π reaction partner to afford bicyclooctenone derivative 12 or cis-decalin product 13. Reaction with 1-phenyl substituted 1,3-butadiene 11a (entry 1) cleanly afforded a mixture of bicyclooctenones endo-12aα and exo-12aβ in a 4:1 ratio favoring the endo-adduct. When the 2-subsituted-1,3-butadiene β-myrcene (11b, entry 2) was used in the reaction, cis-decalin 13b was found to be the major product.18 Considering the result that a mixture of two compounds was generated at lower reaction temperature (150 °C),2 cycloadduct 13b should be thermodynamically more favored in comparison to its bicyclooctenone isomer obtunone.19 Similarly, reaction with 2,3-disubsituted-1,3-butadiene 11c (entry 3) produced cis-decalin 13c as the major product in excellent yield (94%). While reaction with cyclopentadiene dimer 11d (entry 4) generated cycloadduct 12d as the sole product in almost quantitative isolated yield, use of cyclohexadiene 11e (entry 5) as reaction partner led to a more complex product mixture. Structure elucidation confirmed that compounds 12e and 13e were the major products in this instance along with a small amount of the rearranged bicyclooctene 14 (approx. 10%).
Table 3.
Retro-[4+2]/[4+2] cycloadditions with dienesa
 | |||
|---|---|---|---|
| entry | dienophile (equiv) | cycloadduct | yieldb (%) |
| 1 |

|
|
81 |
| 18 | |||
|
| |||
| 2 |
|
|
57 |
|
| |||
| 3 |
|
|
94 |
|
| |||
| 4 |
|
|
98 |
|
| |||
| 5 |

|
|
57 |
| 25 | |||
| 9 | |||
Reaction conditions: dimer (−)-2, diene, SiC chip, mesitylene, μW, 180 °C, 15 min;
Isolated yield after column chromatography;
Approximately 5% of an inseparable minor product was observed by 1H-NMR analysis (ca. 19:1 isomeric ratio).
In order to probe possible interconversion between adducts 12e and 13e, pure compound 12e was resubjected to the reaction conditions (μW, SiC, mesitylene, 180 °C, 15 min). Product analysis indicated that a mixture of cycloadducts 12e, 13e, and 14 was generated in a 2:1:1 ratio. Extended reaction time (1 h) or elevated temperature (200 °C) did not change the ratio in which case further decomposition was observed. It is plausible that compounds 12e and 13e may interconvert through Cope rearrangement20 (Figure 2, pathway a) or via a retro-[4+2] reaction of 12e followed by recombination of o-quinol and cyclohexadiene (pathway b). To further understand this process, trapping experiments were conducted as shown in Scheme 3. In both experiments, no crossover Diels-Alder cycloadduct was observed; reactions either afforded a mixture of 12e/13e/14 from 12e or recovered starting materials from cycloadduct 7. These results support an intramolecular [3,3] sigmatropic rearrangement mechanism for the interconversion of cycloadducts 12e and 13e.21
Figure 2.

Cope rearrangement vs. a retro-[4+2]/[4+2] process
Scheme 3.

Trapping experiment
The generation of product 14 may be explained by thermal α-ketol rearrangement of hydroxy enone 13e (Figure 2, pathway c).22 A similar ring contraction has been observed in a related α-methyl-α-hydroxylcyclohexanone system under basic conditions.23 However, use of related basic conditions (LiHMDS/THF or Al2O3/hexanes) did not effect ketol rearrangement of 13e.
Based on the results from our reaction screen and follow-up product analyses, we noticed that activated alkenes or alkynes are more reactive in the reaction with the model o-quinol. Moreover, cis-alkenes in general showed better reactivity than trans-alkenes, 1,1-disubstituted, and trisubstituted alkenes. Acyclic 1,3-dienes were found to react as 4π components only if they lacked substitution at the C1 or C4 positions, and displayed reactivity as 2π components in the cases of less substituted alkenes. Taken together, these results suggest that Diels-Alder cycloaddition of o-quinols is an electronically and sterically demanding process. These observations are also in accordance with literature reported examples of 2,4-cyclohexadienones.1,3
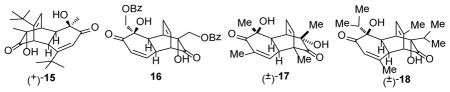
To further evaluate the scope of the microwave-assisted protocol, ortho-quinol dimers (+)-15,11 16,24 17, and 1825 were subjected to optimized reaction conditions (μW, SiC, mesitylene, 170 °C, 15 min, Table 4). In all cases, good to excellent isolated yield of the desired cycloadducts 19 to 25 were obtained as single diastereomers.
Table 4.
Retro-[4+2]/[4+2] cascade using other o-quinol precursorsa
 | ||||
|---|---|---|---|---|
| entry | dimer | dienophile (equiv) | cycloadductb | yieldc (%) |
| 1 | (+)-15 |
 11d |
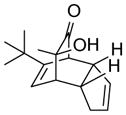 19 |
93 |
| 2 | (+)-15 |
 9e |
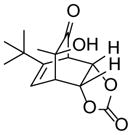 20 |
78 |
| 3 | 16 |
9b |
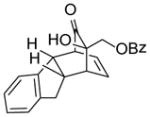 21 |
86 |
| 4 | (±)-17 |
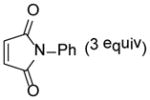 6 |
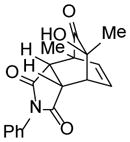 22 |
99 |
| 5 | (±)-17 |
 9b |
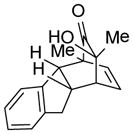 23 |
78 |
| 6 | (±)-17 |
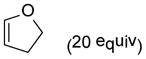 9d |
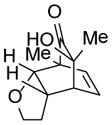 24 |
85 |
| 7 | (±)-18 |
 6 |
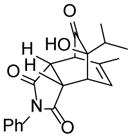 25 |
80 |
Reaction conditions: dimer, dienophile, SiC chip, mesitylene, μW, 170 °C, 15 min;
Single diastereomer isolated;
Isolated yield after silica gel chromatography.
Computational Studies
Previously computational modeling has been employed mainly in studies of MOBs as dienes in [4+2] cycloadditions.3a,26 Quideau and coworkers have also utilized calculations to study the dimerization of ortho-quinols.27 We wished to further study Diels-Alder cycloadditions of o-quinols and dienophiles/dienes computationally to further understand the energetics, regio-, and stereochemical features of the cycloadditions.
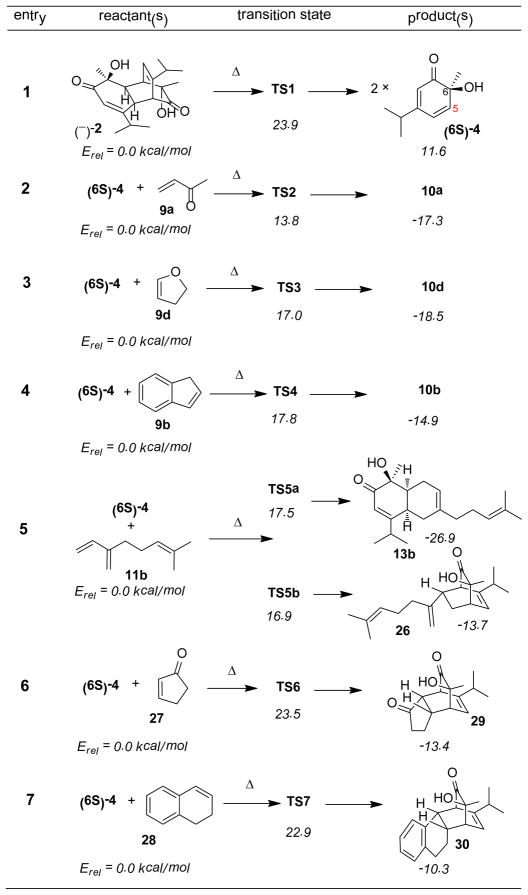
In our studies, B3LYP calculations28 first showed (Table 5, entry 1) that cycloreversion of dimer 2 is modestly endothermic, with forward and reverse barriers of 23.9 and 12.3 kcal/mol, respectively. The lowest energy transition state has C2 symmetry; similar results have been described for related cycloadditions.23 Predicted reaction energetics support a low transient concentration of dienone 4. Cycloaddition of 4 with 9a, 9d, 9b as representative dienophiles is predicted in each case (entries 2 – 4) to have a low reaction barrier, with exothermicity favorable to a unidirectional process. For each of these reactions, we optimized all eight potential transition states; the observed product was correctly assigned by theory in all cases. In every favored cycloaddition, the shorter transition state bond connects to C-5 of dienone 4 (Figure 3). Two failed reactants, 27 and 28, showed unexpectedly higher reaction barriers (entries 6 and 7) when compared to structural analogues 9a and 9b. It is noteworthy that the barriers for two reaction modes of myrcene (11b, entry 5) are nearly equal, consistent with the experimental product distribution (ca. 1:1) at lower reaction temperature (150 °C).2 The lower energy 13b was the sole product generated from the reaction at higher temperature (180 °C, cf. Table 3, entry 2), presumably from cycloadduct 26 via Cope rearrangement.
Table 5.
B3LYP/6-31G(d) Reaction Energetics
|
Figure 3.

Examples of transition state structures.
The observed ensemble of selectivity and the correlation between theory and experiment are striking. In all cases, cycloaddition stereochemistry favors the usual Alder endo rule, with additional facial selectivity syn to the less sterically demanding hydroxyl group. Hyperconjugative effects (Cieplak-Fallis model)29 from the methyl group in the transition state may also contribute to facial selectivity.24, 30 Dienone 4 has a high lying HOMO (−6.9 eV) and a low-lying LUMO (−2.3 eV) and as a consequence reacts with both electron-rich and electron-poor dienophiles. In every case, the preferred regiochemistry follows initial bonding to C5 of the dienone which maximizes resonance stabilization in the asynchronous transition state. In general, reactions with lower barriers showed more asynchronous transition state structures.31
In a side by side comparison of MVK (9a) and cyclopentenone 27, similar dienophiles electronically, the MVK π system shows better secondary-orbital overlap in transition states in reactions with o-quinol 4 (Figure 3, TS2). In contrast, the exo C=O double bond of cyclic ketone 27 does not align well with the o-quinol in the transition state (TS6). Similarly, in TS4 the arene has a more optimal π-π interaction with the 4π partner, while in TS7 the dihydronaphthalene (28) aromatic ring is oriented nearly perpendicular to the cyclohexadienone ring, presumably to minimize steric interactions, thereby raising TS energetics by 5.1 kcal/mol in comparison to the indene case (cf. Table 5, entries 4 and 7). Besides the sterics and electronics of dienophiles, these results suggest that secondary-orbital overlap of dienophiles with ortho-quinols may be another important factor determining dienophile reactivity.
Competition between cycloreversion, [3,3] sigmatropic shifts19 and secondary dienone-phenol 32 or α-ketol21 rearrangements add complexity to these results. For example, DFT calculations show that (−)-2 should undergo a degenerate and homochiral Cope rearrangement with a barrier of 22.4 kcal/mol which should compete with cycloreversion. Figure 4 summarizes predicted energetics for the dienone 4 + 1,3-cyclohexadiene potential surface. No transition state for [4+2] cycloaddition leading directly to 13e (TS9) was identified with DFT calculations. Repeated attempts to locate TS9 instead optimized to TS8 which leads to 12e. Based on these predictions, the most likely pathway to 13e is through a Cope rearrangement of 12e which has a predicted barrier of 29.6 kcal/mol. According to their relative energetics, Cope rearrangement of 12e should be favored over cycloreversion. Our trapping experiments (Scheme 3) supported only a [3,3] sigmatropic rearrangement for interconversion of 12e and 13e, consistent with computations.
Figure 4.

DFT results for cycloaddition and Cope rearrangement.
The rearrangement of 13e to 14 can proceed by thermal isomerization of neutral 13e or through catalysis by acid, base, or lithiation. Computational results are summarized in Scheme 4. The predicted thermal barrier (41.1 kcal/mol) is too high to be operative. Protonation, deprotonation, or lithiation of 13e all decrease the predicted barrier significantly and affect product energetics. Adventitious proton catalysis seems most compatible with our experimental results. For the most favorable case, B3LYP/6-31+G(d) calculations predict that deprotonated 13e should undergo an exothermic rearrangement with a barrier of only 13.1 kcal/mol. The apparent conflict of this result with our LiHMDS-catalyzed experiments (vide infra) was resolved by showing that rearrangement of lithiated 13e is now endothermic by 3.5 kcal/mol because O-Li-O bridging creates product strain.
Scheme 4.

DFT results for α-ketol rearrangement pathways
Rearrangement of 4 to 8 can proceed by similarly diverse mechanisms.21,31 DFT computations for the methyl migration step (Scheme 5) showed low barriers for ionic pathways. Once again, our experimental results are consistent with trace acid catalysis.
Scheme 5.

DFT results for dienone-phenol rearrangement
Biological Studies
Both enantiomers of synthesized bicyclo[2.2.2]octenones and cis-decalins were subjected to screening at 20 μM for inhibition in three biological assays at the National Cancer Institute (NCI): AP-1 (activator protein-1, an oncogenic transcription factor),33 TRAIL (Tumor necrosis factor-α-related apoptosis-inducing ligand) sensitization,34 and HIF-2 (hypoxia inducible factor 2).35 Compound (−)-31 (Figure 5, inset) showed selective inhibition against AP-1 at 4 μM concentration with luciferase reporter assays in HEK293 cells. TPA (12-O-tetradecanoylphorbol-13-acetate) induced AP-1 activity was repressed by more than 50% after pretreatment with the compound at 8 μM. However, the compound did not show inhibition of NF-κB (nuclear factor kappa B) or SRE (serum response element) dependent transcription at the same concentration (Figure 5). SRE was also used as a proliferation control which can measure cytotoxic activity for the compound. Thus, this result suggests that compound (−)-31 targets the events needed for AP-1 activation selectively, rather than other transcription factors such as HIF-2, NF-κB, and SRE. As AP-1 is required for tumor promotion and progression,36 identification of novel and specific AP-1 inhibitors such as bicyclooctenone (−)-31 may be beneficial for cancer prevention and therapy. Although further experiments in the NCI 60-cell screen at 10 μM showed that (−)-31 did not pass the threshold for further evaluation of cell growth inhibition, analogues of compound 31 may be pursued in future studies for SAR analysis.
Figure 5.

(−)-31 shows higher selectivity for AP-1-dependent activity than other transcription factors.
CONCLUSION
We have utilized microwave-assisted parallel screening for evaluation of Diels-Alder reactions of ortho-quinol dimers in an effort to identify reactive reaction partners among a broad panel of compounds. Elucidation of products, analysis of the results, and computational studies allowed us to not only confirm the diastereoselectivity of [4+2] cycloadditions of ortho-quinols, but also determine that sterics and proper secondary orbital alignment are the major governing factors for dienophile reactivity. The high concordance between experimental and calculated results suggest that theoretical calculations of energetics may be used to pre-screen reaction partners for [4+2] cycloadditions of ortho-quinols.37 Furthermore, biological evaluation of a set of compounds generated in the study showed that bicyclo[2.2.2]octenones may be worthwhile scaffolds for development of screening libraries, as novel AP-1 inhibitory activity was observed for one member of the small library. Further studies involving retro-Diels-Alder/Diels-Alder cascade processes to produce natural products and analogues are currently in progress and will be reported in future publications.
EXPERIMENTAL SECTION
Optimization of the retro-[4+2]/[4+2] reaction
A 10 mL microwave tube with a silicon carbide chip was charged with dimer 2 (10.0 mg, 0.030 mmol) and N-phenylmaleimide 6 (15.6 mg, 0.150 mmol).12 0.5 mL of solvent was added under an argon atmosphere and the reaction was irradiated in a microwave reactor (stir off) at the indicated temperature (cf. Table 1),. Reactions in DMF (entry 1) and DMA (entry 2) were directly submitted to UPLC-MS analysis, and crude reactions were diluted with EtOAc, washed with water, brine, dried over MgSO4. The organic fraction was concentrated, dried in vacuo, and purified by column chromatography. Reactions in mesitylene were stopped after indicated reaction time, the crude mixture was passed through a short silica gel plug, flushed using hexanes to remove mesitylene, and finally flushed with EtOAc to elute both the product and excess maleimide 6. The organic fraction was concentrated, dried in vacuo, analyzed by UPLC-MS, and purified by column chromatography to afford cycloadduct 7.
Reaction partner screening
In a microwave reaction vessel (4×48 well SiC block) were charged dimer 2 (3.0 mg, 0.009 mmol), dienophiles/dienes (10 equiv), and 100 μL mesitylene.12 The resulting mixtures were heated in a microwave reactor with 48 well SiC block (stir off) at 180 °C (IR temperature) for 1 h. After the reaction, the crude mixture was passed through a short silica gel plug, flushed using hexanes to remove mesitylene, and finally flushed with EtOAc to elute both the product and excess dienophiles/dienes. The organic fraction was finally concentrated and analyzed by UPLC-MS.
General procedure for microwave reaction of o-quinol dimers (20~30 mg scale)
A 10 mL microwave tube with silicon carbide chip was charged with o-quinol dimer (0.060 mmol) and dienophile (equivalents, cf. Tables 2–4). 0.5 mL of mesitylene was added under an argon atmosphere and the reaction was irradiated in a microwave reactor at 170 or 180 °C (IR temperature) for 15 min (stir off). Reactions were stopped after indicated reaction time, the crude mixture was passed through a short silica gel plug, flushed using hexanes to remove mesitylene (and in a few cases excess nonpolar dienophiles), and finally flushed with EtOAc to elute the crude products. The organic fraction was concentrated, dried in vacuo, and the residue was purified by silica gel column chromatography to afford the corresponding Diels-Alder adducts.
(1R,3S,4S,7S)- 3-Hydroxy-7-(4-methyoxy)phenyl-3-methyl-6-(propan-2-yl)bicyclo[2.2.2]oct-5-en-2-one (10c)
Dimer 2 (20.0 mg, 0.060 mmol) and 4-vinylanisole 9c (81.0 μL, 0.60 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=10:1 to 8:1) followed by preparative TLC (CH2Cl2/EtOAc=10:1) to afford product 10c (35.5 mg, 98%) as a yellow oil. Rf = 0.41 (hexanes/EtOAc=2:1); IR (thin film): νmax 3434, 2959, 1721, 1511, 1248, 1178 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.07 (d, J = 8.0 Hz, 2H), 6.79 (d, J = 8.0 Hz, 2H), 6.18 (d, J = 7.2 Hz, 1H), 3.77 (s, 3H), 3.38 (dd, J = 8.4, 5.6 Hz, 1H), 3.06 (d, J = 0.8 Hz, 1H), 2.94 (ddd, J = 6.0, 3.2, 2.4 Hz, 1H), 2.68 (ddd, J = 12.8, 9.6, 2.4 Hz, 1H), 2.53 (s, 1H), 1.90 (sept., J = 6.8 Hz, 1H), 1.61 (ddd, J = 12.8, 6.0, 2.8 Hz, 1H), 1.28 (s, 3H), 0.89 (d, J = 6.8 Hz, 3H), 0.73 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 213.0, 158.4, 144.7, 135.9, 128.7 (two carbons overlapping), 125.7, 113.6 (two carbons overlapping), 72.1, 59.0, 55.2, 43.1, 39.9, 33.1, 27.6, 25.9, 20.5, 19.7; HRMS–ESI (m/z): [M−H2O+H]+ calcd for C19H23O2, 283.1698; found, 283.1713. [α]22D = +36.4° (c = 0.67, CHCl3).
(1R,2S,6R,7S,9S)- 9-Hydroxy-9-methyl-11-(propan-2-yl)-3,5-dioxa-tricyclo[5.2.2.02,6]undec-10-ene-4,8-dione (10e)
Dimer 2 (20.0 mg, 0.060 mmol) and vinylene carbonate 9e (77.3 μL, 1.20 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=3:1) to afford product 10e (23.1 mg, 76%) as a white solid. Rf = 0.11 (hexanes/EtOAc=3:1); mp 135–137 °C; IR (thin film): νmax 3426, 2964, 1795, 1731, 1164, 1057 cm−1; 1H NMR (CDCl3, 400 MHz): δ 5.97 (d, J = 6.4 Hz, 1H), 5.36 (dd, J = 8.0, 3.6 Hz, 1H), 5.04 (dd, J = 8.0, 3.6 Hz, 1H), 3.64 (dd, J = 3.6, 2.0 Hz, 1H), 3.38 (dd, J = 6.4, 3.6 Hz, 1H), 2.43 (s, 1H), 2.39 (sept., J = 6.8 Hz, 1H), 1.32 (s, 3H), 1.022 (d, J = 6.8 Hz, 3H), 1.016 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 205.5, 154.4, 145.5, 122.2, 74.6, 74.3, 71.9, 54.4, 45.8, 33.1, 25.2, 20.23, 20.18; HRMS–ESI (m/z): [2M+Na]+ calcd for C26H32NaO10, 527.1893; found, 527.1880. [α]22D = +125.6° (c = 0.52, CHCl3).
(1R,4S,7S)-Dimethyl-7-hydroxy-7-methyl-8-oxo-5-(propan-2-yl)bicyclo[2.2.2]octa-2,5-diene-2,3-dicarboxylate (10f)
Dimer 2 (20.0 mg, 0.060 mmol) and dimethyl acetylenedicarboxylate 9f (74.0 μL, 0.60 mmol) in 300 μL of mesitylene were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (CH2Cl2/EtOAc=15:1 to 10:1) to afford product 10f (33.3 mg, 90%) as a yellow oil. Rf = 0.30 (CH2Cl2/EtOAc=5:1); IR (thin film): νmax 3485, 2959, 1726, 1265 cm−1; 1H NMR (CDCl3, 400 MHz): δ 6.03 (d, J = 6.4 Hz, 1H), 4.35 (d, J = 2.0 Hz, 1H), 4.04 (d, J = 6.0 Hz, 1H), 3.81 (s, 3H), 3.77 (s, 3H), 2.54 (s, 1H), 2.48 (doublet of sept., J = 6.8, 1.2 Hz, 1H), 1.32 (s, 3H), 1.02 (d, J = 6.8 Hz, 6H); 13C NMR (CDCl3, 100 MHz): δ 201.9, 166.4, 164.2, 149.8, 143.2, 134.4, 123.7, 67.4, 58.1, 52.7, 52.6, 50.8, 31.9, 26.3, 20.3 (two carbons overlapping); HRMS–ESI (m/z): [M+Na]+ calcd for C16H20NaO6, 331.1158; found, 331.1158. [α]22D = +58.7° (c = 0.51, CHCl3).
(1S,3S,4R)- 3-Hydroxy-3-methyl-6-phenyl-7-(propan-2-yl)bicyclo[2.2.2]octa-5,7-dien-2-one (10g)
Dimer 2 (20.0 mg, 0.060 mmol) and vinylene carbonate 9g (65.9 μL, 1.20 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=8:1) to afford product 14 (25.8 mg, 80%) as a yellow oil. Rf = 0.27 (hexanes/EtOAc=3:1); IR (thin film): νmax 3434, 2963, 1724, 1445, 1068 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.46–7.44 (m, 2H), 7.36–7.32 (m, 2H), 7.29–7.26 (m, 1H), 6.73 (dd, J = 6.0, 2.0 Hz, 1H), 6.08 (dd, J = 6.4, 2.0 Hz, 1H), 4.43 (dd, J = 2.0, 2.0 Hz, 1H), 3.79 (ddd, J = 6.4, 6.4, 2.0 Hz, 1H), 2.53 (sept., J = 6.8 Hz, 1H), 2.30 (s, 1H), 1.35 (s, 3H), 1.07 (d, J = 6.8 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 205.3, 149.2, 141.1, 136.0, 129.2, 128.6 (two carbons overlapping), 127.8, 125.3 (two carbons overlapping), 125.1, 69.5, 60.3, 48.5, 32.1, 26.4, 20.7 (two carbons overlapping); HRMS–ESI (m/z): [M+Na]+ calcd for C18H20NaO2, 291.1361; found, 291.1363. [α]22D = +27.6° (c = 0.81, CHCl3).
(1R,3S,4S,7R)-3-Hydroxy-3-methyl-7-[(E)-2-phenylethenyl]-6-(propan-2-yl)bicyclo[2.2.2]oct-5-en-2-one (12aα)
Dimer 2 (20.0 mg, 0.120 mmol) and 1-phenyl-1,3-butadiene38 (11a, 76.1 mg, 1.20 mmol) were thermolyzed following the general procedure. The crude mixture was purified by preparative TLC (hexanes/EtOAc=4:1) to afford product 12aα (28.8 mg, 81%) as a white solid. Rf = 0.22 (hexanes/EtOAc=5:1); mp 68–70 °C; IR (thin film): νmax 3439, 2962, 2932, 1725, 1449, 1126 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.27~7.26 (m, 4H), 7.20~7.17 (m, 1H), 6.37 (dd, J = 16.0, 1.0 Hz, 1H), 6.11 (ddd, J = 7.0, 1.5, 1.5 Hz, 1H), 5.86 (dd, J = 16.0, 8.5 Hz, 1H), 3.09 (dd, J = 2.0, 2.0 Hz, 1H), 3.00~2.94 (m, 1H), 2.83 (ddd, J = 7.0, 3.0, 2.5 Hz, 1H), 2.53 (ddd, J = 12.5, 8.5, 3.0 Hz, 1H), 2.50 (bs, 1H), 2.30 (doublet of sept., J = 7.0, 1.0 Hz, 1H), 1.26 (s, 3H), 1.21 (ddd, J = 13.0, 5.0, 2.5 Hz, 1H), 1.02 (d, J = 7.0 Hz, 3H), 0.98 (d, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 212.9, 144.4, 137.1, 132.6, 129.7, 128.5 (two carbons overlapping), 127.2, 126.2, 126.0 (two carbons overlapping), 72.3, 56.6, 42.9, 39.2, 33.1, 27.3, 25.9, 20.9, 20.5; HRMS–ESI (m/z): [M−H2O+H]+ calcd for C20H23O, 279.1749; found, 279.1748. [α]22D = +119.5° (c = 0.91, CHCl3).
(1R,3S,4S,7S)-3-Hydroxy-3-methyl-7-[(E)-2-phenylethenyl]-6-(propan-2-yl)bicyclo[2.2.2]oct-5-en-2-one (12aβ)
Utilizing the same protocol as compound 12aα, product 12aβ (19.8 mg, 97%) was isolated as a white solid. Rf = 0.32 (hexanes/EtOAc=5:1); mp 104–106 °C; IR (thin film): νmax 3440, 2962, 2871, 1726, 1449, 1142, 1081 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.33~7.26 (m, 4H), 7.21~7.17 (m, 1H), 6.40 (d, J = 15.6 Hz, 1H), 6.10 (ddd, J = 15.6, 8.8, 1.6 Hz, 1H), 6.04 (dd, J = 6.8, 1.2 Hz, 1H), 3.03 (dd, J = 1.6, 1.6 Hz, 1H), 2.85~2.82 (m, 1H), 2.65~2.58 (m, 1H), 2.40 (sept., J = 6.8 Hz, 1H), 2.30 (s, 1H), 2.12 (ddd, J = 12.8, 5.2, 2.0 Hz, 1H), 1.77~1.70 (m, 1H), 1.28 (s, 3H), 1.05 (d, J = 6.8 Hz, 3H), 1.04 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 213.4, 147.8, 137.1, 132.1, 130.3, 128.5 (two carbons overlapping), 127.2, 126.3 (two carbons overlapping), 125.5, 73.5, 56.6, 43.1, 42.5, 32.6, 26.4, 26.3, 20.54, 20.50; HRMS–ESI (m/z): [M+H]+ calcd for C20H25O2, 297.1855; found, 297.1867. [α]22D = +137.8° (c = 0.57, CHCl3).
(1S,4aR,8aS)-1-Hydroxy-1-methyl-6-[4-methylpent-3-en-1-yl]-4-(propan-2-yl)-4a,5,8,8a-tetrahydronaphthalen-2(1H)-one (13b)
Dimer 2 (20.0 mg, 0.060 mmol) and β-myrcene 11c (103.3 μL, 0.60 mmol) were thermolyzed following the general procedure. The crude mixture was purified and 13b was isolated as a colorless oil (20.8 mg, 57%). 1H-NMR, 13C-NMR, and IR spectra for compound 13b were found to be identical to literature reported data.17 Rf = 0.38 (hexanes/EtOAc=5:1); [α]22D = +217.9° (c = 1.01, CHCl3).
(1S,4aR,8aS)-1-Hydroxy-1,6,7-trimethyl-4-(propan-2-yl)-4a,5,8,8a-tetrahydronaphthalen-2(1H)-one (13c)
Dimer 2 (20.0 mg, 0.060 mmol) and 2,3-dimethyl-1,3-butadiene 11c (67.9 μL, 0.60 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=20:1 to 15:1) to afford product 13c (27.3 mg, 92%) as an oil. Rf = 0.38 (hexanes/EtOAc=8:1); IR (thin film): νmax 3491, 2968, 2926, 1722, 1671, 1242, 1152 cm−1; 1H NMR (CDCl3, 400 MHz): δ 5.85 (d, J = 2.8 Hz, 1H), 3.55 (s, 1H), 2.94~2.91 (m, 1H), 2.38 (sept., J = 6.8 Hz, 1H), 2.35~2.30 (m, 3H), 2.07 (dd, J = 17.6, 6.8 Hz, 1H), 1.59 (s, 3H), 1.48 (s, 3H), 1.32 (s, 3H), 1.06 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 203.1, 172.8, 126.1, 123.3, 120.0, 76.6, 44.4, 36.8, 33.3, 30.8, 29.4, 24.4, 22.4, 20.6, 19.0, 18.7; HRMS–ESI (m/z): [M+H]+ calcd for C16H24O2, 249.1855; found, 249.1876. [α]22D = +133.6° (c = 0.46, CHCl3).
(1S,2R,7S,8R,10S)-10-Hydroxy-10-methyl-12-(propan-2-yl)tricyclo-[6.2.2.02,7]dodeca-5,11-dien-9-one (12e)
Dimer 2 (60.0 mg, 0.180 mmol) and 1,3-cyclohexadiene 11e (343.0 μL, 3.60 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel column (hexanes:EtOAc=20:1 to 15:1) to afford 12e (51.8 mg, 57%) as a white solid. Rf = 0.32 (hexanes/EtOAc=5:1); mp 46–48 °C; IR (thin film): νmax 3443, 2960, 2928, 1725, 1366, 1137 cm−1; 1H NMR (CDCl3, 400 MHz): δ 5.89 (d, J = 7.6 Hz, 1H), 5.87~5.82 (m, 1H), 5.42 (ddd, J = 9.6, 2.4, 2.4 Hz, 1H), 3.00 (dd, J = 2.0, 2.0 Hz, 1H), 2.79~2.72 (m, 2H), 2.64~2.62 (m, 1H), 2.52 (s, 1H), 2.22 (doublet of sept., J = 7.2, 0.8 Hz, 1H), 1.93~1.86 (m, 1H), 1.84~1.75 (m, 1H), 1.61 (dddd, J = 12.4, 10.0, 4.8, 4.8 Hz, 1H), 1.23 (s, 3H), 1.21~1.16 (m, 1H), 0.94 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 213.6, 144.8, 130.2, 128.4, 125.3, 72.9, 56.3, 49.2, 35.5, 32.9, 31.8, 26.3, 25.9, 23.5, 20.8, 20.5; HRMS–ESI (m/z): [M+H]+ calcd for C16H23O2, 247.1698; found, 247.1721. [α]22D = +160.4° (c = 1.17, CHCl3).
(1R,2S,3S,7R,8S)-3-Hydroxy-3-methyl-6-(propan-2-yl)tricyclo-[6.2.2.02,7]dodeca-5,9-dien-4-one (13e)
Utilizing the same protocol for compound 12e, product 13e (22.5 mg, 25%) was isolated as a white solid. Rf = 0.39 (hexanes: EtOAc = 5:1); mp 52–53 °C; IR (thin film) νmax: 3463, 2963, 2867, 1673, 1376, 1170 cm−1; 1H NMR (CDCl3, 400 MHz): δ 6.04 (dd, J = 3.2, 3.2 Hz, 1H), 5.84 (s, 1H), 5.77 (dd, J = 3.2, 3.2 Hz, 1H), 3.99 (s, 1H), 2.96 (dddd, J = 4.4, 3.2, 2.0, 2.0 Hz, 1H), 2.83 (dd, J = 8.0, 2.8 Hz, 1H), 2.78 (ddd, J = 4.0, 2.0, 2.0 Hz, 1H), 2.44 (sept., J = 6.8 Hz, 1H), 2.26 (dd, J = 8.4, 1.6 Hz, 1H), 1.64~1.59 (m, 1H), 1.52 (dddd, J = 12.4, 9.2, 2.8, 2.8 Hz, 1H), 1.40 (dddd, J = 12.4, 12.4, 3.2, 3.2 Hz, 1H), 1.27~1.20 (m, 1H), 1.19 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 202.5, 170.8, 133.8, 132.7, 119.2, 73.9, 47.9, 45.7, 35.4, 32.8, 32.0, 29.9, 26.1, 24.5, 23.2, 20.9; HRMS–ESI (m/z): [M + H]+ calcd for C16H23O2, 247.1698; found, 247.1707. [α]22D = −128.1° (c = 0.52, CHCl3).
1-[(1R,2S,3S,6R,7S)-3-hydroxy-5-(propan-2-yl)tricyclo-[5.2.2.02,6]-undeca-4,8-dien-3-yl]ethanone (14)
Utilizing the same protocol for compound 12g, product 14 (8.2 mg, 9%) was isolated as a white solid. Rf = 0.46 (hexanes: EtOAc = 5:1); mp 98–100 °C ; IR (thin film) νmax: 3450, 2952, 2936, 2915, 1698, 1367, 1102 cm−1; 1H NMR (CDCl3, 500 MHz): δ 6.27 (dd, J = 7.2, 7.2 Hz, 1H), 5.89 (dd, J = 7.2, 7.2 Hz, 1H), 5.03 (dd, J = 1.5, 1.5 Hz, 1H), 3.92 (s, 1H), 2.98 (ddd, J = 8.5, 2.2, 2.2 Hz, 1H), 2.77~2.75 (m, 1H), 2.58~2.55 (dd, J =2.0 Hz, 1H), 2.34 (dd, J = 8.2, 2.2 Hz, 1H), 2.29 (sept., J = 7.0 Hz, 1H), 2.09 (2, 3H), 1.49 (dddd, J = 12.0, 9.5, 4.5, 2.2 Hz, 1H), 1.41 (dddd, J = 12.0, 9.0, 3.0, 3.0 Hz, 1H), 1.33 (dddd, J = 11.5, 11.5, 3.5, 3.5 Hz, 1H), 1.26~1.20 (m, 1H), 1.06 (d, J = 7.0 Hz, 3H), 1.04 (d, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 210.5, 159.5, 134.0, 129.2, 124.8, 90.8, 53.5, 48.7, 32.7, 30.9, 27.7, 25.1, 24.2, 23.6, 21.7, 20.8; HRMS–ESI (m/z): [M−H2O+H]+ calcd for C16H21O, 229.1592; found, 229.1597. [α]22D = +210.4° (c = 0.30, CHCl3).
Gradifloracin (16)
Oxidation of salicyl alcohol benzoate (50.0 mg, 0.17 mmol) according to the reported procedure11 (1.2 equiv DIEA, 1.5 equiv [(−)-sparteine]2Cu2O2(PF6)2, 3Å MS, O2, CH2Cl2, −78 °C, 40 h) generated dimer 16 as a grey solid (15.0 mg, 28%, 61% brsm) after silica gel chromatography. The 1H, 13C NMR, IR, and HRMS of 16 were identical to literature reported data.24,39 [α]22D = −5.1° (c = 0.57, CHCl3; lit.S5 [α]22D = −13.6°, c = 0.728, CHCl3). The ee for 16 (24%) was determined based on chiral HPLC analysis of its derivative 23.
Dimer (±)-17
Oxidation of 2,6-dimethylphenol (20.0 mg, 0.164 mmol) according to the reported procedure11 [1.0 equiv LiOH·H2O, 1.2 equiv (N,N′-di-tert-butylethylene-diamine)2Cu2O2(PF6)2, 3Å MS, O2, CH2Cl2, −78 °C, 16 h] generated dimer 17 as a light yellow solid (16.2 mg, 72%) after silica gel chromatography. 1H, 13C NMR, IR, and HRMS of 17 were identical to literature reported data.24
(1R,2S,6R,7S,9R)-11-tert-Butyl-9-hydroxy-9-methyltricyclo[5.2.2.02,6]-undeca-4,10-dien-8-one (19)
According to the general procedure, dimer (+)-15 (30.0 mg, 0.083 mmol) and dicyclopentadiene 18 (54.9 mg, 0.415 mmol) were thermolyzed according to General Procedure C. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=15:1) to afford product 19 (38.0 mg, 93%) as a white solid. Rf = 0.26 (hexanes/EtOAc=7:1); mp 92–93 °C; IR (thin film): νmax 3425, 2963, 2928, 1721, 1364 cm−1; 1H NMR (CDCl3, 400 MHz): δ 5.85 (dd, J = 6.8, 2.0 Hz, 1H), 5.62~5.59 (m, 1H), 5.34~5.32 (m, 1H), 3.25 (dd, J = 2.0, 2.0 Hz, 1H), 3.20–3.10 (m, 2H), 2.83 (dddd, J = 9.2, 4.8, 2.4, 2.4 Hz, 1H), 2.63 (bs, 1H), 2.46 (ddddd, J = 16.8, 9.2, 2.0, 2.0, 2.0 Hz, 1H), 1.94 (ddddd, J = 16.8, 6.0, 2.0, 2.0, 2.0 Hz, 1H), 1.21 (s, 3H), 0.92 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 214.9, 148.7, 132.8, 130.4, 122.2, 73.2, 52.6, 50.0, 47.5, 38.2, 34.2, 33.2, 28.2 (three carbons overlapping), 26.3; HRMS–ESI (m/z): [M−H2O+H]+ calcd for C16H21O, 229.1592; found, 229.1604. [α]22D = −90.0° (c = 0.65, CHCl3).
(1R,2S,6R,7S,9S)-11-tert-Butyl-9-hydroxy-9-methyl-3,5-dioxatricyclo-[5.2.2.02,6]undec-10-ene-4,8-dione (20)
Dimer (+)-15 (30.0 mg, 0.083 mmol) and methyl vinyl ketone 9 (105.4 μL, 1.66 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=5:1 to 3:1) to afford product 20 (34.8 mg, 78%) as a colorless oil. Rf = 0.13 (hexanes/EtOAc=3:1); IR (thin film): νmax 3423, 2965, 1786, 1731, 1367, 1162, 1058 cm−1; 1H NMR (CDCl3, 400 MHz): δ 5.99 (d, J = 6.8, 1.2 Hz, 1H), 5.36 (dd, J = 8.0, 3.6 Hz, 1H), 5.03 (dd, J = 8.0, 3.2 Hz, 1H), 3.81 (dd, J = 3.6, 2.0 Hz, 1H), 3.39 (dd, J = 6.8, 3.2 Hz, 1H), 2.56 (bs, 1H), 1.31 (s, 3H), 1.04 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 205.8, 154.4, 147.9, 121.2, 74.5, 74.2, 71.8, 52.9, 45.6, 34.9, 27.8 (three carbons overlapping), 25.3; HRMS–ESI (m/z): [2M+H]+ calcd for C28H37O10, 533.2387; found, 533.2399. [α]22D = −176.9° (c = 1.09, CHCl3).
(12-Hydroxy-13-oxotetracyclo-[9.2.2.02,10.03,8]pentadeca-3,5,7,14-tetraen-12-yl)methyl benzoate (21)
Gradifloracin 16 (10.0 mg, 0.020 mmol) and indene 9b (23.3 μL, 0.20 mmol) in mesitylene were irradiated in microwave reactor at 170 °C for 15 min. Following the general procedure the obtained crude mixture was purified by silica gel chromatography (hexanes/EtOAc=10:1 to 8:1) to afford product 21 (11.8 mg, 80%) as a light yellow oil. Rf = 0.38 (hexanes/EtOAc, 3:1). IR (thin film) νmax: 3443, 2917, 2849, 1726, 1692, 1272 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.08 (dd, J = 8.4, 1.2 Hz, 2H), 7.60–7.56 (m, 1H), 7.45 (dd, J = 8.4, 8.4 Hz, 2H), 7.19–7.13 (m, 3H), 7.12–7.09 (m, 1H), 6.35 (dd, J = 7.2, 7.2 Hz, 1H), 5.94 (dd, J = 7.2, 7.2 Hz, 1H), 4.45 (d, J = 12.0 Hz, 1H), 4.28 (d, J = 12.0 Hz, 1H), 3.83 (dd, J = 9.2, 2.0 Hz, 1H), 3.57 (ddd, J = 6.4, 2.8, 1.2 Hz, 1H), 3.50–3.43 (m, 1H), 3.29 (ddd, J = 6.4, 2.8, 1.2 Hz, 1H), 3.23 (dd, J = 16.8, 10.4 Hz, 1H), 3.13 (s, 1H), 2.69 (dd, J = 16.8, 4.4 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 209.9, 166.6, 144.3, 142.4, 133.3, 133.2, 129.8 (two carbons ovrlp), 129.6, 128.6, 128.5 (two carbons ovrlp), 127.3, 126.7, 124.4, 124.0, 74.3, 68.4, 53.1, 50.1, 45.4, 37.7, 33.9; HRMS–ESI (m/z): [M + Na]+ calcd for C23H20NaO4, 383.1259; found, 383.1252. [α]22D = −22.3° (c = 0.24, CHCl3). The ee for 21 (24%) was determined using a Waters Breeze HPLC System (ChiralPak AD-H, 150 × 4.6 mm, 10% isopropanol in hexane, 1.0 mL/min, retention time 12.3 min (minor enantiomer) and 14.6 min (major enantiomer)) using UV detection at 254 nm.
9-Hydroxy-7,9-dimethyl-4-phenyl-4-azatricyclo[5.2.2.02,6]-undec-10-ene-3,5,8-trione (22)
Dimer 17 (20.0 mg, 0.072 mmol) and N-phenylmaleimide 6 (37.4 mg, 0.216 mmol) were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=2:1) to afford product 2 (44.6 mg, 99%) as a light yellow solid. Rf = 0.08 (hexanes/EtOAc=3:1); mp 161–163 °C; IR (thin film): νmax 3444, 2977, 1703, 1698, 1385, 1186 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.45~7.40 (m, 2H), 7.39~7.34 (m, 1H), 7.17 (dd, J = 7.6, 1.6 Hz, 2H), 6.42 (dd, J = 8.4, 6.4 Hz, 1H), 5.95 (dd, J = 8.4, 1.2 Hz, 1H), 3.85 (dd, J = 8.4, 3.6 Hz, 1H), 3.54 (ddd, J = 6.4, 3.2, 1.6 Hz, 1H), 2.96 (d, J = 8.0 Hz, 1H), 2.95 (s, 1H), 1.57 (s, 3H), 1.32 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 210.3, 177.1, 174.4, 133.9, 132.9, 131.6, 129.1 (two carbons overlapping), 128.7, 126.4 (two carbons overlapping), 70.8, 50.5, 45.4, 44.9, 40.9, 25.6, 14.8; HRMS–ESI (m/z): [M+H]+ calcd for C18H18NO4, 312.1236; found, 312.1249.
12-Hydroxy-12-methyl-14-(propan-2-yl)-tetracyclo[9.2.2.02,10.03,8]pentadeca-3,5,7,14-tetraene-13-one (23)
Dimer 17 (20.0 mg, 0.072 mmol) and indene 11 (83.9 μL, 0.72 mmol) were thermolyzed following the general procedure. The crude mixture was purified by preparative TLC (hexanes/EtOAc=3:1) to afford product 23 (28.7 mg, 78%) as a light yellow solid. The 1H, 13C NMR, IR, and HRMS of 16 were identical to literature reported data.1a
8-hydroxy-1,8-dimethyl-3-oxatricyclo[5.2.2.02,6]undec-10-en-9-one (24)
Dimer 17 (20.0 mg, 0.072 mmol) and 2,3-dihydrofuran 9d (111.9 μL, 1.48 mmol) were thermolyzed following the general procedure. The concentrated mixture was dissolved in 2 mL of CH2Cl2 followed by addition of MP-TsOH resin (100 mg), and the reaction was shaken for 30 min. The MP-TsOH resin was filtered away and the filtrate was concentrated. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=8:1 to 7:1) to afford product 24 (25.6 mg, 85%) as a white solid. Rf = 0.27 (hexanes/EtOAc=2:1); mp 117–119 °C; IR (thin film): νmax 3429, 2973, 2933, 1726, 1451, 1366, 1080 cm−1; 1H NMR (CDCl3, 400 MHz): δ 6.33 (dd, J = 7.2, 7.2 Hz, 1H), 5.75 (dd, J = 8.4, 1.6 Hz, 1H), 3.89~3.82 (m, 2H), 3.53 (ddd, J = 8.4, 8.4, 8.4 Hz, 1H), 3.18 (ddd, J = 8.4, 8.4, 8.4 Hz, 1H), 2.92~2.90 (m, 1H), 2.50 (s, 1H), 2.10~2.00 (m, 1H), 1.64~1.55 (m, 1H), 1.32 (s, 3H), 1.24 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 213.4, 134.1, 132.6, 84.5, 71.9, 69.1, 55.1, 46.4, 39.0, 30.9, 26.5, 14.5; LRMS m/z (% relative intensity): 231.1 [(M+Na)+, 31], 191.1 (100), 163.1 (70), 141.1 (34).
9-hydroxy-11-methyl-4-phenyl-9-(propan-2-yl)-4-azatricyclo[5.2.2.02,6]undec-10-ene-3,5,8-trione (25)
Dimer 18 (30.0 mg, 0.090 mmol) and N-phenylmaleimide 6 (46.9 mg, 0.271 mmol) in mesitylene were thermolyzed following the general procedure. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc=6:1 to 5:1) to afford product 25 (49.1 mg, 80%) as a white solid. Rf = 0.31 (hexanes/EtOAc=2:1); mp 169–170 °C; IR (thin film): νmax 3480, 2966, 2924, 1708, 1500, 1383, 1187 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.45~7.41 (m, 2H), 7.38~7.35 (m, 1H), 7.14 (dd, J = 7.2, 1.2 Hz, 2H), 6.03 (ddd, J = 6.4, 1.6, 1.6 Hz, 1H), 3.68 (dd, J = 8.0, 3.2 Hz, 1H), 3.59 (dd, J = 3.2, 1.6 Hz, 1H), 3.56 (dd, J = 6.4, 3.2 Hz, 1H), 3.36 (dd, J = 8.0, 3.2 Hz, 1H), 2.46 (s, 1H), 1.87 (sept., J = 6.8 Hz, 1H), 1.84 (d, J = 1.6 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 209.4, 177.5, 175.1, 136.0, 131.6, 129.1 (two carbons overlapping), 128.8, 126.3 (two carbons overlapping), 125.0, 75.9, 53.5, 42.9, 42.2, 39.8, 33.2, 20.7, 17.8, 16.2; HRMS–ESI (m/z): [M+H]+ calcd for C20H22NO4, 340.1549; found, 340.1553.
Computational Methods
All calculations were carried out with Gaussian 03 Revision E.01,40 Spartan 06 or Spartan 08.41 Structures were optimized and characterized by frequency analysis at the B3LYP/6-31G(d) level of theory with all transition states showing a single imaginary vibrational mode corresponding to the expected reaction.42 Unscaled zero point vibrational energy (ZPVE) corrections have been applied to total energies. Energies and geometries for stationary points are summarized in the Supporting Information. Solvation was not included in these calculations because it is expected that our nonpolar solvents would not significantly affect reaction energetics.
Supplementary Material
Acknowledgments
This work was generously supported by the NIGMS CMLD Initiative (P50 GM067041 J.A.P., Jr.), the National Science Foundation (CHE-0910826 R.P.J.), and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. We thank Dr. Emil Lobkovsky (Cornell University) for X-ray crystal structure analysis and Anton Paar Corporation, CEM Corporation, and Waters Corporation for assistance with instrumentation.
Footnotes
Supporting Information Available Experimental procedures and characterization data for all new compounds, including X-ray structure analysis of compound 10e, X-ray crystallographic data (CIF), and detailed biological methods and results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For use of ortho-quinol dimers in retro-[4+2]/[4+2] cascades, see: Singh VK, Deota PT, Bedekar AV. J Chem Soc Perkin Trans 1. 1992:903.Singh V, Samanta B. Tetrahedron Lett. 1999;40:1807.
- 2.Dong S, Hamel E, Bai R, Covell DG, Beutler JA, Porco JA., Jr Angew Chem, Int Ed. 2009;48:1494. doi: 10.1002/anie.200805486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For use of MOB-derived dimers, see: Liao CC, Peddinti RK. Acc Chem Res. 2002;35:856. doi: 10.1021/ar000194n.Chittimalla SK, Shiao HY, Liao CC. Org Biomol Chem. 2006;4:2267. doi: 10.1039/b602928k.Snyder SA, Kontes F. J Am Chem Soc. 2009;131:1745. doi: 10.1021/ja806865u.
- 4.For reviews on ortho-quinol and MOB-derived [4+2] cyclodimers, see: Magdziak D, Meek SJ, Pettus TRR. Chem Rev. 2004;104:1383. doi: 10.1021/cr0306900.Quideau S, Pouysegu L, Deffieux D. Synlett. 2008:467.
- 5.Kappe CO. Chem Soc Rev. 2008;37:1127. doi: 10.1039/b803001b. [DOI] [PubMed] [Google Scholar]
- 6.For a recent review on use of graphite in microwave reactions, see: Besson T, Thiery V, Dubac J. Microwaves in Organic Synthesis. 2. Vol. 1. Wiley-VCH; Weinheim, Germany: 2006. p. 416.For a recent example employing graphite, see: Cho HY, Ajaz A, Himali D, Waske PA, Johnson RP. J Org Chem. 2009;74:4137. doi: 10.1021/jo900245v.
- 7.Kremsner JM, Kappe CO. J Org Chem. 2006;71:4651. doi: 10.1021/jo060692v.Razzaq T, Kremsner JM, Kappe CO. J Org Chem. 2008;73:6321. doi: 10.1021/jo8009402.For silicon carbide, see: Harris GL, editor. Properties of Silicon Carbide. Institute of Electrical Engineers; London: 1995. Choyke WJ, Matsunami H, Pensl G, editors. Silicon Carbide: Recent Major Advances. Springer; Berlin: 2004. Saddow SE, Agarwal A, editors. Advances in Silicon Carbide Processing and Applications. Artech House Inc; Norwood, MA: 2004.
- 8.Damm M, Kappe CO. Mol Diversity. 2009;13:529. doi: 10.1007/s11030-009-9167-3. [DOI] [PubMed] [Google Scholar]
- 9.Kremsner JM, Stadler A, Kappe CO. J Comb Chem. 2007;9:285. doi: 10.1021/cc060138z. [DOI] [PubMed] [Google Scholar]; (b) Damm M, Kappe CO. J Comb Chem. 2009;11:460. doi: 10.1021/cc900007w. [DOI] [PubMed] [Google Scholar]
- 10.Beeler AB, Su S, Singleton CA, Porco JA., Jr J Am Chem Soc. 2007;129:1413. doi: 10.1021/ja0674744. [DOI] [PubMed] [Google Scholar]
- 11.(a) Li C, Johnson RP, Porco JA., Jr J Am Chem Soc. 2003;125:5095–5106. doi: 10.1021/ja021396c. [DOI] [PubMed] [Google Scholar]; (b) Lei X, Johnson RP, Porco JA., Jr Angew Chem, Int Ed. 2003;42:3913. doi: 10.1002/anie.200351862. [DOI] [PubMed] [Google Scholar]
- 12.Dong S, Zhu J, Porco JA., Jr J Am Chem Soc. 2008;130:2738. doi: 10.1021/ja711018z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.See Supporting Information for complete experimental details.
- 14.For UPLC, see: Mazzeo JR, Neue UD, Kele M, Plumb RS. Anal Chem. 2005;77:460 A.Nováková L, Matysová L, Solich P. Talanta. 2006;68:908. doi: 10.1016/j.talanta.2005.06.035.
- 15.For a recent review on the dienone-phenol rearrangement, see: Whiting DA. In: Comp Org Synth. Trost BM, Fleming I, editors. Vol. 3. Pergamon Press; Oxford: 1991. pp. 803–821.
- 16.Carman RM, Van Dongen JMAM. Aust J Chem. 1984;37:2607. [Google Scholar]
- 17.Wannere CS, Paul A, Herges R, Houk KN, Schaefer HF, III, Schleyer PvR. J Comput Chem. 2007;28:344. doi: 10.1002/jcc.20532. [DOI] [PubMed] [Google Scholar]
- 18.Snider BB, Chen Z. Org Prep Proced Int. 1999;31:537. [Google Scholar]
- 19.Kuo YH, Chen CH, Huang SL. Chem Pharm Bull. 1999;46:181. [Google Scholar]
- 20.Competition between Cope rearrangements and retro-Diels-Alder reactions in similar bicyclic structures has been reported: Su KJ, Mieusset JL, Arion VB, Brecker L, Brinker UH. Org Lett. 2007;9:113. doi: 10.1021/ol0626793.Jeong JP, Lee OS, Yang K. Bull Korean Chem Soc. 2002;23:829.Singh V, Sharma U, Prasanna V, Porinchu M. Tetrahedron. 1995;51:6015.
- 21.For [3,3]-sigmatropic rearrangement of bridged 1,5-dienes, see: Bultman MS, Ma J, Gin DY. Angew Chem, Int Ed. 2008;47:6821. doi: 10.1002/anie.200801969.and references cited therein.
- 22.Paquette LA, Hofferberth JE. Org React. 2003;62:477. [Google Scholar]
- 23.(a) Nagahama S, Tazaki M. Bull Chem Soc Jpn. 1987;60:4175. [Google Scholar]; b) Shimizu T, Hiranuma S, Qian ZH, Yoshioka H. Synlett. 1991:725. [Google Scholar]
- 24.Liao YH, Xu LZ, Yang SL, Dai J, Zhen YS, Zhu M, Sun NJ. Phytochemistry. 1997;45:729. [Google Scholar]
- 25.Gagnepain J, Castet F, Quideau S. Angew Chem, Int Ed. 2007;46:1533. doi: 10.1002/anie.200604610. [DOI] [PubMed] [Google Scholar]
- 26.Dory YL, Roy AL, Soucy P, Deslongchamps P. Org Lett. 2009;11:1197. doi: 10.1021/ol8026768. [DOI] [PubMed] [Google Scholar]
- 27.Gagnepain J, Mereau R, Dejugnac D, Leger JM, Castet F, Deffieux D, Pouysegu L, Quideau S. Tetrahedron. 2007;63:6493. [Google Scholar]
- 28.Details of computational methods and results are presented in the Supporting Information.
- 29.(a) Cieplak AS. J Am Chem Soc. 1981;103:4540. [Google Scholar]; (b) Macaulay JB, Fallis AG. J Am Chem Soc. 1990;112:1136. [Google Scholar]
- 30.Hou HF, Peddinti RK, Liao CC. Org Lett. 2002;4:2477. doi: 10.1021/ol020087o. [DOI] [PubMed] [Google Scholar]
- 31.In each case, we find that the transition state DFT wavefunction is stable relative to becoming open shell. Thus, these reactions may be described as asynchronous but concerted. Further examples of transition state structures are shown in the Supporting Information.
- 32.Miller B. Acc Chem Res. 1975;8:245. and references therein. [Google Scholar]
- 33.Ruocco KM, Goncharova EI, Young MR, Colburn NH, McMahon JB, Henrich CJ. J Biomol Screen. 2007;12:133. doi: 10.1177/1087057106296686. [DOI] [PubMed] [Google Scholar]
- 34.Booth NL, Sayers TJ, Brooks AD, Thomas CF, Jacobsen K, Goncharova EI, McMahon JB, Henrich CJ. Cancer Immunol Immunother. 2009;58:1229. doi: 10.1007/s00262-008-0637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woldemichael GM, Vasselli JR, Gardella RS, McKee TC, Linehan WM, McMahon JB. J Biomol Screen. 2006;11:678. doi: 10.1177/1087057106289234. [DOI] [PubMed] [Google Scholar]
- 36.Young MR, Li J, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker R. Proc Natl Acad Sci USA. 1999;96:9827. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez AM, Prieto P, de la Hoz A, Díaz-Ortiz A. Org Biomol Chem. 2011;9:2371. doi: 10.1039/c0ob01037e. [DOI] [PubMed] [Google Scholar]
- 38.Lebel H, Paquet V. J Am Chem Soc. 2004;126:320. doi: 10.1021/ja038112o. [DOI] [PubMed] [Google Scholar]
- 39.Liao Y, Xu L, Yang S, Dai J, Zhen Y, Zhu M, Sun N. Phytochemistry. 1997;45:729. [Google Scholar]
- 40.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 0.3, Revision E.01. Gaussian Inc; Wallingford, CT: 2004. [Google Scholar]
- 41.Spartan 06 and Spartan 08. Wavefunction Inc; Irvine, CA: [Google Scholar]
- 42.(a) Kohn W, Becke AD, Parr RG. J Phys Chem. 1996;100:12974. [Google Scholar]; (b) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (c) Lee C, Yang Y, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.