

Abstract
General, high-yielding MAOS protocols for the expedient synthesis of functionalized 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazines are described amenable to an iterative analog library synthesis strategy for the lead optimization of an M1 antagonist screening hit. Optimized compounds proved to be highly selective M1 antagonists.
In the course of our program in small molecule probe development for the Molecular Library Screening Center Network (MLSCN),1 a high-throughput screen identified the 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazine scaffold 1 as an attractive hit for a CNS target (Fig. 1). While numerous reports describe syntheses of 1, yields are typically moderate (<50%) with prolonged reaction times at high temperatures (steps requiring 18- to >60 h at reflux).2–5 In order to employ an iterative analog library synthesis approach for the lead optimization of 2, a weak, but selective muscarinic acetylcholine receptor antagonist (M1 IC50 = 22 μM, M4 IC50 > 150 μM), significant refinements were required in the synthetic protocols for delivering analogs 1, with diversity at both C3 and C6.2–5
Figure 1.

Generic structure of 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazine 1 and our M1 antagonist screening lead 2.
As many of the leads identified from HTS campaigns are small heterocyclic compounds, our laboratory has devoted significant effort to develop efficient protocols for the preparation of diverse heterocyclic templates employing microwave-assisted organic synthesis (MAOS).6–12 In recent reports, we have described general, high-yielding MAOS protocols for the expedient synthesis of 1,2,4-triazines 3,6 imidazoles 4,7 quinoxalines 5,8 pyrazinone 6,9 5-aminooxazoles 7,10 quinoxalinones 8,11 pyrazolo[1,5-a]pyrimidines 9,12 and pyrazolo[3,4-d]pyrimidines 1012 from simple starting materials (Fig. 2). Therefore, application of MAOS to develop a general, high-yielding, and expedient synthesis of the 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazine scaffold 1 seems warranted (see Scheme 1).
Figure 2.

Heterocyclic templates accessed through MAOS in our laboratory.
Scheme 1.

Classical synthesis of 3,6-disubstituted-[1,2,4]triazolo 4,3-b]pyridazines 1.
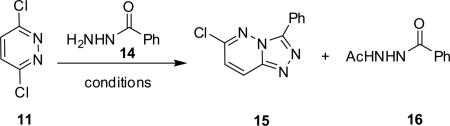
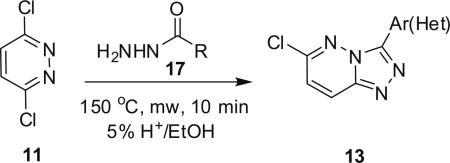
Classical conditions for the synthesis of 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazines 1 involve refluxing 3,6-dichloropyridazine 11 with an acylhydrazide 12 in toluene for 16 h, or more typically for 60 h, to provide the 3-aryl-6-chloro-[1,2,4]triazolo[4,3-b]pyridazine 13 in yields less than 50%.3–5 Introduction of the amino moiety in the 6-position was accomplished through an SNAr reaction employing either neat or steel bomb conditions at 100–140 °C for 8–30 h to deliver analogs 1 in yields ranging from 40% to 70%.3–6 Moreover, previous efforts were focused on traditional medicinal chemistry approaches and the development of structure–activity relationships (SARs), with little concern for achieving high chemical yields or reaction generality for either the heterocycle synthesis or the SNAr reaction. Indeed, the 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazine scaffold 1 has been an important pharmacophore for the development GABAA receptor agonists at the α2/α3-subunit.3–5 Interestingly, microwave-assisted organic synthesis has never before been applied to this heterocyclic system, and even more surprising when one considers a 1–6 day reaction time to deliver a single derivative of 1.
By varying solvent and temperature parameters, microwave conditions were rapidly developed to accelerate and generalize the synthesis of the 3-phenyl-6-chloro-[1,2,4]triazolo[4,3-b]pyridazine 15 core employing 3,6-dichloropyridazine 11 and acylhydrazide 14 (Table 1). When HOAc was employed as a solvent or catalyst, a corresponding acetylated phenyl acylhydrazide 16 was obtained in varying quantities. Optimal HOAc conditions employed 5% HOAc/EtOH at 150 °C for 10 min to afford the desired 15, along with 16 in an 85:15 ratio (Table 1, entry 9). Despite the side product, the conversion to 15/16 was quantitative and isolated yields of 15 exceeded 82%. Application of the same MAOS conditions, but replacement of HOAc with 5% 4 N HCl in dioxane, afforded 100% conversion to 15 in 95% isolated yield without producing 16 (Table 1, entry 13). Thus, a reaction that previously required up to 60 h of conventional heating to provide <50% yield,3–5 now afforded 95% yield of the desired product 15 in 10 min by virtue of MAOS—a 360-fold reduction in reaction time.13
Table 1.
Optimization of MAOS conditions to produce 15
 | ||||
|---|---|---|---|---|
| Entry | T (°C) | Solvent | Time (min) | 15:16a |
| 1 | 140 | HOAc | 10 | 42:58 |
| 2 | 160 | HOAc | 10 | 28:72 |
| 3 | 180 | HOAc | 10 | 24:76 |
| 4 | 200 | HOAc | 10 | 13:87 |
| 5 | 150 | 50% HOAc/EtOH | 10 | 78:22 |
| 6 | 170 | 50% HOAc/EtOH | 10 | 64:36 |
| 7 | 150 | 10% HOAc/EtOH | 10 | 79:21 |
| 8 | 170 | 10% HOAc/EtOH | 10 | 74:26 |
| 9 | 150 | 5% HOAc/EtOH | 10 | 85:15 |
| 10 | 170 | 5% HOAc/EtOH | 10 | 77:23 |
| 11 | 135 | 5% HOAc/EtOH | 20 | 80:20 |
| 12 | 135 | EtOHb | 10 | – |
| 13 | 150 | 5% HCI/EtOHc | 10 | 100:0 |
Ratio determined by analytical LC-MS and 1H NMR; conversion >95%.
No product formed without acid catalysis.
5% 4 N HCl/dioxane.
Attention was now directed at the application of these new MAOS conditions to a diverse array of acylhydrazides to ensure that this new protocol would indeed be general. As shown in Table 2, the MAOS protocol, employing either catalytic HOAc (Method A) or HCl (Method B), proved to be general with respect to a wide range of electron-rich (entry 13g), electron-deficient (entry 13f), and hindered acylhydrazides (entry 13a) 17 as well as heterocyclic congeners (entries 13h, 13i) affording the desired 3-aryl/heteroaryl-6-chloro-[1,2,4]triazolo[4,3-b]pyridazines 13 in isolated yields ranging from 74% to 97% in 10 min at 150 °C.
Table 2.
Generality of the MAOS protocol to deliver analogs 13
 | |||
|---|---|---|---|
| Compound | R | Yielda (%) | Yieldb (%) |
| 13a |
|
79 | 92 |
| 13b |
|
77 | 93 |
| 13c |
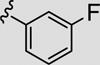
|
81 | 91 |
| 13d |
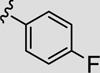
|
87 | 96 |
| 13e |
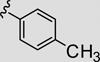
|
80 | 95 |
| 13f |
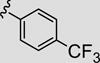
|
75 | 97 |
| 13g |
|
74 | 96 |
| 13h |
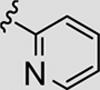
|
70 | 88 |
| 13i |
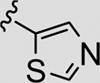
|
72 | 86 |
5% HOAc/EtOH, remaining mass balance congeners or 16.
5%4N HCI/dioxane. All yields for isolated, analytically pure materials.
Developing a general MAOS-mediated SNAR protocol for the reaction of diverse amines with analogs 13 to deliver 3-aryl, 6-amino-[1,2,4]triazolo[4,3-b]pyridazines 1 proved more difficult. Nucleophilic amines (benzyl, aliphatic, piperidines, and piperazines) reacted smoothly in EtOH at 170 °C for 10 min to produce analogs 1 in yields ranging from 73% to 92% (Scheme 2). Less nucleophilic amines, such as anilines, required K2CO3 in DMF with micorwave irradiation for 15 min at 180 °C to produce analogs 1 in yields exceeding 65%.14 Furthermore, analogs 13 readily participated in general microwave-assisted Sonogashira and Suzuki couplings to afford analogs 18 and 19 in yields exceeding 80% in every case examined (Scheme 2).
Scheme 2.

MAOS protocols to functionalize the 3-aryl-6-chloro-[1,2,4]triazolo[4,3-b]pyridazine 13.
Utilizing these new MAOS protocols, we resynthesized the M1 versus M4 selective antagonist HTS hit 2 (Scheme 3). Beginning with 15, delivered in 95% yield (Table 1), an SNAr reaction with Boc-piperazine provided 20, which was then deprotected using 1:1 TFA:DCM to afford 21 in 80% yield for the two steps. 21 was then acylated employing standard polymer-supported reagents and scavengers to generate the original HTS hit 2 in 70% yield.15 Evaluation of 2 against M1–M5 indicated that 2 was indeed a selective M1 antagonist (M1 IC50 = 23 μM, M2–M5 IC50 >> 50 μM). Prior to this discovery there was only one other M1 selective small molecule antagonist,16 and prior to its discovery, the only M1 selective antagonist was MT7, a 71 amino acid peptide toxin from the green mamba snake.17 Encouraged by this result, we employed an iterative parallel synthesis approach, employing our new MAOS protocols, to rapidly develop structure–activity relationships in an attempt to improve the M1 antagonist potency while maintaining selectivity for M2–M5.
Scheme 3.

Application of MAOS protocols for the resynthesis of the M1 antagonist HTS hit 2.
As shown in Figure 3, we simultaneously varied the substituents at the C-3 and C-6 positions, synthesizing small 12- to 24-member libraries employing the synthetic routes depicted in Schemes 2 and 3. Analogs of 2 were triaged in a single point 10 μM screen for the compound's ability to decrease an EC80 concentration of acetylcholine.
Figure 3.

Synthetic plan to optimize 2 for M1 antagonist potency, while maintaining selectivity versus M2–M5.
SAR for this series was rather ‘flat’, with subtle changes leading to a complete loss of M1 inhibitory activity. Out of ~60 analogs, only four demonstrated significant M1 antagonism; however, we managed to improve upon HTS hit 2. As shown in Figure 4, exploration of the C3 position identified both the 3-OMe phenyl derivative 22 and the 4-Me phenyl congener 23 as engendering more potency (M1 IC50 = 3.59 μM and 4.09 μM, respectively), while maintaining selectivity (M2–M5 IC50 >> 50 μM). When holding the 3-OMe phenyl moiety constant at C3 and exploring alternatively functionalized piperazines for the bromofuranoic amide at C6, we identified two piperazinyl piperazine analogs, 24 and 25, which maintained M1 antagonism (M1 IC50 = 3.99 μM and 6.64 μM, respectively) and selectivity (M2–M5 IC50 >> 50 μM). Moreover, these latter analogs, with basic amines, afforded improved solubility and physiochemical characteristics.
Figure 4.

Optimized analogs of 2 as highly selective M1 antagonists with improved M1 inhibitory activity as compared to HTS hit 2.
In summary, we have applied MAOS to the preparation of 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazines 1, and developed general and high-yeilding protocols with over a 360-fold acceleration in reaction rate. For both the heterocyclic synthesis and the subsequent SNAr steps, reaction time, yield, and overall reaction generality were dramatically improved under these MAOS protocols; more importantly, these new protocols allow for an iterative analog library synthesis approach for lead optimization to be employed for the rapid synthesis of large numbers of analogs of 1. Employing these new MAOS protocols, a lead optimization campaign centered on the selective, but weak M1 antagonist hit 2 (M1 IC50 = 23 μM) delivered two analogs, 22 and 24, with over a 6-fold increase in M1 inhibitory activity (M1 IC50 = 3.99 μM and 6.64 μM, respectively) while maintaining selectivity versus M2– M5 (IC50 >> 50 μM). These compounds represent a novel chemo-type of selective, small molecule M1 antagonists, and hold promise as leads for potential new therapeutic agents for Parkinson's Disease and dystonia.
Acknowledgments
The authors warmly thank Department of Pharmacology and the NIH/MLSCN (3U54 MH074427-02S1, Chemistry Supplement) for support of this research. Vanderbilt is a member of the production phase of the MLCSN, termed the MLPCN, and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development (1U54MH084659-01).
References and notes
- 1.For information on the Molecular Library Screening Center Network (MLSCN) see: http://nihroadmap.nih.gov/molecularlibraries.
- 2.Druey J, Ringler BH. Helv. Chim. Acta. 1951;34:195. [Google Scholar]
- 3.Carling RW, Moore KW, Street LJ, Wild D, Isted C, Leeson PD, Thomas S, O'Connor D, McKernan RM, Quirk K, Cook SM, Atack JR, Wafford KA, Thompson SA, Dawson GR, Ferris P, Castro JL. J. Med. Chem. 2004;47:1807–1822. doi: 10.1021/jm031020p. [DOI] [PubMed] [Google Scholar]
- 4.Cox JM, Harper B, Mastracchio A, Leiting B, Roy RS, Patel RA, Wu JK, Lyons KA, He H, Xu S, Zhu B, Thornberry NA, Weber AE, Edmondson SD. Bioorg. Med. Chem. Lett. 2007;17:4579–4583. doi: 10.1016/j.bmcl.2007.05.087. [DOI] [PubMed] [Google Scholar]
- 5.Tarzia G, Occelli E, Toja E, Barone D, Corsico N, Gallico L, Luzzani F. J. Med. Chem. 1988;31:1115–1123. doi: 10.1021/jm00401a010. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Z, Leister WH, Strauss KA, Wisnoski DD, Lindsley CW. Tetrahedron Lett. 2003;44:1123–1127. [Google Scholar]
- 7.Wolkenberg SE, Wisnoski DD, Leister WH, Zhao Z, Wang Y, Lindsley CW. Org. Lett. 2004;6:1453–1456. doi: 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Z, Wisnoski DD, Wolkenberg SE, Leister W, Wang Y, Lindsley CW. Tetrahedron Lett. 2004;45:4873–4876. doi: 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]
- 9.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Bioorg. Med. Chem. Lett. 2005;15 doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]
- 10.Nolt MB, Smiley MA, Varga SL, McClain RT, Wolkenberg SE, Lindsley CW. Tetrahedron. 2006;62:4698–4704. [Google Scholar]
- 11.Shipe WD, Yang F, Zhao Z, Wolkenberg SE, Nolt MB, Lindsley CW. Heterocycles. 2006;70:665–689. [Google Scholar]
- 12.Daniels RN, Kim K, Hughes MA, Lebois EP, Muchalski H, Lindsley CW. Tetrahedron Lett. 2008;49:305–310. [Google Scholar]
- 13.Typical MAOS experimental for 6-chloro-3-p-tolyl-[1,2,4]triazolo[4,3-b]-pyridazine (13e). (Method A) To a 5 mL microwave reaction vessel were added 3,6-dichloropyridazine (100 mg, 0.671 mmol) and p-toluic hydrazide (111 mg, 0.738 mmol) in a 3 mL solution of 5% AcOH/EtOH. The vial was irradiated in a microwave synthesizer at 150 °C for 10 min. LC–MS (single peak, 2.91 min, m/e, 245.1 (M+1)) indicated that all starting material had been consumed affording 131 mg (80%) of 6-chloro-3-p-tolyl-[1,2,4]triazolo[4,3-b]-pyridazine as a white solid following column purification. (Method B) To a 5 mL microwave reaction vessel were added 3,6-dichloropyridazine (100 mg, 0.671 mmol) and p-toluic hydrazide (111 mg, 0.738 mmol) in a 3 mL solution of 5% 4 N HCl/EtOH. The vial was irradiated in a microwave synthesizer at 150 °C for 10 min. LC–MS (single peak, 2.91 min, m/e, 245.1 (M+1)) indicated that all starting material had been consumed affording 156 mg (95%) of 6-chloro-3-p-tolyl-[1,2,4]triazolo[4,3-b]pyridazine as a white solid following a silica plug and concentration in vacuo. 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 2.41 (s, 3H), 7.43 (d, J = 8 Hz, 2H), 7.53 (d, J = 9.7 Hz, 1H), 8.19 (d, J = 8.2 Hz, 2H), 8.52 (d, J = 9.7 Hz, 1H); 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 21.1, 122.5, 122.8, 127.1, 127.3, 129.5, 140.3, 143.8, 146.8, 149.1; LC–MS: single peak, 2.91 min, m/e, 245.1 (M+1).
- 14.Typical MAOS experimental for N-(4-methoxybenzyl)-3-p-tolyl-[1,2,4]triazolo[4,3-b]pyridazin-6-amine. To a 5 mL microwave reaction vessel were added 6-chloro-3-p-tolyl- [1,2,4]triazolo[4,3-b]pyridazine (50 mg, 0.205 mmol) and 4-methoxy-benzyl amine (35 μL 6 mmol) in 3 ml of ethanol. The vial was initially heated in a microwave synthesizer to 170 °C for 25 min. Preparative LC–MS afforded 51.6 mg (73%) of N-(4-methoxybenzyl)-3-p-tolyl-[1,2,4]triazolo[4,3- b]pyridazin-6-amine as a white solid. 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 2.38 (s, 3H), 3.71 (s, 3H), 4.41 (d, J = 5.5 Hz, 2H), 6.88 (d, J = 9.9 Hz, 1H), 6.92 (d, J = 8.6 Hz, 2H), 7.33 (d, J = 6.7 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 7.97 (d, J = 9.8 Hz, 1H), 8.21 (d, J = 8.2 Hz, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 21.5, 44.7, 55.5, 114.2, 117.0, 124.4, 124.6, 127.0, 129.3, 129.6, 130.9, 139.4, 143.8, 146.3, 154.1, 158.8; LC–MS: single peak, 3.00 min, m/e, 346.2 (M+1).
- 15.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J. Comb. Chem. 2008;10:345–358. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 16.Lewis LM, Sheffler D, Williams R, Bridges TA, Kennedy JP, Brogan JT, Mulder MJ, Williams L, Nalywajko NT, Niswender C, Weaver CD, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:885. doi: 10.1016/j.bmcl.2007.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradley KN. Pharmacol. Ther. 2000;85:87–109. doi: 10.1016/s0163-7258(99)00064-9. [DOI] [PubMed] [Google Scholar]