

Abstract
Mitochondria are the powerhouses of eukaryotic cells as they feed metabolism with its major substrate. Oxidative-phosphorylation relies on the generation, by an electron/proton transfer chain, of an electrochemical transmembrane potential utilized to synthesize ATP. Although these fundamental principles are not a matter of debate, the emerging picture of the respiratory chain diverges from the linear and fluid scheme. Indeed, a growing number of pieces of evidence point to membrane compartments that possibly restrict the diffusion of electron carriers, and to supramolecular assembly of various complexes within various kinds of supercomplexes that modulate the thermodynamic and kinetic properties of the components of the chain. Here, we describe a method that allows the unprecedented time-resolved study of the respiratory chain in intact cells that is aimed at assessing these hypotheses. We show that, in yeast, cytochrome c is not trapped within supercomplexes and encounters no particular restriction to its diffusion which questions the functional relevance of these supramolecular edifices.
Keywords: bioenergetics, electon transfer, respiration, compartmentalization
Since the discovery of cytochrome by Keilin (1), followed by the elucidation of the sequence of events leading to oxidative-phosphorylation (2) and by the identification of the enzymatic complexes involved in the process (3, 4), the structural and functional organization of the respiratory electron transfer chain (ETC) has been a matter of lively debate and ongoing studies. The first model was a rigid one involving sequential redox reactions. Later, Green and Tzagaloff argued that, on the contrary, ubiquinone and cytochrome c act as mobile electron carriers between immobile membrane-bound complexes (5). In Green and Tzagaloff’s model, supported by Kröger and Klingenberg (6, 7), the respiratory chain is comprised of membrane-bound complexes and electron shuttles that are randomly distributed within the inner membrane and intermembrane space of mitochondria. Extensive studies by Hackenbrock and colleagues of the diffusion properties of the different actors of the respiratory chain, in particular of cytochrome c, brought compelling evidences for a random collision model (reviewed in ref. 8). Rich reviewed these issues and listed four different models ranging from “liquid” to “solid” state, the intermediate cases comprising the partial association of the individual components, or “patches” containing higher concentration of one or more cofactors (9). Among these, Rich favored the “free quinone diffusion and interaction by collisions” model.
Strikingly, the emergence of mild isolation techniques of membrane components, together with flux-control analysis studies, led to another reversal of paradigm. The characterization by Schägger and Pfeiffer of “supercomplexes” containing cytochrome bc1 and cytochrome c oxidase (CcOx), in various stoichiometric ratios (10), fostered the revival of the idea that the functional properties of the respiratory chain are shaped by its ultrastructural organization. Since then, many supercomplex assemblies from a wide variety of organisms or organs have been biochemically characterized, with a large diversity of stoichiometries and complex compositions [see e.g., the recent exhaustive review by Lenaz and Genova (11)]. This culminated in the isolation by Acin-Pérez, et al. of a functional “respirasome” from mammal cells, transferring electrons all the way from NADH or succinate to molecular oxygen (12). In addition, the composition and abundance of the biochemically characterized supercomplexes have been shown to vary with growth or physiological conditions (13, 14), supporting their physiological significance. However, parallel studies on whole cells aimed at understanding the dynamics of the supercomplexes and their functional relevance are lacking. In addition, the question as to whether mobile electron carriers (quinone and cytochrome c) are indeed trapped within assemblies defining “diffusion channels” still remains mainly unanswered (but see ref. 15).
In the photosynthetic counterpart of the respiratory electron transfer chain, functional studies underlined the existence of supercomplexes (16, 17) on the one hand and of domains that restrict the diffusion of mobile electron carriers, on the other hand (16, 18, 19). The purification of a functionally competent cytochrome b6f/Photosystem I supercomplex (20) is one such example. Also, Photosystems I and II segregate in different zones of the thylakoid membrane, the latter being found in the stacked regions and the former in the nonappressed regions (21, 22). This mere observation provides ultrastructural support for the idea that the thylakoid membrane consists of juxtaposed domains with distinct functions.
In contrast to light (the substrate of the photosynthetic ETC), neither NADH nor oxygen (the electron donor and acceptor of the respiratory ETC, respectively) can be delivered as a rapid pulse and this precludes the time-resolved characterization of the respiratory ETC. To probe the function of Complex IV (Cytochrome c Oxidase, hereafter CcOx), Gibson and Greenwood designed a method allowing its activation by light and hence its time-resolved analysis (23). This technique is based on the reversible and competitive inhibition of reduced CcOx by carbon monoxide (CO): CO binds to reduced heme a3, thus restraining O2 accessibility, but can be photo-dissociated, allowing fast O2 binding. This approach has been fruitfully applied to a variety of terminal oxidases and has provided detailed insights into the electron pathway and proton pumping mechanism associated with CcOx turnover [see e.g., (24–29)].
Here we have scaled up this approach to follow, in a time-resolved manner and in intact cells, the sequence of electron transfer not only within CcOx but along the entire respiratory ETC. We have characterized the kinetics of oxidation of cytochrome c in intact Saccharomyces cerevisiae cells as well as its diffusion characteristics. We observed a fast oxidation of cytochrome c consistent with its prebinding to CcOx in both the wild-type and a cardiolipin-lacking mutant of the W303 (W303-1B) strain, although the bc1-CcOx supercomplex stability is notably decreased in the latter (30). This fast oxidation component was absent in the BY (BY4742) strain, indicative of a decreased affinity of cytochrome c to CcOx.
In addition, we show that the oxidation of cytochrome c by a given CcOx is a random process. Thus the intricate topology of the inner membrane does not create domains that limit significantly the diffusion of the soluble cytochrome c electron carrier. Despite the considerable amount of evidence supporting the existence of supercomplexes in the respiratory chain of yeast, the present results question their functional relevance, and the approach described here provides an unprecedented tool to address this issue in vivo as it can be, in principle, extended to other cellular systems.
Results
Carbon Monoxide Inhibited Conditions.
We first validated the method and assessed its feasibility by studying flash-induced absorption changes within intact cells in the presence of CO and absence of O2. As shown in Fig. 1A we observed the characteristic derivative-shaped spectrum reflecting the photo-dissociation of CO and the resulting band shift of the Soret band of the reduced heme a3 from 430 nm to 445 nm (31, 32). We note however that this difference spectrum was slightly red-shifted (by c.a. 2 nm) with respect to that of bovine CcOx. This slight red-shift may stem from the sample being strongly scattering (33) or from differences between species. The CO rebinding kinetics were monitored at 445 nm at various concentrations of CO, yielding a bimolecular rate constant of ca. 5.4 104 M-1 s-1 in the W303 strain (Fig. 1B), in good agreement with that found with purified CcOx (23, 34, 35). In addition we studied the dependence of the amplitude of the flash-induced absorption change at 445 nm on the energy of the flash. As expected, this amplitude followed a monoexponential saturation profile which can be used to assess the relative amount of photo-dissociated CcOx (see Fig. S1).
Fig. 1.

Photolysis of CO in whole cells. (A) Spectrum of CO photolysis from heme a3 in a suspension of yeast (W303) cells (open symbols) under a pure CO atmosphere. A spectrum from the literature (32), red-shifted by two nanometers, is shown for comparison (solid line). (B) Kinetics of CO rebinding to heme a3 after photolysis, at PCO = 1 atm (closed symbols) and PCO = 0.5 atm/PAr = 0.5 atm (open symbols), and monoexponential fits (solid lines). Plots and linear fits of the apparent rate constants against [CO] are shown in the inset.
Light-Activation of the Respiratory Chain in the Presence of Carbon Monoxide and Oxygen.
We proceeded to light-trigger the respiratory chain by addition of oxygen to the gaseous phase in equilibrium with the cell suspension. Fig. 2 shows the spectra of the light-induced absorption changes at various times after a flash applied to a suspension of cells equilibrated with 94% CO and 6% O2. At early times (20 μs) the spectrum was identical in shape and amplitude to that shown in Fig. 1A. Hence, at this oxygen concentration, the state of CcOx prior to photoactivation was identical to that observed in the absence of oxygen. The absorption changes then displayed the characteristic spectroscopic signature of oxidation of hemes a/a3, seen as the troughs at 445 and 605 nm that are observed in similar experiments with purified CcOx. Interestingly, we also observed bleaching at 420 nm, 520 nm, and 550 nm that is characteristic of the oxidation of a c-type heme (either soluble cytochrome c or heme c1 from cytochrome bc1), showing that intercomplex electron transfer indeed occurred. In addition, a trough developed at 434 nm and 560 nm that indicated oxidation of a b-type heme and thus the turnover of cytochrome bc1 complex. An absorption increase around 470 nm also developed at longer times, assigned by Chance to the oxidation of flavoprotein-like compounds (36). This absorption increase can be assigned either to the succinate:ubiquinol oxidoreductase (37) or to the internal (38) and external NADH:ubiquinol oxidoreductases (39) all of which have a FAD prosthetic group. In any case, these absorption changes were lost on addition of cytochrome bc1 inhibitors, as expected if they arose from turnover of complexes located upstream of cytochrome bc1 with respect to CcOx (see Fig. S2).
Fig. 2.

Monitoring by visible spectrophotometry the photoactivated reactions of the respiratory chain with oxygen. Spectra of whole yeast (W303) cells at various times after CO photolysis, under an atmosphere of PCO = 0.94 and PO2 = 0.06. The excitation light was switched from 605 to 430 nm in order to explore the red part of the spectrum (see Materials and Methods). Notable spectrophotometric signals include: CO photolysis from CcOx (see Fig. 1A), oxidation of CcOx (troughs at 445 nm and 605 nm), oxidation of cytochrome c (troughs at 420 nm, 520 nm, and 550 nm), oxidation of flavoproteins (band in the 440–500 nm region), and oxidation of b-type hemes (slight troughs at 430 nm and 560 nm, seen at longer times).
Extensive studies of light-induced oxidation of purified CcOx have led to the characterization of the sequence of complete oxidation starting from the binding of O2 to the fully reduced enzyme. Briefly, and without entering into the mechanistic details which are accurately documented (24–29), at 1 mM [O2], oxygen binds in about 8–12 μs and this triggers the electron and proton transfer sequence. Hemes a and a3 are oxidized in ∼30–40 μs and the formation of the ferryl (F) state follows in ∼100 μs. The overall catalytic process then proceeds through a ∼1–2 ms component which is kinetically limited by proton transfer. Because the triggering event; i.e., oxygen binding, is a second order reaction, its rate is expected to depend on the concentration of oxygen. We thus studied the dependence upon [O2] of the transient absorption changes reflecting the catalytic cycle of CcOx. As shown in Fig. 3 A and B, we observed multiphasic kinetics. By fitting these kinetics with two exponentials, a fast component with a rate constant depending linearly on [O2] was observed. Bimolecular rate constants of 6.8 107 M-1 s-1 and 9.5 107 M-1 s-1 in the W303 and BY strains, respectively were obtained (Fig. 3C), in good agreement with previously published data (23, 34, 40, 41). This binding thus occurs in the submillisecond time range when [O2] is in the 80–160 μM range, fast enough to reveal the slower component with a maximal rate of 400 s-1, in good agreement with the reported rate of the slow, proton transfer limited, components discussed above. Notably, further increase in [O2] resulted in the preoxidation of CcOx that preoxidation likely stems from the spontaneous dark dissociation of CO from the reduced heme a3 which, even though it is a slow process [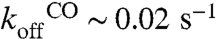 (35)], allows the binding of oxygen in the dark and the subsequent oxidation of a fraction of CcOx, particularly under relatively high [O2]. Importantly, the amplitude of the flash-induced absorption changes at 445 nm, which reflects the photo-dissociation of CO from the reduced heme a3 , allow the assessment of the state of the system prior to photolysis. On this basis we restricted the [O2] limit to 160 μM, a concentration at which oxygen binding occurs with a rate constant of 10,700 s-1 which is faster than the slowest, proton transfer limited, catalytic step of CcOx but in the same time range as formation of the F state. This limited [O2] range likely accounts for the fact that the transient absorption changes reported here did not display the same wealth of kinetic components as those obtained with purified enzyme at higher oxygen concentration (see refs. 42 and 27 for a recent review), and that two kinetic components were adequate to fit these transients.
(35)], allows the binding of oxygen in the dark and the subsequent oxidation of a fraction of CcOx, particularly under relatively high [O2]. Importantly, the amplitude of the flash-induced absorption changes at 445 nm, which reflects the photo-dissociation of CO from the reduced heme a3 , allow the assessment of the state of the system prior to photolysis. On this basis we restricted the [O2] limit to 160 μM, a concentration at which oxygen binding occurs with a rate constant of 10,700 s-1 which is faster than the slowest, proton transfer limited, catalytic step of CcOx but in the same time range as formation of the F state. This limited [O2] range likely accounts for the fact that the transient absorption changes reported here did not display the same wealth of kinetic components as those obtained with purified enzyme at higher oxygen concentration (see refs. 42 and 27 for a recent review), and that two kinetic components were adequate to fit these transients.
Fig. 3.

Oxidation kinetics of CcOx under various oxygen concentrations. (A) Kinetics of CcOx oxidation in the W303 strain at different [O2], monitored at 445 nm, with correction for contributions from flavoproteins and cytochrome c (see Materials and Methods). Solid lines in (A and B) are biexponential fits of the deconvoluted kinetics in the W303 strain. (B) Kinetics of CcOx oxidation in the BY strain at various [O2]. (C) Plots and linear fits of the apparent rate constants of the fast phase of CcOx oxidation against [O2] for the W303 (orange symbols) and BY (blue symbols) strains.
Time-Resolution of Cytochrome c Oxidation.
Despite the kinetic limitation resulting from the limited [O2] which could be used, the possibility to characterize the kinetics of cytochrome c oxidation at 551 nm provides a powerful means to assess the degree of binding to its membrane-bound electron acceptor. Indeed, electron transfer from reduced, bound cytochrome c to oxidized CuA of CcOx has been shown to occur in vitro with rates ranging from 7 103 to 6 104 s-1 (42, 43). As another example, in bacterial caa3-type oxidases which contain a c-type cytochrome subunit, the oxidation of heme c following photoactivation develops in the 30 to 240 μs range (44–46). Thus, a stable complex with a prebound cytochrome c should be evident as a fast 551 nm kinetic component. Fig. 4 shows the transient absorption changes at 551 nm at several [O2] with the W303 (Fig. 4A) and BY (Fig. 4B) strains. Using W303, the kinetics were monophasic at low [O2] and displayed multiple kinetic components as [O2] was increased. The rate of the fastest component was ca. 3.6 103 s-1 at the highest [O2] (Fig. 4C). In the BY case, different results were obtained. Indeed, the maximum cytochrome c oxidation rate saturated to reach 200 s-1 at high [O2]. Notably, such [O2] were high enough to promote a 10-fold faster oxidation of hemes a/a3 (compare e.g., the blue traces in Figs. 4B and 3B), showing that the sluggishness of the cytochrome c oxidation does not stem from a kinetic limitation within CcOx.
Fig. 4.

Kinetics of cytochrome c oxidation at different [O2], monitored at 551 nm. (A) W303 strain. (B) BY strain. (C) kinetics for [O2] = 156 μM of cytochrome c oxidation in the W303 strain, with monoexponential (gray line) and biexponential fits (orange line) of the data; the corresponding residuals are shown below.
Assessing the Restriction to Diffusion of Cytochrome c.
Rather than attempting to time-resolve the sequential oxidation of CcOx and cytochrome c, we then aimed at probing the size of the cytochrome c pool oxidizable by turnover of one CcOx. The rationale behind this aim was to probe possible restrictions to the interaction between the membrane-bound enzyme and its pool of soluble electron donors. Such restrictions may be a consequence of the intricate topology of the mitochondrial inner membrane or of the supramolecular association of the respiratory complexes. To do so, the influx of electrons into the cytochrome c pool needs to be blocked, thereby allowing the assessment of the entire fraction of cytochrome c accessible to the CcOx-mediated oxidation. CO binds to reduced heme a3 and may spontaneously dissociate (see above). Thus cytochrome bc1 inhibiting conditions which significantly slow down the reduction of cytochrome c and, consequently, the reduction of CcOx, required that oxygen concentration was kept low enough to make oxygen binding a poor competitor with CO binding. In these conditions, a single laser flash did not lead to the complete oxidation of the cytochrome c pool, despite the slow reduction of cytochrome c (Fig. 5). This limited oxidation yield may result either from a limited access to the cytochrome c pool which would stem from constraints on the diffusion of cytochrome c, or from a limited number of turnovers sustained by the photo-dissociated CcOx owing to the low amount of CcOx that binds O2 and performs turnover after photoactivation. We favor the latter hypothesis. Indeed, the fraction of cytochrome c oxidized after the first flash of the series decreased with [O2], while the total amplitude (reached after several flashes) remained the same (Fig. S3), suggesting that the oxidation yield is limited by O2 binding.
Fig. 5.

Progressive oxidation of cytochrome c by a series of laser flashes after inhibition of cytochrome bc1. Relative amount of cytochrome c oxidized on a series of flashes at different laser intensities (ΦCO), monitored at 470 nm (see Materials and Methods) in the W303 strain. Gray dashed lines are fits of the data with a geometrical probability law, constraining the value of α = 0.08 (see text).
This possible limitation can be estimated quantitatively: in the absence of O2, the decay of the light-induced absorption at 445 nm yields the pseudofirst order rate constant, kCO, for the rebinding of CO (Fig. 1B). When measured in the presence of oxygen this decay yields the overall rate constant reflecting O2 and CO binding; i.e., the sum of the individual pseudofirst order rate constants of the two processes, (kCO + kO2). We obtained 53 and 80 s-1 for kCO and kCO + kO2, respectively so that kO2 = 27 s-1. The probability for a photoactivated CcOx to bind O2 rather than CO is thus 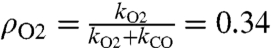 . Once photoactivated, the 34% fraction of CcOx having bound O2 may undergo multiple turnovers as the competition between CO and O2 continues until CO binds to the reduced heme a3. These successive turnovers of one photooxidized CcOx thus constitute a series of independent events where O2 competes with CO for binding, with the probability ρO2. This series corresponds to a sequential repeat of independent Bernoulli trials; i.e., an experiment with only two possible random outcomes, here O2 or CO binding. Quantitatively, the sequence is as follows: 34% of the photoactivated CcOx bind O2 rather than CO, ∼12% (0.342) undergo a second turnover, ∼4% (0.343) a third one etc. On average, each photooxidized CcOx thus undergoes slightly more than a single-turnover, in the conditions of the experiments.
. Once photoactivated, the 34% fraction of CcOx having bound O2 may undergo multiple turnovers as the competition between CO and O2 continues until CO binds to the reduced heme a3. These successive turnovers of one photooxidized CcOx thus constitute a series of independent events where O2 competes with CO for binding, with the probability ρO2. This series corresponds to a sequential repeat of independent Bernoulli trials; i.e., an experiment with only two possible random outcomes, here O2 or CO binding. Quantitatively, the sequence is as follows: 34% of the photoactivated CcOx bind O2 rather than CO, ∼12% (0.342) undergo a second turnover, ∼4% (0.343) a third one etc. On average, each photooxidized CcOx thus undergoes slightly more than a single-turnover, in the conditions of the experiments.
By using such conditions where a limited fraction of cytochrome c is oxidized by the light-induced turnovers of CcOx, we can address the issue as to whether the probability to oxidize a given cytochrome c is independent of the redox state of the other cytochromes c in the pool, as expected in a fully stochastic system with no diffusion constraints. Alternatively this probability may evolve as the cytochrome c pool gets progressively oxidized and would reflect a wide distribution in the size of the cytochrome c pool accessible to the light-induced oxidation burst. Fig. 5 shows the progressive cytochrome c oxidation in response to a series of flashes at various intensities. The lines shown in Fig. 5 are fits of the data with a geometric probability law, indicative of the homogeneous, pool-like relationship between cytochrome c and CcOx.
Discussion
Photoactivation of CcOx has been most fruitfully carried out to characterize in detail its electron and proton transfer reactions. The present study extends this approach to the entire respiratory chain in intact cells. Although, as mentioned above, the equilibration of the sample with the gaseous phase together with the spontaneous dissociation of CO in the dark limits the range of [O2] which can be used and thus limits the time resolution of the technique, we successfully time-resolved the sequential electron transfer along the entire chain starting from hemes a/a3 to the NADH: and succinate: dehydrogenases via cytochrome bc1 complex (Fig. 1B). This methodological extension might pave the way to the functional analysis of the different complexes in their native environments, and possibly to the study of their regulation in response to physiological changes. However, stimulated by the growing evidence for the existence of supramolecular complexes formed from several components, or perhaps even from the whole respiratory electron transfer chain, we first aimed at characterizing the functional consequences of the supramolecular organization of the respiratory chain. Supercomplexes have been found in a wide variety of organisms ranging from bacteria and unicellular eukaryotes to multicellular organisms including fungi, higher plants, and mammals (11). In addition, the abundance and/or stability of these supramolecular edifices have been shown to respond to physiological changes (13, 14), thus pointing to their physiological relevance.
Yet, to our knowledge, functional data supporting these views are scarce. Respirasomes; i.e., assemblies comprising the entire electron transfer chain, have been isolated and shown to be able to promote electron transfer from NADH to oxygen (12), providing evidence that such a supramolecular structure can sustain electron transfer. Also, Boumans, et al. concluded that, in isolated yeast mitochondria, the ETC is a “single functional unit” as neither cytochromes c nor ubiquinones showed the characteristics expected for a pool, at least in low phosphate conditions (15). However, experimental support for a role of supercomplexes in situ; i.e., in intact cells, is still lacking. According to current views, the purpose, if any, of such supramolecular assemblies is to shuttle the electron so as to avoid possible kinetic limitations which can arise from diffusion controlled processes. To be functionally relevant these assemblies thus require the mobile electron carriers to be trapped within supercomplexes that are formed from membrane-bound enzymes. In principle, diffusion-limited electron transfer reactions can be distinguished from electron transfer reactions between two bound complexes kinetically, as illustrated in the photosynthetic chain by the kinetic characteristics of electron transfer from plastocyanin to Photosystem I (47).
Interaction Between Soluble Cytochrome c and Cytochrome c Oxidase.
In the W303 strain, the overall oxidation rate of cytochrome c increased with [O2] (Fig. 3A). Thus, in the [O2] range used, the oxidation of at least a fraction of the cytochrome c pool is rate-limited by the binding of oxygen to CcOx. Fig. 4C shows the oxidation kinetics of cytochrome c at the highest [O2] studied here. The quality of the fit was clearly improved when using two exponentials rather than one, pointing to the occurrence of distinct, kinetically limiting, steps for the fast and slow components as the diffusion-limited oxidation of cytochrome c has been shown to be a pseudofirst order process (43, 48). This multiphasic behavior thus discloses at least two different types of cytochrome c, the fastest fraction being oxidized with a rate constant of ∼3.6 103 s-1 (time constant ∼280 μs). Although slower than the rate of O2 binding to the CcOx at this [O2] (104 s-1), we assign this fast oxidation to electron transfer from prebound cytochrome c to oxidized CcOx. Indeed, time-resolved studies of the oxidation of cytochrome c in caa3-type oxidases have shown that this reaction is in fact a multiphasic process. Although the first phase of cytochrome c oxidation can be as fast as 3–6 104 s-1, the major kinetic component occurs with a rate constant in the 1 to 4 103 s-1 time range (44, 46). The value found here for the fastest step in the oxidation of cytochrome c is thus similar to the caa3 case where cytochrome c is an integral subunit of the oxidase and thus bound to its electron acceptor. Notably, the relative amplitude of this fast component which, in the present framework, reflects the fraction of prebound cytochrome c, accounts for 16% of the entire cytochrome c, so that, with a stoichiometry of 4–5 cytochrome c per CcOx, a large majority of CcOx would have a prebound cytochrome c. In the Δcrd1 strain, impaired in the cardiolipin biosynthesis pathway and having a much lower abundance and stability of the cytochrome bc1—CcOx supercomplex (30, 49, 50), the cytochrome c oxidation kinetics were similar to those observed in the WT (Wild-type) (Fig. S4). However, the amplitude of the fast component was slightly lower, decreasing from 16% of the total amplitude in the WT to 12 % in the Δcrd1, showing that the lack of cardiolipin hardly affects the association of CcOx and cytochrome c.
The Kinetics of Electron Transfer in the BY Strain.
The other yeast strain used in this study, BY, derived from the S288c strain, which bears a mutation in the HAP1 gene (51). HAP1 encodes a transcription factor involved, on the one hand, in the expression of aerobic genes in response to [O2] increase, and, on the other hand, in the activation of ROX1, a gene involved in the down-regulation of anaerobic genes under normoxic conditions. Interestingly, completely different results were obtained with this strain. The kinetics of cytochrome c oxidation, under the same conditions as for W303, were independent of [O2], and strictly monoexponential with a rate constant of 150 s-1. This slow cytochrome c oxidation is all the more striking as the binding rate constant of O2 to heme a3 was found to be slightly higher in the BY strain, 1 104 s-1 at [O2] = 130 μM. The millisecond kinetic component of CcOx oxidation (also found in W303), with a rate constant of 250 s-1, was also faster than the cytochrome c oxidation. Thus, in this case, the fraction of prebound cytochrome c must be too low to detect and the oxidation of cytochrome c is kinetically limited by a process which is slower than the slowest reactions occurring within CcOx.
Both the increased O2 binding to CcOx and the kinetically limiting binding of cytochrome c, likely stem from the genetic background of the BY strain. Among the various consequences of the hap1 mutation, the decreased accumulation of cytochrome c (as confirmed by the absorption spectra of whole cells, see Fig. S5), and the enhanced expression of iso-2-cytochrome c, and of CcOx subunit Vb (52) are particularly relevant here. CcOx containing the isoform Vb has been reported to have a larger maximal turnover rate (53) than CcOx with isoform Va. This finding is in line with our observations of a significantly higher O2 binding rate constant in BY than in W303. In addition, upregulation of the iso-2-cytochrome c decreases the rate of electron transfer to oxidized CcOx (54), although the structural rationale for this change remains unknown. In combination with the lower relative abundance of cytochrome c, which should translate into a decreased probability of binding to the CcOx, this accounts for the absence of any detectable prebound cytochrome c and for the relatively slow electron transfer capability of BY. The rate-limiting step in the overall oxidation of cytochrome c oxidation in BY would then be its diffusion towards its binding site. Incidentally, this difference between the W303 and By strains provides a proof of concept for the possible extension of our approach to investigate fine regulations of the respiratory chain and its constituents.
Cytochrome c Freely Diffuses in the Intermembrane Space.
Another way to probe the ultrastructure of the respiratory chain is to estimate the size of the cytochrome c pool able to donate electrons to oxidized CcOx. Indeed, in addition to supercomplexes, the topology of the inner membrane may define local compartments acting as barriers to the diffusion of mobile electron carriers. According to recent three-dimensional (3D) models of mitochondria derived from tomography imaging, the topology of the inner membrane defines domains of variable size, communicating with the intermembrane space through pores called “cristae junctions” of 14 nm inner diameter (55). Although the cytochrome c molecule is smaller than these pores [approximately 3 nm in diameter (56)], this has led to the hypothesis that the cristae might define local domains that limit diffusion.
Both supercomplex assembly and cristae formation have been proposed to “channel” the soluble carriers such as cytochrome c thereby improving their kinetic efficiency. If such channels do exist, irrespectively of their origin -supercomplexes or compartmentalization- one expects the amount of cytochrome c accessible to oxidation upon one flash to be significantly smaller than the overall cytochrome c pool, in particular when the flash intensity is subsaturating. Assuming that the progressive oxidation of all the cytochromes c in the pool is a succession of independent events having the same probability of occurrence, the fraction χ(n) of oxidized cytochrome c upon a series of flashes (Fig. 5), should increase with the flash number, n, following the geometric probability law: 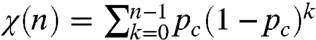 , where pc is the fraction of oxidized cytochrome upon the first flash. In addition to this stochastic framework, one need take into account the fact that, with successive flashes being spaced in time and despite the presence of cytochrome bc1 inhibitors, a fraction of the cytochrome c oxidized upon the nth flash in the series is rereduced when flash (n + 1) is triggered. We experimentally determined this fraction, α, as 8%. The above geometric probability law must thus be modified according to:
, where pc is the fraction of oxidized cytochrome upon the first flash. In addition to this stochastic framework, one need take into account the fact that, with successive flashes being spaced in time and despite the presence of cytochrome bc1 inhibitors, a fraction of the cytochrome c oxidized upon the nth flash in the series is rereduced when flash (n + 1) is triggered. We experimentally determined this fraction, α, as 8%. The above geometric probability law must thus be modified according to: 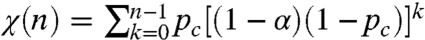 .
.
The dashed lines in Fig. 5 are fits of the data with such a function, constraining α to 0.08 and allowing pc to vary with the flash intensity. The satisfying agreement between the fits and data shows that the entire dataset, comprising several such experiments at different flash energies, can be described by this model which assumes only that the progressive oxidation of the cytochrome c pool is a succession of independent and thus equiprobable events. In other words, the individual cytochromes c are all equivalent with respect to their oxidation probability, ruling out the existence of a widespread distribution of cytochrome c to CcOx stoichiometric ratios. Indeed, restriction of the soluble electron carrier would necessarily result in the existence of different compartments with individual cytochrome c to CcOx stoichiometric ratios being higher and lower than the average value. In a compartment with a low stoichiometric ratio the relative extent of the flash-induced cytochrome c oxidation would be larger than the average and hence the characteristic parameter pc of the geometric law in this particular compartment would be larger than the average one, 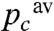 . Conversely, in compartments with a larger cytochrome c to CcOx ratio the relative amount of oxidizing equivalent generated by one flash would be lower than the average, henceforth pc would be lower than
. Conversely, in compartments with a larger cytochrome c to CcOx ratio the relative amount of oxidizing equivalent generated by one flash would be lower than the average, henceforth pc would be lower than 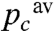 . In both cases the progressive oxidation of the cytochrome c in these two compartments would deviate from the geometric law with parameter
. In both cases the progressive oxidation of the cytochrome c in these two compartments would deviate from the geometric law with parameter 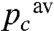 and, importantly, so would their weighted average (as illustrated in Fig. S6). Interestingly, this observation indicates that under oxygen-limiting conditions, restraining the diffusion of the soluble cofactors may result in a suboptimal efficiency in the electron transfer, because compartments with low cytochrome c to CcOx ratios would have their cytochrome c pools overoxidized.
and, importantly, so would their weighted average (as illustrated in Fig. S6). Interestingly, this observation indicates that under oxygen-limiting conditions, restraining the diffusion of the soluble cofactors may result in a suboptimal efficiency in the electron transfer, because compartments with low cytochrome c to CcOx ratios would have their cytochrome c pools overoxidized.
Consistent with our conclusion on the absence of a significant limitation to the diffusion of cytochrome c, we obtained similar results with the Δatp20 mutant (see Fig. S7), which lacks the usual cristae (57), confirming that the ultrastructure of the mitochondrial inner membrane doesn’t significantly impede the free diffusion of cytochrome c.
Conclusion
As tempting as it may be to derive functional implications from ultrastructural features of the respiratory chain observed in purified systems, we demonstrate here that, in intact yeast, the respiratory ETC essentially behaves as a homogeneous system on the time scale of hundreds of milliseconds. Cytochrome c may be prebound to the CcOx, but this interaction is determined by the bimolecular interactions between these two proteins and their stoichiometric ratio, rather than by an effective trapping of the soluble carrier within a particular supramolecular structure. Even though the possibility remains that quinones are trapped within supercomplexes, as regards to cytochrome c, the present conclusions are in excellent agreement with the random collision model previously described by Hackenbrock, et al. (8).
Importantly, destabilization of either the mitochondrial cristae structure or supercomplex association of the components of the ETC did not result in obvious phenotypes in our assay of flash-induced respiration. On the other hand, we observed significant differences between the W303 and BY control strains, which we ascribed to the specific genetic background of the latter, implying expression of some respiratory proteins at modified levels and/or as different isoforms. This difference provides a proof of concept for the possible extension of the present approach to probe regulation of the respiratory chain and to its extension to other cellular systems, such as plant or mammal cells.
Materials and Methods
Yeast Strains.
The Saccharomyces cerevisiae strains used in this study were derived form BY4742 (from Euroscarf) and from W303-1B (58). In all strains, the gene YHB1 encoding a flavohemoprotein was deleted to avoid any possible interference coming from the spectral signal from this enzyme. Mutant strains were generated by deleting the CRD1 or the ATP20 gene. None of the deletions affected respiratory growth of the strains.
The set of strains used was thus as follows:
BY4742 yhb1∷LEU2 (called BY in this report)
BY4742 yhb1∷LEU2 crd1∷kanMX4
BY4742 yhb1∷LEU2 atp20∷kanMX4
W303-1B yhb1∷LEU2 (called W303 in this report)
W303-1B yhb1∷LEU2 crd1∷URA3
W303-1B yhb1∷LEU2 atp20∷URA3
Culture Medium and Sample Preparation.
Yeasts cells were grown on YPD0.5 medium, composed of 1% Yeast Extract, 2% Peptone, and 0.5% Glucose. In such a medium, growth displays two distinct phases, the glucose being initially consumed in fermentative pathways, while the ethanol produced during this first phase triggers obligatory respiratory growth once the glucose is depleted (50). The cells were grown in erlenmeyer flasks (filled at 1/5 with cell culture medium) at 28 °C under vigorous shaking (200 rpm) and harvested in this second exponential growth phase, shortly after the diauxic transition happened. Cells were spun down (1,500 g, 5 min), the pellet was washed in distilled water and, after a second centrifugation, resuspended in MES 40 mM, pH 6.5. Nigericin and valinomycin were added at 10 μM, and the cells were incubated aerobically for at least 10 min. Oxygen consumption assays using a Clark electrode (Qubit Systems) showed that the uncoupled respiration rate of the samples was similar before and after the time-resolved absorption experiments. When specified, 10 μM Tri-Decyl Stigmatellin (TDS, a kind gift of F. Giusti, IBPC), a cytochrome bc1 inhibitor, was added.
Optical and Spectrophotometric Setup.
The experimental setup has been described elsewhere (59). Detecting and actinic flashes were both provided by OPO resonant cavities (respectively Panther and Slopo, Continuum) pumped by the third harmonic of pulsed Nd:YAG lasers. The excitation wavelength was 590 nm or 605 nm. When spectral information in the red part of the spectrum was required, excitation was switched to 430 nm, and its intensity was adjusted based on the kinetics and amplitude of the cytochrome c signal at 551 nm.
A specific cuvette was designed for the purpose of the experiment. A reservoir (volume c.a. 6 mL) is connected to a cubic cuvette of 8 mm optical pathlength, with three quartz windows. The actinic flash is orthogonal to the detecting beam. The sample was equilibrated with the gaseous phase in the reservoir and transferred to the cuvette by a piston operated magnetically. A single actinic flash was then fired after a few seconds and the sample was renewed. Equilibration of the gaseous phase was achieved by bubbling the reservoir, using a peristaltic pump that connected the reservoir to a 500 mL bottle filled with CO and oxygen (or argon) at the desired partial pressures.
Deconvolution of Spectroscopic Signals.
Cytochrome c redox changes were monitored at 551 nm, a wavelength which is free of significant contributions from other electron carriers in the electron transfer chain. However, in the blue region of the spectra, the absorption changes associated with the binding of oxygen or CO and/or with the redox changes of hemes a/a3, monitored at 445 nm, had to be corrected for overlapping spectral contributions from cytochrome c and flavoproteins. According to the cytochrome c difference spectra found in the literature (60), and assuming that the reduced minus oxidized flavoprotein extinction is roughly constant from 445 to 470 nm, a deconvolution for the CcOx signal is given by:
| [1] |
where s445, s470, and s551 are the raw kinetics measured at the corresponding wavelengths. This deconvolution was validated by comparing such results with the light-induced absorption changes measured at 605 nm after correction for the small (10%) cytochrome c contribution at this wavelength (Fig. S8).
When cytochrome bc1 inhibitors were added, the contribution from flavoproteins was prevented, as demonstrated by the equivalence of the kinetics at 470 and 551 nm (Fig. S2). The correction for cytochrome c redox change contributions to Soret band of CcOx was then calculated as follows:
Data Processing.
Spectrophotometric data were acquired with lab-written software developed under Microsoft Visual Basic, and processed with Origin 8.1 software (OriginLab).
Supplementary Material
Acknowledgments.
We warmly thank Claire Lemaire, Francis Haraux, and Mickael Cohen for their support and insightful advices. Pierre Joliot, Francis-André Wollman and Peter Rich are strongly acknowledged for fruitful discussions and critical reading of the paper. This work has been supported by CNRS, Université Pierre et Marie Curie and by Agence Nationale de la Recherche (ANR-07-BLAN-0360-01).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.W. is a guest editor invited by the Editorial Board.
See Author Summary on page 18203.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109510108/-/DCSupplemental.
References
- 1.Keilin D. On cytochrome, a respiratory pigment, common to animal, yeast and higher plants. Proc R Soc Lond B Bio. 1925;98:312–339. [Google Scholar]
- 2.Chance B, Williams GR. A method for the localization of sites for oxidative phosphorylation. Nature. 1955;176:250–254. doi: 10.1038/176250a0. [DOI] [PubMed] [Google Scholar]
- 3.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J Biol Chem. 1955;217:395–407. [PubMed] [Google Scholar]
- 4.Hatefi Y, Haavik AG, Fowler LR, Griffiths DE. Studies on the electron transfer system. XLII. Reconstitution of the electron transfer system. J Biol Chem. 1962;237:2661–2669. [PubMed] [Google Scholar]
- 5.Green DE, Tzagoloff A. The mitochondrial electron transfer chain. Arch Biochem Biophys. 1966;116:293–304. doi: 10.1016/0003-9861(66)90036-1. [DOI] [PubMed] [Google Scholar]
- 6.Kroger A, Klingenberg M. Further evidence for the pool function of ubiquinone as derived from the inhibition of the electron transport by antimycin. Eur J Biochem. 1973;39:313–323. doi: 10.1111/j.1432-1033.1973.tb03129.x. [DOI] [PubMed] [Google Scholar]
- 7.Kroger A, Klingenberg M. The kinetics of the redox reactions of ubiquinone related to the electron-transport activity in the respiratory chain. Eur J Biochem. 1973;34:358–368. doi: 10.1111/j.1432-1033.1973.tb02767.x. [DOI] [PubMed] [Google Scholar]
- 8.Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembr. 1986;18:331–368. doi: 10.1007/BF00743010. [DOI] [PubMed] [Google Scholar]
- 9.Rich PR. Electron and proton transfers through quinones and cytochrome bc complexes. Biochim Biophys Acta. 1984;768:53–79. doi: 10.1016/0304-4173(84)90007-7. [DOI] [PubMed] [Google Scholar]
- 10.Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. Embo J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenaz G, Genova ML. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid Redox Sign. 2010;12:961–1008. doi: 10.1089/ars.2009.2704. [DOI] [PubMed] [Google Scholar]
- 12.Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Rosca MG, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez LA, Monette JS, Chavez JD, Maier CS, Hagen TM. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 2009;490:30–35. doi: 10.1016/j.abb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boumans H, Grivell LA, Berden JA. The respiratory chain in yeast behaves as a single functional unit. J Biol Chem. 1998;273:4872–4877. doi: 10.1074/jbc.273.9.4872. [DOI] [PubMed] [Google Scholar]
- 16.Lavergne J, Joliot P. Restricted diffusion in photosynthetic membranes. Trends Biochem Sci. 1991;16:129–134. doi: 10.1016/0968-0004(91)90054-y. [DOI] [PubMed] [Google Scholar]
- 17.Lavergne J, Joliot P, Verméglio A. Partial equilibration of photosynthetic electron carriers under weak illumination: a theoretical and experimental study. Biochim Biophys Acta. 1989;975:346–354. [Google Scholar]
- 18.Joliot P, Lavergne J, Beal D. Plastoquinone compartmentation in chloroplasts. 1. Evidence for domains with different rates of photo-reduction. Biochim Biophys Acta. 1992;1101:1–12. [Google Scholar]
- 19.Kirchhoff H, Horstmann S, Weis E. Control of the photosynthetic electron transport by PQ diffusion microdomains in thylakoids of higher plants. Biochim Biophys Acta. 2000;1459:148–168. doi: 10.1016/s0005-2728(00)00143-2. [DOI] [PubMed] [Google Scholar]
- 20.Iwai M, et al. Isolation of the elusive supercomplex that drives cyclic electron flow in photosynthesis. Nature. 2010;464:1210–1213. doi: 10.1038/nature08885. [DOI] [PubMed] [Google Scholar]
- 21.Andersson B, Anderson JM. Lateral heterogeneity in the distribution of chlorophyll-protein complexes of the thylakoid membranes of spinach chloroplasts. Biochim Biophys Acta. 1980;593:427–440. doi: 10.1016/0005-2728(80)90078-x. [DOI] [PubMed] [Google Scholar]
- 22.Vallon O, Wollman F-A, Olive J. Lateral distribution of the main protein complexes of the photosynthetic appartus in Chlamydomonas reinhardttii and in spinach: an immunocytochemical study using inteact thylakoid membranes and PS II enriched membrane preparation. Photobioch Photobiop. 1986;12:203–220. [Google Scholar]
- 23.Gibson QH, Greenwood C. Reactions of cytochrome oxidase with oxygen and carbon monoxide. Biochem J. 1963;86:541–554. doi: 10.1042/bj0860541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babcock GT, Wikstrom M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 25.Einarsdottir O, Szundi I. Time-resolved optical absorption studies of cytochrome oxidase dynamics. Biochim Biophys Acta. 2004;1655:263–273. doi: 10.1016/j.bbabio.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Wikstrom M, Verkhovsky MI. Towards the mechanism of proton pumping by the haem-copper oxidases. Biochim Biophys Acta. 2006;1757:1047–1051. doi: 10.1016/j.bbabio.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Belevich I, Verkhovsky MI. Molecular mechanism of proton translocation by cytochrome c oxidase. Antioxid Redox Sign. 2008;10:1–29. doi: 10.1089/ars.2007.1705. [DOI] [PubMed] [Google Scholar]
- 28.Brzezinski P, Gennis RB. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaila VR, Verkhovsky MI, Wikstrom M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 30.Pfeiffer K, et al. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 31.Vanneste WH. The stoichiometry and absorption spectra of components a and a-3 in cytochrome c oxidase. Biochemistry. 1966;5:838–848. doi: 10.1021/bi00867a005. [DOI] [PubMed] [Google Scholar]
- 32.Einarsdottir O, Georgiadis KE, Sucheta A. Intramolecular electron transfer and conformational changes in cytochrome c oxidase. Biochemistry. 1995;34:496–508. doi: 10.1021/bi00002a014. [DOI] [PubMed] [Google Scholar]
- 33.Bryant FD, Seiber BA, Latimer P. Absolute optical cross sections of cells and chloroplasts. Arch Biochem Biophys. 1969;135:97–108. doi: 10.1016/0003-9861(69)90520-7. [DOI] [PubMed] [Google Scholar]
- 34.Brunori M, Giuffre A, Sarti P. Cytochrome c oxidase, ligands and electrons. J Inorg Biochem. 2005;99:324–336. doi: 10.1016/j.jinorgbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 36.Chance B. Spectrophotometry of intracellular respiratory pigments. Science. 1954;120:767–775. doi: 10.1126/science.120.3124.767. [DOI] [PubMed] [Google Scholar]
- 37.Lemire BD, Oyedotun KS. The Saccharomyces cerevisiae mitochondrial succinate:ubiquinone oxidoreductase. Biochim Biophys Acta. 2002;1553:102–116. doi: 10.1016/s0005-2728(01)00229-8. [DOI] [PubMed] [Google Scholar]
- 38.de Vries S, Grivell LA. Purification and characterization of a rotenone-insensitive NADH:Q6 oxidoreductase from mitochondria of Saccharomyces cerevisiae. Eur J Biochem. 1988;176:377–384. doi: 10.1111/j.1432-1033.1988.tb14292.x. [DOI] [PubMed] [Google Scholar]
- 39.Joseph-Horne T, Hollomon DW, Wood PM. Fungal respiration: a fusion of standard and alternative components. Biochim Biophys Acta. 2001;1504:179–195. doi: 10.1016/s0005-2728(00)00251-6. [DOI] [PubMed] [Google Scholar]
- 40.Oliveberg M, Brzezinski P, Malmstrom BG. The effect of pH and temperature on the reaction of fully reduced and mixed-valence cytochrome c oxidase with dioxygen. Biochim Biophys Acta. 1989;977:322–328. doi: 10.1016/s0005-2728(89)80087-8. [DOI] [PubMed] [Google Scholar]
- 41.Verkhovsky MI, Morgan JE, Wikstrom M. Oxygen binding and activation: early steps in the reaction of oxygen with cytochrome c oxidase. Biochemistry. 1994;33:3079–3086. doi: 10.1021/bi00176a042. [DOI] [PubMed] [Google Scholar]
- 42.Hill BC. The reaction of the electrostatic cytochrome c-cytochrome oxidase complex with oxygen. J Biol Chem. 1991;266:2219–2226. [PubMed] [Google Scholar]
- 43.Geren LM, et al. Design of a ruthenium-cytochrome c derivative to measure electron transfer to the initial acceptor in cytochrome c oxidase. J Biol Chem. 1995;270:2466–2472. doi: 10.1074/jbc.270.6.2466. [DOI] [PubMed] [Google Scholar]
- 44.Hirota S, et al. A flash-photolysis study of the reactions of a caa3-type cytochrome oxidase with dioxygen and carbon monoxide. J Bioenerg Biomembr. 1996;28:495–501. doi: 10.1007/BF02110439. [DOI] [PubMed] [Google Scholar]
- 45.Brzezinski P, Wilson MT. Photochemical electron injection into redox-active proteins. Proc Natl Acad Sci USA. 1997;94:6176–6179. doi: 10.1073/pnas.94.12.6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sigurdson H, Namslauer A, Pereira MM, Teixeira M, Brzezinski P. Ligand binding and the catalytic reaction of cytochrome caa(3) from the thermophilic bacterium Rhodothermus marinus. Biochemistry. 2001;40:10578–10585. doi: 10.1021/bi010344h. [DOI] [PubMed] [Google Scholar]
- 47.Farah J, Rappaport F, Choquet Y, Joliot P, Rochaix JD. Isolation of a psaf-deficient mutant of chlamydomonas reinhardtii: efficient interaction of plastocyanin with the photosystem i reaction center is mediated by the psaf subunit. EMBO J. 1995;14:4976–4984. doi: 10.1002/j.1460-2075.1995.tb00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hazzard JT, Rong SY, Tollin G. Ionic strength dependence of the kinetics of electron transfer from bovine mitochondrial cytochrome c to bovine cytochrome c oxidase. Biochemistry. 1991;30:213–222. doi: 10.1021/bi00215a031. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280:29403–29408. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rigoulet M, Mourier A, Galinier A, Casteilla L, Devin A. Electron competition process in respiratory chain: regulatory mechanisms and physiological functions. Biochim Biophys Acta. 2010;1797:671–677. doi: 10.1016/j.bbabio.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 51.Gaisne M, Becam AM, Verdiere J, Herbert CJ. A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1) Curr Genet. 1999;36:195–200. doi: 10.1007/s002940050490. [DOI] [PubMed] [Google Scholar]
- 52.Ter Linde JJ, Steensma HY. A microarray-assisted screen for potential Hap1 and Rox1 target genes in Saccharomyces cerevisiae. Yeast. 2002;19:825–840. doi: 10.1002/yea.879. [DOI] [PubMed] [Google Scholar]
- 53.Waterland RA, Basu A, Chance B, Poyton RO. The isoforms of yeast cytochrome c oxidase subunit V alter the in vivo kinetic properties of the holoenzyme. J Biol Chem. 1991;266:4180–4186. [PubMed] [Google Scholar]
- 54.Allen LA, Zhao XJ, Caughey W, Poyton RO. Isoforms of yeast cytochrome c oxidase subunit V affect the binuclear reaction center and alter the kinetics of interaction with the isoforms of yeast cytochrome c. J Biol Chem. 1995;270:110–118. doi: 10.1074/jbc.270.1.110. [DOI] [PubMed] [Google Scholar]
- 55.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–324. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- 56.Chan SK, Tulloss I, Margoliash E. Primary structure of the cytochrome c from the snapping turtle, Chelydra serpentina. Biochemistry. 1966;5:2586–2597. doi: 10.1021/bi00872a016. [DOI] [PubMed] [Google Scholar]
- 57.Paumard P, et al. The ATP synthase is involved in generating mitochondrial cristae morphology. Embo J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 59.Beal D, Rappaport F, Joliot P. A new high-sensitivity 10-ns time-resolution spectrophotometric technique adapted to in vivo analysis of the photosynthetic apparatus. Rev Sci Instrum. 1999;70:202–207. [Google Scholar]
- 60.Margoliash E, Frohwirt N. Spectrum of horse-heart cytochrome c. Biochem J. 1959;71:570–572. doi: 10.1042/bj0710570. [DOI] [PMC free article] [PubMed] [Google Scholar]
