

Abstract
Membrane–bound phosphodiesterase 6 (PDE6) plays an important role in visual signal transduction by regulating cGMP levels in rod photoreceptor cells. Our understanding of PDE6 catalysis and structure suffers from inadequate characterization of the α and β subunit catalytic core, interactions of the core with two intrinsically–disordered, proteolysis–prone inhibitory PDEγ (Pγ) subunits, and binding of two isoprenyl–binding proteins δ, called PrBP/δ, to the isoprenylated C–termini of the catalytic core. Structural studies of native PDE6 have been also been hampered by lack of a heterologous expression system for the holo–enzyme. In this work, we purified PDE6 in the presence of PrBP/δ and screened for additives and detergents that selectively suppress PDE6 basal activity while sparing that of the trypsin–activated enzyme. Some detergents removed PrBP/δ from the PDE complex, separating it from the holo–enzyme after PDE6 purification. Additionally, selected detergents also significantly reduced dissociation of PDE6 subunits, increasing its homogeneity, and stabilizing the holo–enzyme by substituting for its native membrane environment.
Phosphodiesterase 6 (PDE6) plays an important role in visual signal transduction by regulating cGMP levels in rod photoreceptor cells (1). This protein belongs to the phosphodiesterase (PDE) super–family comprised of 11 members which hydrolyze cAMP and cGMP and thus regulate cellular levels of these second messengers (2). All PDEs possess a highly conserved C–terminal catalytic domain and various N–terminal domains that regulate enzyme activity. PDE6 is a structurally unique heterotetramer as compared with other PDE types, consisting of PDE6α (~99 kDa), PDE6β (~98 kDa), and two inhibitory PDE6 γ subunits (Pγ) (~10 kDa) (3–5). Although Pγ inhibits the catalytic domains of both PDE6α and PDE6β (6–8), PDEαβγ2 still displays basal cGMP hydrolytic activity (4–6, 9).
The catalytic subunits PDE6α and PDE6β are posttranslationally farnesylated and geranylgeranylated, respectively, at their C–termini (10). Presumably these modifications anchor PDE6 to the membranes of rod outer segments (ROS) (3, 10–12). PDE6 can be removed from ROS membranes by the 17–kDa prenyl–binding protein δ (PrBP/δ) which, through binding to the isoprenylated carboxyl–termini of the catalytic subunits (13), has become a novel method for PDE6 isolation (14). The resulting PDE6 complex then was highly purified, enabling its structural study by single particle analysis (14).
Significant structural information about PDEs is available. For example, a nearly full–length structure of PDE2 (15) provided insights into PDE topology and domain arrangement in addition to the regulation and function of this enzyme. The structures of several catalytic domains of the 11 PDE families have also been elucidated (15–17). However, no high–resolution crystal structure is yet available for full–length holo–PDE6. The catalytic core of PDE6 is most structurally similar to that of PDE5 and to a lesser degree to those of PDE2, 4, 10 and 11 (2). The catalytic domain of a PDE5/6 chimeric protein complexed with the inhibitory Pγ peptide provided a structural basis for the Pγ regulatory mechanism (18). The crystal structure of PrBP/δ in complex with Arl2–GTP showed that this protein has a fold similar to a modulator of rho family G proteins called RhoGDI (19) and the protein UNC119 (20) both of which form a hydrophobic pocket for binding the geranylgeranyl moiety. Finally, the overall topology of PDE6 generated from single particle analysis of negatively stained electron microscopy images at 30 Å (21) and 18 Å resolution (14), displayed domain arrangements similar to those seen in PDE2. Failure to obtain PDE6 functional expression in various heterologous systems has impeded understanding the structural basis of PDE6 catalysis and regulation (11, 22, 23). Moreover, PDE6α and PDE6β expressed in vitro did not form heterodimers as observed in vivo. Folding of this protein may require specific chaperone machinery that only exists in photoreceptor cells (24).
Previous use of classical detergents used to facilitate protein extraction failed to improve PDE6 purification because this also solubilized rhodopsin and other membrane proteins present in ROS (25). But an extensive list of reports in the literature also documents the effectiveness of including detergents in crystallization trials of soluble proteins, suggesting stabilization of a unique conformation of these proteins and possibly that the detergent also inhibits non–specific interactions between protein monomers (26–29). Here we characterized PDE6 purified in the presence of PrBP/δ and investigated the effects of different additives/detergents on the subunit integrity and catalytic activity of this enzyme.
EXPERIMENTAL PROCEDURES
Protein Preparations: holo–PDE6
Holo–PDE6 was prepared from frozen bovine retinas (W. L. Lawson Co., Lincoln, NE) essentially as described previously (14). First, bovine ROS membranes were isolated from 200 frozen retinas under dim red light as specified by Papermaster (30) and then PDE6 was purified by the procedure reported by Goc et al (14). Gel filtration chromatography of crude protein was carried out on a Superdex 200 10/300 GL column (GE Healthcare) at a flow rate of 0.4 ml/min in gel filtration buffer containing 10 mM HEPES, pH 7.5, containing 100 mM NaCl, 2 mM MgCl2 and 1 mM DTT. Protein samples were concentrated to 10 mg/ml using a 30 kDa MWCO filter (Millipore) and filtered through a 0.22 μM PVDF filter and 250 μl was loaded on the column. All SEC experiments were performed at 4 °C. Fractions containing PDE6 were pooled and concentrated to 10 mg/ml with a 30 kDa MWCO filter (Millipore). All purification steps were performed without detergent.
Protein Preparations: PDE6–PrBP/δ(PDE6/δ) and PDE6–PrBP/δ–GST (PG)
To free PG, ROS membranes were incubated with PrBP/δ–GST at a ratio of 10:1 mg of total protein for 60 min at room temperature and the complex was extracted with 10 ml of isotonic buffer (20 mM HEPES, 5 mM MgCl2, 1 mM DTT, and 100 mM NaCl, pH 7.5). The supernatant was separated from membranes by centrifugation at 15,000 rpm at 4 °C for 20 min. This procedure was performed three times. The supernatants were combined and centrifuged at 15,000 rpm for 60 min at 4°C to remove remaining ROS membrane particles, followed by overnight dialysis at 4 °C against 11.9 mM potassium phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4, containing 1 mM DTT. The resulting extract was applied onto a 5–ml GSTrap column (GE) at a flow rate 2.5 ml/min and washed with 10 column volumes of equilibrating buffer (11.9 mM potassium phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4). Bound proteins were eluted with 10 ml of 50 mM Tris–HCl, pH 8.0, containing 10 mM glutathione and 0.5 mM DTT. To obtain PG, fractions containing PDE6–PrBP/δ–GST were pooled and further purified by gel filtration as described for the holo–enzyme.
For the PDE6/δ preparation, fractions containing PDE6–PrBP/δ–GST were pooled and treated with thrombin at room temperature overnight. Then the sample was concentrated from 30 ml to 500 μl by using a 30 kDa MWCO filter (Millipore) and further purified by gel filtration as described for the holo–enzyme. For each run, concentrated protein (~2–5 mg/ml in 250 μl) was loaded into the column. Fractions containing PDE6/δ were pooled and concentrated to 10 mg/ml. The resulting protein was homogeneous as assessed by SDS–PAGE, native–PAGE and immunoblotting (14). Protein concentration was determined spectrophotometrically at 280 nm and also by the Bradford method (31).
From 200 bovine retinas yields of PG, PDE6/δ and PDE6 were about 1.5, 1 and 1.5 mg, respectively.
Trypsin–treated PDE6 (PDE6t)
To prepare trypsin–treated PDE6 (PDE6t), holo–PDE6, and PDE6–GST–PrBP/δ were mixed with trypsin–agarose (Princeton Separations, Adelphia, NJ) at a ratio of 30 μg protein to 1 μl of resin and incubated at room temperature for 30 min. Trypsin–agarose was removed by passage through a 0.45 μM PVDF filter. PDE6t was further purified by using either buffer–exchange with a 50,000 MWCO filter (Millipore) or gel filtration to remove proteolytic Pγ fragments (3). Briefly, protein was concentrated to 100 μl (~2–5 mg/ml) with a 50 kDa MWCO Amicon Ultra filter and diluted with size exclusion chromatography (SEC) buffer (20 mM HEPES, 100 mM NaCl, 2 mM MgCl2, 1 mM DTT, pH 7.5) to 500 μl. This process was repeated 6 times. Proteolytic fragments of small molecular weight passed through the 50 kDa MWCO membrane whereas the remaining PDE6 catalytic core of 200 kDa was retained in the supernatant. Alternatively, the trypsin–treated protein was separated on a gel filtration column as described below; PDE6t eluted earlier and separated from smaller proteolytic fragments that eluted later.
For activity assays, PDE6t was prepared from holo–PDE6 in three different ways. First, PDE6 was mixed with trypsin at room temperature for a specified time period. Then soybean trypsin inhibitor was added to stop the reaction. Second, PDE6 was mixed with trypsin–agarose at room temperature for a specified time period after which the trypsin–agarose was removed by membrane filtration. To remove proteolytic fragments buffer was exchanged at least 6 times with a 50 kDa MWCO membrane (Millipore). Third, after removal of trypsin–agarose from PDE6t by membrane filtration, PDE6t was further purified by size exclusion chromatography. PDE6t eluted from the column as a single sharp peak at a position identical to untreated PDE6.
PDE6 Activity Assay
The pH–sensitive fluorescent dye SNAFL–1 (C–1270, Invitrogen) was used to monitor proton release due to PDE6–catalyzed hydrolysis of cGMP and was measured fluorometrically (32) with a NovoSTAR (BMG Labtech) micro–plate spectrofluorometer reader. The assay was performed at 30°C in assay buffer consisting of 20 mM MOPS, pH 8.0, 150 mM KC1, 10 mM MgC12 and 1 mM DDT. The protein (2 nM final concentration) was pre–incubated with the assay buffer for 10 min and the reaction was initiated by addition of cGMP (2 mM) and SNAFL–1 (20 μM) to achieve a final assay volume of 100 μl. The excitation wavelength was 480 nm and emission was recorded at 540 nm with a gain value set at 1800. In the presence of PDE6, a steady increase in fluorescence intensity was observed that achieved a maximum in ~60 min. The percentage of cGMP hydrolysis was plotted as the indicator of enzymatic activity versus time. For PDE6, the data were fitted by linear regression with the slope determined from the data. For PDE6t, the data were fitted by nonlinear regression. The half–life (T1/2) to reach a maximum fluorescence was calculated from the time–dependent curve by fitting the data to the Boltzmann sigmoid equation with Prism 3.0. Reported data are presented as the means and standard deviations from at least three independent experiments.
Detergent Concentrations
The concentration of PDE6 in the assays was ≤ 10 nM. To determine the tested concentrations for each detergent, detergent concentrations relative their critical micelle concentrations for PDE6 were initially calculated as follows:
where 2 accounts for the two catalytic subunits of PDE6 and 1.2~3.0 stands for the ratio of micelles to protein. For a detergent with a low CMC value, a 3–fold ratio was used and for a detergent with a high CMC value, this ratio was 1.2. C8E4 is a typical high–CMC detergent with an aggregation number of ~82 and a CMC of ~8 mM. For this detergent, the calculated micellar concentration required for PDE6 (2 nM) is 2 nM × 82 × 2 × 1.5 or ~ 0.6 μM. Thus, at the 10 mM concentration selected for testing, C8E4 was sufficiently above its CMC to form micelles around PDE6. Anapoe X–100 is a typical low–CMC detergent (aggregation number ~200, CMC 0.3 mM). For this detergent, the micellar concentration required for PDE6 is 2 nM × 200 × 2 × 3 or ~ 2.4 μM. At the tested concentration of 0.8 mM, Anapoe X–100 was also sufficiently above its CMC to form micelles around PDE6.
Measuring PDE6 Activity Directly by Product Formation
PDE6 degrades cyclic GMP to GMP and releases protons into solution. Accordingly, PDE6 activity was measured here by monitoring the proton release accompanying cGMP hydrolysis in a weakly buffered solution containing the pH–sensitive fluorescent dye SNARF–1. This method requires a certain amount of substrate to generate the pH changes that can be reflected by the dye. We tested PDE6t hydrolysis activity at various cGMP concentrations. For PDE6 activity measurements, the useful range of cGMP was about 1–10 mM. Below 1 mM, the resulting pH change was too small to generate a fluorescence signal much over background and at concentrations above 10 mM, cGMP significantly reduced the fluorescence intensity of SNARF–1.
Thus as an alternative, PDE6 activity was measured directly during hydrolysis of cGMP to GMP through isolating both substrate and product by HPLC, essentially as described in (33) with modifications. Protein was first incubated in assay buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 1 mM DTT) for 15 min at room temperature. To initiate the reaction, cGMP was added to a final concentration 0.5 mM. The total volume for the assay was 100 μl. The reaction was performed at 37 °C and terminated at indicated time intervals by rapid protein denaturation at 95 °C for 5 min. Control experiments were performed without cGMP or enzyme added. In a typical assay, 2 nM PDE6t and 5 nM PDE6 were used for time course studies. To determine the apparent Km value for cGMP, the activity of 0.4 nM PDE6t was measured at concentrations of cGMP ranging from 5 – 200 μM and the resulting samples were further processed for nucleotide content.
cGMP and GMP were separated by a Zorbax ODS 5 μm, 4.6 × 250 mm reverse–phase HPLC column equilibrated with 7.5% CH3CN/H2O containing 10 mM tetrabutylammonium bromide and 100 mM K2HPO4/KH2PO4, pH 6.5. Nucleotides were eluted at a flow rate of 1.0 ml/min and identified by their UV spectra and elution positions relative to authentic standards. Reaction samples totaling 100 μl were centrifuged at 16,100g for 10 min to remove denatured proteins. Supernatants were collected, and 30 μl of each sample supernatant was diluted five times with the equilibrating buffer (100 mM K2HPO4/KH2PO4, pH 6.5, containing 10 mM tetrabutylammonium bromide and 7.5% acetonitrile). An aliquot of each supernatant (100 μL) was injected onto a Zorbax ODS 5 μm, 4.6 × 250 mm reverse–phase HPLC column (Agilent, Santa Clara, CA) and monitored at 254 nm. Nucleotides were eluted isocratically at a flow rate of 1.0 ml/min at room temperature.
Apparent Km values were calculated from the concentration–dependent curve with Prism 3.0 software. Reported data are presented as the means and standard deviations (S.D.s) from at least three independent experiments.
Electron Microscopy with Negative Staining
Purified PDE6 samples with or without detergents were adsorbed for 45 s onto glow–discharged carbon–coated copper grids (Quantifoil, Micro Tools, Ochtrup, Germany). Grids then were either washed with two 7 μl drops of water or directly stained with two drops of 1% uranyl formate. Electron micrographs were recorded with a T12 transmission electron microscope (FEI Tecnai) operated at 100 kV.
RESULTS
Isolation of Different Forms of PDE6
First, we tested whether the Pγ subunit could be removed through activation of membrane bound PDE6 by first light–bleaching the ROS and then purifying the activated enzyme. However, the presence of Pγ was still detected in samples of the resulting purified active PDE6 by SDS–PAGE and immunoblots with an anti–Pγ antibody (data not shown).
Next, Pγ subunits were subjected to limited trypsin proteolysis to generate active PDE6t. Digestion conditions were optimized by altering protein/trypsin weight ratios, incubation times and temperatures, and methods of tryptic treatment as described in Experimental Procedures. Under optimal conditions, the digestion yielded a stable product with a size slightly smaller than untreated PDE6, as judged by SDS–PAGE (Figure 1A). Enzymatic activity was measured by using the SNARF–1 fluorescence assay. Although tryptic digestion produced fully activated PDE6, this treatment also led to protein heterogeneity because there are multiple trypsin digestion sites in the PDE6 catalytic core. Under controlled conditions, trypsin can effectively degrade the Pγ–subunits (8, 34, 35), but limited trypsin digestion also removes the C–termini from PDE6 complex with a different cleavage ratio for PDE6α and PDE6β (8). Thus, even though the SDS–PAGE and gel filtration profiles showed high apparent sample homogeneity after trypsin digestion, these procedures could not provide the resolution needed to reveal unique sites and heterogeneity resulting from such digestion. After longer digestion, a 70–kDa polypeptide fragment(s) appeared (Figure 1A). Gel–filtration analysis showed that this 70–kDa fragment(s) could still form a higher molecular weight complex (Figure 1B), probably by combining with truncated PDEαβ subunits.
Figure 1.
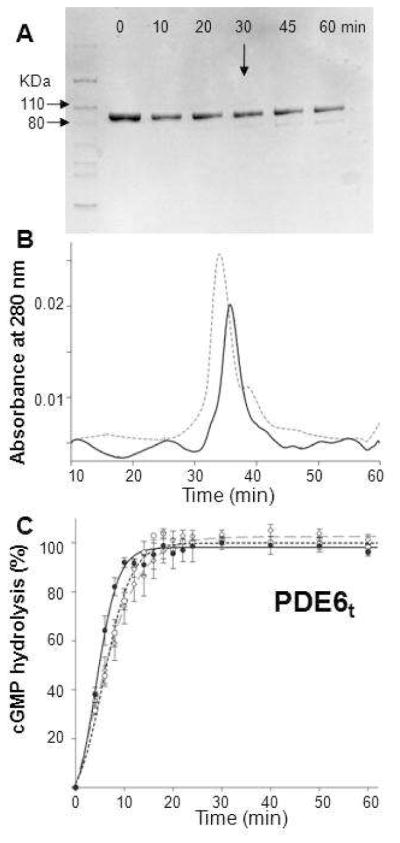
Preparation of trypsinized PDE6. A. The time course of trypsin digestion monitored by SDS–PAGE is shown. Purified PDE6 was mixed with trypsin at 100:1 (weight ratio) and incubated at room temperature for 10, 20, 30, 45 and 60 min. Protein without trypsin was included as a control (Lane 0). At 30 min (arrow), a weak band with a smaller molecular weight appeared. B. Size exclusion chromatography profile of PDE6t (30 min, room temperature). PDE6t (solid line) eluted at nearly the same position as untreated PDE6 (dashed line). C. PDE6t activity with cGMP (2 mM) as substrate. Enzyme activities monitored with the pH–sensitive fluorescent dye SNARF–1were positively correlated with increasing fluorescence intensity over time. PDE6t was generated as follows: Purified PDE6 was first mixed with trypsin at a 100:1 weight ratio for 10 min. Soybean trypsin inhibitor was then added at a 1:10 mass ratio to stop the reaction (dashed line, –○–). Alternatively, PDE6 was mixed with trypsin–agarose at 30 μg protein per 1 μl agarose for 30 min and the reaction was stopped by passing the sample through a filter to remove the trypsin–agarose. Trypsinized peptides then were removed either by gel filtration (dashed line, –◇–) or buffer exchange with a 50 kDa MWCO filter (Millipore) (solid line, –•–). Activities for PDE6t prepared by all three methods were identical.
Purified PDE6 Activity Measurements with SNARF–1
PDE6 degrades cyclic GMP to GMP, releasing a proton into the bulk solution (Figure 2A). Accordingly, PDE6 activity was measured by monitoring the proton release accompanying cGMP hydrolysis in a weakly buffered solution containing the pH–sensitive fluorescent dye SNARF–1 (32). The pH–dependent fluorescence intensity profile of SNARF–1 is shown in Figure 2B. This assay allowed rapid screening for effects of many additives on the PDE6 activity (Figure 2C).
Figure 2.

PDE6 activity measurements with the pH-sensitive fluorescent dye SNARF-1. A. Diagram presenting PDE6-catalyzed hydrolysis of cyclic GMP to GMP with release of a proton. B. PDE6 activity measurements with the pH-sensitive fluorescent dye SNARF-1 in 0.1 M HEPES over a pH range from 6.8 to 8.2. The fluorescence intensity of SNARF-1 decreased with increasing pH (–•–) whereas the buffer used showed no fluorescence (–○–). C. Enzyme activity measurements of PDE6 and PDE6t with SNARF-1. PDE6t achieved maximal cGMP hydrolysis in about 5 min (–•–), whereas PDE6 exhibited much lower residual activity that resulted in total cGMP hydrolysis in about 60 min (-○-). The percentage of cGMP hydrolysis is plotted as a function of time.
PDE6t rapidly hydrolyzed cGMP with the fluorescence intensity achieving maximum in approximately 5 min with a T1/2 of ~2 min (Figure 2C, bold black line –•–). The change in pH during the assay was ~0.1 pH unit for the whole time course. Initial rates for holo–PDE were not affected by an increased substrate concentration, implying that the assay operated at Vmax. Indeed, the initial rate of cGMP hydrolysis also was proportional to the amount of enzyme added. In contrast, purified PDE6 displayed basal cGMP hydrolytic activity as demonstrated by slower hydrolysis of cGMP with T1/2 of 30 min (Figure 2C, fine line –○–). This result indicates that the initial rate of PDE6 activity was about 7% of PDE6t and was not completely suppressed by the inhibitory Pγ subunit under these assay conditions.
In control experiments, 5 mM EDTA nearly abolished PDE6t activity, probably by chelating Mg2+ or other metal ions and preventing them from interacting with PDE6t catalytic sites (Figure 3), whereas Ca2+ ions just reduced PDE6 activity (Figure 3A). Chaotropic additives were not effective in removing Pγ from the PDE6 complex. Urea at 2M concentration had no effect on enzymatic activity of PDE6t, and arginine had only a minor effect (Figure 3A).
Figure 3.

Effects of additives and detergents on PDE6t and PDE6 activity. A. Effects of reagents on PDE6t and PDE6 activity. Activities of purified PDE6t (upper panel) and PDE6 (lower panel) were measured in the absence (solid lines) or in the presence (dashed lines) of designated reagents. Urea (1 M, –▲–), Arginine (3 mM, –◆–), CaCl2 (0.1M, –X–) and EDTA (5 mM, –◇–). The percentage of maximum PDE6t activity vs. time is shown. B. Effects of selected detergents on PDE6 activity. The activities of PDE6t (upper panel) were measured in the absence (solid line) or in the presence of specified detergents (dashed lines). C8E4 (10 mM, –○–), Anapoe X–100 (0.8 mM, –◇–), CHAPS (25 mM, –Δ–), n–octyl–β–D–glucopyranoside (20 mM, –◆–), n–dodecyl–N,N–dimethylamine–N-oxide (1 mM, –
 –) and dimethyldecylphosphine oxide (5 mM, –×–). Those detergents that did not alter PDE6t activity were then tested for their ability to suppress PDE6 activity (lower panel). PDE6 without detergent (solid line), with detergents (dashed lines). N–undecyl–β–D–maltopyranoside (1 mM, –Δ–), C8E4 (10 mM, –○–), Anapoe X–100 (0.8 mM, –◇–). The percentage of cGMP hydrolyzed is plotted vs. time.
–) and dimethyldecylphosphine oxide (5 mM, –×–). Those detergents that did not alter PDE6t activity were then tested for their ability to suppress PDE6 activity (lower panel). PDE6 without detergent (solid line), with detergents (dashed lines). N–undecyl–β–D–maltopyranoside (1 mM, –Δ–), C8E4 (10 mM, –○–), Anapoe X–100 (0.8 mM, –◇–). The percentage of cGMP hydrolyzed is plotted vs. time.
Effects of Detergents on PDE6 Stabilization
Because Pγ subunits could not be quantitatively removed, we then tried to identify conditions whereby these subunits could be retained with the catalytic core of PDE6. To explore the effects of detergents on purified PDE6 stability, we measured basal PDE6 activity in the presence of several representatives selected to cover a broad range of non–ionic, zwitterionic, ionic and specialty detergents. PDE6t activity measurements served as controls for the effects of such additives on catalytic activity. One detergent from each class was chosen with a CMC in the 0.1 to 10 mM range. Purified PDE6 was pre–incubated in assay buffer with the ‘test’ detergent and then cGMP and SNARF–1 were added to initiate the reaction. Fluorescence intensities were recorded over 60 min.
As shown in Figure 3B, the selected detergents had various effects on PDE6t activity. While CHAPS and OG, reduced the rate of cGMP hydrolysis, others, including dimethyldecylphosphine oxide and n–dodecyl–N,N–dimethylamine–N–oxide, abolished PDE6t activity altogether. Others such as Anapoe X–100 and C8E4 preserved PDE6t enzymatic activity. The T1/2 for PDE6t to reach maximum activity was determined for all detergents selected for testing (Table 1), and those with T1/2 values close to T1/2 values calculated in their absence were then subjected to further analysis for their effects on PDE6 activity. The rationale behind selection of the detergent concentrations used in these experiments is explained in the Experimental Procedures section.
Table 1.
PDE6t activity in the presence of degergentsa measured by the SNARF–1 assay.
| Detergent Name | Concentration | T1/2 (min) |
|---|---|---|
| None (control) | – | 2.5 ± 0.1 |
| Anapoe–35 | 0.3 mM | 1.1 ±0.3 |
| Anapoe X–100 | 0.8 mM | 2.3 ±0.1 |
| Cymal–6 | 2.0 mM | 3.0 ±0.1 |
| 2,6–dimethyl–4–heptyl–β–D–maltopyranoside | 10 mM | 3.2 ±0.2 |
| MEGA–8 | 16 mM | 3.2 ±1.0 |
| PMAL–C8 | 0.1 mM | 2.5 ±0.1 |
| n–undecyl–β–D–maltopyranoside | 2 mM | 2.8 ±0.8 |
| n–dodecyl–α–D–maltopyranoside | 0.5 mM | 2.1 ±0.2 |
| n–undecyl–β–D–thiomaltopyranoside | 0.6 mM | 2.1 ±0.4 |
| C8E12 | 0.3 mM | 0.7 ±0.3 |
| C4E8 | 10 mM | 2.1 ±0.2 |
| sucrose monododecanoate | 1 mM | 3.0 ±0.3 |
Chemical names of the indicated detergents are provided in the Abbreviations section. SNARF–1 fluorescence intensity was plotted vs. time. The T1/2 for reaching maximum fluorescence was determined by a non–linear regression analysis. For detergents with no effect on PDE6t activity, T1/2 values for PDE6t were similar in the presence and absence of detergent.
Detergents found not to alter PDE6t activity but to reduce purified PDE6 activity are indicated in Table 2. All such detergents were non–ionic. Anapoe X–100 and C8E4 significantly repressed PDE6 activity (Figure 3B (lower panel)), but other detergents only moderately repressed PDE6 activity, e.g. n–undecyl–β–D–maltopyranoside. Polyoxyethylene glycols, such as Anapoe detergents, had the greatest effect on PDE6 basal activity suppression, whereas some maltosides and glucosides had only a moderate effect. For example, CYGLU–3, a non–ionic glucoside, failed to suppress PDE6 activity even at the high concentration of 30 mM, whereas Anapoe X–100 and Anapoe–35 at a relatively low concentration (0.3–0.8 mM) efficiently repressed PDE6 activity. At less than 1 mM concentration, Anapoe X–100 reduced PDE6 activity ~12–fold, and Anapoe–35 reduced it nearly 30–fold.
Table 2.
Holo–PDE6 activity measured using the SNARF–1 assay in the presence of detergentsa.
| Name | Concentration | cGMP hydrolysis (%/min) |
|---|---|---|
| Control (no detergent) | – | 1.47±0.05 |
| Anapoe–35 | 0.3 mM | 0.04 ± 0.01 |
| Anapoe X–100 | 0.8 mM | 0.15 ± 0.05 |
| Cymal–6 | 2.0 mM | 0.36 ± 0.05 |
| 2,6–dimethyl–4–heptyl–β–D–maltopyranoside | 10 mM | 0.31 ± 0.03 |
| MEGA–8 | 16 mM | 0.36 ± 0.06 |
| PMAL–C8 | 0.2% | 0.33 ± 0.03 |
| n–undecyl–beta–D–maltopyranoside | 2 mM | 0.43 ± 0.05 |
| n–dodecyl–α–D–maltopyranoside | 0.5 mM | 0.36 ± 0.05 |
| n–undecyl–β–D–thiomaltopyranoside | 0.6 mM | 0.16 ± 0.06 |
| C8E12 | 0.3 mM | 0.15 ± 0.05 |
| C4E8 | 10 mM | 0.04 ±0.01 |
| sucrose monododecanoate | 1 mM | 0.17 ± 0.06 |
Holo–PDE6 activity in the presence of detergents was measured by the SNARF–1 assay (see Experimental Procedures). Some detergents significantly reduced PDE6 activity by 20–fold or more (i.e. C8E4 and Anapoe–35 and some exhibited more moderate repression by a 5–fold reduction (i.e. DDM and MEGA–8). Data represent the means and S.D.s of at least three separate experiments.
Stabilizing Effects of C8E4 on PDE6
As shown in Figure 4, the fluorescence intensity of purified PDE6 in the presence of C8E4 remained constant during the 60 min test period (Figure 4A, dashed line), but steadily increased in the absence of this detergent. SNARF–1 also retained its sensitivity to pH changes in the presence of C8E4 (Figure 4B). When PDE6t was pre–incubated for 10 min in assay buffer containing C8E4 or purified PDE6 was digested by trypsin and the resultant PDE6t enzyme was added to the assay mixture containing C8E4, full activity was observed. This result could be ascribed to steric hindrance of Pγ by C8E4 micelles, preventing Pγ release from the catalytic site of PDE6. Thus, the C8E4–induced suppression of PDE6 basal activity could be due to a stabilized interaction of Pγ with PDE6, with C8E4 forming a complex with PDE6 through an interaction with Pγ.
Figure 4.

Effect of C8E4 on PDE6 activity. A. Measurements of PDE6t (bold black line, –•–) and PDE6 (dashed line, –○–) activity in the presence of C8E4. PDE6 activity without C8E4 is shown as a solid grey line (–○–). In the presence of C8E4, PDE6t was fully functional, whereas PDE6 activity was completely abolished. B. In the presence of C8E4, the fluorescence intensity of SNARF–1decreased with increasing pH, indicating that the dye responded appropriately to changes in pH. C. PDE6t activity in the presence of C8E4. Protein was pre–incubated in the assay buffer with C8E4 for 10 min before PDE6t activity was monitored (solid line). Alternatively, PDE6 was digested with trypsin in the presence of C8E4 before soybean trypsin inhibitor was added to stop the protease reaction and the resulting PDE6t was added to assay buffer along with C8E4 for activity measurements (dashed line). Both preparations of PDE6t were fully active.
PrBP/δ binding to PDE6
PrBP/δ can extract PDE6 from ROS membranes by binding to the isoprenylated carboxyl–termini of the catalytic subunits (13). But based on EM observations, PrBP/δ failed to form a stable complex with PDE6 and the protein solution contained a mixed population of complexes with one or two PrBP/δ proteins bound. Instead, it was observed that PG (PDE6–PrBP/δ–GST) was more stable (14).
The elution profiles of three forms of PDE6 in the absence of detergents are shown in Figure 5A. PDE6 and PDE6/δ eluted from the column as single sharp peaks, whereas a large fraction of PG formed a high molecular weight aggregate. In the presence of PrBP/δ–GST, PDE6 tended to aggregate, but these aggregates broke apart upon proteolytic removal of GST. Based on SEC analysis, the aggregation level of PG also increased with its protein concentration (data not shown). To reduce PG aggregation, various buffer conditions were tested. PG was buffer exchanged or dialyzed against the test media, and the resulting protein conformation was examined by SEC. Among conditions tested, changes in pH (6.5 to 9.0) and NaCl (0 – 0.5 M) did not affect the elution profile of PG, i.e., the protein remained as a mixture of the catalytic core dimer and aggregates. Addition of reagents and detergents had various effects. Glycerol allowed more aggregation but glucose did not change the elution profile of PG. Whereas DDM, CHAPS, and Anapoe detergents partially reduced PG aggregation, virtually no aggregation occurred in the presence of C8E4 (Figure 5C). Instead the protein sample displayed a single sharp peak followed by a second peak with a lower molecular weight. SDS–PAGE analysis identified PDE6 in the first peak (peak 1) and PrBP/δ–GST in the second peak (peak 2), indicating dissociation of PrBP/δ–GST in the presence of C8E4. A similar phenomenon was observed with PDE6/δ. When this protein was incubated with C8E4, two peaks were observed upon SEC (Figure 5D), indicating that PrBP/δ had disassociated from the PDE6–PrBP/δ complex.
Figure 5.

SEC analysis of detergent effects on reducing PG aggregation. A. Elution profiles of three forms of PDE6 in the absence of detergent. PDE6 (short dashed line) and PDE6/δ (solid line) eluted as single peaks, whereas PG (large dashed line) eluted as two major peaks indicating a mixture of a high molecular mass aggregate and a monomer. Insert: SDS–PAGE analysis of SEC fractions from the two PG peaks. The top band is PDE6 and lower band is GST–δ. Some detergents, such as Anapoe–35 (B), reduced aggregation of PG. Insert: SDS–PAGE analysis of SEC fractions (indicated with dotted line). In the presence of C8E4 (C), PG aggregation was almost completely abolished. PDE6 was found mainly in Peak 1 (insert, labeled as 1). The second major peak eluting after the PDE6 peak represented GST– PrBP/δ as indicated by SDS–PAGE analysis (insert, labeled as 2). D. SEC analysis of the PDE6/δ conformation in the presence of C8E4; 0.5 ml fractions were collected SDS–PAGE of fractions from the two major peaks is shown in the insert. The left band (labeled with 1) shows the fraction from the main peak with an estimated MW of 200 kDa whereas the right band illustrates the fraction from the last peak with an estimated MW of 15 kDa (as indicated with an arrow). For all inserts, analysis of the protein sample before SEC is shown next to the protein marker. The top band corresponded to PDE6 and lower band to GST–δ.
Because PrBP/δ did not form a tight complex with PDE6, it could be argued that PrBP/δ dissociation from PDE6 was spontaneous rather than due to the presence of C8E4. Thus, we determined if C8E4 could deplete PrBP/δ–GST from the PG complex. PG was incubated with GST–resin and unbound proteins were removed by washing with buffer. PG without detergent was included as control to monitor PrBP/δ–GST binding to the resin. As shown in Figure 6A, no protein was detected after several washes. C8E4 (10 mM) was then added to the resin and left to incubate for 1 hr. Then the resin was washed and subsequently eluted with reduced glutathione (10 mM); fractions were analyzed by SDS–PAGE. As shown in Figure 6A, PDE6 released from the resin was mainly found in the C8E4–flow through whereas PrBP/δ–GST was mainly present in the elution fraction. Thus, C8E4 dissociated PrBP/δ–GST from PDE6 possibly by C8E4 micelles embedding the lipid moieties of PDE6.
Figure 6.

SDS–PAGE analysis of detergent–depleted GST–PrBP/δ dissociated from the PG complex. A. C8E4 removed GST–PrBP/δ from the PG complex. PG was mixed with GST–FF resin and unbound proteins were removed by washing the resin with PBS. Then the resin was incubated with PBS plus 10 mM C8E4 for 1 hr at room temperature. The flow through was collected (Lane F, under C8E4) and the resin was washed again with PBS plus C8E4. Elution was accomplished with reduced glutathione (10 mM). F: flow through; W: wash fractions; E: Elution fraction. B. Dissociation of the PG complex was decreased by incubation with CnEm detergents with longer alkyl chains. CnEm: Cn = CH3(CH2)n–1 alkyl chain, Em = (OCH2CH2)mOH oligoethyleneglycol. In the presence of C8E4 or C8E6, PDE6 was identified mainly in SEC flow through fractions as shown by SDS–PAGE. But in the presence of C12E8, the PG complex remained intact in the elution fraction. Lanes F and E: flow through and elution fractions in the presence of CnEm.
We tested other polyoxyethylene detergents, including CnEm, to determine their effects on PG dissociation. For CnEm detergents, the length of alkyl chain determines the detergent CMC and aggregation number, whereas the head group has a strong influence upon interactions with proteins. As shown in Figure 6B, PDE6 in the presence of C8E4 and C8E6 was mainly present in the flow through fractions, indicating its disassociation from PrBP/δ–GST. But in the presence of C10E5, PDE6 was found in the flow through fraction (strong) as well as in elution fraction (weak). In the presence of C12E8, however, the PG complex remained intact and was found mainly in the elution fractions. Thus, detergents with longer chain lengths were identified that did not compete with PrBP/δ–GST for binding to the lipid–like heads of PDE6 C–termini, probably due to their more hydrophobic hydrocarbon chain “tails”. Alternatively, perhaps such detergents do not insert well into the prenyl–binding pocket of PrBP.
Verification of suppression of PDE6 basal activity by C8E4 detergent in a different enzymatic assay
The SNARF–1 assay is only useful for initial screening because it has a relatively low sensitivity and utilizes almost all the substrate during the course of the reaction. Therefore, the catalytic properties of PDE6 and PDE6t were also examined by direct HPLC analysis of cGMP hydrolysis (Supplementary Figure 1). Trypsin–activated PDE6t rapidly hydrolyzed cGMP whereas PDE6 still displayed a certain level of activity. The catalytic parameters for PDE6t were determined at 31 ± 4 μM for Km and 5122 ± 285 mol cGMP/mol·sec for Vmax, in agreement with previous studies (18, 36–40). In the presence of C8E4, PDE6t retained full activity (Figure 7C), rapidly hydrolyzing cGMP with the GMP level achieving a maximum in ~1 min and a T1/2 of about 0.8 min without detergent. In the presence of C8E4, PDE6t rapidly hydrolyzed cGMP with a T1/2 of about 1.2 ± 0.2 min in the presence of C8E4.
Figure 7.

Catalytic properties of purified PDE6t and PDE6 assessed directly by product formation. A. Percentages of cGMP hydrolysis by PDE6t and PDE6 are plotted as a function of time. PDE6t: solid line. PDE6: dashed lines (2 mM, –○– and 5 nM–◇–). B. The rate of cGMP hydrolysis by PDE6t is plotted as a function of cGMP concentration. The Km value is 31 ± 4 μM and Vmax is 5122 ± 285 mol cGMP/mol·sec. C: PDE6t activity in the presence and absence of C8E4. The percentage of cGMP hydrolysis by PDE6t is plotted as a function of time. PDE6t without C8E4: solid line. PDE6t with C8E4: dashed line. D. cGMP hydrolysis in the presence and absence of C8E4. The percentage of cGMP hydrolysis by PDE6 is plotted as a function of time. PDE6 without C8E4: solid line. PDE6 with C8E4: dashed line.
PDE6 also displayed cGMP hydrolytic activity in an enzyme–concentration dependent manner. At 5 nM, it slowly hydrolyzed cGMP (Figure 7A), but in the presence of C8E4, this basal activity was markedly depressed (Figure 7D); PDE6 hydrolysis reduced approximately 10–fold from 1.4 to 0.13. Thus, both the SNARF–1 and direct product–based enzymatic assays showed that activated PDE6 remained fully functional in C8E4 detergent and purified PDE6 itself retained its inhibitory subunits (Supplementary Figure 2). The activity of PDE6 was significantly suppressed in the presence of C8E4.
Electron Microscopy of PDE6 in Detergents
Taken together the forgoing data have demonstrated that certain types of detergents tend to blunt the activity of PDE6, presumably by masking its isoprenylated C–termini and stabilizing the Pγ subunits. Thus, the resulting PDE6 should be more suitable for structural analysis.
Negative staining EM allows rapid screening of protein homogeneity with only small amounts of sample. Because PDE6/δ was highly purified, we studied its conformation in detergents by this procedure. In most detergents tested, such as C8E4 (Figure 8, left panel of bottom layer) and DDM (Figure 8, middle panel of top layer), PDE6/δ appeared as one major population of particles exhibiting an elongated shape with a wide top and a narrower end. The shapes of these particles were similar to those without detergent (Figure 8, top left panel), and identical to those seen in our previous EM study (14). Samples with all tested detergents except CHAPS appeared fully homogeneous, indicating compatibility with PDE6/δ. But in CHAPS, most of the particles were smaller than those observed in DDM, C8E4 or without detergent. Many displayed a V–shaped structure (Figure 8, CHAPS right panel of bottom layer), indicating that CHAPS might have caused disassociation of the PDE6 complex, thereby providing a possible explanation for the reduced activity of PDE6t in its presence.
Figure 8.

EM images of negatively stained PDE6/δ complexes in the absence and presence of indicated detergents. The protein retained homogeneity in all detergents tested except CHAPS in which the complex seems to have dissociated into particles of smaller size. Inset, higher magnification of a single particle. Scale bars, 100 nm.
DISCUSSION
PDE6 is an important enzyme in phototransduction. However, structural information on full–length PDE6 is lacking. As a prelude to detailed structural studies, we first measured the enzymatic activity of our purified bovine ROS PDE6 preparation and observed a basal level of cGMP hydrolytic activity. While being firmly attached to PDEα and β subunits, Pγ subunits only weakly block the catalytic activities of the PDEα and β core. Gtα effectively displaces Pγ during the activation process, but this inhibitory subunit is not removed because a larger complex with other proteins involved in termination of the activation process is formed, including the G protein transducin (41, 42). Because this basal activity could be a ‘marker’ for structural variability, we screened for additives/detergents that would completely suppress such activity while preserving the intrinsic activity of the PDE6 holo–enzyme.
Based on our activity assays, the modest levels of basal activity exhibited by PDE6 purified from bovine ROS probably indicated that Pγ was not tightly bound to the catalytic sites of PDE6. The C–terminus of Pγ may only loosely associate with the catalytic site of PDE6, or perhaps only one Pγ is tightly bound, with the other loose or free, allowing this basal PDE6 activity. Meanwhile, the whole Pγ inhibitory unit could still be bound to PDE6 via a high ionic affinity interaction with the central region of Pγ. Indeed, Pγ is known to inhibit PDE6 with the resulting Pγ–PDE6 interactions having been defined by many biochemical studies. The central region of Pγ (residues 24 to 45) is rich in Pro residues and positively charged amino acids and binds to the GAF–A domain of PDE6 with high affinity (Kd ~ 50 pmol) (32). The C–terminus of Pγ (residues 74 to 87) binds to the catalytic domain of PDE6 and blocks substrate entry into the catalytic site (3, 6, 22, 43–46). An NMR study indicates that W70 plays a key role in this interaction (9). Therefore, the protein may exhibit different dynamic conformations that result in sample heterogeneity. Pγ could have different association affinities for PDE6α and PDE6β. This dynamic conformation of Pγ along with its intrinsically disordered structure further complicates the possibility of crystallizing PDE6.
Here we have shown that certain types of detergents could help to stabilize PDE6. In the presence of C8E4, the basal activity of PDE6 was almost completely abolished, whereas PDE6t remained fully active. The fluorescence–based PDE6 activity measurements provided a quick and effective way to screen for detergents that stabilize PDE6. Fortunately, this method required only small amounts of protein, allowing rapid screening of samples with additives and detergents along with appropriate replicates and controls. But the same method requires high concentrations of substrate (cGMP) to generate pH changes sufficient for quantification the SNARF-1 dye. Therefore, to determine catalytic parameters of PDE6 in the presence of detergents such as C8E4, we used the more rigorous methodology directly measuring of the hydrolysis product, GMP, by HPLC. In agreement with the SNARF–1 assay results, the presence of C8E4 did not alter the PDE6t activity, and the basal activity of PDE6 was greatly suppressed.
It is still unclear why some detergents repress PDE6 activity whereas others do not. Pγ certainly plays an inhibitory role in the PDE6 structure. The amino acid sequence of Pγ may explain its preference for interaction with polyoxyethylene glycols such as C8E4 and Anapoe X–100. Based on our enzymatic activity assay results, only non–ionic detergents stabilized PDE6; zwitterionic and ionic detergents decreased or abolished PDE6t activity, indicating a major conformational change or denaturation. Among the non–ionic detergents tested, polyoxyethylene detergents had the greatest stabilizing effect on the interaction with Pγ.
The isoprenylated carboxyl–termini of PDE6αβ pose one of the major challenges in structural characterization of the enzyme purified from ROS. In the absence of membranes, hydrophobic isoprenyl groups are excluded from the aqueous phase and likely form a myriad of conformations when their tails are wedged non–specifically into the protein core. The conformational heterogeneity of the protease–prone Pγ subunit and its possible heterogeneous truncation represent two of several major challenges for PDE6 subunit integrity. Amino acid analyses and biochemical studies of Pγ indicated that it is an intrinsically disordered protein (IDP)(46, 47) whereas an NMR study revealed that, despite being intrinsically disordered in solution, Pγ also exhibits transient secondary structure (9). Generally, IDPs undergo a disorder–to–order transition upon interaction with their functional partners. With C8E4 having the most significant effect on PDE6, not only stabilizing Pγ, but also interacting with the isoprenyl groups of PDE6 and either displacing PrBP/δ–GST away from the complex or interfering with the binding to PrBP/δ. Micelles of C8E4 may restrict the movement of Pγ and help to keep it in place. The stabilizing effect of detergents was more detergent–specific than concentration–dependent. The presence of polyoxylethylene micelles could be useful in masking the isoprenylated C–terminal regions of PDE6 and reducing PDE6 heterogeneity.
However, we did not observe a direct interaction between detergents and PDE6. Based on SEC analysis, elution profiles of PDE6 were the same in the presence or absence detergents. There were no obvious size changes conferred by the presence of detergents. So how do detergents stabilize PDE6? Pγ is rich in Pro residues and positively charged amino acid residues in its central region. This property may allow the peptide to interact more favorably with certain types of detergents such as C8E4. It is likely that detergent micelles can effectively surround hydrophobic patches on Pγ, restricting its movements and stabilizing its blockage of entry sites into PDE6 catalytic domains.
Hydrophobic interaction chromatography succeeded in quantitatively separating glutamic acid-rich protein (GARP2) from the PDE6 holoenzyme (48). Moreover, PrBP/δ also releases PDE6 from ROS without solubilization of GARP2 (48). Consistently with this report, we have not observed GARP2 in our preparations when PDE6 was released from rod membranes with PrBP/δ–GST. Based on these observations, we speculate that C8E4 likely would dissociate GARP2 from PDE6 when added to ROS membranes, but this detergent will also solubilize a fraction of rhodopsin causing problems with further investigation of this enzyme in biochemical acceptable form. The effect of this detergent on transducin will require additional study if the goal is to isolate the complex of PDE6-transducin. Another unexplored area to investigate is to determine if blocking the active site with filmy attached Pγ-C8E4 will prevent or allow binding of nucleoside inhibitors.
In summary, we have demonstrated that certain types of detergents suppress the basal enzymatic activity of PDE6, presumably by stabilizing its structure. These detergents did not decrease the catalytic ability of activated PDE6. Moreover, the same detergents interacted with the C–termini of PDE6 and disassociated PrBP/δ–GST from the ternary complex. An extensive body of literature demonstrates the effectiveness of including detergents in crystallization trials to prevent non–specific aggregation due to hydrophobic interactions. This is further supported by our EM studies which illustrate a strategy to screen detergents quickly that allow PDE6 to retain its homogeneity. Detergents may play a critical role in the eventual structural characterization of the PDE6 complex.
Supplementary Material
Acknowledgments
We thank Dr. Leslie T. Webster, Jr. and the Palczewski’s laboratory for critical comments on the manuscript. The authors would like to thank Susan Farr and Dr. Beata Jastrzebska for helping with PDE6 preparation, Professor John Mieyal for comments on PDE6 activity assays and using the instrument and Heather Holdaway for technical assistance with electronic microscopy.
ABBREVIATIONS
- Anapoe–35
polyethylene glycol monododecyl ether (23)
- Bis–Tris
bis(2–hydroxyethyl)–amino–tris(hydroxymethyl)–methane)
- C12E8
octyloxyethylene(8)dodecyl ether
- C8E4
tetraethyleneglycol monooctylether
- cGMP
cyclic guanosine 3′,5′–monophosphate
- CHAPS
3–[(3–cholamidopropyl)dimethylammonio]–1–propanesulfonate
- CMC
critical micelle concentration
- CnEm
Cn = CH3(CH2)n–1 alkyl chain, Em = (OCH2CH2)m OH oligoethyleneglycol
- CYGLU
3, 3–cyclohexyl–1–propyl–β–D–glucoside
- Cymal–6
6–cyclohexyl–1–hexyl–β–D–maltoside
- DDM
n–dodecyl–α–D–maltopyranoside
- GST
glutathione S–transferase
- IDP
intrinsically disordered protein
- MEGA–8
octanoyl–N–methylglucanide
- PG
PDE6–PrBP/δ–GST
- PMAL–C8
(poly(maleic anhydride–alt–1–decene) substituted with 3–(dimethylamino) propylamine)
- PrBP/δ
isoprenyl–binding protein δ
- Pγ
inhibitory γ subunit of PDE6
- ROS
rod outer segment(s)
- SEC
size exclusion chromatography
- T1/2
half–life
Footnotes
This research was supported, in whole or in part, by National Institutes of Health Grants EY009339, and P30 EY11373. KP is John H. Hord Professor of Pharmacology.
SUPPORTING INFORMATION AVAILABLE
Descriptions of PDE6 activity measurements using the pH–sensitive fluorescent dye SNARF–1 and HPLC and size exclusion chromatography of PDE6 conformation in the presence of C8E4. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Polans A, Baehr W, Palczewski K. Turned on by Ca2+! The physiology and pathology of Ca(2+)-binding proteins in the retina. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- 2.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 3.Artemyev NO, Surendran R, Lee JC, Hamm HE. Subunit structure of rod cGMP-phosphodiesterase. J Biol Chem. 1996;271:25382–25388. doi: 10.1074/jbc.271.41.25382. [DOI] [PubMed] [Google Scholar]
- 4.Baehr W, Devlin MJ, Applebury ML. Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J Biol Chem. 1979;254:11669–11677. [PubMed] [Google Scholar]
- 5.Deterre P, Bigay J, Forquet F, Robert M, Chabre M. cGMP phosphodiesterase of retinal rods is regulated by two inhibitory subunits. Proc Natl Acad Sci U S A. 1988;85:2424–2428. doi: 10.1073/pnas.85.8.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artemyev NO, Hamm HE. Two-site high-affinity interaction between inhibitory and catalytic subunits of rod cyclic GMP phosphodiesterase. Biochem J. 1992;283(Pt 1):273–279. doi: 10.1042/bj2830273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artemyev NO, Rarick HM, Mills JS, Skiba NP, Hamm HE. Sites of interaction between rod G-protein alpha-subunit and cGMP-phosphodiesterase gamma-subunit. Implications for the phosphodiesterase activation mechanism. J Biol Chem. 1992;267:25067–25072. [PubMed] [Google Scholar]
- 8.Hurley JB, Stryer L. Purification and characterization of the gamma regulatory subunit of the cyclic GMP phosphodiesterase from retinal rod outer segments. J Biol Chem. 1982;257:11094–11099. [PubMed] [Google Scholar]
- 9.Song J, Guo LW, Muradov H, Artemyev NO, Ruoho AE, Markley JL. Intrinsically disordered gamma-subunit of cGMP phosphodiesterase encodes functionally relevant transient secondary and tertiary structure. Proc Natl Acad Sci U S A. 2008;105:1505–1510. doi: 10.1073/pnas.0709558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anant JS, Ong OC, Xie HY, Clarke S, O’Brien PJ, Fung BK. In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J Biol Chem. 1992;267:687–690. [PubMed] [Google Scholar]
- 11.Qin N, Baehr W. Expression and mutagenesis of mouse rod photoreceptor cGMP phosphodiesterase. J Biol Chem. 1994;269:3265–3271. [PubMed] [Google Scholar]
- 12.Christiansen JR, Kolandaivelu S, Bergo MO, Ramamurthy V. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2011;108:8862–8866. doi: 10.1073/pnas.1103627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook TA, Ghomashchi F, Gelb MH, Florio SK, Beavo JA. Binding of the delta subunit to rod phosphodiesterase catalytic subunits requires methylated, prenylated C-termini of the catalytic subunits. Biochemistry. 2000;39:13516–13523. doi: 10.1021/bi001070l. [DOI] [PubMed] [Google Scholar]
- 14.Goc A, Chami M, Lodowski DT, Bosshart P, Moiseenkova-Bell V, Baehr W, Engel A, Palczewski K. Structural Characterization of the Rod cGMP Phosphodiesterase 6. J Mol Biol. 2010;401:363–373. doi: 10.1016/j.jmb.2010.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pandit J, Forman MD, Fennell KF, Dillman KS, Menniti FS. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc Natl Acad Sci U S A. 2009;106:18225–18230. doi: 10.1073/pnas.0907635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu RX, Hassell AM, Vanderwall D, Lambert MH, Holmes WD, Luther MA, Rocque WJ, Milburn MV, Zhao Y, Ke H, Nolte RT. Atomic structure of PDE4: insights into phosphodiesterase mechanism and specificity. Science. 2000;288:1822–1825. doi: 10.1126/science.288.5472.1822. [DOI] [PubMed] [Google Scholar]
- 17.Sung BJ, Hwang KY, Jeon YH, Lee JI, Heo YS, Kim JH, Moon J, Yoon JM, Hyun YL, Kim E, Eum SJ, Park SY, Lee JO, Lee TG, Ro S, Cho JM. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature. 2003;425:98–102. doi: 10.1038/nature01914. [DOI] [PubMed] [Google Scholar]
- 18.Barren B, Gakhar L, Muradov H, Boyd KK, Ramaswamy S, Artemyev NO. Structural basis of phosphodiesterase 6 inhibition by the C-terminal region of the gammasubunit. EMBO J. 2009;28:3613–3622. doi: 10.1038/emboj.2009.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grizot S, Faure J, Fieschi F, Vignais PV, Dagher MC, Pebay-Peyroula E. Crystal structure of the Rac1-RhoGDI complex involved in nadph oxidase activation. Biochemistry. 2001;40:10007–10013. doi: 10.1021/bi010288k. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Constantine R, Vorobiev S, Chen Y, Seetharaman J, Huang YJ, Xiao R, Montelione GT, Gerstner CD, Davis MW, Inana G, Whitby FG, Jorgensen EM, Hill CP, Tong L, Baehr W. UNC119 is required for G protein trafficking in sensory neurons. Nat Neurosci. 2011;14:874–880. doi: 10.1038/nn.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kajimura N, Yamazaki M, Morikawa K, Yamazaki A, Mayanagi K. Three-dimensional structure of non-activated cGMP phosphodiesterase 6 and comparison of its image with those of activated forms. J Struct Biol. 2002;139:27–38. doi: 10.1016/s1047-8477(02)00502-6. [DOI] [PubMed] [Google Scholar]
- 22.Granovsky AE, Natochin M, McEntaffer RL, Haik TL, Francis SH, Corbin JD, Artemyev NO. Probing domain functions of chimeric PDE6alpha’/PDE5 cGMP-phosphodiesterase. J Biol Chem. 1998;273:24485–24490. doi: 10.1074/jbc.273.38.24485. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Bulgakov OV, Wen XH, Woodruff ML, Pawlyk B, Yang J, Fain GL, Sandberg MA, Makino CL, Li T. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 2004;101:13903–13908. doi: 10.1073/pnas.0405160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolandaivelu S, Huang J, Hurley JB, Ramamurthy V. AIPL1, a protein associated with childhood blindness, interacts with alpha-subunit of rod phosphodiesterase (PDE6) and is essential for its proper assembly. J Biol Chem. 2009;284:30853–30861. doi: 10.1074/jbc.M109.036780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada T, Le Trong I, Fox BA, Behnke CA, Stenkamp RE, Palczewski K. X-Ray diffraction analysis of three-dimensional crystals of bovine rhodopsin obtained from mixed micelles. J Struct Biol. 2000;130:73–80. doi: 10.1006/jsbi.1999.4209. [DOI] [PubMed] [Google Scholar]
- 26.Wang G. NMR of membrane-associated peptides and proteins. Curr Protein Pept Sci. 2008;9:50–69. doi: 10.2174/138920308783565714. [DOI] [PubMed] [Google Scholar]
- 27.Dobrovetsky E, Menendez J, Edwards AM, Koth CM. A robust purification strategy to accelerate membrane proteomics. Methods. 2007;41:381–387. doi: 10.1016/j.ymeth.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Caffrey M. Membrane protein crystallization. J Struct Biol. 2003;142:108–132. doi: 10.1016/s1047-8477(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 29.Cudney R, Patel S, Weisgraber K, Newhouse Y, McPherson A. Screening and optimization strategies for macromolecular crystal growth. Acta Crystallogr D Biol Crystallogr. 1994;50:414–423. doi: 10.1107/S0907444994002660. [DOI] [PubMed] [Google Scholar]
- 30.Papermaster DS. Preparation of retinal rod outer segments. Methods Enzymol. 1982;81:48–52. doi: 10.1016/s0076-6879(82)81010-0. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Brown RL. Functional regions of the inhibitory subunit of retinal rod cGMP phosphodiesterase identified by site-specific mutagenesis and fluorescence spectroscopy. Biochemistry. 1992;31:5918–5925. doi: 10.1021/bi00140a031. [DOI] [PubMed] [Google Scholar]
- 33.Goc A, Angel TE, Jastrzebska B, Wang B, Wintrode PL, Palczewski K. Different properties of the native and reconstituted heterotrimeric G protein transducin. Biochemistry. 2008;47:12409–12419. doi: 10.1021/bi8015444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pentia DC, Hosier S, Collupy RA, Valeriani BA, Cote RH. Purification of PDE6 isozymes from mammalian retina. Methods Mol Biol. 2005;307:125–140. doi: 10.1385/1-59259-839-0:125. [DOI] [PubMed] [Google Scholar]
- 35.Catty P, Pfister C, Bruckert F, Deterre P. The cGMP phosphodiesterase-transducin complex of retinal rods. Membrane binding and subunits interactions. J Biol Chem. 1992;267:19489–19493. [PubMed] [Google Scholar]
- 36.Gillespie PG, Beavo JA. Inhibition and stimulation of photoreceptor phosphodiesterases by dipyridamole and M&B 22,948. Mol Pharmacol. 1989;36:773–781. [PubMed] [Google Scholar]
- 37.Gillespie PG, Beavo JA. cGMP is tightly bound to bovine retinal rod phosphodiesterase. Proc Natl Acad Sci U S A. 1989;86:4311–4315. doi: 10.1073/pnas.86.11.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillespie PG, Prusti RK, Apel ED, Beavo JA. A soluble form of bovine rod photoreceptor phosphodiesterase has a novel 15-kDa subunit. J Biol Chem. 1989;264:12187–12193. [PubMed] [Google Scholar]
- 39.Mou H, Grazio HJ, 3rd, Cook TA, Beavo JA, Cote RH. cGMP binding to noncatalytic sites on mammalian rod photoreceptor phosphodiesterase is regulated by binding of its gamma and delta subunits. J Biol Chem. 1999;274:18813–18820. doi: 10.1074/jbc.274.26.18813. [DOI] [PubMed] [Google Scholar]
- 40.Muradov H, Boyd KK, Artemyev NO. Rod phosphodiesterase-6 PDE6A and PDE6B subunits are enzymatically equivalent. J Biol Chem. 2010;285:39828–39834. doi: 10.1074/jbc.M110.170068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malinski JA, Wensel TG. Membrane stimulation of cGMP phosphodiesterase activation by transducin: comparison of phospholipid bilayers to rod outer segment membranes. Biochemistry. 1992;31:9502–9512. doi: 10.1021/bi00154a024. [DOI] [PubMed] [Google Scholar]
- 42.Wensel TG, Stryer L. Reciprocal control of retinal rod cyclic GMP phosphodiesterase by its gamma subunit and transducin. Proteins. 1986;1:90–99. doi: 10.1002/prot.340010114. [DOI] [PubMed] [Google Scholar]
- 43.Skiba NP, Artemyev NO, Hamm HE. The carboxyl terminus of the gammasubunit of rod cGMP phosphodiesterase contains distinct sites of interaction with the enzyme catalytic subunits and the alpha-subunit of transducin. J Biol Chem. 1995;270:13210–13215. doi: 10.1074/jbc.270.22.13210. [DOI] [PubMed] [Google Scholar]
- 44.Takemoto DJ, Hurt D, Oppert B, Cunnick J. Domain mapping of the retinal cyclic GMP phosphodiesterase gamma-subunit. Function of the domains encoded by the three exons of the gamma-subunit gene. Biochem J. 1992;281(Pt 3):637–643. doi: 10.1042/bj2810637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muradov KG, Granovsky AE, Schey KL, Artemyev NO. Direct interaction of the inhibitory gamma-subunit of Rod cGMP phosphodiesterase (PDE6) with the PDE6 GAFa domains. Biochemistry. 2002;41:3884–3890. doi: 10.1021/bi015935m. [DOI] [PubMed] [Google Scholar]
- 46.Guo LW, Assadi-Porter FM, Grant JE, Wu H, Markley JL, Ruoho AE. One-step purification of bacterially expressed recombinant transducin alpha-subunit and isotopically labeled PDE6 gamma-subunit for NMR analysis. Protein Expr Purif. 2007;51:187–197. doi: 10.1016/j.pep.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 47.Uversky VN, Permyakov SE, Zagranichny VE, Rodionov IL, Fink AL, Cherskaya AM, Wasserman LA, Permyakov EA. Effect of zinc and temperature on the conformation of the gamma subunit of retinal phosphodiesterase: a natively unfolded protein. J Proteome Res. 2002;1:149–159. doi: 10.1021/pr0155127. [DOI] [PubMed] [Google Scholar]
- 48.Pentia DC, Hosier S, Cote RH. The glutamic acid-rich protein-2 (GARP2) is a high affinity rod photoreceptor phosphodiesterase (PDE6)-binding protein that modulates its catalytic properties. J Biol Chem. 2006;281:5500–5505. doi: 10.1074/jbc.M507488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.