

Abstract
The nonmevalonate pathway (NMP) of isoprene biosynthesis is an exciting new route toward novel antibiotic development. Inhibitors against several enzymes in this pathway are currently under examination. A significant liability of many of these agents is poor cell penetration. To overcome and improve our understanding of this problem, we have synthesized a series of lipophilic, prodrug analogs of fosmidomycin and FR900098, inhibitors of the NMP enzyme Dxr. Several of these compounds show improved antibacterial activity against a panel of organisms relative to the parent compound, including activity against Mycobacterium tuberculosis (Mtb). Our results show that this strategy can be an effective way for improving whole cell activity of NMP inhibitors.
There continues to be a pressing need for drugs with novel mechanisms of action. Most clinically useful anti-infectives target only a few of the hundreds of potential targets in metabolism, and global rates of drug resistant bacteria are on the rise.1,2 For example, in 2010, 2 million people died from tuberculosis, caused by M. tuberculosis (Mtb), including approximately 150,000 individuals who died from one of several multi-drug resistant strains.3,4 Without new therapeutics working through unique targets, drug resistance and decreased drug susceptibility will continue to be a public health concern.1,2
Recently, the nonmevalonate pathway (NMP) has been examined as a novel route against bacteria and parasites.5–10 The role of the NMP is to synthesize activated five carbon units that the cell will elaborate into more complex structures. Humans use the mevalonate pathway to biosynthesize the same isoprenoid units. As such, the enzymes found in the NMP are not found in humans, leading to the interest in this pathway for antibacterial drug targeting. While many of the enzymes in the NMP have been examined, 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Dxr or IspC) has been studied to the greatest extent.5,6 This enzyme is responsible for reducing and isomerizing 1-deoxy-D-xylulose 5-phosphate (DXP or DOXP) to 2-C-methyl-D-erythritol 3-phosphate (MEP). Several crystal structures of Dxr from various bacteria have been reported.11,12 Most of the work developing inhibitors against Dxr has been in the context of Plasmodium falciparum13–30 although more recent studies have targeted Dxr from Mtb.31,32
Two well-known inhibitors of Dxr, fosmidomycin (1) and FR900098 (2), are natural products and mimic Dxr’s substrate DXP (Figure 1). Both compounds are quite potent inhibitors against Dxr from a variety of organisms.33,34 Interestingly, however, while fosmidomycin is active against some bacteria and P. falciparum, it is inactive against Mtb.35,36 Mtb relies on Dxr for isoprene production35 but does not have a GlpT transporter used by many organisms for fosmidomycin uptake.37 This, combined with the polar nature of fosmidomycin and the lipophilic tuberculosis cell wall, is thought to be the underlying reason for the lack of effect of fosmidomycin against Mtb.35,36 Our work aims to develop novel inhibitors of Dxr and use them as tools to examine this enzyme for its potential as an antibacterial target. As a proof of this concept and expanding on prior work from the malaria field,13–30 we present a series of analogs of compounds 1 and 2 designed to understand if added lipophilicity increases cell penetration and antibacterial activity. It is expected that these prodrugs enter the cell and are hydrolyzed by nonspecific, cellular esterases to reveal the phosphonate which then inhibits Dxr.29 The compounds have the general structure shown in Figure 1.
Figure 1.

Structures of parent Dxr inhibitors (1 and 2) and lipophilic esters.
There are several published synthetic routes for fosmidomycin derivatives.13–22,24–28,30–32 We combined and modified these routes to yield the best overall synthetic schemes. The syntheses of the target compounds are shown in Schemes 1–3. Diethyl fosmidomycin (10)25 was prepared using the route shown in Scheme 1. Diethyl phosphite (3) was combined with O-benzylhydroxylamine (4) to yield phosphoramidate (5).38 This compound was combined with diethyl 3-bromopropylphosphonate (6) to yield compound 7.39 The phosphoramide of 7 was cleaved using acidic conditions to yield compound 8.39 Compound 8 was formylated21 and debenzylated25 to give compounds 9 and 10, respectively.
Scheme 1.

Reagents and conditions: (a) TBABr, CCl4, KHCO3, K2CO3, CH2Cl2; (b) NaH, TBABr, NaI, THF; (c) HCl, EtOH; (d) (CH3CO)2O, HCO2H; (e) H2, 10% Pd/C.
Scheme 3.

Reagents and conditions: (a) HCl, 80 °C, 1 h; (b) i: BnONH2, MeOH, reflux, 1 h; ii: NaBH3CN, MeOH, HCl, rt, 1 h; (c) AcCl, TEA, CH2Cl2, rt, 18 h; (d) i: TMSBr, CH2Cl2, 0 °C, 30 min; ii: H2O, rt, 18 h; (e) RBr or RCl, TEA, DMF or DMPU, 60 °C, 6 h; (f) H2, 10% Pd/C, MeOH.
Two routes were used in the preparation of lipophilic esters of FR900098 (Schemes 2 and 3). Scheme 2 begins with the lipophilic moieties installed on the phosphonate (compounds 12). To prepare ethyl analog 15, O-benzylhydroxylamine was acetylated40 and combined with 12a (prepared using microwave conditions41) to yield compound 14a.25 For isopropyl analog 16, Boc-protected 11 was alkylated with compound 12b to give 13. Removal of the Boc group, followed by acetylation yielded compound 14b. Hydrogenation to remove the benzyl group gave target compounds 1542 and 16.43
Scheme 2.

Reagents and conditions: (a) NaH, NaI, DMF; (b) i: TFA, CH2Cl2, ii: TEA, AcCl, CH2Cl2, 18 h; (c) H2, 10% Pd/C, MeOH, 2 h.
Most of the lipophilic esters were prepared using the route shown in Scheme 3.25 This scheme uses a shorter, but more costly, path to compound 8. Diethyl acetal-protected diethyl phosphonate ester 17 was deprotected to give aldehyde 18. This compound underwent reductive amination with O-benzylhydroxylamine and NaBH3CN to yield compound 8. Acetylation yielded compound 14a. Removal of the diethyl ester using TMSBr gave common intermediate 19. Interestingly, despite its use by many groups, the TMSBr deprotection step is not well described. In our hands, the addition of water after deprotection does not yield a solid. The solid, deprotected product is isolated only after complete removal of water or with the addition of THF to facilitate precipitation. Intermediate 19 was alkylated with several different alkyl groups to give compounds 20a–g. Subsequent hydrogenation yielded target compounds 21–27. The analytical data for compounds 22, 24, and 27 matches literature values.25 Experimental details are given for new compounds 2144, 2345, 2546, and 2647.
The compounds were examined for antibacterial and antitubercular activities using methods previously reported.48,49 The activities are shown in Table 1. The two parent compounds, fosmidomycin (1) and FR900098 (2) show activity against Gram (−) strains but this activity is less pronounced against the panel of Gram (+) organisms. The exception is fosmidomycin’s potent activity against B. anthracis (0.78 μg/mL). As has been demonstrated by others35,36, fosmidomycin does not have antitubercular activity (MIC >500 μg/mL) and this is also the case for its acetyl derivative, FR900098, which is inactive.
Table 1.
Antibacterial activities of compounds 1, 2, 10, 15, 16 and 21–27.a
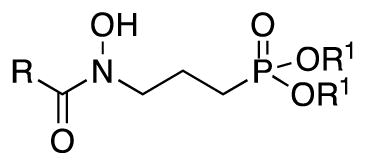
| Compound | R | R1 | Gram (+)
|
Gram (−)
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B. anthracis | E. faecalis | S. aureus (MSSA) | S. aureus (MRSA) | M. tuberculosis (H37Rv) | Acineto-bacter | E. coli k12 | E. coli tolc | |||
| Fosmidomycin (1) | H | H/Nab | 0.78 | >200 | >200 | 200 | >500 | 100 | 12.5 | 6.25 |
| FR900098 (2) | CH3 | H/Nab | 50 | >200 | >200 | 50 | >500 | 50 | 200 | 12.5 |
| 10 | H | Et | >200 | >200 | >200 | >200 | 400 | >200 | >200 | >200 |
| 15 | CH3 | Et | 200 | >200 | >200 | 200 | 200–400 | >200 | >200 | >200 |
| 16 | CH3 | iPr | >200 | >200 | >200 | >200 | 400 | >200 | >200 | >200 |
| 21 | CH3 |
|
NDc | ND | ND | ND | 100 | ND | ND | ND |
| 22 | CH3 |
|
>200 | >200 | >200 | >200 | 400 | >200 | >200 | 200 |
| 23 | CH3 |
|
>200 | >200 | 100 | 25 | 400 | >200 | >200 | >200 |
| 24 | CH3 |
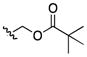
|
200 | >200 | 100 | 50 | 50–100 | >200 | >200 | 100 |
| 25 | CH3 |
|
200 | >200 | 100 | 50 | 400 | >200 | >200 | >200 |
| 26 | CH3 |
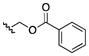
|
100 | >200 | >200 | >200 | 25–100 | 200 | >200 | 50 |
| 27 | CH3 |
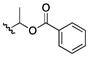
|
50 | >200 | >200 | >200 | 100–200 | 100 | 50 | 50 |
Minimum Inhibitory Concentrations (MIC) in μg/mL.
Compounds 1 and 2 used as the monosodium salts.
ND = not determined.
Several of the lipophilic esters showed improved activity relative to fosmidomycin, particularly for Gram (+) bacteria. While diethyl fosmidomycin (10) showed little activity against the panel, as the size of the lipophilic ester increased, activity against these organisms generally increased as well. For example, 24 and 26 showed better antitubercular activity compared with compounds 10, 15 and 16. Interestingly, analogs with a secondary ester (22, 25 and 27) did not outperform their primary counterparts. A possible explanation for this could be that the cellular esterases needed to reveal the phosphonate do not tolerate branching within the ester. Lack of activity of this family of esters in certain strains may be due to efflux mechanisms or inefficient cleavage of the ester.
Our results suggest that lipophilic esters of fosmidomycin and FR900098 improve cell activity and antibacterial activity for certain organisms. While fosmidomycin takes advantage of GlpT for cell entry, these lipophilic esters are not reliant on this transporter.50 As glpT mutation is the only documented path toward fosmidomycin resistance, we expect these compounds to avoid such a resistance pathway. Dxr inhibitors optimized for both cell penetration and the enzyme’s active site could provide an important tool for target validation on the road toward development of a novel therapeutic.
Acknowledgments
This work was supported by funding from the GWU Department of Chemistry, the GWU University Facilitating Fund, the Intramural Research Program of the NIAID (NIH), the American Lebanese Syrian Associated Charities (ALSAC), and NIH (AI086453 to CSD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat Rev Drug Discov. 2007;6:29. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 2.Silver LL. Clin Microbiol Rev. 2011;24:71. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 4.http://www.who.int/mediacentre/factsheets/fs194/en/index.html.
- 5.Eoh H, Brennan PJ, Crick DC. Tuberculosis (Edinb) 2008 doi: 10.1016/j.tube.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Proteau PJ. Bioorg Chem. 2004;32:483. doi: 10.1016/j.bioorg.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Rohdich F, Bacher A, Eisenreich W. Bioorg Chem. 2004;32:292. doi: 10.1016/j.bioorg.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Rohdich F, Bacher A, Eisenreich W. Biochem Soc Trans. 2005;33:785. doi: 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- 9.Singh N, Cheve G, Avery MA, McCurdy CR. Current Pharmaceutical design. 2007;13:1161. doi: 10.2174/138161207780618939. [DOI] [PubMed] [Google Scholar]
- 10.Testa CA, Brown MJ. Curr Pharm Biotechnol. 2003;4:248. doi: 10.2174/1389201033489784. [DOI] [PubMed] [Google Scholar]
- 11.Henriksson LM, Unge T, Carlsson J, Aqvist J, Mowbray SL, Jones TA. J Biol Chem. 2007;282:19905. doi: 10.1074/jbc.M701935200. [DOI] [PubMed] [Google Scholar]
- 12.Mac Sweeney A, Lange R, Fernandes RP, Schulz H, Dale GE, Douangamath A, Proteau PJ, Oefner C. J Mol Biol. 2005;345:115. doi: 10.1016/j.jmb.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 13.Devreux V, Wiesner J, Goeman JL, Van der Eycken J, Jomaa H, Van Calenbergh S. J Med Chem. 2006;49:2656. doi: 10.1021/jm051177c. [DOI] [PubMed] [Google Scholar]
- 14.Devreux V, Wiesner J, Jomaa H, Rozenski J, Van der Eycken J, Van Calenbergh S. J Org Chem. 2007;72:3783. doi: 10.1021/jo0700981. [DOI] [PubMed] [Google Scholar]
- 15.Devreux V, Wiesner J, Jomaa H, Van der Eycken J, Van Calenbergh S. Bioorg Med Chem Lett. 2007;17:4920. doi: 10.1016/j.bmcl.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 16.Haemers T, Wiesner J, Busson R, Jomaa H, Van Calenbergh S. Eur J Org Chem. 2006:3856. [Google Scholar]
- 17.Haemers T, Wiesner J, Van Poecke S, Goeman J, Henschker D, Beck E, Jomaa H, Van Calenbergh S. Bioorg Med Chem Lett. 2006;16:1888. doi: 10.1016/j.bmcl.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 18.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Science. 1999;285:1573. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 19.Kuntz L, Tritsch D, Grosdemange-Billiard C, Hemmerlin A, Willem A, Bach TJ, Rohmer M. Biochem J. 2005;386:127. doi: 10.1042/BJ20041378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurz T, Behrendt C, Kaula U, Bergmann B, Walter RD. Aust J Chem. 2007;60:154. doi: 10.1002/ardp.200700107. [DOI] [PubMed] [Google Scholar]
- 21.Kurz T, Behrendt C, Pein M, Kaula U, Bergmann B, Walter RD. Arch Pharm (Weinheim) 2007;340:661. doi: 10.1002/ardp.200700107. [DOI] [PubMed] [Google Scholar]
- 22.Kurz T, Schluter K, Kaula U, Bergmann B, Walter RD, Geffken D. Bioorg Med Chem. 2006;14:5121. doi: 10.1016/j.bmc.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Kurz T, Schluter K, Pein M, Behrendt C, Bergmann B, Walter RD. Arch Pharm (Weinheim) 2007;340:339. doi: 10.1002/ardp.200700013. [DOI] [PubMed] [Google Scholar]
- 24.Ortmann R, Wiesner J, Reichenberg A, Henschker D, Beck E, Jomaa H, Schlitzer M. Bioorg Med Chem Lett. 2003;13:2163. doi: 10.1016/s0960-894x(03)00354-8. [DOI] [PubMed] [Google Scholar]
- 25.Ortmann R, Wiesner J, Reichenberg A, Henschker D, Beck E, Jomaa H, Schlitzer M. Arch Pharm (Weinheim) 2005;338:305. doi: 10.1002/ardp.200500976. [DOI] [PubMed] [Google Scholar]
- 26.Ortmann R, Wiesner J, Silber K, Klebe G, Jomaa H, Schlitzer M. Arch Pharm (Weinheim) 2007;340:483. doi: 10.1002/ardp.200700149. [DOI] [PubMed] [Google Scholar]
- 27.Reichenberg A, Wiesner J, Weidemeyer C, Dreiseidler E, Sanderbrand S, Altincicek B, Beck E, Schlitzer M, Jomaa H. Bioorg Med Chem Lett. 2001;11:833. doi: 10.1016/s0960-894x(01)00075-0. [DOI] [PubMed] [Google Scholar]
- 28.Schluter K, Walter RD, Bergmann B, Kurz T. Eur J Med Chem. 2006;41:1385. doi: 10.1016/j.ejmech.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 29.Schultz C. Bioorg Med Chem. 2003;11:885. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- 30.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. Arch Pharm (Weinheim) 2007;340:667. doi: 10.1002/ardp.200700069. [DOI] [PubMed] [Google Scholar]
- 31.Andaloussi M, Henriksson LM, Wieckowska A, Lindh M, Bjorkelid C, Larsson AM, Suresh S, Iyer H, Srinivasa BR, Bergfors T, Unge T, Mowbray SL, Larhed M, Jones TA, Karlen A. Journal of medicinal chemistry. 2011;54:4964. doi: 10.1021/jm2000085. [DOI] [PubMed] [Google Scholar]
- 32.Deng L, Diao J, Chen P, Pujari V, Yao Y, Cheng G, Crick DC, Prasad BV, Song Y. Journal of medicinal chemistry. 2011;54:4721. doi: 10.1021/jm200363d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kojo H, Shigi Y, Nishida M. The Journal of antibiotics. 1980;33:44. doi: 10.7164/antibiotics.33.44. [DOI] [PubMed] [Google Scholar]
- 34.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot (Tokyo) 1980;33:13. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- 35.Brown AC, Parish T. BMC Microbiol. 2008;8:78. doi: 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhiman RK, Schaeffer ML, Bailey AM, Testa CA, Scherman H, Crick DC. J Bacteriol. 2005;187:8395. doi: 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakamoto Y, Furukawa S, Ogihara H, Yamasaki M. Biosci Biotechnol Biochem. 2003;67:2030. doi: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- 38.Zwierzak A, Osowska K. Synthesis. 1984:223. [Google Scholar]
- 39.Blazewska K, Gajda T. Tetrahedron. 2003;59:10249. [Google Scholar]
- 40.Haemers T, Wiesner J, Giessmann D, Verbrugghen T, Hillaert U, Ortmann R, Jomaa H, Link A, Schlitzer M, Van Calenbergh S. Bioorganic & medicinal chemistry. 2008;16:3361. doi: 10.1016/j.bmc.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Villemin D, Simeon F, Decreus H, Jaffres PA. Phosphorus, Sulfur and Silicon. 1998;133:209. [Google Scholar]
- 42.Fokin AA, Yurchenko AG, Rodionov VN, Gunchenko PA, Yurchenko RI, Reichenberg A, Wiesner J, Hintz M, Jomaa H, Schreiner PR. Organic letters. 2007;9:4379. doi: 10.1021/ol702082k. [DOI] [PubMed] [Google Scholar]
- 43.Ohler E, Kanzler S. Synthesis. 1995:539. [Google Scholar]
- 44.21. A solution of compound 20a (0.29 g, 0.63 mmol) in dry EtOH (50mL) was hydrogenated over 10% Pd/C (112.3 mg) overnight. After filtration and evaporation, the crude product was purified by column chromatography using ethyl acetate to give 0.18 g (0.48 mmol, 76%) of the desired product as a light yellow viscous oil. 1H-NMR (CDCl3, 200MHz), δ (ppm): 0.90 (t, 6H); 1.22–1.50 (m, 12H); 1.57–2.12 (m, 8H); 2.17 (s, 3H); 3.76 (t, 2H); 3.98 (q, 4H). ESI-MS m/z 366 ([M+H]+), 388 ([M+Na]+).
- 45.23. Prepared from 20c in 65% yield as described above. 1H-NMR (CDCl3, 200MHz), δ (ppm): 1.49 (s, 18H); 1.99–2.13 (m, 4H); 2.17 (s, 3H); 3.77 (t, 2H); 4.51 (d, 4H); 8.93 (bs, 1H). ESI-MS m/z 426 ([M+H]+).
- 46.25. Prepared from 20e in 43% yield as described above. 1H-NMR (CDCl3, 200MHz), δ (ppm): 1.50 (s, 18H); 1.58 (d, 6H); 1.73–1.94 (m, 4H); 1.98 (s, 3H); 3.33 (q, 2H); 6.27–6.42 (m, 2H). ESI-MS m/z 470([M-CH3]+).
- 47.26. Prepared from 20f in 59% yield as described above. 1H-NMR (CDCl3, 200MHz), δ (ppm): 1.85–2.10 (m, 4H); 2.15 (s, 3H); 3.62–3.77 (m, 2H); 5.88 (s, 2H); 5.94 (s, 2H); 7.40–7.67 (m, 6H); 7.98–8.20 (m, 4H). ESI-MS m/z 466 ([M+H]+).
- 48.Kim P, Zhang L, Manjunatha UH, Singh R, Patel S, Jiricek J, Keller TH, Boshoff HI, Barry CE, 3rd, Dowd CS. Journal of medicinal chemistry. 2009;52:1317. doi: 10.1021/jm801246z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yendapally R, Hurdle JG, Carson EI, Lee RB, Lee RE. Journal of medicinal chemistry. 2008;51:1487. doi: 10.1021/jm701356q. [DOI] [PubMed] [Google Scholar]
- 50.McKenney ES, Sargent M, Khan H, Couch RD, Uh E, Jackson ER, San Jose G, Dowd CS, Van Hoek ML. doi: 10.1371/journal.pone.0038167. manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]