

Abstract
The M2 protein of influenza A virus forms a proton-selective channel that is required for viral replication; it is also the target of the anti-influenza drugs, amantadine and rimantadine. Widespread drug-resistant mutants, however, has greatly compromised the effectiveness of these drugs. Here, we report the solution NMR structure of the highly pathogenic, drug resistant mutant V27A. The structure reveals subtle structural differences from wildtype that maybe linked to drug resistance. The V27A mutation significantly decreases hydrophobic packing between the N-terminal ends of the transmembrane helices, which explains the looser, more dynamic tetrameric assembly. The weakened channel assembly can resist drug binding either by destabilizing the rimantadine-binding pocket at Asp44, in the case of the allosteric inhibition model, or by reducing hydrophobic contacts with amantadine in the pore, in the case of the pore blocking model. Moreover, the V27A structure shows a substantially increased channel opening at the N-terminal end, which may explain the faster proton conduction observed for this mutant. Furthermore, due to the high quality NMR data recorded for the V27A mutant, we were able to determine the structured region connecting the channel domain to the C-terminal amphipathic helices that was not determined in the wildtype structure. The new structural data show that the amphipathic helices are packed much more closely to the channel domain and provide new insights into the proton transfer pathway.
Keywords: influenza M2, V27A mutant, flu drug resistance, proton channel
Introduction
Matrix protein 2, M2, forms a highly selective proton channel that is an important constituent of the influenza virus. It equilibrates pH across the viral membrane during viral entry and across the trans-Golgi membrane of infected cells during viral maturation [1; 2; 3]. It is necessary for viral replication and therefore remains an attractive target for ongoing studies aiming at developing anti-influenza drugs. In fact, two similar M2 inhibitors, amantadine and rimantadine, had been successfully used for treating flu A infections [4], but emergence of drug resistant strains has severely compromised the effectiveness of these compounds [5]. Single amino acid substitutions at positions 26, 27, 30, 31, and 34 have been reported to confer drug resistance [1; 6; 7]. Recent studies indicate that the resistance is rising and now exceeds 90%, with S31N being the most frequent substitution [5; 8; 9]. Another common drug resistant mutant is V27A, which sometimes coexists with S31N mutation [10; 11]. It has been suggested that the mechanism of V27A resistance may be different than that of S31N [12]. Therefore, in order to fully understand the resistance, it is of high importance to obtain structural data on the V27A mutant.
Early structural characterization of the M2 TM peptide by solid-state NMR (ssNMR) have converged on a model of the channel domain [13; 14]. In this model, the TM peptides form a left-handed four-helix bundle with a well defined hydrophilic pore. The model shows that the two key gating residues, His37 and Trp41, are inside the pore, and that they physically occlude the C-terminal end of the channel. Recently, high resolution structures of the M2 channel have been determined by X-ray crystallography [15] and solution NMR [16]. The crystal structures of the TM peptide M222-46 were determined at pH 5.3 and 7.3 [15]. Unlike the previous models, the crystal structures show very wide opening at the C-terminal end of the channel, which was interpreted as the open state [15]. The solution NMR structure was determined for a longer construct M218-60 at pH 7.5, which shows tight assembly of the TM helices (residues 24-46) and the AP helices (residues 52-60) that is consistent with being in the closed state at the experimental pH [16].
The major controversy between the M222-46 crystal structure and the M218-60 solution structure resides in the location of drug binding. In the M222-46 structure, amantadine binds inside of the channel pore, where the hydrophobic adamantyl cage is coordinated by serine hydroxyls and the amine group of the drug does not appear to form any short-range, inter-molecular interactions [15]. The crystal structure led to the proposal that the drug directly blocks proton conduction by physically obstructing the pore [15]. In the M218-60 structure, rimantadine binds to the external face of the channel, between two adjacent TM helices, where the amine group of rimantadine is within hydrogen bond distance from the carboxyl of Asp44, and the adamantyl cage interacts with the hydrophobic side chains of Leu40, Ile42, and Leu43 [16]. This lipid-facing binding site suggests an allosteric inhibition mechanism, wherein drug binding stabilizes the closed state.
A recent ssNMR study of M222-46 in lipid bilayer reported that both drug sites are present, with the pore site having higher affinity for amantadine than the lipid-facing site [17]. This study also suggests that, upon binding to the channel pore, amantadine tumbles isotropically and does not appear to form any specific interaction with the pore-lining residues [17]. In contrast, another ssNMR study of M218-60, also carried out in a lipid bilayer, showed that rimantadine binding exerts a long-range structural effect on the channel that is more consistent with the allosteric inhibition mechanism [18]. It is not clear if the discrepancy between the different studies is due to the highly truncated construct used, experimental artifacts, or other factors.
In this study, we determined the solution NMR structure of the V27A18-60 drug resistant mutant. The similarities and differences in the structure and dynamic properties between the wildtype (WT), V27A, and S31N variants allowed an in-depth analysis of possible modes of drug resistance. Distinct feature of the V27A channel pore also provides an explanation for its faster rate of proton conduction. Furthermore, owing to the higher quality NMR data acquired for the C-terminal region of V27A18-60 (as compared to that of WT), the mutant structure shows more clearly the structured region that connects the channel domain to the C-terminal AP helices. Using the overall better defined structure, we propose a proton transfer pathway during M2 proton conduction.
Materials and Methods
Protein expression, purification, labeling, and NMR sample preparation
V27A18–60 (RSNDSSDPLAVAASIIGILHLILWILDRLFFKSIYRFFEHGLK) was cloned, expressed and purified as previously described [16]. Briefly, the protein was expressed into inclusion bodies as a fusion to His9–trpLE. The M2 peptide was cleaved using cyanogen bromide in 70% formic acid. The digest was dialyzed against water, lyophilized, and loaded onto a C4 column (Grace–Vydac) in 2:1:2 hexafluoroisopropanol:formic acid:water and separated using a gradient of 3:2 isopropanol:acetonitrile. For the NMR study, the lyophilized peptide was then refolded at 250 mM by dissolving in 6 M guanidine and 150 mM DHPC and dialyzing against the final NMR buffer containing 40 mM sodium phosphate and 30 mM glutamate. The sample was concentrated to a final concentration of 0.75 mM (monomer). Rimantadine was added after concentrating.
Liposomal proton flux assay
Liposome assay for M2 channels was established based on works from the Schroeder, Miller and Busath labs [25; 26; 27] as described earlier [19]. Briefly, in this assay, a proton gradient was used to drive proton conduction. Liposomes were made with identical pH and ion concentrations inside and outside, but highly buffered inside and only weakly buffered outside. Protein-mediated conductance of protons from the external bath into the liposome interior was initiated by adding hydrochloric acid under continuous rapid mixing. Proton flux was monitored as an increase in pH of the external bath.
NMR spectroscopy
NMR experiments were conducted at 30°C on spectrometers equipped with cryogenic probes (Bruker, Billerica, MA). Sequence specific assignment of backbone 1HN, 15N, and 13Cα chemical shifts were accomplished using a combination of the available WT(18-60) resonance assignments and a pair of tr-HNCA and tr-HNCOCA experiments [28; 29] recorded with a 15N-, 13C-, and 85% 2H-labeled protein. NOEs involving both backbone and sidechain protons were assigned using the 3D 15N-edited and 13C-edited NOESYs recorded with NOE mixing times of 110 and 150 ms, respectively, on a sample containing 15N-, 13C-labeled protein, rimantadine, and deuterated DHPC (D22-DHPC) (Avanti Polar Lipids, Inc.).
Structure determination of the V27A mutant
The M218-60 structure was obtained by refining a homology model derived from the WT18-60 structure against V27A NMR restraints (including 948 intra- and 60 inter-subunit NOE-derived distance restraints). A simulated annealing protocol was implemented in the program XPLOR-NIH [30], in which the bath is cooled from 1000 to 20 K with a temperature step of 20 K, and 6.7 ps of Verlet dynamics at each temperature step, using a time step of 3 fs. The force constant for distance restraints is ramped from 25 to 50 kcal mol-1 Å-2, whereas that for backbone dihedral angles is ramped from 10 to 30 kcal mol-1 rad-2. The structural restraints yielded an ensemble of 15 structures with rmsd of 0.92 Å for backbone and 1.37 Å for all heavy atoms (refinement statistics are given in Table 1). The structure of the V27A amantadine-resistant mutant has been deposited to the Protein Data Bank with PDB accession code 2KWX.
Table 1. NMR and refinement statistics for the V27A mutant of M2.
| Residues 24 – 60 | |
|---|---|
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 252 × 4 |
| Intra-residue | 68 × 4 |
| Inter-residue | 169 × 4 |
| Sequential (|i − j|= 1) | 100 × 4 |
| Medium-range (|i −j| < 4) | 64 × 4 |
| Long-range (|i − j| > 5) | 5 × 4 |
| Intermolecular | 15 × 4 |
| Hydrogen bonds | 42 × 4 |
| Total dihedral angle restraints | 60 × 4 |
| φ (TALOS) | 30 × 4 |
| ψ (TALOS) | 30 × 4 |
| Structure statistics | |
| Violations (mean ± s.d.) | |
| Distance constraints (Å) | 0.049 ± 0.002 |
| Dihedral angle constraints (°) | 1.877 ± 0.121 |
| Max. dihedral angle violation (°) | 6.740 |
| Max. distance constraint violation (Å) | 0.171 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.003 ± 0.000 |
| Bond angles (°) | 0.497 ± 0.005 |
| Impropers (°) | 0.322 ± 0.005 |
| Average pairwise r.m.s. deviation** (Å) | |
| Heavy | 1.370 |
| Backbone | 0.923 |
Pairwise r.m.s. deviation was calculated among 15 refined structures.
Results and Discussion
Structure of the V27A drug resistant mutant
In order to directly compare the structure of the V27A mutant with that of the WT and the S31N mutant, we examined the V27A18-60 construct under the same conditions as those used in the NMR studies of WT18-60 [16] and S31N18-60 [19]. Under these conditions the V27A18-60 mutant yields NMR spectrum that is similar to those of WT and S31N, although demonstrates overall better spectral quality (Figure 1A). Using a previously established liposomal proton flux assay [19], we showed that the V27A18-60 peptide reconstituted in liposomes conduct protons ∼ 2 times faster than WT at pH ∼6 and that addition of 50 μM rimantadine only inhibited conductance by ∼1%, compared to near complete inhibition of WT18-60 by 50 μM rimantadine (Supplementary Figure S1).
Figure 1.
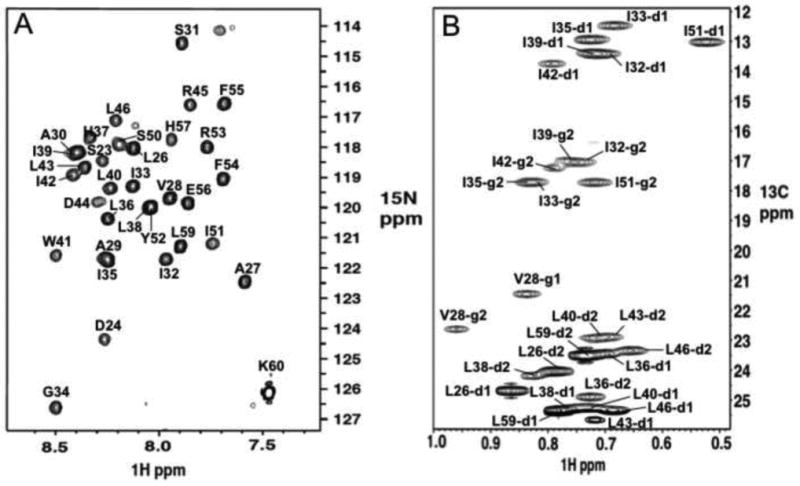
Amide and methyl spectra of the V27A18-60 mutant display residue specific assignment of backbone amine and sidechain methyl groups, respectively. a. 1H-15N tr-HSQC spectrum of 15N-, 13C, and 85% 2H-labeled protein. b. 1H-13C HSQC spectrum of methyl groups of uniformly 15N-, 13C-labeled protein. Both spectra were recorded at 30°C, pH 7.5, and 1H frequency of 600MHz.
Figure 1A shows significantly better resonance homogeneity (than that for WT [16]) that allowed the assignment of nuclear Overhauser enhancements (NOEs) between the TM helix residues 46 and residues 51 and 54 of the AP helices. These long-range NOEs were not observed in the previous structural study of WT18-60 due to resonance broadening. The near absence of distance restraints between the TM and AP helices resulted in a “detachment” of the AP helices from the channel domain in the WT (Figure 2A&C). The NMR structure of V27A18-60 was determined by a total of 1008 NOE restraints (see Table 1 for structural statistics). Compared to WT, the structure ensemble of V27A shows much better defined arrangement of the AP helices relative to the pore domain (Figure 2B&D).
Figure 2. Structures of WT and the V27A mutant.
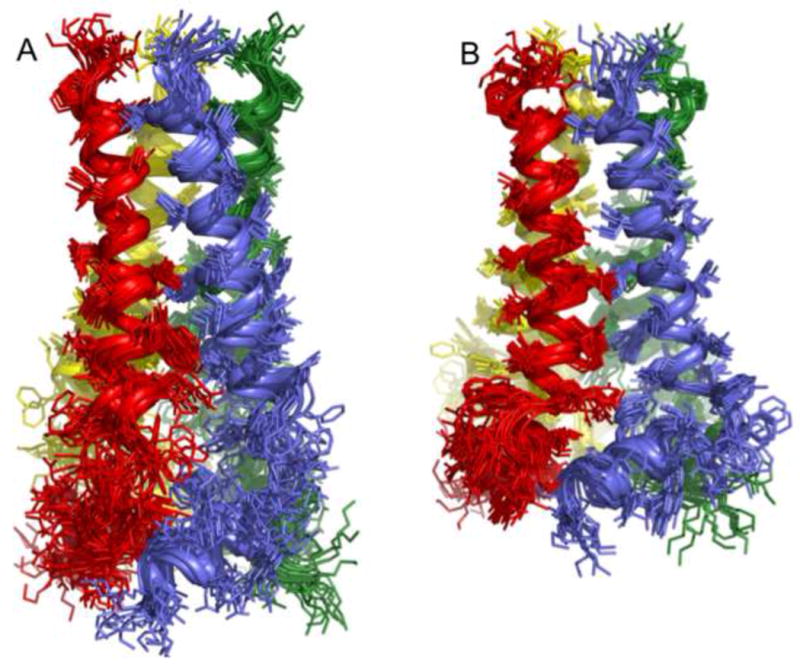
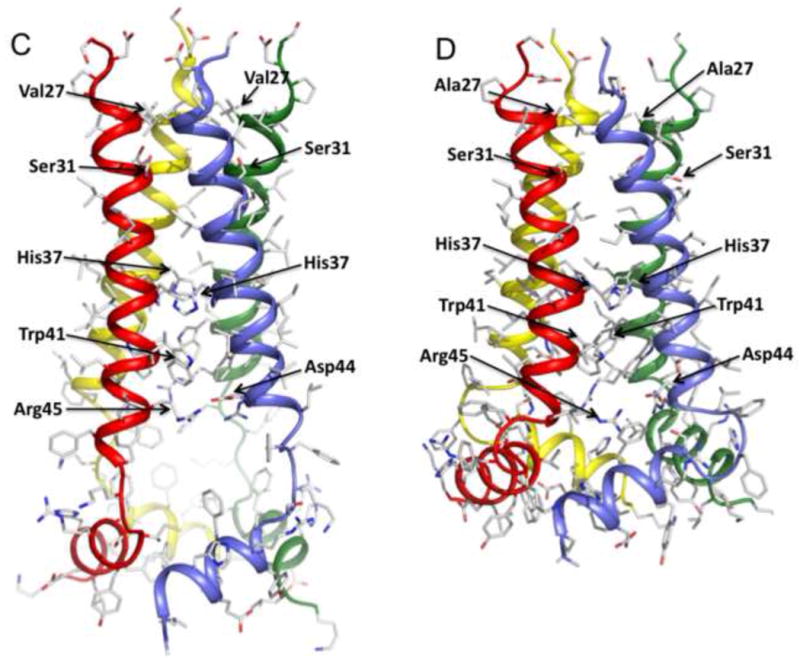
a. Superposition of 15 low energy structures of WT18-60 (2RLF)[16] and b. V27A18-60 (2KWX). The V27A structures were calculated using restraints summarized in Table 1. c. Ribbon representation of the WT structure (2RLF) and d. the V27A structure (2KWX). Compared to WT, the structure ensemble of V27A shows better-defined arrangement of the AP helices relative to the pore domain due to more long-range NOEs.
While the C-terminal region (residues 47– 60) of the V27A structure is more compact due to the newly acquired NOE restraints, the channel pore is overall very similar to that of WT and the S31N mutant. As in the closed channel of WT18-60, the TM helices of V27A form a four-helix bundle with a left-handed twist similar to WT. The N-terminal channel entrance, which in WT is constricted to ∼3 Å by Val27 methyl groups, becomes much wider (∼8 Å) when Val27 is replaced by the smaller alanines in the mutant (Figure 3). This wider opening may facilitate water entrance, thus explaining the faster conductance than the WT [19]. It is widely accepted that water molecules are needed inside the channel pore for supporting proton conduction. Water exchange/NOE cross-peaks in the NOESY spectra indicate the presence of water molecules near Ser31, even in the closed state. The pore widens after Ser31 and becomes the widest at Gly34 position with an inner diameter of ∼ 7 Å. The channel then narrows towards the C terminus as the sidechains of His37 and Trp41 constrict the channel to ∼1.5 Å (Figure 3). Similar to WT, the arrangement of the Trp41 indoles with respect to Asp44 indicates that they form inter-subunit hydrogen bonds that lock the channel gate in the closed conformation. Arg45 is in the vicinity for forming inter-subunit salt bridge with Asp44.
Figure 3.
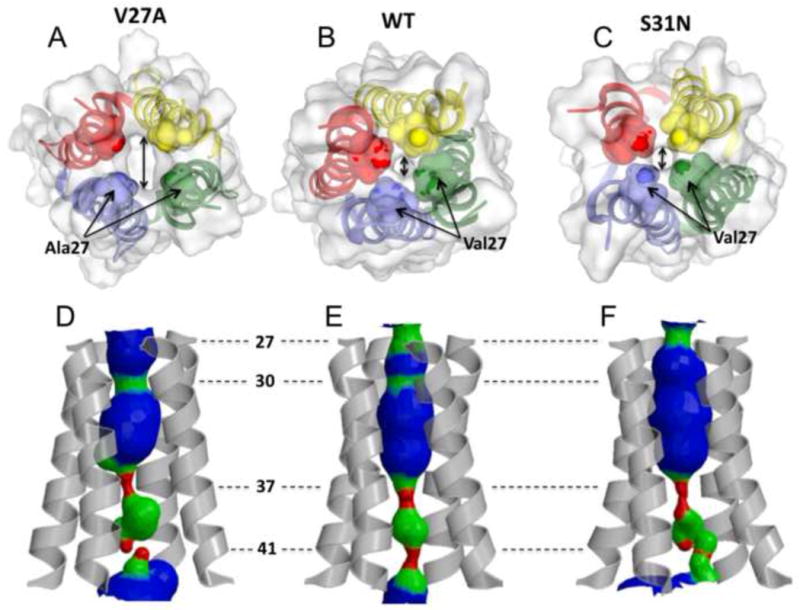
The size of the channel N-terminal entrance of a. the V27A mutant (2KWX), b. WT (2RLF), and c. the S31N mutant (2KIH) indicated by double headed arrow. The side chains at residue position 27 constrict the channel entrance. Mutating valine at this position to alanine doubles the diameter of the channel opening (∼2.5 Å in WT and S31N mutant, ∼5 Å in the V27A mutant). d,e,f The pore surfaces calculated using the program HOLE. d. The WT structure displays two constrictions in the N terminus at the positions 27 and 30. e. The V27A mutant displays one constriction at position 30. f. The S31N mutant is constricted at the position 27, but due to the serine to asparagine substitution at the position 31 the channel forms looser tetramer that result in somewhat larger diameter around Ser30. All of the structures have their C-termini tightly constricted to ∼1.5 Å by side chains of His37 and Trp41.
The C-terminal end of the TM helix extends into a tight turn (residues 47-49) that connects the channel domain to the C-terminal AP helix (residues 50 –60). Within the individual subunits, the AP helix is almost perpendicular to the TM helix. Moreover, the AP helix C-terminal end (around Gly58) of one subunit packs closely against the AP helix N-terminal end (around Ile51) of the adjacent subunit (Figure 2D). The inter-subunit interaction between the AP helices, in addition to those between the TM helices, explains why the C-terminal sequence (47 –60) beyond the TM domain is required for the stable assembly of the M2 tetramer [16; 19; 20]. Another interesting detail revealed by the V27A structure is the position of the Ser50 sidechain. The native residue at position 50 is cysteine, which is palmitoylated in the virus [21]. This cysteine was mutated to serine in our structural investigation to avoid problems associated with potential disulfide bond formation. The V27A structure shows that side chains of Ser50 are pointing along the axial direction of the channel, which would allow, upon palmitoylation, the fatty acid chains to partition naturally into the lipid bilayer of the membrane.
Implications to the mechanism of resistance
Since there have been published results that are consistent with either of the two proposed sites of inhibition, the pore-blocking site [15; 17; 22] and the external site for allosteric inhibition [16; 17; 19; 23], we discuss possible mechanism of drug resistance of the V27A mutant separately for the two different inhibition sites.
We previously proposed a mechanism of inhibition by rimantadine based on the external drug binding site for the S31N mutant [19]. This external, lipid-facing drug pocket is formed by Trp41, Ile42, and Arg45 from one TM helix and Leu40, Leu43, and Asp44 from the adjacent TM helix, and shows a unique amphipathic property. In this pocket, the amino group of rimantadine is in contact with the polar sidechain of Asp44. The poly-cyclic hydrocarbon cage of the drug forms hydrophobic interactions with Ile42 from one TM helix and Leu40 and Leu43 from the adjacent helix. Since the pocket is composed of residues from two adjacent TM helices, the stability and physical properties of the pocket depend on the dynamics and conformation of helical packing. For example, a small change in the dynamics of helix-helix packing could cause large disruption of the external drug pocket and thus reduced binding affinity. Indeed, structural and biochemical studies of the S31N mutant showed that replacing Ser31, which is located in the helix-helix interface, with the bulkier asparagine results in substantially weaker helix-helix packing [19].
The same mechanism in principle can be used to explain drug resistance conferred by the V27A mutation. We previously showed by dithiobis[succinimidylpropionate] (DSP) crosslinking experiment that the V27A variant forms looser, more dynamic tetramer than WT [19]. The weakening of the assembly can be explained by comparing the WT and V27A structures. In WT, the Val27 methyls provide a substantial amount of inter-helical van der Waals interactions that contribute to channel assembly. These interactions result in the formation of a tight hydrophobic ring that constricts the N-terminal channel opening. In the V27A structure, the sidechain of Ala27 is too small to form strong inter-helical packing, which may be the cause of the weakened tetrameric assembly. Moreover, as in the case of S31N, we could not detect protein-rimantadine NOEs for the external drug pocket of the V27A mutant, suggesting that again a mutation in the N-terminal TM region can dramatically decrease drug binding to the external pocket through a long-range, allosteric effect.
Explaining the mechanism of drug resistance in the context of the pore-blocking model is more challenging due to the fact that at least three distinct pore-binding sites have been proposed (reviewed in [24]). Most recently, yet another new pore-blocking concept emerged: upon binding to the channel pore, amantadine tumbles isotropically, i.e. does not have any preferable orientation, and does not appear to form any specific interaction with the pore-lining residues [17]. This concept infers that mutations that confer resistance should alter the properties of the pore. A comparison of the pores of the WT, V27A, and S31N structures (Figure 3) show that the overall architectures of the pores are similar among the three variants except for local changes around the mutated residues. An obvious difference in the V27A structure is that the channel entrance is much wider than in WT. This altered feature should facilitate both entrance and exit of amantadine, and it is unclear how this effect could be linked to drug resistance. It is possible, however, that the adamantly cage of amantadine interacts with the hydrophobic valine sidechains and the drug's amino group forms polar contacts with the Ser31 hydroxyl groups. Thus the V27A mutation would be expected to reduce the hydrophobic interactions with the drug.
Implications to the proton transfer pathway
In addition to drug inhibition, a major area of interest in the M2 field is the mechanism of proton conduction. The higher rate of conductance of V27A suggests reduction in an energy barrier for the proton transport for this mutant. In the WT, the N-terminal channel opening is guarded by Val27 sidechains, which hinder water entrance. Thus channel breathing may be required to admit water. The V27A structure shows that replacing Val27 with alanine more than doubles the channel opening (Figure 3), thus it is expected that this mutation will increase proton flux rate by decreasing energy barrier for water molecules to enter.
Inside the pore, water molecules are localized around Ser31 that may mediate proton passage to His37. What is unclear, however, is how protons may exit the channel. The new V27A structure suggests that the protons may exit the channel near Asp44 or Arg45 (Figure 4a&b). Asp44 and Arg45 are the only polar residues in the C-terminal region of the channel, and they are conveniently positioned at the lipid headgroups where they are accessible by water molecules. In particular, Asp44 is located at the lateral opening of the channel (Figure 4a), thus it is capable of mediating the C-terminal water entrance. The C-terminal base of the channel is sealed by the hydrophobic side chains of Phe55 (Figure 4c), making it impermeable to either water molecules or protons. Assuming that the base does not change conformation during proton conduction, the only possible proton exit is the lateral opening at Asp44.
Figure 4.
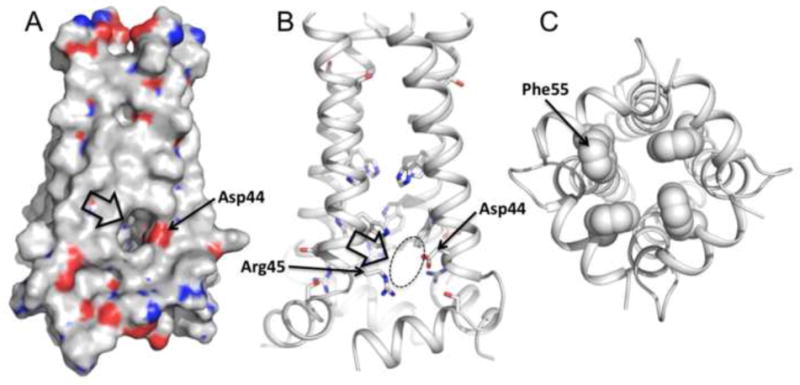
The proton exit suggested by the structure. a. Van der Waals surface and b. ribbon representations of the V27A structure show a lateral opening around Asp44 and Arg45. Asp44 is positioned near the lipid headgroups of presumed bilayer where it is accessible by water molecules, thus is capable of mediating C-terminal proton or hydronium exit. c. The C-terminal base of the channel is sealed by the hydrophobic side chains of Phe55, making it impermeable to either water molecules or protons.
In addition to water near Ser31, water crosspeaks are also present near the Trp41 indol amine in WT [16], the S31N [19], and the V27A mutants. The water molecules around Ser31 may mediate proton transfer to His37, and the water molecules around Trp41 may mediate proton transfer from His37 to the channel exit at Asp44. Further experimental investigation is required to understand how protons pass the His37/Trp41 gate of the channel.
Conclusion
The solution NMR structure of the V27A amantadine-resistant mutant reveals a number of interesting features that have not been observed in any of the known experimental structures and models of the M2 protein. The structure shows a substantially increased channel opening at the N-terminal end, which may explain the faster proton conduction observed for the V27A mutant. The C-terminal region of the structure also strongly suggests that Asp44 and Arg45 facilitate lateral proton exit to the polar region of the membrane during proton conduction.
Supplementary Material
Acknowledgments
We thank Stephen Blacklow, Donald Coen, James Hogle, Kirill Oxenoid, and Jessica Williamson for helpful discussion. This work was supported by grants from the NIH (AI054520 to JJC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. Embo J. 1985;4:3021–4. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helenius A. Unpacking the incoming influenza virus. Cell. 1992;69:577–578. doi: 10.1016/0092-8674(92)90219-3. [DOI] [PubMed] [Google Scholar]
- 3.Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–28. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 4.Davies WL, Grunert RR, Haff RF, McGahen JW, Neumayer EM, Paulshock M, Watts JC, Wood TR, Hermann EC, Hoffmann CE. Antiviral Activity of 1-Adamantanamine (Amantadine) Science. 1964;144:862–3. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- 5.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. Jama. 2006;295:891–4. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 6.Hay AJ, Zambon MC, Wolstenholme AJ, Skehel JJ, Smith MH. Molecular basis of resistance of influenza A viruses to amantadine. J Antimicrob Chemother. 1986;18 B:19–29. doi: 10.1093/jac/18.supplement_b.19. [DOI] [PubMed] [Google Scholar]
- 7.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol. 1993;67:5585–94. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–81. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 9.Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196:249–57. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 10.Krumbholz A, Schmidtke M, Bergmann S, Motzke S, Bauer K, Stech J, Durrwald R, Wutzler P, Zell R. High prevalence of amantadine resistance among circulating European porcine influenza A viruses. J Gen Virol. 2009;90:900–8. doi: 10.1099/vir.2008.007260-0. [DOI] [PubMed] [Google Scholar]
- 11.Anton A, Marcos MA, Martinez MJ, Ramon S, Isanta R, de Molina P, de Anta MT, Pumarola T. Double (V27A/S31N) mutant 2009 pandemic influenza A (H1N1) virus isolated from adamantane non-treated inmunocompetent child. Diagn Microbiol Infect Dis. 2010;67:114–5. doi: 10.1016/j.diagmicrobio.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Astrahan P, Kass I, Cooper MA, Arkin IT. A novel method of resistance for influenza against a channel-blocking antiviral drug. Proteins. 2004;55:251–7. doi: 10.1002/prot.20018. [DOI] [PubMed] [Google Scholar]
- 13.Kovacs FA, Denny JK, Song Z, Quine JR, Cross TA. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J Mol Biol. 2000;295:117–25. doi: 10.1006/jmbi.1999.3322. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H(+) channel. Protein Sci. 2001;10:2241–50. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–9. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–5. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–92. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreas LB, Eddy MT, Pielak RM, Chou J, Griffin RG. Magic angle spinning NMR investigation of influenza A M2(18-60): support for an allosteric mechanism of inhibition. J Am Chem Soc. 2010;132:10958–60. doi: 10.1021/ja101537p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci U S A. 2009;106:7379–84. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pielak RM, Chou JJ. Influenza M2 proton channels. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamem.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugrue RJ, Belshe RB, Hay AJ. Palmitoylation of the influenza A virus M2 protein. Virology. 1990;179:51–6. doi: 10.1016/0042-6822(90)90272-s. [DOI] [PubMed] [Google Scholar]
- 22.Ohigashi Y, Ma C, Jing X, Balannick V, Pinto LH, Lamb RA. An amantadine-sensitive chimeric BM2 ion channel of influenza B virus has implications for the mechanism of drug inhibition. Proc Natl Acad Sci U S A. 2009;106:18775–9. doi: 10.1073/pnas.0910584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du QS, Huang RB, Wang CH, Li XM, Chou KC. Energetic analysis of the two controversial drug binding sites of the M2 proton channel in influenza A virus. J Theor Biol. 2009;259:159–64. doi: 10.1016/j.jtbi.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Pielak RM, Chou JJ. Flu channel drug resistance: a tale of two sites. Protein and Cell. 2010;1:246–258. doi: 10.1007/s13238-010-0025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schroeder C, Ford CM, Wharton SA, Hay AJ. Functional reconstitution in lipid vesicles of influenza virus M2 protein expressed by baculovirus: evidence for proton transfer activity. J Gen Virol. 1994;75(Pt 12):3477–84. doi: 10.1099/0022-1317-75-12-3477. [DOI] [PubMed] [Google Scholar]
- 26.Nguitragool W, Miller C. Uncoupling of a CLC Cl-/H+ exchange transporter by polyatomic anions. J Mol Biol. 2006;362:682–90. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Moffat JC, Vijayvergiya V, Gao PF, Cross TA, Woodbury DJ, Busath DD. Proton transport through influenza A virus M2 protein reconstituted in vesicles. Biophys J. 2008;94:434–45. doi: 10.1529/biophysj.107.109082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzmann M, Wider G, Pervushin K, Wuthrich K. Improved sensitivity and coherence selection for [N-15,H-1]-TROSY elements in triple resonance experiments. J Biomol NMR. 1999;15:181–184. doi: 10.1023/a:1008358030477. [DOI] [PubMed] [Google Scholar]
- 29.Kay LE, Ikura M, Tschudin R, Bax A. Three-dimensional triple resonance NMR spectroscopy of isotopically enriched proteins. J Magn Reson. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Schwieters CD, Kuszewski J, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2002;160:66–74. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.