

Abstract
The discovery of numerous non-coding RNA (ncRNA) transcripts in species from yeast to mammals has dramatically altered our understanding of cell biology, especially disease biology such as cancer. In humans, the identification of abundant long ncRNA (lncRNAs) >200 bp in length has catalyzed their characterization as critical components of cancer biology. Recently, roles for lncRNAs as drivers of tumor suppressive and oncogenic functions have appeared in prevalent cancer types, such as breast and prostate cancer. In this review, we will highlight the emerging impact of ncRNAs in cancer research, with a particular focus on the mechanisms and functions of lncRNAs.
Keywords: long noncoding RNA, lncRNA, cancer, epigenetics
Introduction
The question of which regions of the human genome constitute its functional elements—those expressed as genes or serving as regulatory elements—has long been a central topic in biology. While early cloning-based methods revealed more than 7000 human genes in the 1970s and 1980s (1), large-scale analyses of expressed sequence tags (ESTs) in the 1990s suggested that the estimated number of human genes lay range from 35,000 to 100,000 (2). The completion of the human genome project narrowed the focus considerably by highlighting the surprisingly small number of protein-coding genes, which is now conventionally cited as less than 25,000 (3).
While the number of protein-coding genes (20,000–25,000) has maintained broad consensus, recent studies of the human transcriptome have revealed an astounding number of non-coding RNAs (ncRNAs). These transcribed elements, which lack the capacity to code for a protein, are bafflingly abundant in all organisms studied to date, from yeast to humans (4-6). Yet, over the past decade, numerous studies have demonstrated that ncRNAs have distinct biological functions and operate through defined mechanisms. Still, their sheer abundance—some reports estimate that up to 70% of the human genome is transcribed into RNA (4)—has sparked debates as to whether ncRNA transcription reflects true biology or byproducts of a leaky transcriptional system. Encompassed within these studies are the broad questions of what constitutes a human gene, what distinguishes a gene from a region that is simply transcribed, and how we interpret the biological meaning of transcription.
These developments have been matched by equally insightful discoveries analyzing the role of ncRNAs in human diseases, especially cancer, lending support to the importance of their cellular functions (7, 8). Initial evidence suggests that ncRNAs, particularly long ncRNAs (lncRNAs), have essential roles in tumorigenesis (7), and that lncRNA-mediated biology occupies a central place in cancer progression (9). With the number of well-characterized cancer-associated lncRNAs growing, the study of lncRNAs in cancer is now generating new hypotheses about the biology of cancer cells. Here, we review the current understanding of ncRNAs in cancer, with particular focus on lncRNAs as novel drivers of tumorigenesis.
ncRNA: a new kind of gene
ncRNAs are RNA transcripts that do not encode for a protein. In the past decade, a great diversity of ncRNAs has been observed. Depending on the type of ncRNA, transcription can occur by any of the three RNA polymerases (RNA Pol I, RNA Pol II, or RNA Pol III). General conventions divide ncRNAs into two main categories: small ncRNAs less than 200 bp and long ncRNAs greater than 200 bps (10). Within these two categories, there are also many individual classes of ncRNAs (Table 1), although the degree of biological and experimental support for each class ranges substantially and should be evaluated individually.
Table 1.
Types of ncRNAs known in humans
| Category | Name | Quality of supporting data | Specific role in carcinogenesis | Aberration in cancer | Reference |
|---|---|---|---|---|---|
| Housekeeping RNAs | Transfer RNAs | High | No | No | 10, 11 |
| Ribosomal RNAs | High | No | No | 10, 11 | |
| Small nucleolar RNAs | High | No | No | 10, 11 | |
| Small nuclear RNAs | High | No | No | 10, 11 | |
| Small ncRNAs (200 bp or less in size) | MicroRNAs | High | Yes | Amplification, deletion, methylation, gene expression | 12, 13 |
| Tiny transcription initiation RNAs | High | Not known | Not known | 11 | |
| Repeat associated small interfering RNAs | High | Not known | Not known | 11 | |
| Promoter-associated short RNAs | High | Not known | Not known | 4, 6, 11 | |
| Termini-associated short RNAs | High | Not known | Not known | 4, 6, 11 | |
| Antisense termini associated short RNAs | High | Not known | Not known | 6, 10 | |
| Transcription start site antisense RNAs | Moderate | Not known | Not known | 10 | |
| Retrotransposon-derived RNAs | High | Not known | Not known | 15 | |
| 3’UTR-derived RNAs | Moderate | Not known | Not known | 10 | |
| Splice-site RNAs | Poor | Not known | Not known | 11 | |
| Long ncRNA (over 200 bp in size) | Long or large intergenic ncRNAs | High | Yes | Gene expression, translocation | 7-9, 25, 101 |
| Transcribed ultraconserved regions | High | Yes | Gene expression | 18, 19 | |
| Pseudogenes | High | Yes | Gene expression, deletion | 15, 81 | |
| Enhancer RNAs | High | Yes | Not known | 17, 29 | |
| Repeat-associated ncRNAs | High | Not known | Not known | 15 | |
| Long intronic ncRNAs | Moderate | Not known | Not known | 10, 11 | |
| Antisense RNAs | High | Yes | Gene expression | 14 | |
| Promoter-associated long RNAs | Moderate | Not known | Not known | 4 | |
| Long stress-induced non-coding transcripts | Moderate | Yes | Gene expression | 10, 11 |
Small ncRNAs
The diversity of small ncRNAs has perhaps grown the most, where several dozen classes of small ncRNAs have been proposed (10, 11). These include well-characterized housekeeping ncRNAs (transfer RNA (tRNA) and some ribosomal RNA (rRNA)) essential for fundamental aspects of cell biology, splicing RNAs (small nuclear RNAs (snRNAs)), and a variety of recently-observed RNAs associated with protein-coding gene transcription, such as tiny transcription-initiation RNAs, promoter-associated short RNAs, termini-associated short RNAs, 3’UTR-derived RNAs, and antisense termini-associated short RNAs (10).
To date, the most extensively studied small RNAs in cancer are microRNAs (miRNAs). Elegant studies over the past 15 years have defined an intricate mechanistic basis for miRNA-mediated silencing of target gene expression through the RNA-induced silencing complex (RISC), which employs Argonaute family proteins (such as AGO2) to cleave target mRNA transcripts or inhibiting the translation of that mRNA (Figure 1A) (12). Aberrant expression patterns of miRNAs in cancer have been well documented in most tumor types (Figure 1B), and detailed work from many labs have shown that many miRNAs, including miR-10b, let-7, miR-101, and the miR-15a-16-1 cluster, possess oncogenic or tumor suppressive functions (Figure 1C) (12, 13).
Figure 1. MicroRNA-mediated pathways in cancer.
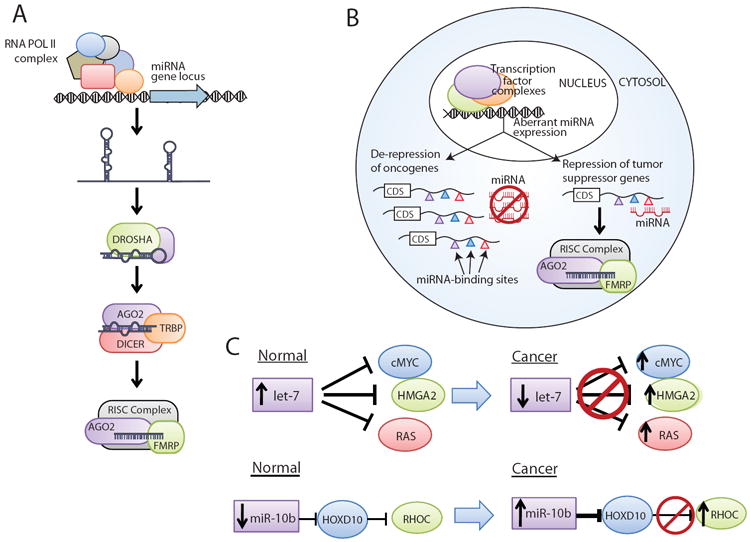
(A) MicroRNA (miRNA) transcription usually occurs by RNA Polymerase II, generating a primary pri-miRNA transcript. The pri-miRNA is processed by DROSHA and cleaved by DICER to generate a mature miRNA, which then associates with Argonaute family proteins in the RNA-induced silencing complex (RISC) to achieve gene expression control. (B) In cancer, aberrant miRNA expression levels can lead either to the repression of tumor suppressor (typically when miRNA levels are upregulated) or de-repression of oncogenes (typically when miRNA levels are downregulated). The colored triangles indicate different miRNA binding sites in the 3’ untranslated region (UTR) of a protein-coding mRNA. Abbreviation: CDS, coding sequence. (C) Two examples of aberrant miRNA signaling in cancer are let-7, which is downregulated in cancer and regulates oncogenes such as cMYC, and miR-10b, which is upregulated in cancer metastases and indirectly upregulates RHOC.
Long ncRNAs
Recent observations of novel long ncRNA species has led to a complex set of terms and terminologies used to describe a given long ncRNA. These include antisense RNAs, which are transcribed on the opposite strand from a protein-coding gene and frequently overlap that gene (14), transcribed ultraconserved regions (T-UCRs), which originate in regions of the genome showing remarkable conservation across species, and ncRNAs derived from intronic transcription.
Although many RNA species are >200 bp in length, such as repeat or pseudogene-derived transcripts (15), the abbreviated term lncRNA (also referred to as lincRNA, for long intergenic ncRNA) does not uniformly apply to all of these (Box 1). While the nomenclature is still evolving, lncRNA typically refers to a polyadenylated long ncRNA that is transcribed by RNA polymerase II and associated with epigenetic signatures common to protein-coding genes, such as trimethylation of histone 3 lysine 4 (H3K4me3) at the transcriptional start site (TSS) and trimethylation of histone 3 lysine 36 (H3K36me3) throughout the gene body (16). This description also suits many T-UCRs and some antisense RNAs, and the overlap between these categories may be substantial. lncRNAs also commonly exhibit splicing of multiple exons into a mature transcript, as do many antisense RNAs but not RNAs transcribed from gene enhancers (eRNAs) or T-UCRs (17-19). Transcription of lncRNAs occurs from an independent gene promoter and is not coupled to the transcription of a nearby or associated parental gene, as with some classes of ncRNAs (promoter/termini-associated RNAs, intronic ncRNAs) (10). In this review we will use the term lncRNA in this manner. When the data is supportive, we include specific T-UCRs and antisense RNAs under the lncRNA umbrella term, and we distinguish other long ncRNAs, such as eRNAs, where appropriate.
Box 1: Defining lncRNAs as distinct transcripts.
Long noncoding RNAs (lncRNAs) are now emerging as a fundamental aspect of biology. However, recent estimates that up to 70% of the human may be transcribed have complicated the interpretation of the act of transcription. While some have argued that many of the transcribed RNAs may reflect a “leaky” transcriptional system in mammalian cells, lncRNAs have largely avoided these controversies due to their strongly defined identity. Below, we have indicated several common features of lncRNAs that confirm their biological robustness:
Epigenetic marks consistent with a transcribed gene (H3K4me3 at the gene promoter, H3K36me3 throughout the gene body
Transcription via RNA polymerase II
Polyadenylation
Often exhibit splicing of multiple exons via canonical genomic splice site motifs
Regulation by well-established transcription factors
Frequently expressed in a tissue-specific manner
Identification of long ncRNAs
Many initial lncRNAs, such as XIST and H19, were discovered in the 1980s and 1990s by searching cDNA libraries for clones of interest (20, 21). In these studies, the intention was generally to identify new genes important in a particular biological process—X chromosome inactivation in the example of XIST—by studying their expression patterns. At the time, most genes uncovered were protein-coding, and this tended to be the assumption, with a handful exceptions, such as XIST, which were subsequently determined to be noncoding as a secondary observation (20).
In the past decade, however, large-scale analyses have focused on identifying ncRNA species in a comprehensive fashion. This paradigm shift has been mediated by dramatic advances in high-throughput technologies, including DNA tiling arrays and next generation RNA sequencing (RNA-Seq) (9, 22-25). These platforms provide systems with which RNA transcription can be observed in an unbiased manner, and have thereby highlighted the pervasive transcription of ncRNAs in cell biology (Box 2). Moreover, whereas conventional cDNA microarrays detected only the transcripts represented by probes on the array, the introduction and popularization of RNA-Seq as a standard tool in transcriptome studies has removed many barriers to detecting all forms of RNA transcripts (9, 26). RNA-Seq studies now suggest that several thousand uncharacterized lncRNAs are present in any given cell type (9, 16), and elegant, large-scale analyses of lncRNAs in stem cells suggest that lncRNAs may be an integral component of lineage-specificity and stem cell biology (27). Observations that many lncRNAs demonstrate tissue-specific expression therefore enables speculations that the human genome may harbor nearly as many lncRNAs as protein-coding genes (perhaps ~15,000 lncRNAs), though only a fraction are expressed in a given cell type.
lncRNAs in cancer
Emerging evidence suggests that lncRNAs constitute an important component of tumor biology (Table 2). Dysregulated expression of lncRNAs in cancer marks the spectrum of disease progression (9) and may serve as an independent predictor for patient outcomes (28). Mechanistically, most well-characterized lncRNAs to date show a functional role in gene expression regulation, typically transcriptional rather than post-transcriptional regulation. This can occur by targeting either genomically local (cis-regulation) or genomically distant (trans-regulation) genes. Recently, a new type of long ncRNAs at gene enhancers, termed eRNAs, have also been implicated in transcriptional regulation (29).
Table 2.
Examples of lncRNAs in cancer
| lncRNA | Function | Cancer Type | Cancer Phenotype | Molecular Interactors | Reference |
|---|---|---|---|---|---|
| HULC | Biomarker | Hepatocellular | Not known | Unknown | 10 |
| PCA3 | Biomarker | Prostate | Not known | Unknown | 82, 83 |
| ANRIL/p15AS | Oncogenic | Prostate, Leukemia | Suppression of senescence via INK4A | Binds PRC1 and PRC2 | 46-48, 68 |
| HOTAIR | Oncogenic | Breast, hepatocellular | Promotes metastasis | Binds PRC2 and LSD1 | 28, 55, 56 |
| MALAT1/NEAT2 | Oncogenic | Lung, prostate, breast, colon | Unclear | Contributory to nuclear paraspeckle function | 76-79 |
| PCAT-1 | Oncogenic | Prostate | Promotes cell proliferation; inhibits BRCA2 | Unknown | 9 |
| PCGEM1 | Oncogenic | Prostate | Inhibits apoptosis; promotes cell proliferation | Unknown | 7, 10 |
| TUC338 | Oncogenic | Hepatocellular | Promotes cell proliferation and colony formation | Unknown | 19 |
| uc.73a | Oncogenic | Leukemia | Inhibits apoptosis; promotes cell proliferation | Unknown | 18 |
| H19 | Oncogenic; Tumor suppressive | Breast, hepatocellular | Promotes cell growth and proliferation; activated by cMYC; downregulated by prolonged cell proliferation | Unknown | 30, 34-36 |
| GAS5 | Tumor suppressive | Breast | Induces apoptosis and growth arrest; Prevents GR-induced gene expression | Binds GR | 57, 58 |
| linc-p21 | Tumor suppressive | Mouse models of lung, sarcoma, lymphoma | Mediates p53 signaling; induces apoptosis | Binds hnRNP-k | 73 |
| MEG3 | Tumor suppressive | Meningioma, hepatocellular, leukemia, pituitary tumors | Mediates p53 signaling; inhibits cell proliferation | Unknown | 69-72 |
| PTENP1 | Tumor suppressive | Prostate, colon | Binds PTEN-suppressing miRNAs | Unknown | 81 |
Abbreviations: Polycomb Repressive Complex 1, PRC1; Polycomb Repressive Complex 2, PRC2; Glucocorticoid Receptor, GR
cis-Regulatory lncRNA
cis-regulation by lncRNAs contributes to local control of gene expression by recruiting histone modification complexes to specific areas of the genome (Figure 2). This effect can either be highly specific to a particular gene, such as the regulation of IGF2 by lncRNAs (30); or, it can encompass a wide chromosomal region, such as X-chromosome inactivation in women through XIST. Historically, cis-regulation through lncRNAs was studied earlier than trans-regulation, as several cis-regulatory lncRNAs, including H19, AIR, KCNQ1OT1, and XIST were earlier discoveries (20, 21, 31). Several cis-regulatory lncRNAs, including H19, AIR and KCNQ1OT1, are also functionally related through their involvement in epigenetic imprinting regions.
Figure 2. Gene expression regulation by lncRNAs.
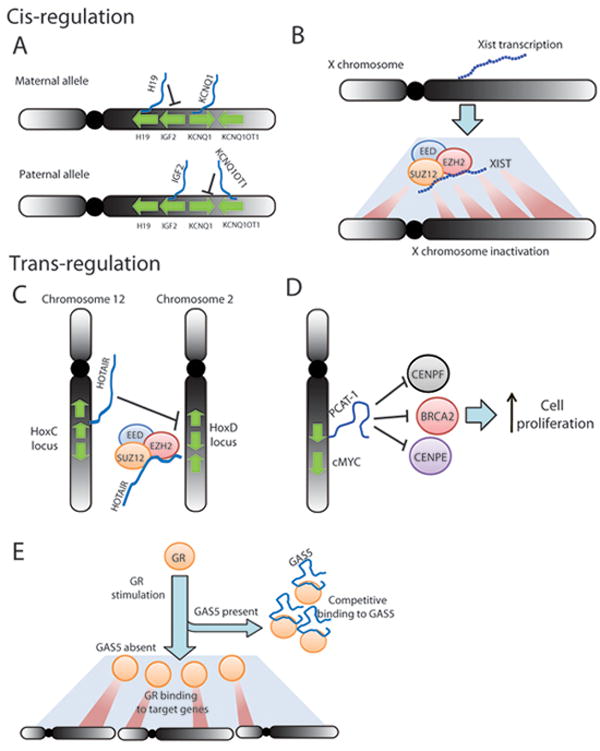
(A) and (B): cis-regulation of gene expression results in local control of genes neighboring, or on the same chromosome as, lncRNA transcription. (A) H19 and KCNQ1OT1 are imprinted lncRNAs on chromosome 11 associated with allele-specific expression of IGF2 and KCNQ1. (B) XIST transcription facilitates inactivation of an individual X chromosome in women by recruiting the Polycomb Repressive Complex 2 (PRC2). (C) and (D) trans-regulation of gene expression results in control of genomically-distant genes. (C) HOTAIR is transcribed from the HoxC cluster on chromosome 12 but represses the HoxD locus via PRC2-mediated epigenetic modifications. (D) PCAT-1 is transcribed from chromosome 8 but regulates target genes such as BRCA2, CENPE, and CENPF, thereby impacting cell proliferation. (E) The GAS5 lncRNA binds Glucocorticoid Receptor (GR) and sequesters it, preventing upregulation of GR-target genes across the genome. The blue line represents the GAS5 transcript.
Imprinting lncRNAs
lncRNA involvement in imprinted regions of the genome is critical for maintaining parent-of-origin-specific gene expression. In particular, an imprinted region of human chromosome 11 (orthologous to mouse chromosome 7) has been extensively studied for the role of lncRNAs. In humans, most well-known are the H19 and KCNQ1OT1 lncRNAs (21, 31), which are expressed on the maternal and paternal alleles, respectively, and maintain silencing of the IGF2 and KCNQ1 genes on those alleles (Figure 2A) (32).
Of the imprinting-associated ncRNAs, H19 has been most extensively studied in cancer. Aberrant expression of H19 is observed in numerous solid tumors, including hepatocellular and bladder cancer (30, 33). The functional data on H19 point in several directions, and it has been linked to both oncogenic and tumor suppressive qualities (34). For example, there is evidence for its direct activation by cMYC (35) as well as its downregulation by p53 and during prolonged cell proliferation (36). In model systems, siRNA knockdown of H19 expression impairs cell growth and clonogenicity in lung cancer cell lines in vitro (35) and decreased xenograft tumor growth of Hep3B hepatocellular carcinoma cells in vivo (30). Together, these data support a general role for H19 in cancer, although its precise biological contributions are still unclear.
Other imprinting-associated lncRNAs are only tangentially associated with cancer. Although loss of imprinting is observed in many tumors, the role for lncRNAs in this process is not well defined. For example, Beckwith-Wiedemann syndrome (BWS), a disorder of abnormal development with an increased risk for cancer, displays aberrant imprinting patterns of KCNQ1OT1 (32, 37); but a direct association or causal role for KCNQ1OT1 in cancer is not described (37). Conversely, aberrant H19 methylation in BWS appears to predispose to cancer development more strongly (37)
XIST
XIST, perhaps the most well studied lncRNA, is transcribed from the inactivated X chromosome, in order to facilitate that chromosome’s inactivation, and manifests as multiple isoforms (38, 39). On the active X allele, XIST is repressed by its antisense partner ncRNA, TSIX (39). XIST contains a double-hairpin RNA motif in the RepA domain, located in the first exon, which is crucial for its ability to bind Polycomb Repressive Complex 2 (PRC2) and propagate epigenetic silencing of an individual X chromosome (Figure 2B) (40).
Despite the body of research on XIST, a precise role for XIST in cancer has remained elusive (41). Some evidence initially suggested a role for XIST in hereditary BRCA1-deficient breast cancers (42, 43), where data indicated that BRCA1 was not required for XIST function in these cells (44). Others have reasoned that XIST may be implicated in the X chromosome abnormalities observed in some breast cancers. There have also been surprising accounts of aberrant XIST regulation in other cancers, including lymphoma and male testicular germ-cell tumors, where XIST hypomethylation is, unexpectedly, a biomarker (45). Yet, it remains unclear whether these observations reflect a passenger or driver status for XIST, as a well-defined function for XIST in cancer has yet to attain a board consensus.
ANRIL
Located on Ch9p21 in the INK4A/ARF tumor suppressor locus, ANRIL was initially described by examining the deletion of this region in hereditary neural system tumors, which predispose for hereditary cutaneous malignant melanoma (46). ANRIL was subsequently defined as a polyadenylated lncRNA antisense to the CDKN2A and CDKN2B genes. In vitro data have suggested that ANRIL functions to repress the INK4A/INK4B isoforms (47), but not ARF. This repression is mediated through direct binding to CBX7 (47), a member of Polycomb Repressive Complex 1 (PRC1), and SUZ12 (48), a member of PRC2, which apply repressive histone modifications to the locus. However, these studies were performed in different cell types and it is not known whether ANRIL binds both complexes simultaneously.
ANRIL also displays a highly complicated splicing pattern, with numerous variants, including circular RNA isoforms (49). Currently it is unclear whether these isoforms have tissue-specific expression patterns or unique functions, which may suggest a biological basis for this variation. Through GWAS, ANRIL has also been identified by single nucleotide polymorphisms (SNPs) correlated with a higher risk of atherosclerosis and coronary artery disease (50), and ANRIL expression has been noted in many tissues. The function and isoform-level expression of ANRIL in these tissue types is not yet elucidated, but may shed light onto its role in diverse disease processes.
HOTTIP and HOTAIRM1
An intriguing theme emerging in developmental biology is the regulation of HOX gene expression by lncRNAs. Highly conserved among metazoan species, HOX genes are responsible for determining tissue patterning and early development, and in humans HOX genes reside in four genomic clusters. Within these clusters, HOX genes display intriguing anterior-posterior and proximal-distal expression patterns that mirror their genomic position 5’ to 3’ in the gene cluster.
Two recently-discovered lncRNAs, termed HOTTIP and HOTAIRM1, may help to explain this co-linear patterning of HOX gene expression. HOTTIP and HOTAIRM1 are located at opposite ends of the HoxA cluster, and each helps to enhance gene expression of the neighboring HoxA genes (51, 52). HOTAIRM1, located at the 3’ end, coordinates HOXA1 expression and has tissue-specific expression patterns identical to HOXA1 (51). HOTTIP, by contrast, is at the 5’ end of the cluster and similarly enhances expression of the 5’ HoxA genes, most prominently HOXA13 (52). Mechanistic studies of HOTTIP suggest that it binds WDR5 and recruits the MLL H3K4 histone methyltransferase complex to the HoxA cluster to support active chromatin confirmation (52). These observations distinguish HOTTIP and HOTAIRM1, as most lncRNAs to date facilitate gene repression.
While HOTAIRM1 and HOTTIP have not been extensively studied in cancer, expression of these may have important roles in the differentiation status of cancer cells. For example, differentiation of myeloid cancer cell lines, such as K562 and NB4, by treatment with small molecule drugs led to an increase in HOTAIRM1 expression, implicating it in myeloid differentiation (51). Moreover, HoxA genes are broadly known to be important for many cancers, particularly HOXA9, which is essential for oncogenesis in leukemias harboring MLL rearrangements. Thus, HOTAIRM1 and HOTTIP also suggest a potential role for lncRNAs in MLL-rearranged leukemias.
trans-Regulatory lncRNAs
Like most cis-acting lncRNAs, trans-acting lncRNAs typically facilitate epigenetic regulation of gene expression. However, because trans-acting lncRNAs may operate at geographically distant locations of the genome, it is generally thought that the mature lncRNA transcript is the primary actor in these cases, as opposed to cis-regulating lncRNAs like H19, AIR and KCNQ1OT1 which may function through the act of transcription itself (34, 53, 54).
HOTAIR
trans-Regulatory lncRNAs were brought to widespread attention by the characterization of HOTAIR. First described in fibroblasts, HOTAIR is located in the HoxC cluster; but unlike HOTTIP and HOTAIRM1, HOTAIR was found to regulate HoxD cluster genes in a trans-regulatory mechanism (Figure 2C) (55). These observations raise the question of whether all Hox clusters are regulated by lncRNAs, either by a cis-regulatory or a trans-regulatory mechanism.
In cancer, HOTAIR is upregulated in breast and hepatocellular carcinomas (10), and in breast cancer overexpression of HOTAIR is an independent predictor of overall survival and progression-free survival (28). Work by Howard Chang and colleagues has further defined a compelling mechanistic basis for HOTAIR in cancer. HOTAIR has two main functional domains, a PRC2-binding domain located at the 5’ end of the RNA, and a LSD1/CoREST1-binding domain located at the 3’ end of the RNA (55, 56). In this way, HOTAIR is thought to operate as a tether that links two repressive protein complexes in order to coordinate their functions. In breast cancer, HOTAIR overexpression facilitates aberrant PRC2 function by increasing PRC2 recruitment to the genomic positions of target genes. By doing so, HOTAIR mediates the epigenetic repression of PRC2 target genes, and profiling of repressive (H3K27me3) and active (H3K4me3) chromatin marks shows widespread changes in chromatin structure following HOTAIR knockdown (28).
Furthermore, HOTAIR dysregulation results in a phenotype in both in vitro and in vivo models. Ectopic overexpression of HOTAIR in breast cancer cell lines increases their invasiveness both in vitro and in vivo. Supporting this, in benign immortalized breast cells overexpressing EZH2, a core component of PRC2, knockdown of HOTAIR mitigated EZH2-induced invasion in vitro (28). Taken together, these data provide the most thorough picture of a lncRNA in cancer.
PCAT-1
Using RNA-Seq (i.e. transcriptome sequencing) on a large panel of tissue samples, our lab recently described approximately 1,800 lncRNAs expressed in prostate tissue, including 121 lncRNAs that are transcriptionally dysregulated in prostate cancer (9). These 121 Prostate Cancer-Associated Transcripts (PCATs) may represent an unbiased list of potentially functional lncRNAs associated with prostate cancer. Among these, we focused on PCAT-1, a 1.9 kb, polyadenylated lncRNA comprised of two exons and located in the Chr8q24 gene desert (9).
PCAT-1 demonstrates tissue-specific expression and is selectively upregulated only in prostate cancer. Interestingly, PCAT-1, unlike HOTAIR, is repressed by PRC2, and PCAT-1 overexpression may define a molecular subtype of prostate that is not coordinated by PRC2 (9). In vitro and in vivo experiments demonstrated that PCAT-1 supports cancer cell proliferation (J.R.P. and A.M.C., unpublished data). Like HOTAIR, PCAT-1 functions predominantly as a transcriptional repressor by facilitating trans-regulation of genes preferentially involved in mitosis and cell division, including known tumor suppressor genes such as BRCA2 (Figure 2D). Intriguingly, because loss of BRCA2 function is known to increase cell sensitivity to small molecule inhibitors of PARP1, these data may suggest that PCAT-1 may impact cellular response to these drugs as well.
The discovery of PCAT-1 highlights the power of unbiased transcriptome studies to explore a rich set of lncRNAs associated with cancer. While PCAT-1 is the first cancer lncRNA to be discovered by this method, we anticipate that many additional studies will employ this approach.
GAS5
GAS5, first identified in murine NIH-3T3 cells, is a mature, spliced lncRNA manifesting as multiple isoforms up to 12 exons in size (57). Using HeLa cells engineered to express GAS5, Kino et al. recently described an intriguing mechanism by which GAS5 modulates cell survival and metabolism by antagonizing the glucocorticoid receptor (GR). The 3’ end of GAS5 both interacts with the GR DNA-binding domain (DBD) and is sufficient to repress GR-induced genes, such as cIAP2, when cells are stimulated with dexamethasone. By binding to the GR, GAS5 serves as a decoy that prevents GR binding to target DNA sequences (Figure 2E) (57).
In cancer, GAS5 induces apoptosis and suppresses cell proliferation when overexpressed in breast cancer cell lines, and in human breast tumors GAS5 expression is downregulated (58). Although it is unclear whether this phenotype is due to an interaction with GR, it is intriguing that GAS5 may also be able to suppress signaling by other hormone receptors, such as androgen receptor (AR), though this effect was not seen with estrogen receptor (ER) (57).
Other long ncRNAs
eRNAs
eRNAs are transcribed by RNA polymerase II at active gene enhancers (17). But unlike lncRNAs, they are not polyadenylated and are marked by a H3K4me1 histone signature denoting enhancer regions (17), rather than the H3K4me3/H3K36me3 signature classically associated with lncRNAs. While research on eRNAs is still in the earliest phases, an emerging role for them in hormone signaling is already being explored. Nuclear hormone receptors, such as AR and ER, are critical regulators of numerous cell growth pathways and are important in large subsets of prostate (AR), breast (ER), and thyroid (PPARγ) cancers. To date, eRNAs have been most directly implicated in prostate cancer, where they assist in AR-driven signaling and are maintained by FOXA1, a transcription factor that mediates cell lineage gene expression in several cell types (29).
T-UCRs
Ultraconserved regions in the genome were initially described as stretches of sequence >200 bp long with 100% conservation between humans and rodents but harboring no known gene (59). As high levels of sequence conservation are hallmarks of exonic sequences in protein-coding genes, ultraconserved regions strongly suggest the presence of either a gene or a regulatory region, such as an enhancer. Subsequently, numerous ultraconserved sequences were found to be transcriptionally active, defining a class of T-UCRs as ncRNAs (18). Many transcripts from T-UCRs are polyadenylated and associated with H3K4me3 at their transcriptional start sites (TSSs), indicating that many are likely lncRNAs according to our definition (60).
Aberrant expression of T-UCRs has been noted in several cancer types, including neuroblastoma (60), leukemia (18), and hepatocellular carcinoma (19). Most notably, one T-UCR gene, termed TUC338, has been shown to promote both cell proliferation and anchorage-independent growth in hepatocellular carcinoma cell lines (19), and TUC338 transcript is localized to the nucleus, suggesting a role in regulation of expression (19). Calin et al. further demonstrated that T-UCRs are targets for miRNAs (18). While T-UCRs remain poorly characterized as a whole, further exploration of the role and mechanism of these ncRNAs will likely elucidate novel aspects of tumor biology.
Functions and mechanisms of long ncRNAs
Like protein-coding genes, there is considerable variability in the function of long ncRNAs. Yet, clear themes in the data suggest that many long ncRNAs contribute to associated biological processes. These processes typically relate to transcriptional regulation or mRNA processing, which is reminiscent of miRNAs and may indicate a similar sequence-based mechanism akin to miRNA binding to seed sequences on target mRNAs. However, unlike miRNAs, long ncRNAs show a wide spectrum of biological contexts that demonstrate greater complexity to their functions.
Epigenetic transcriptional regulation
The most dominant function explored in lncRNA studies relates to epigenetic regulation of target genes. This typically results in transcriptional repression, and many lncRNAs were first characterized by their repressive functions, including ANRIL, HOTAIR, H19, KCNQ1OT1, and XIST (10, 47, 55). These lncRNAs achieve their repressive function by coupling with histone modifying or chromatin remodeling protein complexes.
The most common protein partners of lncRNAs are the PRC1 and PRC2 polycomb repressive complexes. These complexes transfer repressive post-translational modifications to specific amino acid positions on histone tail proteins, thereby facilitating chromatin compaction and heterochromatin formation in order to enact repression of gene transcription. PRC1 may be comprised of numerous proteins, including BMI1, RING1, RING2 and Chromobox (CBX) proteins, which act as a multi-protein complex to ubiquitinate histone H2A at lysine 119 (61). PRC2 is classically composed of EED, SUZ12, and EZH2, the latter of which is a histone methyltransferase enzymatic subunit that trimethylates histone 3 lysine 27 (61). Both EZH2 and BMI1 are upregulated in numerous common solid tumors, leading to tumor progression and aggressiveness (13, 61).
Indeed, ANRIL, HOTAIR, H19, KCNQ1OT1, and XIST have all been linked to the PRC2 complex, and in all except H19, direct binding has been observed between PRC2 proteins and the ncRNA itself (40, 48, 55, 62, 63). Binding of lncRNAs to PRC2 proteins, however, is common and observed for ncRNAs, such as PCAT-1, which do not appear to function through a PRC2-mediated mechanism. It is estimated that nearly 20% of all lncRNAs may bind PRC2 (64), though the biological meaning of these observations remains unclear. It is possible that PRC2 promiscuously binds lncRNAs in a non-specific manner. However, if lncRNAs are functioning in a predominantly cis-regulatory mechanism—such as ANRIL, KCNQ1OT1, and XIST—then numerous lncRNAs may bind PRC2 to facilitate local gene expression control throughout the genome. Relatedly, studies of PRC2-ncRNA binding properties have been able to determine a putative PRC2-binding motif that includes a GC-rich double hairpin, indicating a structural basis for PRC2-ncRNA binding in many cases (40).
Similarly, PRC1 proteins, particularly CBX proteins, have been implicated in ncRNA-based biology. For example, ANRIL binds CBX7 in addition to PRC2 proteins, and this interaction with CBX7 recruits PRC1 to the INK4A/ARF locus to mediate transcriptional silencing (47). More broadly, work with mouse polycomb proteins demonstrated that treatment with RNAse abolished CBX7 binding to heterochromatin on a global level, supporting the notion that ncRNAs are critical for PRC1 genomic recruitment (65).
While PRC1 and PRC2 are perhaps the most notable partners of lncRNAs, numerous other epigenetic complexes are implicated in ncRNA-mediated gene regulation. For example, the 3’ domain of HOTAIR contains a binding site for the LSD1/CoREST, a histone deacetylase complex that facilitates gene repression by chromatin remodeling (Figure 3A) (56). AIR is similarly reported to interact with G9a, a H3K9 histone methyltransferase (66). KCNQ1OT1 has been shown to interact with PRC2 (63), G9a (63), and DNMT1, which methylates CpG dinucleotides in the genome. More rarely, lncRNAs have been observed in activating epigenetic complexes. In a recent example, HOTTIP interacts with WDR5 to mediate recruitment of the MLL histone methyltransferase to the distal HoxA locus. MLL transfers methyl groups to H3K4me3, thereby generating open chromatin structures that promote gene transcription (52).
Figure 3. Mechanisms of lncRNA function.
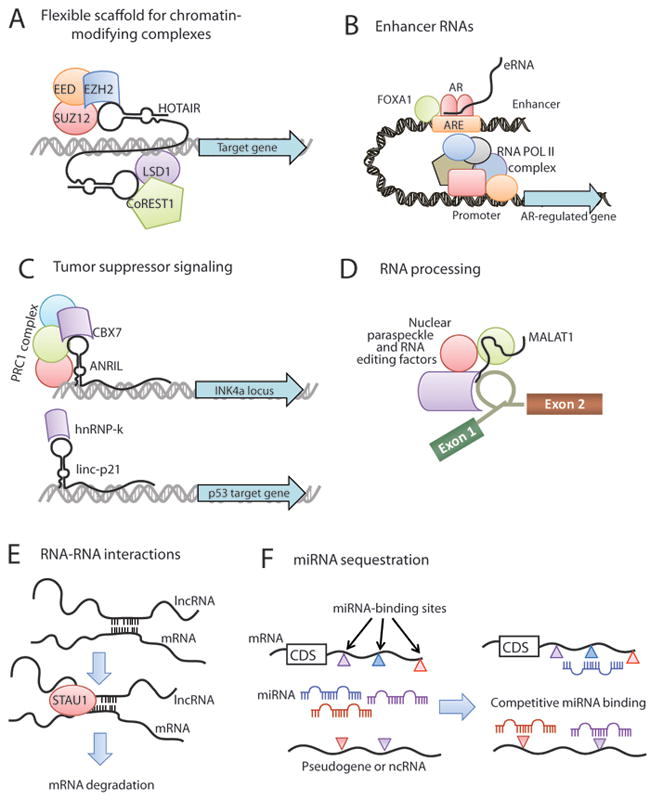
(A) lncRNAs, such as HOTAIR, may serve as a scaffolding base for the coordination of epigenetic or histone-modifying complexes, including Polycomb repressive complexes and LSD1/CoREST. (B) Enhancer RNAs (eRNAs) transcribed from gene enhancers may facilitate hormone signaling by cooperating with lineage-specific complexes such as FOXA1 and Androgen Receptor. (C) lncRNAs may directly impact tumor suppressor signaling either by transcriptional regulation of tumor suppressor genes through epigenetic silencing (e.g. ANRIL, upper) or by mediating activation of tumor suppressor target genes (e.g. linc-p21, lower). (D) MALAT1 and NEAT2 lncRNAs may be integral components of the nuclear paraspeckle and contribute to post-transcriptional processing of mRNAs. (E) Gene expression regulation may occur through direct lncRNA-mRNA interactions which arise from hybridization of homologous sequences and can serve as a signaling for STAU1-mediated degradation of the mRNA. (F) RNA molecules, including mRNAs, pseudogenes, and ncRNAs, can serve as molecular sponges for miRNAs. This generates an environment of competitive binding of miRNAs to achieve gene expression control based upon the degree of miRNA binding to each transcript. The colored triangles represent different miRNA binding sites in a transcript. Abbreviation: CDS, coding sequence.
In some cases, the mere act of lncRNA transcription is critical for the recruitment of protein complexes. Studies for both H19, KCNQ1OT1 and AIR suggest that transcriptional elongation of these genes is an important component of their function (34, 53, 54). By contrast, other lncRNAs, including HOTTIP as well as many trans-regulatory ones, do not show this relationship (52). For these lncRNAs, biological function may be centrally linked to their role as flexible scaffolds. In this model, lncRNAs serve as tethers that rope together multiple protein complexes through a loose arrangement. Supporting this model are the multiple lncRNAs found to bind multiple protein complexes, such as ANRIL (binding PRC1 and PRC2) and HOTAIR (binding PRC2 and LSD1/CoREST) (Figure 3A).
Enhancer-associated long ncRNAs
In addition to facilitating epigenetic changes that impact gene transcription, emerging evidence suggests that some ncRNAs contribute to gene regulation by influencing the activity of gene enhancers. For example, HOTTIP is implicated in chromosomal looping of active enhancers to the distal HoxA locus (52), though knockdown and overexpression of HOTTIP is not sufficient to alter chromosomal confirmations (52). There is also a report of local enhancer-like ncRNAs that typically lack the H3K4me1 enhancer histone signature, but possess H3K4me3, and function to potentiate neighbor gene transcription in a manner independent of sequence orientation (67).
A major recent development has been the discovery of eRNAs, which are critical for the proper coordination of enhancer genomic loci with gene expression regulation. While the mechanism of their action is still unclear, in prostate cancer cells, induction of AR signaling increased eRNA synthesis at AR-regulated gene enhancers, suggesting that eRNAs facilitate active transcription upon induction of a signaling pathway (29). Using chromatin conformation assays, Wang et al. showed that eRNAs are also important for the establishment of enhancer-promoter genomic proximity by chromosomal looping. Moreover, eRNAs work in conjunction with cell lineage-specific transcription factors, such as FOXA1 in prostate cells, thereby creating a highly specialized enhancer network to regulate transcription of genes in individual cell types (Figure 3B) (29). Future work in this area will likely provide insight into signaling mechanisms important in cancer.
Modulating tumor suppressor activity
The role of many lncRNAs as transcriptional repressors lends itself to inquiry as a mechanism for suppression of tumor suppressor genes. Here, one particular hotspot is the chromosome 9p21 locus, harboring the tumor suppressor genes CDKN2A and CDKN2B, which give rise to multiple unique isoforms, such as p14, p15, and p16, and function as inhibitors of oncogenic cyclin dependent kinases. Expression of this region is impacted by several repressive ncRNAs, such as ANRIL (Figure 3C, upper), and the p15-Antisense RNA, the latter of which also mediates heterochromatin formation through repressive histone modifications and was observed in leukemias (47, 68).
Moreover, several lncRNAs are implicated in the regulation of p53 tumor suppressor signaling. MEG3, a maternally-expressed imprinted lncRNA on Chr14q32, has been shown to activate p53 and facilitate p53 signaling, including enhancing p53 binding to target gene promoters (69). MEG3 has also been linked to p53 signaling in meningioma (70), and MEG3 overexpression suppresses cell proliferation in meningioma and hepatocellular carcinoma cell lines (70, 71). In human tumors, MEG3 downregulation is widely noted, with frequent hypermethylation of its promoter observed in pituitary tumors (10) and leukemias (72). Taken together, these data implicate MEG3 as a putative tumor suppressor.
A recently described murine lncRNA located near the p21 gene, termed linc-p21, has also emerged as a promising p53-pathway gene. In murine lung, sarcoma, and lymphoma tumors, linc-p21 expression is induced upon activation of p53 signaling and represses p53 target genes through a physical interaction with hnRNP-K, a protein that binds the promoters of genes involved in p53 signaling (Figure 3C, lower) (73). linc-p21 is further required for proper apoptotic induction (73). These data highlight linc-p21 as a candidate tumor suppressor gene. However, due to sequence differences between species, it is currently unclear whether the human homologue of linc-p21 plays a similarly important role in human tumor development.
Regulation of mRNA processing and translation
While many lncRNAs operate by regulating gene transcription, post-transcriptional processing of mRNAs is also critical to gene expression. A primary actor in these processes is the nuclear paraspeckle, a sub-cellular compartment found in the interchromatin space within a nucleus and characterized by PSP1 protein granules (74). While nuclear paraspeckle functions are not fully elucidated, this structure is known to be involved in a variety of post-transcriptional activities, including splicing and RNA editing (74). Paraspeckles are postulated to serve as storage sites for mRNA prior to its export to the cytoplasm for translation, and one study discovered a paraspeckle-retained, polyadenylated nuclear ncRNA, termed CTN-RNA, that is a counterpart to the protein-coding murine CAT2 (mCAT2) gene (75). CTN-RNA is longer than mCAT2, and under stress conditions, cleavage of CTN-RNA to the mCAT2 coding transcript resulted in increased mCAT2 protein (75).
In cancer, two ncRNAs involved in mRNA splicing and nuclear paraspeckle function, MALAT1 and NEAT1, are overexpressed. MALAT1 and NEAT1 are genomic neighbors on Chr11q13, and both are thought to contribute to gene expression by regulating mRNA splicing, editing, and export (Figure 3D) (76, 77). MALAT1 may further serve as a precursor to a small, 61-base-pair ncRNAs that is generated by RNase P cleavage of the primary MALAT1 transcript and exported into the cytoplasm (78). Although a unique role for MALAT1 in cancer is not yet known, its overexpression in lung cancer predicts for aggressive, metastatic disease (79).
Regulatory RNA-RNA interactions
Recent work on mechanisms of RNA regulation has highlighted a novel role for RNA-RNA interactions between ncRNAs and mRNA sequences. These interactions are conceptually akin to miRNA regulation of mRNAs, as sequence homology between the ncRNA and the mRNA is important to the regulatory process.
This sequence homology may be derived from ancestral repeat elements that contribute sequence to either the untranslated sequences of a protein-coding gene, or, less frequently, the coding region itself. For example, STAU1-mediated mRNA decay involves the binding STAU1, a RNA degradation protein, to protein-coding mRNAs that interact with lncRNAs containing ancestral Alu repeats. In this model, sequence repeats, typically Alus, in lncRNAs and mRNAs partially hybridize, forming double-stranded RNA complexes that then recruit STAU1 to implement RNA degradation (Figure 3E) (80). A related concept is found with XIST, which contains a conserved repeat sequence, termed RepA, in its first exon. RepA is essential for XIST function and the RepA sequence is necessary to recruit PRC2 proteins for X-chromosome inactivation (40).
Another model for mRNA regulation was recently posited by Pandolfi and colleagues, who suggested that transcribed pseudogenes serve as decoy for miRNAs that target the protein-coding mRNA transcripts of their cognate genes (81). Sequestration of miRNAs by the pseudogene then regulates the gene expression level of the protein-coding mRNA indirectly (Figure 3F). In addition to pseudogenes, this model more broadly suggests that all long ncRNAs, as well as other protein-coding mRNAs, may function as molecular “sponges” that bind and sequester miRNAs in order to control gene expression indirectly. In their study, Pandolfi and colleagues demonstrate that pseudogenes of two cancer genes, PTEN and KRAS, may be biologically active, and that PTENP1, a pseudogene of PTEN that competes for miRNA binding sites with PTEN, itself functions as a tumor suppressor in in vitro assays and may be genomically lost in cancer (81). This intriguing hypothesis may shed new light onto the functions or ncRNAs, pseudogenes, and even the UTRs of a protein-coding gene.
Implications of ncRNAs for cancer management
lncRNA diagnostic biomarkers
For clinical medicine, lncRNAs offer several possible benefits. lncRNAs, such as PCAT-1, commonly demonstrate restricted tissue-specific and cancer-specific expression patterns (9). This tissue-specific expression distinguishes lncRNAs from miRNAs and protein-coding mRNAs, which are frequently expressed from multiple tissue types. While the underlying mechanism for this is unclear, recent studies of chromatin confirmation show tissue-specific patterns, which may impact ncRNA transcription (29, 52). Given this specificity, ncRNAs may be superior biomarkers than many current protein-coding biomarkers, both for tissue-of-origin tests as well as cancer diagnostics.
A prominent example is PCA3, a lncRNA that is a prostate-specific gene and markedly overexpressed in prostate cancer. Although the biological function of PCA3 is unclear, its utility as a biomarker has led to the development of a clinical PCA3 diagnostic assay for prostate cancer, and this test is already being employed for clinical uses (82, 83). In this test, PCA3 transcript is detected in prostate cancer patient urine samples, which contain prostate cancer cells shed into the urethra. Thus, monitoring PCA3 does not require invasive procedures (Figure 4A) (82). The PCA3 test represents the most effective clinical translational of a cancer-associated ncRNA gene, and the rapid timeline these developments—only 10 years from between its initial description and a clinical test—suggests that the use of ncRNAs in clinical medicine is only beginning. Non-invasive detection of other aberrantly expressed lncRNAs, such as upregulation of HULC, which occurs in hepatocellular carcinomas, has also been observed in patient blood sera (10); however other lncRNA-based diagnostics have not been developed for widespread use.
Figure 4. Clinical implications of lncRNAs.
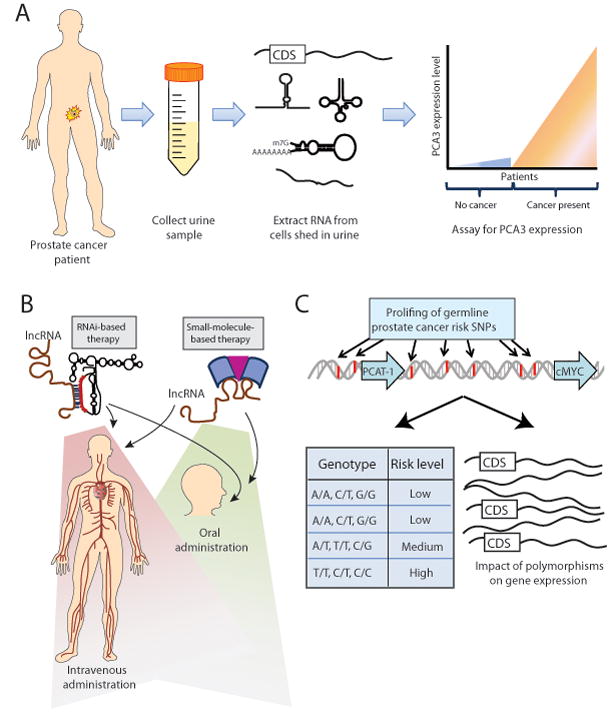
(A) The PCA3 urine biomarker test for prostate cancer employs a non-invasive approach to disease diagnosis by collecting patient urine samples, isolating nucleic acids from cells in the urine sediment, and quantifying PCA3 expression. (B) lncRNA-based therapies may target the lncRNA by utilizing either RNA interference (RNAi), which uses sequence homology between the lncRNA and the RNAi therapeutic molecule, or a small molecule therapy that interacts with the lncRNA. These therapeutic avenues may be appropriate for systemic therapy by either intravenous or oral administration. (C) Genome-wide association studies (GWAS) may provide germline polymorphisms that predict an individual patient’s clinical risk for disease development, response to therapy, or disease aggressiveness, while also providing molecular information through the impact of polymorphisms on gene expression of key genes. Abbreviation: CDS, coding sequence.
lncRNA-based therapies
The transition from ncRNA-based diagnostics to ncRNA-based therapies is also showing initial signs of development. Although the implementation of therapies targeting ncRNAs is still remote for clinical oncology, experimental therapeutics employing RNA interference (RNAi) to target mRNAs have been tested in mice, cynomolgus monkeys, and humans (84), as part of a phase I clinical trial for patients with advanced cancer (Figure 4B). Davis and colleagues found that systemic administration of RNAi-based therapy was able to effectively localize to human tumors and reduce expression of its target gene mRNA and protein (84). Currently, ongoing clinical trials are further evaluating the safety and efficacy of RNAi-based therapeutics in patients with a variety of diseases, including cancer (85), and these approaches could be adapted to target lncRNA transcripts.
Other studies investigate an intriguing approach that employs modular assembly of small molecules to adapt to aberrant RNA secondary structure motifs in disease (86). This approach could potentially target aberrant ncRNAs, mutant mRNAs, as well as nucleotide triplet-repeat expansions seen in several neurological diseases (such as Huntington’s disease). However, most RNA-based research remains in the early stages of development, and the potential for RNAi therapies targeting lncRNAs in cancer is still far from use in oncology clinics.
lncRNAs in genomic epidemiology
In the past decade, genome-wide association studies (GWAS) have become a mainstream way to identify germline SNPs that may predispose to myriad human diseases. In prostate cancer, over 20 GWAS have reported 31 SNPs with reproducible allele-frequency changes in prostate cancer patients compared to men without prostate cancer (87), and these 31 SNPs cluster into 14 genomic loci (87). In principle, profiling of these SNPs could represent an epidemiological tool to assess patient populations with a high risk of prostate cancer.
Of the 14 genomic loci, the most prominent by far is the “gene desert” region upstream of the cMYC oncogene on chromosome 8q24, which harbors 10 of the 31 reproducible SNPs associated with prostate cancer (Figure 4C). Several SNPs in the 8q24 region have been studied for their effect on enhancers (88), particularly for enhancers of cMYC (89), and chromosome looping studies have shown that many regions within 8q24 may physically interact with the genomic position of the cMYC gene (90).
Recently, our identification of PCAT-1 as a novel chr8q24 gene implicated in prostate cancer pathogenesis further highlights the importance and complexity of this region (Figure 4C) (9). Although the relationship between PCAT-1 and the 8q24 SNPs is not clear at this time, this discovery suggests that previously-termed “gene deserts” may, in fact, harbor critical lncRNA genes, and that SNPs found in these regions may impact uncovered aspects of biology. Relatedly, GWAS analyses of atherosclerosis, coronary artery disease, and type 2 diabetes have all highlighted ANRIL on chr9p21 as a ncRNA gene harboring of disease-associated SNPs (50).
Clinically, the use of GWAS data may identify patient populations at risk for cancer and may stratify patient disease phenotypes, such as aggressive versus indolent cancer, and patient outcomes (91). SNP profiles may also be use to predict a patient’s response to a given therapy (92). As such, the clinical translation of GWAS data remains an area of interest for cancer epidemiology.
Future directions
Defining the lncRNA component of the human genome
Going forward, it is clear that the systematic identification and annotation of lncRNAs, and their expression patterns in human tissues and disease, is important to clarifying the molecular biology underlying cancer. These efforts will be facilitated by large-scale RNA-Seq studies followed by ab initio or de novo sequence data assembly to discover lncRNAs in an unbiased manner (9, 26).
However, it is increasingly appreciated that a number of annotated but uncharacterized transcripts are important lncRNAs—HOTTIP is one such example (52). Similarly, the STAU1-interacting lncRNAs described by Gong and colleagues were also found by screening for annotated transcripts that contained prominent Alu repeats (80). While these examples were annotated as non-coding genes, it is also possible that other annotated genes, enumerated in early studies as protein-coding but not studied experimentally, are mislabeled ncRNA genes. These may include the generic “open-reading frame” genes (such as LOCxxx or CxxORFxx genes) that have not received detailed study.
Supporting this, Dinger et al. recently argued that bioinformatically distinguishing between protein-coding and non-coding genes can be difficult and that traditional computational methods for doing this may have been inadequate in many cases (93). For example, XIST was initially identified as a protein-coding gene because it has a potential, unused open reading frame (ORF) of nearly 300 amino acids (94). Additional complications further include an increasing appreciation of mRNA transcripts that function both by encoding a protein and at the RNA level, which would support miRNA sequestration hypotheses posited by Pandolfi and colleagues (81), and of very small ORFs (encoding peptides <10kDA) (95).
Elucidating the role of lncRNA sequence conservation
In general, most protein-coding exons are highly conserved and most lncRNAs are poorly conserved. This is not always true, as T-UCRs are prime examples of conserved ncRNAs. However, the large majority of lncRNAs exhibit substantial sequence divergence among species, and lncRNAs that do show strong conservation frequently only exhibit this conservation in a limited region of the transcript, and not the remainder of the gene.
This conundrum has sparked many hypotheses, many of which have merit. Small regions of conservation could indicate function domains of a given ncRNA, such as a binding site for proteins, microRNAs, mRNAs, or genomic DNA. Development of abundant ncRNA species could also suggest evolutionary advancement as species develop. In support of this latter proposition, many have commented that complex mammalian genomes (such as the human genome) have a vastly increased non-coding DNA component of their genome compared to single-celled organisms and nematodes, whereas the complement of protein-coding genes varies less throughout evolutionary time (96).
For lncRNAs, the issue of sequence conservation is paramount. However, it is now well established that poorly-conserved lncRNAs can be biologically important, but it is unclear whether these represent species-specific evolutionary traits or whether functional homologs have simply not been found. For example, AIR was initially described in mice in the 1980s, but a human homolog was not identified until 2008 (97).
Moreover, even lncRNAs with relatively high conservation, such as HOTAIR, may have species-specific function. Indeed, a study of murine HOTAIR (mHOTAIR) showed that mHOTAIR did not regulate the HoxD locus and did not recapitulate the functions observed in human cells (98). Other ncRNAs observed in mice, such as linc-p21, also show only limited sequence homology to their human forms and may have divergent functions as well. This may support hypotheses of rapid evolution of lncRNAs during the course of mammalian development. Moreover, this may suggest either that lncRNAs may have functions independent of conserved protein complexes (which have comparatively static functions throughout evolution) or that lncRNAs may adapt to cooperate with different protein complexes in different species.
Determining somatic alterations of lncRNAs in cancer
To date, somatic mutation of lncRNAs in cancer is not well explored. While numerous lncRNAs display altered expression levels in cancer, it is unclear to what extent cancers specifically target lncRNAs for genomic amplification/deletion, somatic point mutations, or other targeted aberrations.
In several examples, data suggest that lncRNAs may be a target for somatic aberrations in cancer. For example, approximately half of prostate cancers harbor gene fusions of the ETS family transcription factors (ERG, ETV1, ETV4, ETV5), which generally result in the translocation of an androgen-regulated promoter to drive upregulation of the ETS gene (99). One patient was initially found to have an ETV1 translocation to an intergenic androgen-regulated region (100) which was subsequently found to encode a prostate-specific lncRNA (PCAT-14) (9), thereby creating a gene fusion between the lncRNA and ETV1. Similarly, a GAS5-BCL6 gene fusion, resulting from a chromosomal translocation and retaining the full coding sequence of BCL6, has been reported in a patient with B-cell lymphoma (101). Finally, Poliseno and colleagues demonstrated that the PTEN pseudogene, PTENP1, is genomically deleted in prostate and colon cancers, leading to aberrant expression levels of these genes (81).
These initial data suggest that somatic aberrations of lncRNAs do contribute to their dysregulated function in cancer, although most studies to date identify gene expression changes as the primary alteration in lncRNA function. Yet, the study of mutated lncRNAs in cancer will be an area of high importance in future investigations, as several prominent oncogenes, such as KRAS, show no substantial change in protein expression level in mutated compared to non-mutated cases.
Characterizing RNA structural motifs
Just as protein-coding genes harbor specific domains of amino acids that mediate distinct functions (e.g. a kinase domain), RNA molecules also have intricate and specific structures. Among the most well-known RNA structures is the stem-loop-stem design of a hairpin, which is integral for miRNA generation (12). RNA structures are also known to be essential for binding to proteins, particularly PRC2 proteins (40). However, global profiles of lncRNA structures are poorly understood. While it is clear that lncRNA structure is important to lncRNA function, few RNA domains are well-characterized. Moreover, it is likely that RNA domains occur at the level of secondary structure, as lncRNA sequences are highly diverse yet may form similar secondary structures following RNA folding (102).
To this end, both computational and experimental advancements are beginning to address these topics. While numerous computational algorithms have been proposed to predict RNA structures (102), perhaps the most dramatic advance in this area has been the development of RNA-Seq methods to interrogate aspects of RNA structure globally. Recently, Frag-Seq and PARS-Seq have demonstrated the unbiased evaluation of RNA structures by treating RNA samples with specific RNAses that cleave RNA at highly selective structural positions (103, 104). These RNA fragments are then processed and sequenced to determine the nucleotide sites where RNA transcripts were cleaved, indirectly implying a secondary structure. This area of research promises to yield tremendous insight into the overall mechanics of lncRNA function.
Conclusions
In the past decade, the rapid discovery of ncRNA species by high-throughput technologies has accelerated current conceptions of transcriptome complexity. While a biological understanding of these ncRNAs has proceeded more slowly, increasing recognition of lncRNAs has defined these genes as critical actors of numerous cellular processes. In cancer, dysregulated lncRNA expression characterizes the entire spectrum of disease and aberrant lncRNA function drives cancer through disruption of normal cell processes, typically by facilitating epigenetic repression of downstream target genes. lncRNAs thus represent a novel, poorly-characterized layer of cancer biology. In the near term, clinical translation of lncRNAs may assist biomarker development in cancer types without robust and specific biomarkers, and in the future RNA-based therapies may be a viable option for clinical oncology.
Box 2: Discovery and validation of novel transcripts.
With the advent of high-throughput technologies, more and more ncRNA species are being discovered and characterized in mammalian systems. In this way, advancing technological achievements have dramatically impacted the field of ncRNA research, in large part due to the ability to detect and monitor ncRNA expression in a global and unbiased manner. Yet, because the processing and interpretation of high-throughput data can be challenging, extensive validation by wet-lab assays is still an important part of confirming initial nominations. Below, we have listed the most commonly used methods to discover ncRNAs and validate them.
| Discovery methods | Validation methods |
|
|
Statement of significance.
Long non-coding RNAs represent the leading edge of cancer research. Their identity, function, and dysregulation in cancer are only beginning to be understood, and recent data suggest that they may serve as master drivers of carcinogenesis. Increased research on these RNAs will lead to a greater understanding of cancer cell function and may lead to novel clinical applications in oncology.
Acknowledgments
We thank Sameek Roychowdhury, Matthew Iyer, and members of the Chinnaiyan lab for helpful discussions and comments on this manuscript. Robin Kunkel assisted with figure preparation. We would further acknowledge the numerous labs, authors, and publications that we were unable to cite in this review due to space restrictions.
Grant Support This work was supported by the Department of Defense grants PC100171 (to A.M.C.) and PC094290 (to J.R.P), NIH Prostate Specialized Program of Research Excellence grant P50CA69568 (to A.M.C.), the Early Detection Research Network grant U01 CA 11275 (to. A.M.C.). A.M.C. is supported by the Doris Duke Charitable Foundation Clinical Scientist Award, a Burroughs Welcome Foundation Award in Clinical translational Research, the Prostate Cancer Foundation, the American Cancer Society, and the Howard Hughes Medical Institute. J.R.P. is a Fellow of the University of Michigan Medical Scientist Training Program. A.M.C. is a Taubman Scholar of the University of Michigan.
Footnotes
Disclosure of Potential Conflicts of Interest A.M.C. serves as an advisor to Gen-Probe, Inc., who has developed diagnostic tests using PCA3 and TMPRSS2-ERG. A.M.C. serves on the Scientific Advisory Board of Wafergen, Inc. Neither company was involved in the writing or approval of this manuscript.
References
- 1.Matsubara K, Okubo K. Identification of new genes by systematic analysis of cDNAs and database construction. Curr Opin Biotechnol. 1993;4:672–7. doi: 10.1016/0958-1669(93)90048-2. [DOI] [PubMed] [Google Scholar]
- 2.Liang F, Holt I, Pertea G, Karamycheva S, Salzberg SL, Quackenbush J. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat Genet. 2000;25:239–40. doi: 10.1038/76126. [DOI] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Dijk EL, Chen CL, d’Aubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–7. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- 6.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 7.Huarte M, Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet. 2010;19:R152–61. doi: 10.1093/hmg/ddq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pauli A, Rinn JL, Schier AF. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet. 2011;12:136–49. doi: 10.1038/nrg2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011 doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. 2010;220:126–39. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 12.Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–79. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 13.Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, et al. Coordinated Regulation of Polycomb Group Complexes through microRNAs in Cancer. Cancer Cell. 2011;20:187–99. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW. The antisense transcriptomes of human cells. Science. 2008;322:1855–7. doi: 10.1126/science.1163853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–71. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 16.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calin GA, Liu CG, Ferracin M, Hyslop T, Spizzo R, Sevignani C, et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell. 2007;12:215–29. doi: 10.1016/j.ccr.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 19.Braconi C, Valeri N, Kogure T, Gasparini P, Huang N, Nuovo GJ, et al. Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2011;108:786–91. doi: 10.1073/pnas.1011098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349:38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- 21.Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–5. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 22.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 23.Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–54. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 24.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–10. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011 doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–4. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, et al. The H19 non-coding RNA is essential for human tumor growth. PLoS One. 2007;2:e845. doi: 10.1371/journal.pone.0000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M, et al. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci U S A. 1999;96:5203–8. doi: 10.1073/pnas.96.9.5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989–3000. doi: 10.1093/hmg/10.26.2989. [DOI] [PubMed] [Google Scholar]
- 33.Lottin S, Adriaenssens E, Dupressoir T, Berteaux N, Montpellier C, Coll J, et al. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis. 2002;23:1885–95. doi: 10.1093/carcin/23.11.1885. [DOI] [PubMed] [Google Scholar]
- 34.Gabory A, Jammes H, Dandolo L. The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays. 2010;32:473–80. doi: 10.1002/bies.200900170. [DOI] [PubMed] [Google Scholar]
- 35.Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, et al. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006;66:5330–7. doi: 10.1158/0008-5472.CAN-06-0037. [DOI] [PubMed] [Google Scholar]
- 36.Pantoja C, de Los Rios L, Matheu A, Antequera F, Serrano M. Inactivation of imprinted genes induced by cellular stress and tumorigenesis. Cancer Res. 2005;65:26–33. [PubMed] [Google Scholar]
- 37.Bliek J, Maas SM, Ruijter JM, Hennekam RC, Alders M, Westerveld A, et al. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS. Hum Mol Genet. 2001;10:467–76. doi: 10.1093/hmg/10.5.467. [DOI] [PubMed] [Google Scholar]
- 38.Chow J, Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol. 2009;21:359–66. doi: 10.1016/j.ceb.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 39.Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–42. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–6. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sirchia SM, Tabano S, Monti L, Recalcati MP, Gariboldi M, Grati FR, et al. Misbehaviour of XIST RNA in breast cancer cells. PLoS One. 2009;4:e5559. doi: 10.1371/journal.pone.0005559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–32. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Ganesan S, Silver DP, Greenberg RA, Avni D, Drapkin R, Miron A, et al. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell. 2002;111:393–405. doi: 10.1016/s0092-8674(02)01052-8. [DOI] [PubMed] [Google Scholar]
- 44.Xiao C, Sharp JA, Kawahara M, Davalos AR, Difilippantonio MJ, Hu Y, et al. The XIST noncoding RNA functions independently of BRCA1 in X inactivation. Cell. 2007;128:977–89. doi: 10.1016/j.cell.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 45.Kawakami T, Okamoto K, Ogawa O, Okada Y. XIST unmethylated DNA fragments in male-derived plasma as a tumour marker for testicular cancer. Lancet. 2004;363:40–2. doi: 10.1016/S0140-6736(03)15170-7. [DOI] [PubMed] [Google Scholar]
- 46.Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007;67:3963–9. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 47.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–74. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–62. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011;25:444–8. doi: 10.1096/fj.10-172452. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X, Lian Z, Padden C, Gerstein MB, Rozowsky J, Snyder M, et al. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood. 2009;113:2526–34. doi: 10.1182/blood-2008-06-162164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006;20:1268–82. doi: 10.1101/gad.1416906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pauler FM, Koerner MV, Barlow DP. Silencing by imprinted noncoding RNAs: is transcription the answer? Trends Genet. 2007;23:284–92. doi: 10.1016/j.tig.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28:195–208. doi: 10.1038/onc.2008.373. [DOI] [PubMed] [Google Scholar]
- 59.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–5. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 60.Mestdagh P, Fredlund E, Pattyn F, Rihani A, Van Maerken T, Vermeulen J, et al. An integrative genomics screen uncovers ncRNA T-UCR functions in neuroblastoma tumours. Oncogene. 2010;29:3583–92. doi: 10.1038/onc.2010.106. [DOI] [PubMed] [Google Scholar]
- 61.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S, et al. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol Cell Biol. 2008;28:6473–82. doi: 10.1128/MCB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–46. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 64.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol. 2006;26:2560–9. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–20. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 67.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, et al. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–6. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282:24731–42. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 70.Zhang X, Gejman R, Mahta A, Zhong Y, Rice KA, Zhou Y, et al. Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res. 2010;70:2350–8. doi: 10.1158/0008-5472.CAN-09-3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S, et al. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011 doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benetatos L, Hatzimichael E, Dasoula A, Dranitsaris G, Tsiara S, Syrrou M, et al. CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leuk Res. 2010;34:148–53. doi: 10.1016/j.leukres.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 73.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bond CS, Fox AH. Paraspeckles: nuclear bodies built on long noncoding RNA. J Cell Biol. 2009;186:637–44. doi: 10.1083/jcb.200906113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, et al. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–63. doi: 10.1016/j.cell.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 76.Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29:3082–93. doi: 10.1038/emboj.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilusz JE, Freier SM, Spector DL. 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008;135:919–32. doi: 10.1016/j.cell.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–41. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 80.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature. 2011;470:284–8. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee GL, Dobi A, Srivastava S. Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol. 2011;8:123–4. doi: 10.1038/nrurol.2011.10. [DOI] [PubMed] [Google Scholar]
- 83.Tomlins SA, Aubin SM, Siddiqui J, Lonigro RJ, Sefton-Miller L, Miick S, et al. Urine TMPRSS2:ERG Fusion Transcript Stratifies Prostate Cancer Risk in Men with Elevated Serum PSA. Sci Transl Med. 2011;3:94ra72. doi: 10.1126/scitranslmed.3001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–33. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee MM, Childs-Disney JL, Pushechnikov A, French JM, Sobczak K, Thornton CA, et al. Controlling the specificity of modularly assembled small molecules for RNA via ligand module spacing: targeting the RNAs that cause myotonic muscular dystrophy. J Am Chem Soc. 2009;131:17464–72. doi: 10.1021/ja906877y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu H, Wang B, Han C. Meta-analysis of genome-wide and replication association studies on prostate cancer. Prostate. 2011;71:209–24. doi: 10.1002/pros.21235. [DOI] [PubMed] [Google Scholar]
- 88.Jia L, Landan G, Pomerantz M, Jaschek R, Herman P, Reich D, et al. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 2009;5:e1000597. doi: 10.1371/journal.pgen.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, et al. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107:3001–5. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A. 2010;107:9742–6. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chung CC, Chanock SJ. Current status of genome-wide association studies in cancer. Hum Genet. 2011;130:59–78. doi: 10.1007/s00439-011-1030-9. [DOI] [PubMed] [Google Scholar]
- 92.Giacomini KM, Brett CM, Altman RB, Benowitz NL, Dolan ME, Flockhart DA, et al. The pharmacogenetics research network: from SNP discovery to clinical drug response. Clin Pharmacol Ther. 2007;81:328–45. doi: 10.1038/sj.clpt.6100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature. 1991;351:325–9. doi: 10.1038/351325a0. [DOI] [PubMed] [Google Scholar]
- 95.Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–9. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- 96.Taft RJ, Pheasant M, Mattick JS. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays. 2007;29:288–99. doi: 10.1002/bies.20544. [DOI] [PubMed] [Google Scholar]
- 97.Yotova IY, Vlatkovic IM, Pauler FM, Warczok KE, Ambros PF, Oshimura M, et al. Identification of the human homolog of the imprinted mouse Air non-coding RNA. Genomics. 2008;92:464–73. doi: 10.1016/j.ygeno.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schorderet P, Duboule D. Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet. 2011;7:e1002071. doi: 10.1371/journal.pgen.1002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Prensner JR, Chinnaiyan AM. Oncogenic gene fusions in epithelial carcinomas. Curr Opin Genet Dev. 2009;19:82–91. doi: 10.1016/j.gde.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 101.Nakamura Y, Takahashi N, Kakegawa E, Yoshida K, Ito Y, Kayano H, et al. The GAS5 (growth arrest-specific transcript 5) gene fuses to BCL6 as a result of t(1;3)(q25;q27) in a patient with B-cell lymphoma. Cancer Genet Cytogenet. 2008;182:144–9. doi: 10.1016/j.cancergencyto.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 102.Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–55. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, et al. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods. 2010;7:995–1001. doi: 10.1038/nmeth.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–7. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]