

Abstract
High density lipoprotein (HDL) possesses important anti-atherogenic properties and this review addresses the molecular mechanisms underlying these functions. The structures and cholesterol transport abilities of HDL particles are determined by the properties of their exchangeable apolipoprotein (apo) components. ApoA-I and apoE, which are the best characterized in structural terms, contain a series of amphipathic α-helical repeats. The helices located in the amino-terminal two-thirds of the molecule adopt a helix bundle structure while the carboxy-terminal segment forms a separately folded, relatively disorganized, domain. The latter domain initiates lipid binding and this interaction induces changes in conformation; the α-helix content increases and the amino-terminal helix bundle can open subsequently. These conformational changes alter the abilities of apoA-I and apoE to function as ligands for their receptors. The apoA-I and apoE molecules possess detergent-like properties and they can solubilize vesicular phospholipid to create discoidal HDL particles with hydrodynamic diameters of ~10 nm. In the case of apoA-I, such a particle is stabilized by two protein molecules arranged in an anti-parallel, double-belt, conformation around the edge of the disc. The abilities of apoA-I and apoE to solubilize phospholipid and stabilize HDL particles enable these proteins to be partners with ABCA1 in mediating efflux of cellular phospholipid and cholesterol, and the biogenesis of HDL particles. ApoA-I-containing nascent HDL particles play a critical role in cholesterol transport in the circulation whereas apoE-containing HDL particles mediate cholesterol transport in the brain. The mechanisms by which HDL particles are remodeled by lipases and lipid transfer proteins, and interact with SR-BI to deliver cholesterol to cells, are reviewed.
Keywords: HDL, Cholesterol, Lipoprotein, apoA-I, apoE
7.1 Introduction
Human serum lipoproteins are soluble complexes of proteins (apolipoproteins) and lipids that represent the major cholesterol transport vehicles in both the intravascular and extravascular compartments. Lipoprotein particles are synthesized by the liver and intestine and mediate lipid transport from the intestine to the liver, and between the liver and cells in the periphery of the body. Mature lipoproteins are microemulsion or emulsion particles (diameter range = 7–600 nm) containing a core of neutral lipids (triacylglycerol (TAG), cholesteryl ester (CE) and cholesterol) stabilized by a surface monomolecular film of phospholipids (PL), cholesterol and apolipoproteins (apo). Lipoprotein particles are traditionally fractionated on the basis of their densities (Jonas and Phillips, 2008).
Apolipoproteins are part of a multi-gene family (Li et al., 1988). ApoB100 is the principal protein constituent of low density lipoprotein (LDL) particles and, by acting as a ligand for the low density lipoprotein receptor, it targets TAG and cholesterol for delivery to cells. Because of this activity, apoB100-containing lipoprotein particles are atherogenic (see Chapter 8 for details). The focus in this chapter is on the anti-atherogenic lipoprotein, high density lipoprotein (HDL), which mediates the efflux of cellular cholesterol. HDL participates in the reverse cholesterol transport process whereby excess cholesterol in cells in the periphery is transported to the liver and ultimately excreted from the body in the feces (Cuchel and Rader, 2006). Apolipoprotein-mediated interactions of HDL particles with cell surface receptors and lipid transporters are critical for this process. HDL contains exchangeable apolipoproteins of the A, C and E families that evolved from a common ancestral gene and are structurally similar. These protein molecules contain 22-amino acid tandem repeats that are often separated by a proline residue (Li et al., 1988). The repeating 22-amino acid segments form amphipathic α-helices (Segrest et al., 1992) thereby enabling these apolipoprotein molecules to bind well to PL-water surfaces and stabilize lipoprotein particles. Lipoprotein particles in the circulation are highly dynamic and, besides acting as ligands for cell surface receptors, apolipoproteins also mediate lipoprotein remodeling. In the case of HDL, the apolipoproteins participate in particle remodeling by interacting with lipid transfer proteins and enzymes that modify lipids (Lund-Katz et al., 2003). These reactions are critical for HDL metabolism and effective reverse cholesterol transport (RCT).
In this consideration of apolipoproteins as cholesterol transport proteins, the focus is on human HDL and two of its constituent proteins, apoA-I and apoE, that are the best characterized in structural terms. The structural basis for the multiple functions of these protein molecules and how they influence the contributions of HDL to RCT are addressed in the following sections.
7.2 Structures of ApoA-I and ApoE in the Lipid-Free State
7.2.1 Primary and Secondary Structures
The genes of apoA-I and apoE contain four exons and three introns, with similar locations of intron–exon boundaries and similar intron and exon lengths for the first three exons. The differences in the total length of the mRNA are due to variations in the length of the fourth exon. Exons three and four in the apoA-I and apoE genes encode the entire mature protein sequence. In both cases, exon 4 codes for a primary structure of 11- and 22-amino acids tandem repeats that span residues 44–243 in apoA-I and 62–299 in apoE (Fig. 7.1A) (Li et al., 1988). Each of these repeats has the periodicity of an amphipathic α-helix and these helices are often separated by a proline residue (Li et al., 1988; Segrest et al., 1992). The amphipathic α-helices have been classified into several distinct classes according to the distribution of charged residues around the axis of the helices (Segrest et al., 1994). The class A amphipathic helix is a major lipid-binding motif in exchangeable apolipoproteins and is characterized by the location of basic residues near the hydrophilic/hydrophobic interface and acidic residues at the center of the polar face (Fig. 7.1B). Class G* and Y amphipathic α-helices are also present in exchangeable apolipoproteins (Segrest et al., 1992). The class G* helix is similar to the amphipathic α-helices present in water-soluble globular proteins and possesses a random radial arrangement of positive and negative amino acids in the polar face. Sometimes the amphipathic α-helix is characterized by the presence of a Y-shaped cluster of basic amino acids in the polar face (Fig. 7.1B) giving a class Y helix (Segrest et al., 1994).
Fig. 7.1.

A Distribution of amphipathic α-helices in the human exchangeable apolipoproteins, apoA-I and apoE. The letter P below the rectangles indicates positions of all proline residues. B Amphipathic helix classes found in the exchangeable apolipoproteins. Classification is based on the distribution of charged residues (see Section 2.1). These figures were adapted from Segrest et al. (1992)
Human apoA-I is a 243 amino acid protein (molecular mass = 28.1 kDa) in which the region coded by exon 4 is predicted to contain eight 22-mer and two 11-mer amphipathic α-helices with most of the helices being punctuated by pro-lines (Fig. 7.1A) (Brouillette et al., 2001; Segrest et al., 1992). The predicted α-helices shown in Fig. 7.1A for human apoA-I include approximately 80% of the amino acids and represent the maximal helix content; for comparison, the lipid-free protein is about 50% α-helical in dilute solution, as revealed by circular dichroism measurements (Saito et al., 2003b). Comparison of sequences between mammals indicates that the N-terminal region of apoA-I is highly conserved while the central and C-terminal regions show conservative substitutions between species (Brouillette et al., 2001; Frank and Marcel, 2000). Studies of synthetic peptides corresponding to each of the 22-residue amphipathic α-helices of human apoA-I have shown that the first (residues 44–65) and last (residues 220–241) repeat helices have the greatest lipid affinity (Palgunachari et al., 1996). Hydropathy analysis of the amino acid sequence indicates that the C-terminal region of human apoA-I is very hydrophobic (Saito et al., 2004b), consistent with this region having significant lipid binding ability. Studies of both natural and engineered mutations in the human apoA-I molecule have revealed that the C-terminal region is indeed important for lipid binding (Brouillette et al., 2001; Saito et al., 2004b) and that the central region corresponding to residues 121–186 is important for activation of the enzyme lecithin: cholesterol acyltransferase (LCAT) (Frank and Marcel, 2000; Sorci-Thomas and Thomas, 2002).
Human apoE is a 299 amino acid protein with a molecular mass of 34.2 kDa (Weisgraber, 1994), that is predicted to contain amphipathic α-helices along its length. In contrast to apoA-I, a high proportion of these amphipathic α-helices are class G* (Fig. 7.1A). The distribution of 22-residue repeats and the proline punctuation are less regular than occurs in the apoA-I molecule (Fig. 7.1A). The predicted α-helices include about 70% of the amino acids which is somewhat higher than the value of ~60% measured by circular dichroism for lipid-free apoE in dilute solution (Morrow et al., 2000). There is a high degree of sequence conservation across species with the exceptions of the N- and C-termini: homology begins in the vicinity of residue 26 in the human sequence and continues to approximately residue 288 (Weisgraber, 1994). Hydropathy analysis of the human apoE sequence shows that the C-terminus as well a central region (residues 192–215) are relatively hydrophobic (Saito et al., 2004b); the C-terminal region plays a critical role in lipid binding (Saito et al., 2004b; Weisgraber, 1994). Human apoE exists as three major isoforms, apoE2, apoE3 and apoE4, each differing by cysteine and arginine at positions 112 and 158. ApoE3, the most common form, contains cysteine and arginine at these positions, respectively, whereas apoE2 contains cysteine and apoE4 contains arginine at both sites (Weisgraber, 1994). These apoE isoforms are associated with different levels of disease risk, most notably for atherosclerosis (Davignon et al., 1988; Getz and Reardon, 2009) and Alzheimer’s disease (Mahley et al., 2009; Mahley and Huang, 1999) (see also Chapter 2). Comparisons of apoE2, apoE3 and apoE4 together with studies of other natural apoE mutations have led to the identification of a cluster of basic amino acids in the regions spanning residues 136–150 as the recognition site responsible for the binding of apoE to the low density lipoprotein (LDL) receptor (Hatters et al., 2006; Weisgraber, 1994).
7.2.2 Tertiary Structure
A variety of studies using protein engineering techniques have provided important insights into the lipid-free structures of apoA-I and apoE (for reviews, see Brouillette et al., 2001; Davidson and Thompson, 2007; Saito et al., 2004b; Weisgraber, 1994). Proteolysis analysis (Roberts et al., 1997) and deletion muta-genesis studies (Davidson et al., 1996; Saito et al., 2003b) have suggested that the lipid-free apoA-I molecule is organized into two structural domains; the N-terminal and central parts form a helix bundle whereas the C-terminal α-helices form a separate, less organized structure. The helix bundle organization in the N-terminal domain is also supported by fluorescence studies of single tryptophan mutants of human (Brouillette et al., 2005; Davidson et al., 1999) and chicken apoA-I (Kiss et al., 1999). The guanidine-induced denaturation curve of apoA-I is monophasic (Reijngoud and Phillips, 1982) whereas that for apoE is biphasic (Morrow et al., 2000) indicating that apoE also adopts a two-domain tertiary structure, and that the N- and C-terminal domains unfold independently. The helix bundle motif of the N-terminal domain is similar in apoA-I and apoE except for it being less organized and less stable in apoA-I. A thermal unfolding study has demonstrated that the apoA-I molecule exhibits a loosely folded, molten globular-like structure (i.e. the α-helices do not occupy fixed positions with respect to one another and are not organized into a unique tertiary structure) (Gursky and Atkinson, 1996).
ApoA-I is the only intact human apolipoprotein in the lipid-free state for which a high resolution structure is available to date. The crystal structure (Ajees et al., 2006) reveals an N-terminal anti-parallel four-helix bundle domain and a separate two-helix C-terminal domain (Fig. 7.2A). Apparently, the conditions used for crystallization induced helix formation because some 80% of the residues in this structure are in α-helices, whereas the helix content is closer to 50% for monomeric apoA-I in dilute solution. Nevertheless, cross-linking/mass spectrometry experiments (Silva et al., 2005) indicate that the apoA-I molecule in solution is still folded into two domains, with residues 1–189 forming the helix bundle and residues 190–243 folding separately. In the case of apoE, the structure of the 22-kDa N-terminal fragment has been solved by crystallographic (Wilson et al., 1991) and NMR (Sivashanmugam and Wang, 2009) methods (Fig. 7.2B). As occurs with apoA-I, this domain forms a globular bundle of four elongated α-helices in which the helices are arranged in an anti-parallel manner with their hydrophobic faces oriented towards the interior of the bundle. This structure shares the same basic architecture as the helix bundle of insect apolipophorin III (Narayanaswami and Ryan, 2000). The N-terminal fragments of all three apoE isoforms adopt such a four-helix bundle motif, but subtle differences in the side-chain conformations and salt-bridge arrangements of the isoforms affect their functions and characteristics (Dong et al., 1994, 1996; Wilson et al., 1994). Studies of guanidine (Morrow et al., 2000) and thermal (Acharya et al., 2002) denaturation revealed that the N-terminal fragments of the apoE isoforms differ in stability (apoE4 < apoE3 < apoE2), indicating that replacing cysteine residues by arginine results in a progressive decrease in hydrophobicity and stability of the helix bundle such that in apoE4 this domain exhibits molten globule characteristics (Morrow et al., 2002).
Fig. 7.2.

Crystal structures of human apolipoproteins in the lipid-free state. A The six α-helices in human apoA-I are shown (from Ajees et al., 2006, with permission). The N-terminal anti-parallel four-helix bundle contains helices A (residues 10–39), B (50–84), C (97–137), and D (147–187). The C-terminal domain is formed by the two α-helices E (residues 196–213) and F (219–242). Hydrophobic residues located in the interior of the helix bundles are shown as sticks. B Ribbon model of the structure of the 22-kDa N-terminal domain fragment of human apoE3 (from Weisgraber (1994), with permission). Four of the five helices are arranged in an anti-parallel four-helix bundle. The residues spanned by each helix, together with the region in helix 4 recognized by the LDL receptor, are indicated
The structural organization of the C-terminal domain in both apoA-I and apoE is not well characterized. Fluorescence measurements with 8-anilino-1-naphthalenesulfonic acid (ANS) have suggested that the C-terminal domain of apoE forms a solvent-exposed, less organized structure (Saito et al., 2003a). A model has been proposed in which the helices in the C-terminal domain form an intermolecular coiled-coil helix structure (Choy et al., 2003). The polymorphism at position 112 in the N-terminal domain of apoE affects the properties of the C-terminal domain because of interactions between the domains (Hatters et al., 2006). In apoE4, the N- and C-terminal domains interact differently than they do in the other isoforms: Arg-112 causes a rearrangement of the Arg-61 side chain in the N-terminal domain of apoE4, allowing it to interact with Glu-255 in the C-terminal domain (Dong et al., 1994; Dong and Weisgraber, 1996). This domain interaction in human apoE4 leads to a less organized structure in the C-terminal domain, leading to preferential association with VLDL rather than HDL, which is contrary to the behavior of apoE3 (Weisgraber, 1990). The domain interaction in apoE4 has been suggested to contribute to the accelerated catabolism of this isoform and, consequently, the increased cholesterol and LDL levels in the plasma of individuals with this genotype (Davignon et al., 1988; Mahley et al., 1999). Fluorescence energy transfer measurements indicate that the N- and C-terminal domains of lipid-free apoE3 are close to one another and interact, probably through weak hydrophobic interaction (Narayanaswami et al., 2001); the two domains are closer together in apoE4 than in apoE3 (Hatters et al., 2005). In the case of apoA-I, an electron paramagnetic resonance spectroscopic study indicated that the helices in the C-terminal domain form a compact anti-parallel alignment with residues 188–205 existing as a flexible loop (Oda et al., 2003), whereas fluorescence resonance energy transfer measurements suggested an extended conformation (Behling Agree et al., 2002). Regardless of its exact conformation, the C-terminal domain appears to be relatively disorganized because it contains an exposed hydrophobic surface that is accessible to ANS binding (Saito et al., 2003b). Analogous to the situation with apoE, the N-and C-terminal domains in the apoA-I molecule interact with each other to maintain the overall structure of the protein (Fang et al., 2003; Tricerri et al., 2000). Human and mouse apoA-I both adopt a two-domain tertiary structure implying that apoA-I from all higher mammals is organized similarly (Tanaka et al., 2008). Human apoA-I functions with a relatively stable N-terminal helix bundle and a hydrophobic C-terminal domain, whereas mouse apoA-I functions with an unstable helix bundle domain and a polar C-terminal domain.
7.2.3 Quaternary Structure
It is well established that lipid-free apoA-I and apoE tend to self-associate in aqueous solution because of hydrophobic interaction between amphipathic α-helices. Lipid-free apoA-I self-associates reversibly as a function of protein concentration and forms oligomers at concentrations > 0.1 mg/ml (Donovan et al., 1987; Vitello and Scanu, 1976). Sedimentation equilibrium ultracentrifugation and gel filtration chromatography data suggest a model of apoA-I self-association that involves equilibration between monomer–dimer–tetramer–octamer states. Studies on C-terminal truncation mutants demonstrate that the oligomerization is less pronounced (Davidson et al., 1996), indicating that self-association of apoA-I is mediated by hydrophobic interactions between α-helices in the C-terminal domain (Section 2.2). Compared to human apoA-I, lipid-free mouse apoA-I undergoes only minimal concentration-dependent self-association (Gong et al., 1994), because its C-terminal domain is relatively polar (Tanaka et al., 2008).
ApoE is known to exist as a tetramer in the lipid-free state (Yokoyama et al., 1985) and this tetramerization is thought to be mediated by the C-terminal domain, because the N-terminal 22-kDa fragment is monomeric whereas the 10-kDa C-terminal fragment is tetrameric in aqueous solution (Aggerbeck et al., 1988). Sedimentation velocity experiments provide direct evidence for heterogeneous solution structures of apoE3 and apoE4 (Perugini et al., 2000). In a lipid-free environment, both proteins exist as a slow equilibrium mixture of monomers, tetramers, octamers and a small proportion of higher oligomers. Experiments with mixtures of apoE and phospholipid micelles indicate that apoE oligomers undergo a phospholipid-induced dissociation, suggesting that the monomeric form predominates on lipoprotein particle surfaces (Perugini et al., 2000). Sedimentation velocity studies using recombinant apoE isoforms fused with an amino-terminal extension of 43 amino acids confirmed that monomers, dimers and tetramers are the major species of lipid-free apoE2, apoE3 and apoE4 (Barbier et al., 2006). Analysis of apoE C-terminal truncation variants indicates that the extreme C-terminal residues 267–299, which are known to be very hydrophobic by hydropathy analysis (Saito et al., 2004b), are responsible for the self-association of apoE (Westerlund and Weisgraber, 1993). A segment containing residues 218–266 in the apoE C-terminal domain possesses a high propensity to form a coiled-coil helix structure and an apoE construct containing C-terminal residues 201–299 gives circular dichroism spectra characteristic of coiled-coil helices (Choy et al., 2003). This apoE C-terminal domain construct predominantly forms dimeric and tetrameric species in aqueous solution. A monomeric, biologically active apoE C-terminal domain mutant has been prepared using protein engineering techniques (Fan et al., 2004). In this model, five bulky hydrophobic residues (F257, W264, V269, L279, and V287) are replaced with either smaller hydrophobic or polar/charged residues. Cross-linking experiments indicate that this mutant is 100% monomeric even at concentrations as high as 5 mg/ml. A monomeric, biologically active, full-length apoE has also been generated in similar fashion (Zhang et al., 2007). This mutant is nearly 95–100% monomeric even at 20 mg/ml, consistent with interactions between hydrophobic residues in the C-terminal domain playing a key role in the self-association process. To understand the molecular basis for the different degrees of self-association of the apoE isoforms, the effects of progressive truncation of the C-terminal domain in human apoE3 and apoE4 on their lipid-free structures have been compared (Sakamoto et al., 2008). In contrast to previously reported findings, gel filtration chromatography experiments demonstrate that the monomer–tetramer distribution is different for the two isoforms with apoE4 being more monomeric than apoE3; removal of the C-terminal helices in both isoforms favors the monomeric state.
Overall, it can be inferred that self-association of apoA-I and apoE promotes stabilization of the potential α-helical segments of the C-terminal domain (Sections 7.2.1 and 7.2.2) that are less organized in the monomeric form.
7.3 Interaction of ApoA-I and ApoE with Lipids
7.3.1 Molecular Mechanism of Lipid-Binding
It is well established that the C-terminal domain in the apoA-I and apoE molecules is critical for lipid-binding (for reviews, see Weisgraber, 1994; Saito et al., 2004b; Hatters et al., 2006). In the case of apoA-I, an early model proposed a multi-step mechanism in which the initial binding to lipid occurs through the C-terminal region followed by a conformational switch of residues 1–43 which unmasks a latent lipid-binding domain comprising residues 44–65 (Rogers et al., 1998). By using a series of deletion mutants that progressively lacked different regions along the molecule, we showed that the binding of apoA-I to lipids is modulated by reorganization of the N-terminal helix bundle structure (Saito et al., 2003b). Recent fluorescence experiments with pyrene-labelled apoA-I indicate the helix bundle can open upon interaction with the surface of a lipid particle (Kono et al., 2008). This concept is consistent with earlier experiments with apoE demonstrating that upon binding to lipids, the four-helix bundle in the N-terminal domain undergoes a conformational reorganization to expose the hydrophobic faces of its amphipathic helices for interaction with lipid molecules (Weisgraber, 1994). Molecular area measurements at an air-water interface indicated that the N-terminal domain occupies a larger surface area than can be accounted for by its globular four-helix bundle conformation, suggesting adoption of an open conformation by the helix bundle (Weisgraber et al., 1992; Weisgraber, 1994). Subsequent studies using infrared spectroscopy (Raussens et al., 1998), fluorescence resonance energy transfer (Fisher and Ryan, 1999), and inter-helical disulfide mutants of the apoE N-terminal domain (Lu et al., 2000) confirmed that the four-helix bundle undergoes conformational opening when apoE is complexed with PL. This conformational rearrangement is associated with changes in functionality (Saito et al., 2004b). The helix bundle can adopt either open (recognized by the LDL receptor) or closed (not recognized by the LDL receptor) conformations, depending upon the concentration of apoE bound to the PL particle surface (Saito et al., 2001). The rate of interfacial rearrangement is affected by the stability of the helix bundle domain. Thus, apoE4 rearranges more rapidly than apoE3 upon binding to a lipoprotein surface (Nguyen et al., 2009) because, as mentioned in Section 7.2.2, the apoE4 N-terminal domain is relatively unstable. The different domain-domain interaction in apoE4 compared to apoE3 (Section 7.2.2) causes the C-terminal domain in apoE4 to be relatively disorganized, leading to a higher lipid affinity for this apoE isoform (Saito et al., 2003a).
The similar two-domain tertiary structures of apoA-I and apoE (Section 7.2.2) led to a common two-step mechanism describing the binding of both proteins to spherical lipid particles (Fig. 7.3) (Saito et al., 2003b, 2004b). In this model, the apolipoprotein initially binds to a lipid surface through amphipathic α-helices in the C-terminal domain; this process is accompanied by an increase in α-helicity in this domain (Oda et al., 2003; Saito et al., 2003b). Subsequently, the helix bundle in the N-terminal domain undergoes a conformational opening, converting hydrophobic helix-helix interactions to helix-lipid interactions. The conformational transition from random coil to α-helix upon lipid-binding provides the energetic source to drive the lipid interaction of apolipoproteins (Massey et al., 1979). Calorimetric measurements of binding of apoA-I (Arnulphi et al., 2004; Saito et al., 2003b), apoE (Saito et al., 2001) and apoA-I model peptides (Gazzara et al., 1997) to lipid particles, indicate that lipid-binding of these proteins is accompanied by a large release of exothermic heat, consistent with the lipid-binding of apolipoproteins being enthalpically driven. The contribution of α-helix formation to the enthalpy of binding of apoA-I to egg PC small unilamellar vesicles is −1.1 kcal/α-helical residue (Saito et al., 2004a), is in agreement with a prior estimate (−1.3 kcal/α-helical residue) for plasma apolipoproteins using different membrane systems (Massey et al., 1979). α-Helix formation in apoA-I also contributes to an increase in the favourable free energy of binding to lipid (−42 cal/α-helical residue), leading to an approximately two-order of magnitude increase in the affinity of binding (Saito et al., 2004b). This indicates that the transition of random coil to α-helix plays a critical role in driving apoA-I to interact with a lipid surface. This phenomenon probably explains why many exchangeable apolipoproteins in the lipid-free state contain random coil structure especially in the lipid-binding domain, such as the C-terminal domain of apoA-I and apoE.
Fig. 7.3.

Model of the two-step mechanism of apoA-I binding to a spherical lipid particle. In the lipid-free state, apoA-I is organized into two structural domains in which the N-terminal domain forms a helix bundle whereas the C-terminal domain forms a separate, less organized structure. Initial lipid binding occurs through amphipathic α-helices in the C-terminal domain accompanied by an increase in α-helicity probably in the region including residues 187–220. Subsequently, the helix bundle in the N-terminal domain undergoes a conformational opening, converting hydrophobic helix-helix interactions to the helix–lipid interaction; this second step is only slowly reversible. Reproduced with permission from Saito et al. (2003b)
The binding of apoA-I and apoE to PL is affected by the composition of the lipid particle. For example, addition of either 20–40 mol% cholesterol or 33 mol% egg yolk phosphatidylethanolamine to 100 nm egg yolk phosphatidylcholine (PC) large unilamellar vesicles (LUV) increases the amount of apoA-I binding by increasing the PL polar headgroup space in the relatively flat surface of the LUV (Saito et al., 1997). In contrast, addition of cholesterol to either 100 nm TAG-PC emulsion particles (Saito et al., 1997) or to 20 nm PC small unilamellar vesicles (SUV) (Arnulphi et al., 2004) decreases apoA-I binding; this effect apparently occurs because cholesterol condenses the PL packing and decreases the space available in the highly curved SUV surface.
7.3.2 Apolipoprotein Conformation in Discoidal and Spherical HDL Particles
It is difficult to obtain high resolution structures for HDL particles, because they are not suitable for study by X-ray crystallography and NMR. However, progress has been made by applying alternative methods to study homogeneous, reconstituted, HDL particles containing apoA-I as the sole protein. The study of such particles is relevant because apoA-I is the major protein component of both nascent discoidal and circulating spherical HDL particles (Lund-Katz et al., 2003). The reconstituted HDL discs comprise a segment of PL bilayer surrounded at the edge by apoA-I molecules arranged in a belt-like fashion. The generally accepted double-belt model for the structure of a discoidal HDL particle containing two apoA-I molecules (Segrest et al., 1999) is depicted in Fig. 7.4A. The details of the helix organization in this structure have been resolved by chemical cross-linking and mass spectrometry methods (Davidson and Silva, 2005; Thomas et al., 2006). The two apoA-I molecules are aligned in an anti-parallel fashion so that the amphipathic α-helix spanning residues 121–142 in one apoA-I molecule is opposite the same helix in the other apoA-I molecule. Salt-bridges between the two apoA-I molecules help to stabilize this structure. While there is general agreement about the basic structure shown in Fig. 7.4A, there are uncertainties about the organization of the N- and C-terminal ends of the two apoA-I molecules (Davidson and Thompson, 2007; Thomas et al., 2008). The size of the discoidal HDL particle is determined primarily by the number of apoA-I molecules per particle. However, several discrete-sized particles can be formed in complexes containing a constant number of apoA-I molecules (Li et al., 2004). The conformationally flexible apoA-I molecule (Section 7.3.1) adjusts to different particle sizes by certain segments in the protein desorbing from the particle surface and looping into the aqueous phase. Neither the location within the apoA-I molecule nor the precise nature of the protruding segments are well established (Davidson and Thompson, 2007; Thomas et al., 2008).
Fig. 7.4.
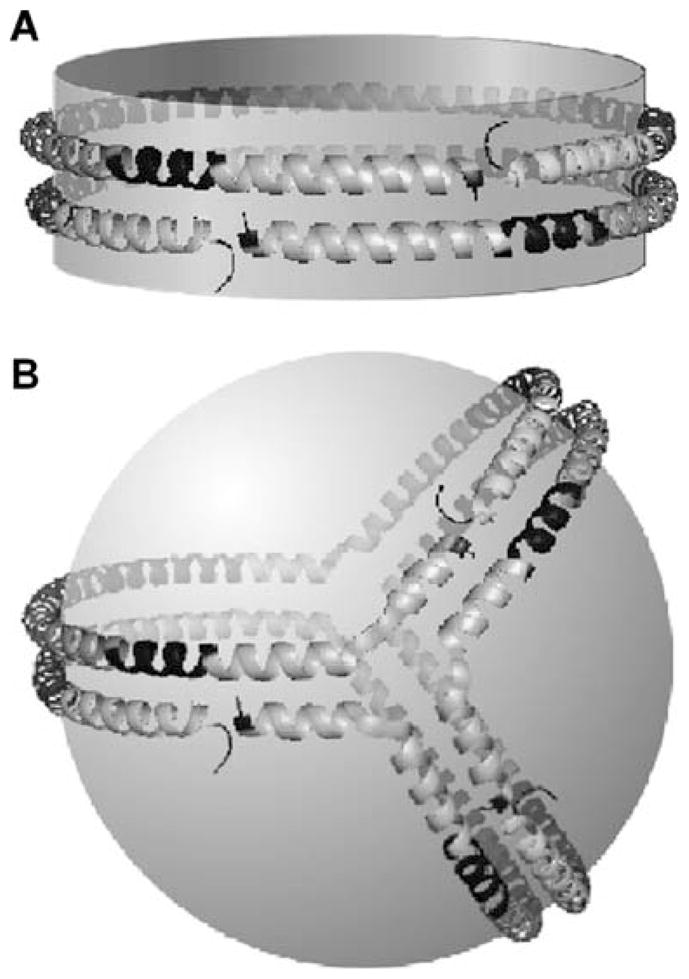
ApoA-I conformation on discoidal and spherical HDL particles. The apoA-I molecules are organized as a double-belt in discoidal particles and as a trefoil in spherical particles. All helix–helix interactions between the two molecules of apoA-I in the disc double-belt arrangement are also present between the three apoA-I molecules in the trefoil organization on the surface of a spherical HDL particle. Reproduced with permission from Silva et al. (2008)
Given the similarities in apoA-I and apoE structure (Section 7.2), it is unsurprising that apoE also complexes with PL to make HDL particles. As occurs in discoidal HDL particles containing apoA-I, the α-helices in apoE molecules also align perpendicular to the acyl chains of the PL molecules (Narayanaswami et al., 2004; Schneeweis et al., 2005). However, the apoE-PL interaction is more complex than that seen with apoA-I in that some apoE helices seem to be situated among the PL polar headgroups on the faces of the disc. X-ray diffraction data for dipalmitoyl PC complexes containing two apoE molecules (Peters-Libeu et al., 2006, 2007) indicate that, unlike apoA-I, apoE does not simply form discoidal particles. Rather, the apoE particles are quasi-spheroidal and the apoE molecules are folded into a helical hairpin and interact primarily with the PL polar headgroups (Hatters et al., 2009; Peters-Libeu et al., 2006, 2007).
In vivo, discoidal nascent HDL particles created by the activity of ABCA1 (Section 7.5.2) are remodelled by various factors (Section 7.3.3) to yield the mature spherical HDL particles found in the circulation (Jonas and Phillips, 2008; Lund-Katz et al., 2003). These spherical HDL particles contain a neutral lipid core composed of CE and TAG surrounded by a monolayer of PL and cholesterol molecules and, unlike a discoidal particle, do not have a particle edge to constrain the apolipoproteins. However, the protein-protein interactions that occur in the double-belt model of a discoidal particle are maintained in a spherical, apoA-I containing, HDL particle (Fig. 7.4B) (Silva et al., 2008). All three apoA-I molecules in the trefoil model of a spherical HDL particle are in identical conformations and the inter-molecular salt bridges are the same as those that exist in the double-belt model of a discoidal particle. The apoA-I molecules in the trefoil arrangement are thought to provide the structural scaffold that stabilizes the spherical HDL particle. However, there is another pool of HDL-associated apoA-I molecules that readily exchanges on and off the particle surface. These apoA-I molecules are likely to be bound with only their C-terminal domain in contact with the particle surface and their N-terminal helix bundle domain in the closed conformation protruding into the aqueous phase (Kono et al., 2008) (cf. Fig. 7.3). ApoE molecules associated with spherical HDL particles also presumably adopt either the helix bundle-closed or -open conformation (cf. Section 7.3.2).
7.3.3 Remodeling of HDL Particles
Remodeling by plasma factors is a critical part of HDL metabolism and underlies the dynamic nature of HDL particles. Thus, the various subpopulations of HDL particles that exist in human plasma (Lund-Katz et al., 2003; Zannis et al., 2006) are continually interconverting due to the lipolytic and lipid transfer activities of the plasma factors listed in Table 7.1. As shown in Fig. 7.7 in Section 7.5.1, the changes in HDL particle shape and size caused by these plasma factors are also central to the participation of HDL in the RCT pathway. The key function of lecithin-cholesterol acyltransferase (LCAT) is to form CE while the other enzymes, hepatic lipase (HL) and endothelial lipase (EL), are involved in releasing fatty acids from PL and TAG. The lipid transfer proteins, cholesterol ester transfer protein (CETP) and phospholipid transfer protein (PLTP), are involved in moving CE, TAG and PL molecules among HDL and other lipoprotein particles (Masson et al., 2009; Rye et al., 2009). As shown in Table 7.1, the interconversion of HDL particles is frequently accompanied by the release of apoA-I molecules into the aqueous phase; rearrangements of HDL particles that are accompanied by a decrease in net surface area lead to desorption of apoA-I molecules and vice versa. Such cycling of apoA-I molecules between HDL particles and the aqueous phase is critical for HDL metabolism (Pownall and Ehnholm, 2006; Rye and Barter, 2004).
Table 7.1.
Plasma factors involved in remodeling of HDL
| HDL conversion | Plasma factors |
|---|---|
| disc → sphere | LCAT |
| large sphere → small sphere + free apo A-I | CETP and HL |
| large sphere → small sphere | EL |
| sphere → larger and smaller spheres + free apo A-I | PLTP |
Fig. 7.7.

Schematic overview of the major pathways involved in HDL-mediated macrophage cholesterol efflux and reverse cholesterol transport to the liver. ApoA-I is produced by the liver and acquires free cholesterol (FC) and phospholipid (PL) from liver and peripheral cells (including macrophages) via the ABCA1 transporter to form nascent (discoidal) HDL particles. Non-lipidated apoA-I is cleared by the kidney. FC efflux from macrophages to HDL particles is also promoted by the ABCG1 transporter and SR-BI. As discussed in Section 7.3.3, the FC in discoidal HDL particles is converted to CE by LCAT activity leading to the formation of mature, spherical HDL particles. PLTP mediates transfer of PL from VLDL into HDL thereby providing PL for the LCAT reaction. Mature HDL particles can be remodeled to smaller particles with the release of apoA-I by the actions of hepatic lipase (HL) and endothelial lipase (EL) which hydrolyze HDL TAG and PL, respectively. In humans, but not rodents, HDL-CE can be transferred to the VLDL/LDL pool by CETP and taken up by endocytosis into hepatocytes via interaction with the LDL receptor (LDLR). HDL-CE and FC are also transferred directly to hepatocytes via SR-BI-mediated selective uptake. Cholesterol taken up by the liver can be recycled back into the ABCA1 pathway, secreted into bile as either FC or bile acids, or assembled into lipoprotein particles that are secreted back into the circulation (not shown)
LCAT is secreted by the liver in humans and is the major enzyme responsible for the esterification of free unesterified cholesterol (FC) present in circulating lipoproteins (Santamarina-Fojo et al., 2000; Zannis et al., 2006). The protein comprises a single polypeptide chain of 416 amino acids and it is glycosylated at four sites giving a molecular mass of ~60 kDa. This lipase contains an α/β-hydrolase fold and an Asp-His-Ser catalytic triad, with Ser 181 being in the active site (Jonas, 2000). The enzyme catalyses a transesterification reaction in which an unsaturated fatty acid is transferred from the sn-2 position of PC to the hydroxyl group of cholesterol via Ser 181, generating CE and lyso-PC. ApoA-I is the major activator of LCAT in plasma so PC and FC molecules in HDL particles are the preferred substrates. Amphipathic α-helices located between residues 143–187 in the apoA-I molecule apparently mediate the binding of LCAT to the HDL particle, with three arginine residues (R149, R153, R160) playing a critical role (Roosbeek et al., 2001). LCAT binds with high affinity to apoA-I-containing discoidal HDL particles (cf. Section 7.3.2) and converts the FC in them to CE. The CE molecules produced are poorly soluble in a PL bilayer and form a neutral lipid core; the resultant spherical, microemulsion-like, particle is stabilized by a mixed PL/apoA-I monolayer (cf. Section 7.3.2). As expected, in LCAT-knockout mice the levels of spherical CE-enriched HDL particles are reduced and the levels of FC-enriched discoidal HDL particles are increased (Santamarina-Fojo et al., 2000).
CETP and PLTP are members of the lipopolysaccharide-binding/lipid transfer protein family and each contains 476 residues in a single polypeptide chain; both proteins are glycosylated and the apparent molecular masses are 74 and 84 kDa, respectively (Masson et al., 2009; Rye et al., 2009). The primary structure of PLTP is ~25% identical to that of CETP and both proteins can be predicted to have a similar tertiary structure. The crystal structure of CETP indicates that the molecule contains a 6 nm-long tunnel that can be filled with two CE molecules and plugged at each end by a PC molecule (Qiu et al., 2007). It is suggested that when CETP binds to a lipoprotein particle, PL molecules bound at the end of the tunnel merge into the PL monolayer at the particle surface and allow neutral lipid molecules to enter and exit the tunnel. Since CETP can bind CE and TAG molecules, it is possible that the protein transfers lipids between lipoprotein particles by a shuttle mechanism. However, CETP can bridge two HDL particles to form a ternary complex and induce particle fusion, thereby changing HDL particle size (Rye et al., 1997) (cf. Table 7.1). CETP-mediated exchange of CE and TAG between HDL and VLDL enriches the HDL particles with TAG, which destabilizes them and promotes dissociation of apoA-I molecules (Rye et al., 2009; Sparks et al., 1995). TAG-enriched HDL particles are also good substrates for HL, the activity of which further reduces the particle size and enhances apoA-I dissociation (Table 7.1). PLTP transfers PL molecules between VLDL and HDL, as well as between different HDL particles (Huuskonen et al., 2001; Van Tol, 2002). As indicated in Table 7.1, PLTP remodels HDL into large and small particles and promotes the dissociation of apoA-I. This remodelling process involves HDL particle fusion and the remodelling is faster with TAG-enriched HDL (Settasatian et al., 2001). The activities of both CETP and PLTP impact significantly on plasma HDL levels. Thus, PLTP-knockout mice exhibit ~50% reductions in plasma HDL levels, whereas inhibition of CETP in both animals and humans increases HDL levels (Masson et al., 2009).
HL and EL are members of the lipase gene family that also includes pancreatic lipase and lipoprotein lipase; members of this family have similar tertiary structures (Wong and Schotz, 2002). Both HL and EL hydrolyze HDL lipids, although the substrate specificities are different. HDL TAG and PL are substrates for HL whereas EL is primarily a phospholipase (Jaye and Krawiec, 2004; Thuren, 2000). Presumably, this variation in substrate specificity is the reason that HL action on TAG-enriched HDL leads to dissociation of apoA-I (Clay et al., 1991), whereas EL action does not (Jahangiri et al., 2005); lipolysis reduces HDL particle size in both cases (Table 7.1). Both lipases affect plasma HDL levels although to different extents; EL-knockout mice have ~50% elevated HDL cholesterol levels (Badellino and Rader, 2004), whereas HL-knockout mice exhibit much smaller increases in HDL cholesterol levels (Homanics et al., 1995).
Plasma HDL consists of particles that contain only apoA-I and particles that contain both apoA-I and apoA-II (Lund-Katz et al., 2003). The latter particles are formed by LCAT-induced fusion of nascent, discoidal HDL particles that contain either apoA-I or apoA-II (Clay et al., 2000). Compared to apoA-I, apoA-II is more lipophilic and dissociates less readily from HDL particles (Pownall and Ehnholm, 2006). The presence of apoA-II stabilizes the HDL particle and reduces its ability to be remodeled. Thus, apoA-II inhibits the ability of CETP to shrink spherical HDL particles and prevents apoA-I dissociation; the latter effect occurs because of stabilizing protein-protein interactions between apoA-I and apoA-II in the HDL particle surface (Rye et al., 2008).
7.4 Lipid Solubilizing Properties of ApoA-1 andApoE
7.4.1 Historical Perspective
The fact that the apolipoproteins of HDL can stabilize ~10 nm lipid microemulsion particles with the structure depicted in Fig. 7.4B for a spherical HDL particle, indicates that an exchangeable apolipoprotein such as apoA-I possesses detergent-like properties and can solubilize lipids. This was first demonstrated directly in the 1960 s by Scanu (1967), who reported that the apoproteins from human HDL can form stable, water-soluble, lipid–protein complexes with HDL PL when the mixture is sonicated. Subsequently, electron microscopic examination (Forte et al., 1971) indicated that such complexes are disc-shaped and circular dichroism measurements (Lux et al., 1972) showed that the interaction with lipid increases the α-helix content of the apolipoprotein. Scanu and colleagues also used the sonication procedure to show that (a) apo HDL can complex with a range of synthetic phospholipids when the lipids are in a liquid-crystalline state (Kruski and Scanu, 1974) and (b) spherical HDL particles can be reassembled in vitro when the HDL neutral lipids (CE and TAG) are included (Hirz and Scanu, 1970).
HDL particles reconstituted with either natural or synthetic lipids have been the subject of extensive investigations since the early 1970s. The first detailed characterization in 1974 was of the disc-shaped complexes formed by dimyristoyl PC (DMPC) and porcine apo HDL (primarily apoA-I). Co-sonication of this mixture under conditions where all the apoprotein is complexed yields lipoprotein discs consisting of two apoprotein molecules and about 200 DMPC molecules; the major diameter is 8–9 nm and the thickness is 4.4 nm, in good agreement with the thickness of a DMPC bilayer (Hauser et al., 1974). The acyl chain melting transition of the DMPC in such a reconstituted HDL particle is broadened, indicating that the apoprotein molecules modify the packing of the lipid molecules (Barratt et al., 1974). The fundamental organization of the discoidal particle as a small segment of DMPC bilayer stabilized by apoprotein molecules was confirmed by small-angle X-ray scattering (Atkinson et al., 1976), and it was found that the apoprotein amphipathic α-helices reduce the cooperativity of DMPC acyl chain motions, but do not bind the lipid molecules tightly (Andrews et al., 1976). Importantly, it was shown in these studies with porcine apo HDL that sonication is not required and that the highly surface-active protein molecules can spontaneously solubilize multilamellar vesicles (MLV) of DMPC to form discoidal HDL particles of the type described above (also see Fig. 7.5) (Hauser et al., 1974). In 1977 it was demonstrated that pure apoA-I, either human or bovine, can form discoidal complexes with DMPC (Jonas et al., 1977) and that the particle size increases with increasing DMPC/apoprotein ratio (Tall et al., 1977); the latter finding is consistent with the predominant location of the apoprotein being an annulus around the perimeter of the disc (Tall et al., 1977) (cf. Fig. 7.4A).
Fig. 7.5.
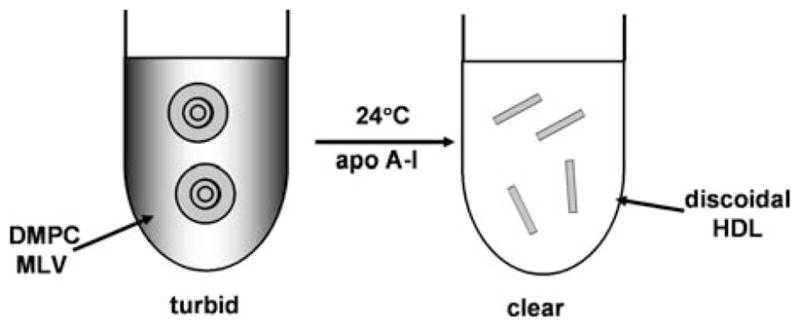
Schematic representation of the spontaneous solubilization of dimyristoyl phosphatidyl-choline (DMPC) multilamellar vesicles (MLV) by apoA-I. When apoA-I is incubated with the turbid MLV suspension at 24°C (the melting temperature of the DMPC acyl chains), the solution becomes optically clear because the DMPC bilayers are solubilized in a few minutes to create discoidal HDL particles that are too small to scatter visible light
7.4.2 Mechanism of Solubilization Reaction
The first detailed study of the kinetics of solubilization of DMPC by human apoA-I was conducted by Pownall and colleagues in 1978 (Pownall et al., 1978). They used turbidimetric measurements (Fig. 7.5) to show that the rate of conversion of DMPC MLV into discoidal apoA-I/DMPC complexes is highest at the gel to liquid-crystal phase transition temperature (24°C) of DMPC. The enhanced rate at the phase transition is due to the presence of lattice defects that create sites to which apoA-I molecules can bind (with an associated increase in α-helix content), thereby destabilizing the DMPC bilayer and promoting rearrangement into discoidal, HDL-like, particles (Pownall et al., 1979, 1981). These fundamental concepts concerning the mechanism of PL solubilization by apolipoproteins are now well accepted. The laboratories of Pownall (Pownall et al., 1987) and Jonas (1992) have contributed much of our detailed understanding of the solubilization process. Generally, the reaction is kinetically controlled. Factors such as the physical state of the PL bilayer, the stability of the bilayer, the curvature of the vesicles and their cholesterol content (see below) are important. Regarding the apolipoprotein component, its molecular weight, hydrophobicity and state of self-association affect the rate of solubilization.
Analysis of the clearance kinetics in longer-term incubations of apoA-I with DMPC MLV at 24°C shows that the reaction is second order and consists of two simultaneous kinetic phases (Segall et al., 2002). As summarized in Fig. 7.6, the two kinetic components are proposed to arise from two distinct types of binding site (with or without lattice defects) for apoA-I on MLV surfaces. If the initial apoA-I/MLV contact occurs at a packing defect in the DMPC bilayer surface, then the reaction proceeds directly to stage 2. Initial contact at a non-defect site requires diffusion of the apoA-I molecule over the surface to a defect site, giving rise to a slow kinetic phase in obtaining stage 2. After adsorption of apoprotein molecules to DMPC bilayer defects, the rate of arrival at stage 3 depends upon the rate at which these molecules can rearrange and insert their α-helices into the defects (Fig. 7.6). As proposed earlier (Pownall et al., 1987), once a critical concentration of α-helices absorbed in the defects is attained, the lipid bilayer becomes unstable and rearranges to form a bilayer disc (stage 3 in Fig. 7.6). Apoprotein molecules at stage 2 of the reaction are bound to the DMPC vesicle surface reversibly whereas the vesicle to disc conversion in stage 3 is irreversible. The reaction of apoA-I with either SUV or MLV of DMPC to form discoidal complexes proceeds similarly. Detailed examination of this system (Jonas et al., 1980; Jonas and Drengler, 1980) has shown that apoA-I binding to saturate the vesicle surface (stage 1) is rapid and that the bilayer disruption is relatively slow. The nature of any intermediates in the vesicle to disc conversion step (stage 3 in Fig. 7.6) is not well understood. In the case of a DMPC SUV (~20 nm in diameter), when the surface is saturated with apoA-I the DMPC/apoA-I stoichiometry is ~1000/1 mol/mol, as compared to a ratio of ~ 100/1 in the discs that are formed (Jonas et al., 1980). Thus, it follows that an entire DMPC SUV cannot convert directly in one step into discoidal product without either incorporation of additional apoA-I molecules or the loss of excess DMPC to other particles. Intermediate complexes have not been detected in the DMPC SUV/apoA-I reaction (Jonas et al., 1980) but the formation of small discs proceeds through the formation of a large disc intermediate when DMPC LUV (~100 nm diameter) are solubilized by apoA-I (Zhu et al., 2007).
Fig. 7.6.

Molecular model explaining the two simultaneous kinetic phases of DMPC solubilization by apolipoproteins. The solubilization of DMPC MLV by apolipoprotein molecules involves four states (as indicated). Completion of the first and second of these stages can each occur by two simultaneous alternative pathways, one more rapid than the other, whereas stages three and four comprise a common pathway. Flexible apolipoprotein molecules react more rapidly than inflexible ones. See Segall et al. (2002) for more details. Reproduced with permission from Segall et al. (2002)
At temperatures other than the DMPC gel to liquid/crystal transition temperature of 24°C, apoA-I reacts slowly with DMPC vesicles because the bilayer contains relatively few packing defects (Pownall et al., 1978, 1987). However, the addition of cholesterol to the DMPC bilayer accelerates the rate of solubilization. The reaction is fastest at 12 mol% cholesterol and it becomes less temperature sensitive (Pownall et al., 1979). The combined effects of cholesterol and temperature can alter the rate of reaction by more than three orders of magnitude because of large variations in the number of lattice defects in the DMPC-cholesterol mixed bilayer. All the cholesterol is solubilized by apoA-I when the DMPC vesicle contains less than about 20 mol% cholesterol but higher levels of cholesterol inhibit the formation of discoidal complexes. The sizes of the HDL particles formed are dependent upon the initial level of cholesterol in the DMPC MLV (Massey and Pownall, 2008); increasing the bilayer cholesterol concentration results in the formation of larger sized discoidal particles that contain more apoA-I molecules.
In contrast to the DMPC system, MLV prepared from a PL such as an egg PC obtained from natural sources, are not solubilized in a matter of minutes at room temperature by apoA-I. The reason for this low reactivity is that the natural PC molecules contain unsaturated acyl chains so that their gel to liquid-crystal phase transition temperatures are well below room temperature and the bilayers contain few lattice defects into which apoA-I molecules can insert (cf. Fig. 7.6). However, a complex lipid mixture representing the lipids of a mammalian plasma membrane forms MLV that are solubilized by apoA-I at 37°C (Vedhachalam et al., 2007a). The rate of ~10% solubilization per hour is much slower than that typically observed with DMPC MLV, presumably because the numbers of lattice defects are fewer in the membrane lipid bilayer. Such a membrane bilayer solubilization process is integral to the mechanism of nascent HDL particle formation by ABCA1 (cf. Section 7.5.2).
7.4.3 Influence of Apolipoprotein Structure
All exchangeable apolipoproteins exhibit some ability to solubilize PL vesicles and create discoidal HDL particles which is consistent with the amphipathic α-helix, rather than a specific amino acid sequence, being the structural motif responsible for this activity (cf. Section 7.3). In agreement with this concept, fragments of the human apoA-I molecule corresponding to residues 1–86 and 149–243 contain amphipathic α-helices and can form similar discoidal DMPC complexes to the intact apoA-I molecule (Vanloo et al., 1991). Since the hydrophobic C-terminal domain (residues 190–243) of human apoA-I molecule is critical for lipid binding (Fig. 7.3), deletion of this segment reduces the ability of the protein to solubilize DMPC MLV (Vedhachalam et al., 2007a). For the same reasons, either deletion or disruption of the C-terminal α-helix (residues 221–243) has the same effect. Deletion of residues 1–43 or 44–65 in the N-terminal helix bundle domain has a less marked effect on the ability of the protein to solubilize DMPC (Vedhachalam et al., 2007a).
Human apoE also solubilizes DMPC vesicles to create discoidal particles (Innerarity et al., 1979). However, apoE reacts more slowly than apoA-I and this is attributed to the less flexible structure of the apoE molecule which reduces the rate of stage 2 in the solubilization reaction (see Fig. 7.6) (Segall et al., 2002). As observed with human apoA-I, removal of the lipid-binding C-terminal domain (residues 192–299) greatly reduces the rate at which apoE solubilizes DMPC MLV. The isolated and flexible C-terminal domain of apoE solubilizes DMPC at a similar rate to apoA-I. The reaction rates of the helix bundle domains of the three commons apoE isoforms vary inversely with the stabilities of these fragments. Overall, it seems that flexibility in an apolipoprotein molecule increases the time-average exposure of hydrophobic surface area, thereby increasing the rate of PL solubilization (see Figs. 7.5 and 7.6; Segall et al. (2002)).
7.5 HDL and Reverse Cholesterol Transport (RCT)
7.5.1 Overview of RCT Pathway – HDL Species and Receptors Involved
Figure 7.7 summarizes the RCT pathway in which HDL mediates the movement of cholesterol from peripheral cells to the liver for excretion from the body (Fielding and Fielding, 1995; Oram and Heinecke, 2005). It is clear from the pathway shown in Fig. 7.7 that apoA-I is involved in all stages of RCT, including the formation of nascent HDL particles, HDL remodeling by LCAT and delivery of HDL cholesterol to the liver via scavenger receptor class B, type 1 (SR-BI). The finding that the severe HDL deficiency associated with the genetic disorder, Tangier disease, is caused by mutations in the ATP-binding cassette transporter A1 (ABCA1) demonstrated that this transporter plays a critical role in HDL production (Oram, 2000). The bulk of the HDL particles in the circulation are produced by ABCA1 expressed in the liver and intestine, with the former being the major contributor (Lee and Parks, 2005). Removal of cholesterol from macrophages in the walls of blood vessels via RCT is critical for preventing the development of atherosclerotic plaque (Cuchel and Rader, 2006).
Studies of macrophage RCT in mice have demonstrated that both apoA-I and ABCA1 play essential roles in promoting RCT; it follows that they are anti-atherogenic proteins (Wang et al., 2007a; Zhang et al., 2003). These two proteins interact to mediate the first step in RCT, the efflux of cellular cholesterol (Fig. 7.7). Efflux of cholesterol from macrophages involves both active and passive processes (Yancey et al., 2003). In addition to the active ABCA1-mediated efflux of cellular PL and FC to lipid-free/poor apoA-I, a related transporter, ABCG1, can promote FC efflux to HDL particles. Passive FC efflux from macrophages also occurs by the so-called aqueous diffusion pathway (Adorni et al., 2007). This simple diffusion process involves desorption of FC molecules from the PL bilayer of the plasma membrane, followed by their diffusion in the aqueous phase and collision-mediated absorption into HDL particles (Phillips et al., 1987). Efflux of cellular FC from macrophages to HDL can also be facilitated by SR-BI (Adorni et al., 2007).
As can be seen from Fig. 7.7, besides being located in macrophages, SR-BI is located on the surface of hepatocytes. SR-BI is abundantly expressed in the liver where it mediates the selective uptake of FC and CE from HDL (Trigatti et al., 2003; van Eck et al., 2005; Zannis et al., 2006). This FC and CE is then released into bile as either FC or bile acid and then, in the last step of RCT, excreted from the body in feces (Fig. 7.7). The hepatic expression of SR-BI positively promotes the flux of cholesterol from macrophages through the RCT pathway (Zhang et al., 2005). In contrast, the SR-BI expressed in macrophages, which can promote efflux of cellular cholesterol (see Section 7.5.3), does not promote macrophage RCT in vivo (Wang et al., 2007b). However, at later stages of atherosclerotic lesion development in mice, SR-BI in macrophages plays an anti-atherogenic role (van Eck et al., 2005). Consistent with this atheroprotective effect, inactivation of the SR-BI gene in apoE-null mice accelerates coronary atherosclerosis (Trigatti et al., 1999). The loss of SR-BI activity is associated with an increase in total plasma cholesterol and the incidence of abnormally large, apoE-enriched HDL particles (Rigotti et al., 1997). The fact that the incidence of atherosclerosis in these animals is associated with higher HDL cholesterol levels indicates that the quality and not the quantity of HDL particles is critical in preventing coronary artery disease. The HDL particles that are present must be able to maintain the appropriate flux of cholesterol through the RCT pathway (Fig. 7.7). Enhancement of RCT from plaque to liver can enhance rapid regression of atherosclerosis (Williams et al., 2007).
7.5.2 ABCA1
ABCA1 is a member of the ATP binding cassette (ABC) family of membrane transporters and its ability to translocate PL across the plasma membrane of cells leads to the formation of nascent HDL particles when lipid-free/poor apoA-I is present in the extracellular medium. The preferred substrate for translocation by ABCA1 has not been established unambiguously, but it is clear that phosphatidylserine can be pumped across the membrane (Alder-Baerens et al., 2005; Chambenoit et al., 2001). ABCA1 is a 2261 amino acid integral membrane protein consisting of two halves of similar structure (Oram and Heinecke, 2005). Each half contains a six-helix transmembrane domain together with a cytosolic nucleotide-binding domain that mediates the ATPase activity. The topology in a cell plasma membrane is predicted to involve a cytosolic N-terminus and some extracellular loops that are heavily glycosylated (Dean et al., 2001). The two transmembrane helical domains form a chamber in which PL molecules are translocated. The molecular mechanism of this pumping action in ABCA1 has not been elucidated, but is likely to be similar to that of a related microbial lipid transporter whose crystal structure is known (Dawson and Locher, 2006).
The expression of ABCA1 is increased by loading cells with cholesterol because the consequent increase in oxysterol level activates the nuclear liver × receptor (LXR) (Oram and Heinecke, 2005). The transcription of ABCA1 is also induced by ligands for the retinoid × receptor and, in the case of murine macrophages, also by cyclic AMP. ABCA1 is degraded rapidly after transcription (half-life of 1–2 h) and its cellular level is sensitive to the presence of an apolipoprotein such as apoA-I, because apoA-I binds to ABCA1 and stabilizes it by modulating its phosphorylation, thereby protecting the transporter from calpain-mediated proteolysis (Wang and Tall, 2003; Yokoyama, 2006). ABCA1 recycles rapidly between the plasma membrane and late endosomal/lysosomal compartments (Neufeld et al., 2001) and is degraded at an intracellular site during this trafficking. When apoA-I is bound to ABCA1 (Oram et al., 2000; Vedhachalam et al., 2007b; Wang et al., 2000), the endocytosis is unaffected whereas the intracellular degradation is reduced which leads to higher levels of the transporter recycling back to the plasma membrane (Lu et al., 2008). This effect leads to enhanced HDL biogenesis because the ABCA1-mediated assembly of nascent HDL particles occurs at the cell surface (Denis et al., 2008; Faulkner et al., 2008).
The nascent HDL products of the apoA-I/ABCA1 reaction are primarily discoidal particles (cf. Section 7.3.2) containing 2, 3 or 4 apoA-I molecules (Duong et al., 2006; Krimbou et al., 2006; Liu et al., 2003). These particles are not only heterogeneous with respect to diameter, but also with respect to lipid composition in that they have different FC contents and PL compositions (Duong et al., 2006). This creation of variable nascent HDL species underlies the heterogeneity in the population of HDL particles present in the plasma compartment (Lund-Katz et al., 2003). The ABCA1/apoA-I reaction also leads to the production of some monomeric apoA-I molecules that are associated with 3–4 PL molecules; this lipid-poor apoA-I (preβ1-HDL) (Chau et al., 2006) is a product of the reaction but also a substrate in that it can react further and be converted into larger discoidal particles (Duong et al., 2008). Different apolipoproteins such as apoE (Krimbou et al., 2004; Vedhachalam et al., 2007c), and peptides containing amphipathic α-h elices (Remaley et al., 2003), can react with ABCA1 to create nascent HDL particles and the particle sizes are dependent upon the protein structure. A striking example of such a protein structural requirement is the observation that removal of the C-terminal α-helix of human apoA-I drastically reduces the level of PL and FC efflux (Vedhachalam et al., 2004), prevents formation of normal size nascent HDL particles and causes formation of very large HDL particles (Liu et al., 2003). As summarized in Sections 7.3 and 7.4.1, the C-terminal α-helix of apoA-I plays a critical role in lipid-binding and lipid-solubilization. It can be inferred that, since this helix is also essential for ABCA1-mediated HDL particle biogenesis, plasma membrane microsolubilization occurs during HDL biogenesis (Gillotte et al., 1999).
As summarized above, much has been learned about the ways in which ABCA1 contributes to the biogenesis of HDL particles. There has been great interest in elucidating the mechanism by which the transporter and apoA-I react to create nascent HDL particles and various models have been proposed (for reviews see Oram and Heinecke, 2005; Yokoyama, 2006; Zannis et al., 2006). Figure 7.8 summarizes a reaction scheme we have proposed recently, that integrates key findings from the literature and our laboratory (Vedhachalam et al., 2007a). A central feature of this mechanism is that membrane PL translocation via ABCA1 induces bending of the membrane bilayer to create high curvature sites to which apoA-I can bind and solubilize membrane PL and FC to create nascent HDL particles. Step 1 involves binding of apoA-I (in a lipid-free/poor state but not in a fully lipidated state) to the ABCA1 molecule probably via interaction of an amphipathic α-helix with a site on an extracellular loop of ABCA1 (Fitzgerald et al., 2002). As mentioned above, this association with apoA-I stabilizes the transporter in the plasma membrane, which leads to enhanced PL translocation and asymmetric PL packing across the bilayer. The resultant membrane strain is relieved by formation of highly curved exovesiculated domains in the plasma membrane to which apoA-I molecules can bind with high affinity (Step 2). The high curvature disorders the molecular packing in the bilayer and creates spaces between the PL polar groups into which apoA-I amphipathic α-helices can penetrate. The binding of apoA-I to the exovesiculated plasma membrane domains creates conditions for the formation of nascent HDL particles, which follows in Step 3.
Fig. 7.8.

Mechanism of interaction of apoA-I with ABCA1 and efflux of cellular phospholipids and cholesterol. The reaction in which apoA-I binds to ABCA1 and membrane lipids to create discoidal nascent HDL particles contains three steps. Step 1 involves the high affinity binding of a small amount of apoA-I to ABCA1 located in the plasma membrane PL bilayer; this regulatory pool of apoA-I up-regulates ABCA1 activity, thereby enhancing the active translocation of membrane PL from the cytoplasmic to exofacial leaflet. This translocase activity leads to lateral compression of the PL molecules in the exofacial leaflet and expansion of those in the cytoplasmic leaflet. Step 2 involves the bending of the membrane to relieve the strain induced by the unequal molecular packing density across the membrane and the formation of an exovesiculated domain to which apoA-I can bind with high affinity. This interaction with the highly curved membrane surface involves apoA-I/membrane lipid interactions and creates a relatively large pool of bound apoA-I. Step 3 involves the spontaneous solubilization by the bound apoA-I of membrane PL and cholesterol in the exovesiculated domains to create discoidal HDL particles containing two, three or four apoA-I molecules/particle. In the diagram, the two transmembrane six-helix domains of ABCA1 are represented as rectangles, whereas the two ATPase domains are shown as ovals. The space between the two rectangles represents the chamber in which translocation of PL molecules occurs. Reproduced with permission from Vedhachalam et al. (2007a)
Step 3 requires the spontaneous solubilization (cf. Section 7.4) of the membrane lipid bilayer to create nascent HDL. We have defined this process, which leads to simultaneous release of cellular PL and FC, as membrane microsolubilization (Gillotte-Taylor et al., 2002; Gillotte et al., 1998, 1999). As discussed in Section 7.4.1, apoA-I can spontaneously solubilize bilayers comprised of the PL typically found in the plasma membrane of mammalian cells to create discoidal HDL particles (Vedhachalam et al., 2007a). The apolipoprotein content of these particles is determined by apoA-I/PL/FC interactions in which ABCA1 is not directly involved and by the structural properties of the apoA-I molecule. Step 3 in the reaction scheme summarized in Fig. 7.8 is the slowest and, therefore, rate-limiting. This fact is exemplified by the observation that apoA-I structural alterations that modulate the rate of model membrane bilayer solubilization have a similar effect on the rate of PL and FC efflux from cells via ABCA1 (Vedhachalam et al., 2007a). Figure 7.9 summarizes a molecular mechanism for Step 3 in which vesiculated membrane bilayers are fragmented into small segments to form discoidal HDL particles. Overall, the mechanism depicted in Fig. 7.8 is consistent with the known properties of ABCA1 and apoA-I.
Fig. 7.9.

Suggested molecular mechanism for the solubilization of PL bilayers by apoA-I to create discoidal HDL particles. This process is envisaged to underlie the solubilization of DMPC MLV depicted in Figs. 7.5 and 7.6, and Step 3 in the formation of nascent HDL particles in the apoA-I/ABCA1 reaction depicted in Fig. 7.8
As shown in the summary of the RCT pathway (Fig. 7.7), the removal of excess cholesterol from macrophages can occur via both ABCA1 and ABCG1 pathways. The nascent HDL particles created by the ABCA1 reaction contain some FC (Duong et al., 2006, 2008), but they are able to acquire additional cholesterol by participation in the ABCG1 pathway. Thus, ABCA1 and ABCG1 can act sequentially to mediate cellular cholesterol export to apoA-I (Gelissen et al., 2008; Vaughan and Oram, 2006). This synergy between lipid transporters is important for regulating cholesterol efflux and maintaining the appropriate cholesterol levels in macrophages (Jessup et al., 2006; Marcel et al., 2008).
7.5.3 SR-BI
SR-BI was the first recognized HDL receptor and it mediates the flux of FC and CE between bound HDL particles and the cell plasma membrane (Acton et al., 1996). Of particular note, SR-BI mediates the selective uptake of CE from HDL, a process whereby HDL lipids are taken up preferentially by cells through a non-endocytic mechanism without either degradation of apolipoproteins or whole particle uptake (Connelly and Williams, 2004; Pittman et al., 1987; Zannis et al., 2006). SR-BI is an 82-kDa membrane glycoprotein containing a large extracellular domain (408 residues) and two transmembrane domains with short cytoplasmic N- and C-terminal domains. The extracellular domain plays a critical role in mediating the selective uptake process (Connelly et al., 1999, 2001; Gu et al., 1998). SR-BI contains a PDZK1 binding motif in its C-terminal domain and interaction with this scaffold protein controls the abundance and localization in the plasma membrane of hepatic SR-BI (Fenske et al., 2008). A minor, alternatively spliced, form called SR-BII has a different C-terminal domain that does not bind to PDZK1, so that this isoform is mostly located in the cell interior (Eckhardt et al., 2004). SR-BI self-associates into dimers and tetramers but this process is not dependent upon the C-terminal domain (Sahoo et al., 2007). SR-BI-mediated lipid uptake does not require endocytosis, indicating that the uptake occurs at the plasma membrane (Harder et al., 2006; Nieland et al., 2005). Furthermore, interactions with other proteins or specific cellular structures are not required for this activity as it has been shown that the purified protein reconstituted into a model membrane is functional (Liu and Krieger, 2002).
As befits a scavenger receptor, SR-BI binds to a range of ligands besides HDL; these include VLDL, LDL, modified LDL and PL vesicles (Connelly and Williams, 2004; Trigatti et al., 2003; Zannis et al., 2006). The amphipathic α-helix is the recognition motif for SR-BI and HDL binds to the receptor via the multiple amphipathic α-helical repeats in the apoA-I molecule (Williams et al., 2000). A specific amino acid sequence in apoA-I is not required for the interaction and there is more than one apolipoprotein binding site on the SR-BI molecule (Thuahnai et al., 2003). The binding of HDL to SR-BI is influenced by the conformation of apoA-I so that larger (~10 nm diameter) HDL particles bind better than smaller (~8 nm) particles (de Beer et al., 2001a; Thuahnai et al., 2004). The presence of apoA-II in HDL particles attenuates the binding to SR-BI (de Beer et al., 2001b). The dependence on HDL concentration of binding to SR-BI and CE selective uptake is similar, indicating that the two processes are linked (Rodrigueza et al., 1999). It is apparent that the mechanism of HDL CE selective uptake by SR-BI involves a two-step process in which the initial binding of an HDL particle to the receptor is followed by the transfer of CE molecules from the bound HDL particle into the cell plasma membrane (Gu et al., 1998; Rodrigueza et al., 1999). The rate of CE selective uptake from the donor HDL particle is proportional to the amount of CE initially present in the particle, indicating a mechanism in which CE moves down its concentration gradient from HDL particles docked on SR-BI into the cell plasma membrane. The activation energy for this process is ~9 kcal/mol, which is consistent with the uptake occurring by a non-aqueous pathway. As depicted in the model shown in Fig. 7.10, HDL binding to SR-BI allows access CE molecules to a channel formed by the extracellular domain of the receptor, from which water is excluded and along which HDL CE molecules move down their concentration gradient into the cell plasma membrane (Rodrigueza et al., 1999). An alternative model proposed a hemi-fusion event between the PL monolayer of an HDL particle and the external leaflet of the plasma membrane, thereby allowing lipid transfer to occur (Gu et al., 1998). However, the fact that the relative rates of selective uptake of CE, FC, PC and sphingomyelin from HDL are different (Rodrigueza et al., 1999; Thuahnai et al., 2001) argues against this model. The efficient transfer of lipid molecules from HDL to the cell plasma membrane requires an optimal alignment of the apoA-I-containing HDL/SR-BI complex (Liu et al., 2002; Thuahnai et al., 2004). The presence of an apolipoprotein such as apoA-I in the donor particle is required for the selective uptake of CE to occur because in the absence of apolipoprotein-mediated binding to SR-BI, the lipid components of the donor fuse with the cell membrane giving stoichiometric lipid uptake (Thuahnai et al., 2001). This phenomenon suggests that there is a fusogenic motif in the extracellular domain of SR-BI.
Fig. 7.10.
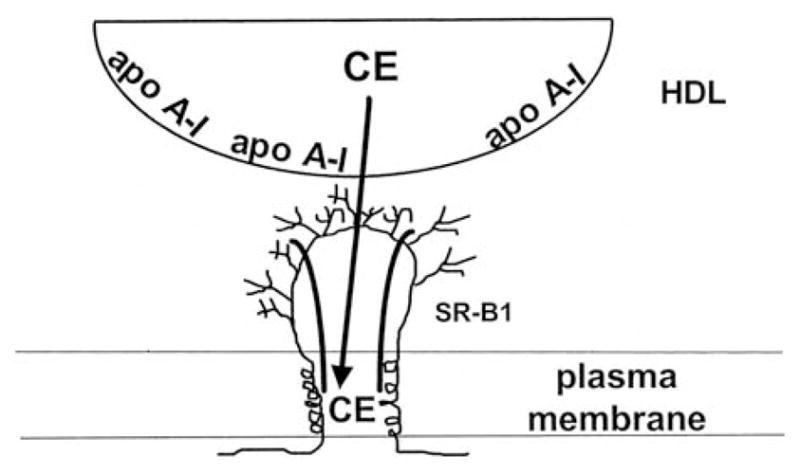
Model of SR-BI-mediated selective uptake of cholesteryl ester (CE) from HDL. This model proposes that SR-BI contains a non-aqueous channel, which excludes water, and serves as a conduit for hydrophobic CE molecules diffusing from bound HDL down their concentration gradient to the cell plasma membrane. The scheme depicts a channel formed by a single SR-BI molecule, but it is possible that self-association of SR-BI is required to create the channel. Reproduced with permission from Rodrigueza et al. (1999)
Because the SR-BI-mediated transfer of lipid molecules between bound HDL and the cell plasma membrane is a passive process, the rate and direction of net transfer are sensitive to the concentration gradient existing between the HDL particle and the plasma membrane. Thus, in the case of FC, SR-BI facilitates the bi-directional flux between HDL and the plasma membrane (Yancey et al., 2003). The concentration gradient for FC is sensitive to the PL content of HDL so that the net transfer of cholesterol out of the cell is promoted by PL-enrichment of HDL (Pownall, 2006; Yancey et al., 2000). Different kinds of cells exhibit large differences in the rate of FC efflux to PL-containing acceptors (Rothblat et al., 1986) and these variations are due to differences in the expression levels of SR-BI (Ji et al., 1997; Jian et al., 1998). The fact that SR-BI-mediated FC efflux to HDL is dependent upon HDL binding to the receptor (Gu et al., 2000) and that the appropriate apoA-I/SR-BI complex must be formed (Liu et al., 2002), point to a similar mechanism operating for SR-BI-mediated HDL CE selective uptake and cellular FC efflux. Depending upon the concentration of HDL present in the extracellular medium, SR-BI-facilitated FC efflux can occur by pathways dependent on and independent of HDL binding to the receptor (Thuahnai et al., 2004). FC efflux is binding-dependent at low concentrations of HDL, where binding to SR-BI is not saturated; under this condition (Fig. 7.10), FC molecules diffuse from the plasma membrane to the bound HDL particle via the hydrophobic channel created by SR-BI. At saturation concentration of HDL, FC efflux is independent of HDL binding to SR-BI. Under this condition, efflux occurs by the aqueous diffusion mechanism whereby FC particles desorb from the plasma membrane and diffuse through the aqueous phase and collide with HDL acceptor particles. The receptor enhances the rate of FC efflux via aqueous diffusion because it perturbs the packing of FC molecules in the plasma membrane; this reorganization and activation of plasma membrane FC molecules is evident from the increased pool of FC accessible to cholesterol oxidase in SR-BI-containing membranes (Kellner-Weibel et al., 2000; Llera-Moya et al., 1999). Similar to CE selective uptake, SR-BI-mediated FC efflux is dependent upon HDL particle size so that, at the same particle concentration, large HDL promotes more FC efflux than small HDL.
7.5.4 ApoE-HDL
In the plasma compartment, apoE influences the lipid concentration because it affects the levels of all lipoproteins by modulating their receptor-mediated clearance into cells. In addition, apoE affects the production of hepatic VLDL and the lipolytic processing of TAG-rich lipoproteins. ApoE, one of the least abundant plasma apolipoproteins, is present on all lipoprotein particles, with the exception of LDL (Mahley and Rall, 1995). The RCT pathway (Section 7.5.1) directs excess cholesterol in peripheral tissues through HDL acceptors to the liver for elimination (Fig. 7.7). Although the major apolipoprotein in HDL2 and HDL3 is apoA-I, HDL2 also contains significant amounts of apoE. ApoE uniquely facilitates RCT by allowing CE-rich core expansion in HDL (Mahley et al., 2006) after LCAT converts FC to CE (Section 7.3.3). ApoA-I-containing HDL can accommodate only a limited amount of CE in its core, resulting in a limited size expansion, whereas the size and CE content of HDL can be increased significantly when apoE is present. Since apoE is a ligand for the LDL receptor, these apoE-enriched HDLs can deliver cholesterol acquired from the periphery to the liver via hepatic LDL receptors (Mahley, 1988). After secretion from the liver as a component of VLDL, apoE redistributes to enrich chylomicrons, remnant lipoprotein particles and HDL (Fazio et al., 2000). ApoE associated with lipoprotein particles serves additional functions such as binding to heparan sulfate proteoglycans to facilitate internalization of the lipoprotein by cells (Mahley et al., 2006). The rapid removal of chylomicron remnant particles requires the presence of a cellular pool of apoE. ApoE internalized by hepatocytes is partially protected from lysosomal degradation and recycles through the Golgi apparatus, suggesting that recycled apoE may have a physiological role in lipoprotein assembly, remnant removal and cholesterol efflux (Fazio et al., 2000). After receptor-mediated endocytosis, the intracellular fate of the TAG-rich lipoproteins is very complex and differs from the degradation pathway of LDL (Heeren et al., 2006). The majority of TAG-rich lipoprotein-derived apoE remains in peripheral recycling endosomes. This pool of apoE is then mobilized by HDL or HDL-derived apoA-I to be recycled back to the plasma membrane; this apoE is secreted into the extracellular medium where it can participate in the formation of apoE-HDL particles. The recycling of apoE in murine macrophages occurs via ABCA1 and, in the presence of apoA-I, recycled apoE exits the cells on HDL-like particles (Hasty et al., 2005). On the other hand, apoA-I stimulates secretion of apoE independently of cholesterol efflux in a human macrophage cell line; this effect reflects a novel, ABCA1-independent, positive feedback pathway for stimulation of apoE secretion (Kockx et al., 2004). Intracellular trafficking of recycling apoE in Chinese hamster ovary cells appears to be linked to cellular cholesterol removal via the endosomal recycling compartment and PL-containing receptors in an alternative pathway to that of the ABCA1/apoA-I route (Braun et al., 2006). The relationships between apoE and ABCA1 for regulating cellular sterol efflux have been examined in macrophages expressing both apoE and ABCA1. ABCA1 expression is required for apoE-mediated cholesterol efflux when endogenously synthesized apoE accumulates extracellularly. However, an ABCA1-independent pathway for lipid efflux that requires the intracellular synthesis and/or transport of apoE also exists (Huang et al., 2006). Overall, it is clear that the secretion of apoE from macrophages is a regulated process and that both ABCA1-dependent and -independent pathways exist (Kockx et al., 2008).
ApoE is the major apolipoprotein in cerebrospinal fluid and it plays a pivotal role in maintaining cholesterol homeostasis in the brain. The most cholesterol-rich organ in the body is the brain, which contains some 25% of total body cholesterol; this cholesterol is a major component of myelin and neuronal and glial cell membranes (Dietschy and Turley, 2001). Nearly all brain cholesterol is synthesized in situ and it is transported on apoE-HDL particles that circulate in the cerebrospinal fluid (Pitas et al., 1987; LaDu et al., 2000). ABCA1 and ABCG1 are involved in regulation of lipid and lipoprotein metabolism in the brain. ABCA1 mediates the initial lipidation of nascent apoE particles (cf. Section 7.5.2), which are secreted from both astrocytes and microglia (Hirch-Reinshagen and Wellington, 2007). These discoidal particles are then thought to acquire further lipids from either neurons or glia through an active transport process mediated by ABCG1 (Kim et al., 2006). This step, together with the subsequent maturation of the circulating lipoprotein, results in the spherical apoE-containing particles observed in cerebrospinal fluid. ApoE has been implicated in the clearance and deposition of amyloid-β in the brain. When complexed with amyloid-β, apoE facilitates the cellular uptake of amyloid-β via apoE receptors (Tokuda et al., 2000). ApoE is required for the extracellular deposition of amyloid-β as amyloid and it delays the clearance of amyloid-β across the blood-brain barrier (Hirch-Reinshagen and Wellington, 2007). It is probable that one of the major functions of apoE in the central nervous system is to mediate neuronal repair, remodelling and protection with apoE4 being less effective than the apoE2 and apoE3 isoforms (Mahley and Huang, 1999). ApoE has been linked to the pathogenesis of Alzheimer’s disease with apoE polymorphism having significant effects (Roses, 1996). The apoE4 allele is a major susceptibility gene that is associated with 40–65% of cases of sporadic and familial Alzheimer’s disease; individuals carrying this isoform have an increased occurrence and reduced age of onset of the disease (Corder et al., 1993). The apoE2 allele may be even more protective than apoE3 against the development of Alzheimer’s disease (Corder et al., 1994). The mechanisms by which apoE is involved in the pathogenesis of Alzheimer’s disease remain incompletely understood. Cholesterol homeostasis and trafficking is currently recognized to participate in aspects of amyloid-β metabolism, but how this may be related to apoE as a risk factor for Alzheimer’s disease requires further clarification.
7.6 HDL and Inflammation
Inflammation, such as is caused by direct tissue injury induces the acute phase (AP) response in which the liver increases the synthesis of a number of particular proteins including serum amyloid A (SAA) (O’Brien and Chait, 2006). Macrophages are mobilized to the injury site where cell death has occurred to remove the cell debris, such as membranes rich in PL and cholesterol. This movement of macrophages to the injury site is assisted by HDL, which becomes proinflammatory in the AP response and can induce monocyte chemotaxis (Ansell et al., 2007). The HDL is also required to mediate RCT and remove the excess cholesterol from the macrophages. The conversion of HDL from its usual anti-inflammatory condition to a proinflammatory state during the AP response is caused by the activity of the AP proteins, SAA and Group IIa secretory phospholipase A2 (Ansell et al., 2007; Kontush and Chapman, 2006; van der Westhuijzen et al., 2007). The enhanced expression of these proteins leads to extensive HDL remodelling and lower circulating levels of HDL cholesterol and apoA-I (Jahangiri et al., 2009; van der Westhuijzen et al., 2007). These changes in HDL are presumably beneficial during the 24 hours or so of the AP response. However, the presence of proinflammatory HDL during the chronic inflammation associated with atherosclerosis (Kontush and Chapman, 2006; O’Brien and Chait 2006) is harmful (Ansell et al., 2007). Although SAA is a major player in the HDL remodeling that occurs during the AP response, current understanding of the role of SAA in the inflammatory response and of how this protein affects HDL metabolism and cholesterol transport is limited.
7.6.1 Serum Amyloid A
SAA consists of four isoforms and, of these, isoforms SAA1 and SAA2 are major AP response proteins whose plasma concentrations increase by some three orders of magnitude after injury. Most of the plasma SAA is bound to HDL so that SAA is the major protein in AP HDL (Tam et al., 2002; van der Westhuijzen et al., 2007). Human SAA1 and SAA2 each contain 122 amino acids and structure prediction analysis suggests that about 80% of these residues are located in an N-terminal helix bundle, with the remainder of the C-terminus being disordered (Stevens, 2004); this structure is analogous to that of apoA-I (see Section 7.2.2). Furthermore, SAA contains amphipathic α-helices and can bind to PL to create SAA-PL complexes in an analogous fashion to apoA-I (Bausserman et al., 1983; Segrest et al., 1976). However, unlike apoA-I, SAA can directly bind cholesterol in solution with high affinity to form an equimolar complex (Liang and Sipe, 1995); the N-terminal region is involved in the binding site (Liang et al., 1996). The lipid-binding capabilities of the SAA molecule allow it to replace apoA-I on HDL particles and thereby modify HDL-mediated cholesterol transport (van der Westhuijzen et al., 2007).
Currently, it is thought that the SAA in AP HDL particles is involved in enhancing removal of cholesterol from macrophages at sites of injury thereby playing a protective role (Tam et al., 2002; van der Westhuijzen et al., 2007). Consistent with this concept, like apoA-I, SAA can participate in the various pathways for FC efflux from macrophages (see Fig. 7.7). Thus, SAA in a lipidated state can mediate cellular FC efflux via SR-BI (Cai et al., 2005; Marsche et al., 2007) (cf. Section 7.5.3) while lipid-free SAA promotes ABCA1-dependent FC and PL efflux and nascent HDL particle assembly (Abe-Dohmae et al., 2006; Stonik et al., 2004) (cf. Section 7.5.2). AP HDL promotes more FC efflux from cholesterol-loaded macrophages than normal HDL and this is thought to occur because SAA2 promotes the availability for export of FC in cells (Tam et al., 2002; van der Westhuijzen et al., 2007). Thus, peptides from SAA2 have been reported to operate inside macrophages to reduce acyl CoA-acyltransferase (ACAT) activity and increase neutral CE hydrolase activity (Tam et al., 2002). These various abilities of SAA to promote cellular cholesterol efflux lead to the retention of cholesterol efflux capacity in AP plasma despite marked decreases in HDL cholesterol and apoA-I levels (Jahangiri et al., 2009).
7.7 Summary and Conclusions
Significant progress is being made in understanding the structures of human exchangeable apolipoproteins, especially apoA-I and apoE. It is established that the amphipathic α-helical repeats in these proteins are the key structural elements responsible for the functions of these molecules. The application of protein engineering techniques together with a range of physical-biochemical methods has shown that apoA-I and apoE adopt two-domain tertiary structures. The α-helices located in the N-terminal two-thirds of the molecule adopt a helix-bundle conformation while the C-terminal region forms a separately folded, relatively disordered, domain. ApoA-I and apoE bind to lipid surfaces with high affinity, and this binding is initiated by the C-terminal domain, which becomes more α-helical upon interaction with the lipid. Subsequently, the N-terminal helix bundle domain can open, allowing helix–helix contacts to be replaced by helix-lipid interactions. ApoA-I and apoE possess detergent-like properties in that their lipid-binding capabilities permit them to solubilize vesicular phospholipid and form discoidal HDL particles. The apoA-I molecules in such particles adopt a ‘double-belt’ conformation with defined protein-protein contacts that are also maintained in spherical HDL particles. These structural models are proving valuable for understanding HDL function, but atomic level resolution structures are required to elucidate the detailed molecular mechanisms underlying the participation of HDL in the RCT pathway. The heterogeneity of HDL with regard to particle size, particle shape, protein composition and lipid composition complicates attempts to derive high resolution structural information. The lipid-binding and lipid-solubilizing properties of apoA-I and apoE underlie their abilities to interact with ABCA1, efflux cellular lipids and create nascent HDL particles. Detailed understanding of why a heterogeneous population of nascent HDL particles is formed will require more knowledge of the structure and lipid translo-case activity of ABCA1, as well as of the membrane microenvironment in which it resides. Good progress is being made in understanding the ways in which apoA-I-containing HDL particles participate in the reverse transport of cholesterol from peripheral cells to the liver. Less is known about how apoE-HDL mediates cholesterol transport in the brain. At this stage, relatively little in structural and mechanistic terms is known about the effects of natural mutations and oxidative modifications on the functionalities of apoA-I, apoE and the HDL particles they form.
Acknowledgments
We are indebted to all our colleagues for their valuable contributions to the studies from our laboratory described here. Our research reported here was supported by NIH Grants HL22633 and HL 56083.
Abbreviations
- ANS
8-anilino-1-napthalenesulfonic acid
- AP
acute phase
- apo
apolipoprotein
- CE
cholesterol ester
- CETP
cholesteryl ester transfer protein
- DMPC
dimyristoyl PC
- EL
endothelial lipase
- FC
free (unesterified) cholesterol
- HDL
high density lipoprotein
- HL
hepatic lipase
- LCAT
lecithin-cholesterol acyltransferase
- LDL
low density lipoprotein
- LUV
large unilamellar vesicle
- MLV
multilamellar vesicle
- PC
phosphatidylcholine
- PL
phospholipid
- PLTP
phospholipid transfer protein
- RCT
reverse cholesterol transport
- SAA
serum amyloid A
- SUV
small unilamellar vesicle
- TAG
triacylglycerol
- VLDL
very low density lipoprotein
References
- Abe-Dohmae S, Kato KH, Kumon Y, Hu W, Ishigami H, Iwamoto N, Okazaki M, Wu CA, Ueda K, Yokoyama S. Serum amyloid A generates high density lipoprotein with cellular lipid in an ABCA1- or ABCA7-dependent manner. J Lipid Res. 2006;47:1542–1550. doi: 10.1194/jlr.M600145-JLR200. [DOI] [PubMed] [Google Scholar]
- Acharya P, Segall ML, Zaiou M, Morrow JA, Weisgraber K, Phillips MC, Lund-Katz S, Snow JW. Comparison of the stabilities and unfolding pathways of human apolipoprotein E isoforms by differential scanning calorimetry and circular dichroism. Biochim Biophys Acta. 2002;1584:9–19. doi: 10.1016/s1388-1981(02)00263-9. [DOI] [PubMed] [Google Scholar]
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, Rothblat GH. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48:2453–2462. doi: 10.1194/jlr.M700274-JLR200. [DOI] [PubMed] [Google Scholar]
- Aggerbeck LP, Wetterau JR, Weisgraber KH, Wu CSC, Lindgren FT. Human apolipoprotein E3 in aqueous solution. II Properties of the amino- and carboxyl-terminal domains. J Biol Chem. 1988;263:6249–6258. [PubMed] [Google Scholar]
- Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy S. Crystal structure of human apolipoprotein A-I: Insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci USA. 2006;103:2126–2131. doi: 10.1073/pnas.0506877103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Alder-Baerens N, Muller P, Pohl A, Korte T, Hamon Y, Chimini G, Pomorski T, Herrmann A. Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J Biol Chem. 2005;280:26321–26329. doi: 10.1074/jbc.M413993200. [DOI] [PubMed] [Google Scholar]
- Andrews AL, Atkinson D, Barratt MD, Finer EG, Hauser H, Henry R, Leslie RB, Owens NL, Phillips MC, Robertson RN. Interaction of apoprotein from porcine high-density lipoprotein with dimyristoyl lecithin. 2 Nature of lipid-protein interaction. Eur J Biochem. 1976;64:549–563. doi: 10.1111/j.1432-1033.1976.tb10335.x. [DOI] [PubMed] [Google Scholar]
- Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high-density lipoprotein. Curr Opin Lipidol. 2007;18:427–434. doi: 10.1097/MOL.0b013e3282364a17. [DOI] [PubMed] [Google Scholar]
- Arnulphi C, Jin L, Tricerri MA, Jonas A. Enthalpy-driven apolipoprotein A-I and lipid bilayer interaction indicating protein penetration upon lipid binding. Biochemistry. 2004;43:12258–12264. doi: 10.1021/bi036118k. [DOI] [PubMed] [Google Scholar]
- Atkinson D, Smith HM, Dickson J, Austin JP. Interaction of apoprotein from porcine high-density lipoprotein with dimyristoyl lecithin. 1 The structure of the complexes. Eur J Biochem. 1976;64:541–547. doi: 10.1111/j.1432-1033.1976.tb10334.x. [DOI] [PubMed] [Google Scholar]
- Badellino KO, Rader DJ. The role of endothelial lipase in high-density lipoprotein metabolism. Curr Opin Lipidol. 2004;19:392–395. doi: 10.1097/01.hco.0000130161.89169.02. [DOI] [PubMed] [Google Scholar]
- Barbier A, Clement-Collin V, Dergunov AD, Visvikis A, Siest G, Aggerbeck LP. The structure of human apolipoprotein E2, E3 and E4 in solution 1. Tertiary and quaternary structure. Biophys Chem. 2006;20:158–169. doi: 10.1016/j.bpc.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Barratt MD, Badley RA, Leslie RB. The interaction of apoprotein from porcine high-density lipoprotein with dimyristoyl phosphatidylcholine. Eur J Biochem. 1974;48:595–601. doi: 10.1111/j.1432-1033.1974.tb03802.x. [DOI] [PubMed] [Google Scholar]
- Bausserman LL, Herbert PN, Forte T, Klausner RD, McAdam KPWJ, Osborne JC, Jr, Rosseneu M. Interaction of the serum amyloid A proteins with phospholipid. J Biol Chem. 1983;258:10681–10688. [PubMed] [Google Scholar]
- Behling Agree AK, Tricerri MA, Arnvig-McGuire K, Tian SM, Jonas A. Folding and stability of the C-terminal half of apolipoprotein A-I examined with a Cys-specific fluorescence probe. Biochim Biophys Acta. 2002;1594:286–296. doi: 10.1016/s0167-4838(01)00317-x. [DOI] [PubMed] [Google Scholar]
- Braun NA, Mohler PJ, Weisgraber KH, Hasty AH, Linton MF, Yancey PG, Su YR, Fazio S, Swift LL. Intracellular trafficking of recycling apolipoprotein E in Chinese hamster ovary cells. J Lipid Res. 2006;47:1176–1186. doi: 10.1194/jlr.M500503-JLR200. [DOI] [PubMed] [Google Scholar]
- Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- Brouillette CG, Dong WJ, Yang ZW, Ray MJ, Protasevich II, Cheung HC, Engler JA. Forster resonance energy transfer measurements are consistent with a helical bundle model for lipid-free apolipoprotein A-I. Biochemistry. 2005;44:16413–16425. doi: 10.1021/bi051018v. [DOI] [PubMed] [Google Scholar]
- Cai L, de Beer MC, de Beer FC, van der Westhuijzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2005;280:2954–2961. doi: 10.1074/jbc.M411555200. [DOI] [PubMed] [Google Scholar]
- Chambenoit O, Hamon Y, Margue D, Rigneult H, Rosseneu M, Chimini G. Specific docking of apolipoprotein A-I at the cell surface requires a functional ABCA1 transporter. J Biol Chem. 2001;276:9955–9960. doi: 10.1074/jbc.M010265200. [DOI] [PubMed] [Google Scholar]
- Chau P, Nakamura Y, Fielding CJ, Fielding PE. Mechanism of prebeta-HDL formation and activation. Biochemistry. 2006;45:3981–3987. doi: 10.1021/bi052535g. [DOI] [PubMed] [Google Scholar]
- Choy N, Raussens V, Narayanaswami V. Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J Mol Biol. 2003;334:527–539. doi: 10.1016/j.jmb.2003.09.059. [DOI] [PubMed] [Google Scholar]
- Clay MA, Newnham HH, Barter PJ. Hepatic lipase promotes a loss of apolipoprotein A-I from triglyceride-enriched human high density lipoproteins during incubation in vitro. Arterioscler Throm. 1991;11:415–422. doi: 10.1161/01.atv.11.2.415. [DOI] [PubMed] [Google Scholar]
- Clay MA, Pyle DH, Rye KA, Barter PJ. Formation of spherical, reconstituted high density lipoproteins containing both apolipoproteins A-I and A-II is mediated by lecithin: cholesterol acyltransferase. J Biol Chem. 2000;275:9019–9025. doi: 10.1074/jbc.275.12.9019. [DOI] [PubMed] [Google Scholar]
- Connelly MA, Klein SM, Azhar S, Abumrad NA, Williams DL. Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J Biol Chem. 1999;274:41–47. doi: 10.1074/jbc.274.1.41. [DOI] [PubMed] [Google Scholar]
- Connelly MA, Llera-Moya M, Monzo P, Yancey P, Drazul D, Stoudt G, Fournier N, Klein SM, Rothblat G, Williams DL. Analysis of chimeric receptors shows that multiple distinct functional activities of scavenger receptor, class B, type I (SR-BI), are localized to the extracellular receptor domain. Biochemistry. 2001;40:5249–5259. doi: 10.1021/bi002825r. [DOI] [PubMed] [Google Scholar]
- Connelly MA, Williams DL. Scavenger receptor BI: A scavenger receptor with a mission to transport high density lipoprotein lipids. Curr Opin Lipidol. 2004;15:287–295. doi: 10.1097/00041433-200406000-00008. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer’s disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines GL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cuchel M, Rader DJ. Macrophage reverse cholesterol transport. Key to the regression of atherosclerosis? Circulation. 2006;113:2548–2555. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Arnvig-McGuire K, Kennedy A, Kosman J, Hazlett T, Jonas A. Structural organization of the N-terminal domains of apolipoprotein A-I: Studies of tryptophan mutants. Biochemistry. 1999;38:14387–14395. doi: 10.1021/bi991428h. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Hazlett T, Mantulin WM, Jonas A. The role of apolipoprotein AI domains in lipid binding. Proc Natl Acad Sci USA. 1996;93:13605–13610. doi: 10.1073/pnas.93.24.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Silva RAG. Apolipoprotein structural organization in high density lipoproteins: Belts, bundles, hinges and hairpins. Curr Opin Lipidol. 2005;16:295–300. doi: 10.1097/01.mol.0000169349.38321.ad. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Thompson TB. The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem. 2007;282:22249–22253. doi: 10.1074/jbc.R700014200. [DOI] [PubMed] [Google Scholar]
- Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- de Beer MC, Durbin DM, Cai L, Jonas A, de Beer FC. Apolipoprotein A-I conformation markedly influences HDL interaction with scavenger receptor BI. J Lipid Res. 2001a;42:309–313. [PubMed] [Google Scholar]
- de Beer MC, Durbin DM, Cai L, Mirocha N, Jonas A, Webb NR, de Beer FC, van der Westhuyzen DR. Apolipoprotein A-II modulates the binding and selective lipid uptake of reconstituted high density lipoprotein by scavenger receptor BI. J Biol Chem. 2001b;276:15832–15839. doi: 10.1074/jbc.M100228200. [DOI] [PubMed] [Google Scholar]
- Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- Denis M, Landry YD, Zha X. ATP-binding cassette A1-mediated lipidation of apolipoprotein A-I occurs at the plasma membrane and not in the endocytic compartments. J Biol Chem. 2008;283:16178–16186. doi: 10.1074/jbc.M709597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Dong LM, Parkin S, Trakhanov SD, Rupp B, Simmons T, Arnold KS, Newhouse YM, Innerarity TL, Weisgraber KH. Novel mechanism for defective receptor binding of apolipoprotein E2 in type III hyperlipoproteinemia. Nat Struct Biol. 1996;3:718–722. doi: 10.1038/nsb0896-718. [DOI] [PubMed] [Google Scholar]
- Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- Donovan JM, Benedek GB, Carey MC. Self-association of human apolipoproteins A-I and A-II and interactions of apolipoprotein A-I with bile salts: Quasi-elastic light scattering studies. Biochemistry. 1987;26:8116–8125. doi: 10.1021/bi00399a015. [DOI] [PubMed] [Google Scholar]
- Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- Duong PT, Weibel GL, Lund-Katz S, Rothblat GH, Phillips MC. Characterization and properties of pre beta-HDL particles formed by ABCA1-mediated cellular lipid efflux to apoA-I. J Lipid Res. 2008;49:1006–1014. doi: 10.1194/jlr.M700506-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt ERM, Cai L, Sun B, Webb NR, van der Westhuyzen DR. High density lipoprotein uptake by scavenger receptor SR-BII. J Biol Chem. 2004;279:14372–14381. doi: 10.1074/jbc.M313793200. [DOI] [PubMed] [Google Scholar]
- Fan D, Li Q, Korando L, Jerome WG, Wang J. A monomeric human apolipoprotein E carboxyl-terminal domain. Biochemistry. 2004;43:5055–5064. doi: 10.1021/bi035958w. [DOI] [PubMed] [Google Scholar]
- Fang Y, Gursky O, Atkinson D. Structural studies of N- and C-terminally truncated human apolipoprotein A-I. Biochemistry. 2003;42:6881–6890. doi: 10.1021/bi034152t. [DOI] [PubMed] [Google Scholar]
- Faulkner LE, Panagotopulos SE, Johnson JD, Woollett LA, Hui DY, Witting SR, Maiorano JN, Davidson WS. An analysis of the role of a retroendocytosis pathway in ABCA1-mediated cholesterol efflux from macrophages. J Lipid Res. 2008;49:1322–1332. doi: 10.1194/jlr.M800048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio S, Linton MF, Swift LL. The cell biology and physiologic relevance of apoE recycling. Trends Cardiovasc Med. 2000;10:23–30. doi: 10.1016/s1050-1738(00)00033-5. [DOI] [PubMed] [Google Scholar]
- Fenske SA, Yesilaltay A, Pal R, Daniels K, Rigotti A, Krieger M, Kocher O. Overexpression of the PDZ1 domain of PDZK1 blocks the activity of hepatic scavenger receptor, class B, type I (SR-BI) by altering its abundance and cellular localization. J Biol Chem. 2008;283:22097–22104. doi: 10.1074/jbc.M800029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228. [PubMed] [Google Scholar]
- Fisher CA, Ryan RO. Lipid binding-induced conformational changes in the N-terminal domain of human apolipoprotein E. J Lipid Res. 1999;40:93–99. [PubMed] [Google Scholar]
- Fitzgerald ML, Morris AL, Rhee JS, Andersson LP, Mendez AJ, Freeman MW. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J Biol Chem. 2002;277:33178–33187. doi: 10.1074/jbc.M204996200. [DOI] [PubMed] [Google Scholar]
- Forte TM, Nichols AV, Gong EL, Lux S, Levy RI. Electron microscopic study on reassembly of plasma high density apoprotein with various lipids. Biochim Biophys Acta. 1971;248:381–386. doi: 10.1016/0005-2760(71)90026-9. [DOI] [PubMed] [Google Scholar]
- Frank PG, Marcel YL. Apolipoprotein A-I: Structure-function relationships. J Lipid Res. 2000;41:853–872. [PubMed] [Google Scholar]
- Gazzara JA, Phillips MC, Lund-Katz S, Palgunachari MN, Segrest JP, Anantharamaiah GM, Snow JW. Interaction of class A amphipathic helical peptides with phospholipid unilamellar vesicles. J Lipid Res. 1997;38:2134–2146. [PubMed] [Google Scholar]
- Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol. 2008;26:534–540. doi: 10.1161/01.ATV.0000200082.58536.e1. [DOI] [PubMed] [Google Scholar]
- Getz GS, Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50:S156–S161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillotte KL, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. Removal of cellular cholesterol by pre-beta-HDL involves plasma membrane microsolubilization. J Lipid Res. 1998;39:1918–1928. [PubMed] [Google Scholar]
- Gillotte KL, Zaiou M, Lund-Katz S, Anantharamaiah GM, Holvoet P, Dhoest A, Palgunachari MN, Segrest JP, Weisgraber KH, Rothblat GH, Phillips MC. Apolipoprotein-mediated plasma membrane microsolubilization. J Biol Chem. 1999;274:2021–2028. doi: 10.1074/jbc.274.4.2021. [DOI] [PubMed] [Google Scholar]
- Gillotte-Taylor KL, Nickel M, Johnson WJ, Francone O, Holvoet P, Lund-Katz S, Rothblat GH, Phillips MC. Effects of enrichment of fibroblasts with unesterified cholesterol on the efflux of cellular lipids to apolipoprotein A-I. J Biol Chem. 2002;277:11811–11820. doi: 10.1074/jbc.M108268200. [DOI] [PubMed] [Google Scholar]
- Gong E, Tan CS, Shoukry MI, Rubin EM, Nichols AV. Structural and functional properties of human and mouse apolipoprotein A-I. Biochim Biophys Acta. 1994;1213:335–342. doi: 10.1016/0005-2760(94)00062-x. [DOI] [PubMed] [Google Scholar]
- Gu X, Kozarsky K, Krieger M. Scavenger receptor class B, type I-mediated [3-H]cholesterol efflux to high and low density lipoproteins is dependent on lipoprotein binding to the receptor. J Biol Chem. 2000;275:29993–30001. doi: 10.1074/jbc.275.39.29993. [DOI] [PubMed] [Google Scholar]
- Gu X, Trigatti B, Xu S, Acton S, Babitt J, Krieger M. The efficient cellular uptake of high density lipoprotein lipid via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J Biol Chem. 1998;273:26338–26348. doi: 10.1074/jbc.273.41.26338. [DOI] [PubMed] [Google Scholar]
- Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-I: implications for a lipid-free molten globular state. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder CJ, Vassiliou G, McBride HM, McPherson R. Hepatic SR-BI-mediated cholesteryl ester selective uptake occurs with unaltered efficiency in the absence of cellular energy. J Lipid Res. 2006;47:492–503. doi: 10.1194/jlr.M500444-JLR200. [DOI] [PubMed] [Google Scholar]
- Hasty AH, Plummer MR, Weisgraber KH, Linton MF, Fazio S, Swift LL. The recycling of apolipoprotein E in macrophages: influence of HDL and apolipoprotein A-I. J Lipid Res. 2005;46:1433–1439. doi: 10.1194/jlr.M400418-JLR200. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Budamagunta MS, Voss JC, Weisgraber KH. Modulation of apolipoprotein E structure by domain interaction. Differences in lipid-bound and lipid-free forms. J Biol Chem. 2005;280:34288–34295. doi: 10.1074/jbc.M506044200. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: Insights into function. Trends Biochem Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Voss JC, Budamagunta MS, Newhouse YN, Weisgraber KH. Insight on the molecular envelope of lipid-bound apolipoprotein E from electron paramagnetic resonance spectroscopy. J Mol Biol. 2009;386:261–271. doi: 10.1016/j.jmb.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser H, Henry R, Leslie RB, Stubbs JM. The interaction of apoprotein from porcine high-density lipoprotein with dimyristoyl phosphatidylcholine. Eur J Biochem. 1974;48:583–594. doi: 10.1111/j.1432-1033.1974.tb03801.x. [DOI] [PubMed] [Google Scholar]
- Heeren J, Beisiegel U, Grewal T. Apolipoprotein E recycling. Implications for dyslipidemia and atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:442–448. doi: 10.1161/01.ATV.0000201282.64751.47. [DOI] [PubMed] [Google Scholar]
- Hirch-Reinshagen V, Wellington CL. Cholesterol metabolism, apolipoprotein E, adenosine triphosphate-binding cassette transporters, and Alzheimer’s disease. Curr Opin Lipidol. 2007;18:325–332. doi: 10.1097/MOL.0b013e32813aeabf. [DOI] [PubMed] [Google Scholar]
- Hirz R, Scanu AM. Reassembly in vitro of a serum high-density lipoprotein. Biochim Biophys Acta. 1970;207:364–367. doi: 10.1016/0005-2795(70)90029-2. [DOI] [PubMed] [Google Scholar]
- Homanics GE, de Silva HV, Osada J, Zhang SH, Wong H, Borensztajn J, Maeda N. Mild dyslipidemia in mice following targeted inactivation of the hepatic lipase gene. J Biol Chem. 1995;270:2974–2980. doi: 10.1074/jbc.270.7.2974. [DOI] [PubMed] [Google Scholar]
- Huang ZH, Fitzgerald ML, Mazzone T. Distinct cellular loci for the ABCA1-dependent and ABCA1-independent lipid efflux mediated by endogenous apolipoprotein E expression. Arterioscler Thromb Vasc Biol. 2006;26:157–162. doi: 10.1161/01.ATV.0000193627.12516.1d. [DOI] [PubMed] [Google Scholar]
- Huuskonen J, Olkkonen VM, Jauhiainen M, Ehnholm C. The impact of phospholipid transfer protein (PLTP) on HDL metabolism. Atherosclerosis. 2001;155:269–281. doi: 10.1016/s0021-9150(01)00447-6. [DOI] [PubMed] [Google Scholar]
- Innerarity TL, Pitas RE, Mahley RW. Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. J Biol Chem. 1979;254:4186–4190. [PubMed] [Google Scholar]
- Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, van der Westhuijzen DR, de Beer FC. HDL remodeling during the acute phase response. Arterioscler Thromb Vasc Biol. 2009;29:261–267. doi: 10.1161/ATVBAHA.108.178681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahangiri A, Rader DJ, Marchadier D, Curtiss LK, Bonnet DJ, Rye KA. Evidence that endothelial lipase remodels high density lipoproteins without mediating the dissociation of apolipoprotein A-I. J Lipid Res. 2005;46:896–903. doi: 10.1194/jlr.M400212-JLR200. [DOI] [PubMed] [Google Scholar]
- Jaye M, Krawiec J. Endothelial lipase and HDL metabolism. Curr Opin Lipidol. 2004;15:183–189. doi: 10.1097/00041433-200404000-00011. [DOI] [PubMed] [Google Scholar]
- Jessup W, Gelissen IC, Gaus K, Kritharides L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr Opin Lipidol. 2006;17:247–257. doi: 10.1097/01.mol.0000226116.35555.eb. [DOI] [PubMed] [Google Scholar]
- Ji Y, Jian B, Wang N, Sun Y, Llera Moya M, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- Jian B, Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- Jonas A. Lipid-binding properties of apolipoproteins. In: Rosseneu M, editor. Structure and Function of Apolipoproteins. CRC Press; Boca Raton: 1992. pp. 217–250. [Google Scholar]
- Jonas A. Lecithin cholesterol acyltransferase. Biochim Biophys Acta. 2000;1529:245–256. doi: 10.1016/s1388-1981(00)00153-0. [DOI] [PubMed] [Google Scholar]
- Jonas A, Drengler SM. Kinetics and mechanism of apolipoprotein A-I interaction with L-alpha-dimyristoylphosphatidylcholine vesicles. J Biol Chem. 1980;255:2190–2194. [PubMed] [Google Scholar]
- Jonas A, Drengler SM, Patterson BW. Two types of complexes formed by the interaction of apolipoprotein A-I with vesicles of L-alpha-dimyristoylphosphatidylcholine. J Biol Chem. 1980;255:2183–2189. [PubMed] [Google Scholar]
- Jonas A, Krajnovich DJ, Patterson BW. Physical properties of isolated complexes of human and bovine A-I apolipoproteins with L-alpha-dimyristoyl phosphatidylcholine. J Biol Chem. 1977;252:2200–2205. [PubMed] [Google Scholar]
- Jonas A, Phillips MC. Lipoprotein Structure. In: Vance DE, Vance JE, editors. Biochemistry of Lipids, Lipoproteins and Membranes. 5. Elsevier; 2008. pp. 485–506. [Google Scholar]
- Kellner-Weibel G, Llera-Moya M, Connelly MA, Stoudt G, Christian AE, Haynes MP, Williams DL, Rothblat GH. Expression of scavenger receptor BI in COS-7 cells alters cholesterol content and distribution. Biochemistry. 2000;39:221–229. doi: 10.1021/bi991666c. [DOI] [PubMed] [Google Scholar]
- Kim WS, Rahmanto AS, Kamili A, Rye KA, Guillemin GJ, Gelissen IC, Jessup W, Hill AF, Garner B. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein-E discs and suppression of amyloid-β peptide generation. J Biol Chem. 2006;282:2851–2861. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- Kiss RS, Kay CM, Ryan RO. Amphipathic alpha-helix bundle organization of lipid-free chicken apolipoprotein A-I. Biochemistry. 1999;38:4327–4334. doi: 10.1021/bi982597p. [DOI] [PubMed] [Google Scholar]
- Kockx M, Jessup W, Kritharides L. Regulation of endogenous apolipoprotein E secretion by macrophages. Arterioscler Thromb Vasc Biol. 2008;28:1060–1067. doi: 10.1161/ATVBAHA.108.164350. [DOI] [PubMed] [Google Scholar]
- Kockx M, Rye KA, Gaus K, Quinn CM, Wright J, Sloane T, Sviridov D, Fu Y, Sullivan D, Burnett JR, Rust S, Assmann G, Anantharamaiah GM, Palgunachari MN, Lund-Katz S, Phillips MC, Dean RT, Jessup W, Kritharides L. Apolipoprotein AI-simulated apolipoprotein E secretion from human macrophages is independent of cholesterol efflux. J Biol Chem. 2004;279:25966–25977. doi: 10.1074/jbc.M401177200. [DOI] [PubMed] [Google Scholar]
- Kono M, Okumura Y, Tanaka M, Nguyen D, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. Conformational flexibility of the N-terminal domain of apolipoprotein A-I bound to spherical lipid particles. Biochemistry. 2008;47:11340–11347. doi: 10.1021/bi801503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontush A, Chapman MJ. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev. 2006;58:342–374. doi: 10.1124/pr.58.3.1. [DOI] [PubMed] [Google Scholar]
- Krimbou L, Denis M, Haidar B, Carrier M, Marcil M, Genest J., Jr Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res. 2004;45:839–848. doi: 10.1194/jlr.M300418-JLR200. [DOI] [PubMed] [Google Scholar]
- Krimbou L, Marcil M, Genest J. New insights into the biogenesis of human high-density lipoprotein. Curr Opin Lipidol. 2006;17:257–267. doi: 10.1097/01.mol.0000226117.43178.7c. [DOI] [PubMed] [Google Scholar]
- Kruski AW, Scanu AM. Interaction of human serum high density lipoprotein apoprotein with phospholipids. Chem Phys Lipids. 1974;13:27–48. doi: 10.1016/0009-3084(74)90040-1. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Reardon C, Van Eldick L, Fagan LM, Bu G, Holtzman D, Getz GS. Lipoproteins in the central nervous system. Ann NY Acad Sci. 2000;903:167–175. doi: 10.1111/j.1749-6632.2000.tb06365.x. [DOI] [PubMed] [Google Scholar]
- Lee JY, Parks JS. ATP-binding cassette transporter AI and its role in HDL formation. Curr Opin Lipidol. 2005;16:19–25. doi: 10.1097/00041433-200502000-00005. [DOI] [PubMed] [Google Scholar]
- Li L, Chen J, Mishra VK, Kurtz JA, Cao D, Klon AE, Harvey SC, Anantharamaiah GM, Segrest JP. Double belt structure of discoidal high density lipoproteins: Molecular basis for size heterogeneity. J Mol Biol. 2004;343:1293–1311. doi: 10.1016/j.jmb.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Li W, Tanimura M, Luo C, Datta S, Chan L. The apolipoprotein multigene family: biosynthesis, structure, structure-function relationships, and evolution. J Lipid Res. 1988;29:245–271. [PubMed] [Google Scholar]
- Liang JS, Schreiber BM, Salmona M, Phillip G, Gonnerman WA, de Beer FC, Sipe JD. Amino terminal region of acute phase, but not constitutive, serum amyloid A (apoSAA) specifically binds and transports cholesterol into aortic smooth muscle and HepG2 cells. J Lipid Res. 1996;37:2109–2116. [PubMed] [Google Scholar]
- Liang JS, Sipe JD. Recombinant human serum amyloid A (apoSSAp) binds cholesterol and modulates cholesterol flux. J Lipid Res. 1995;36:37–46. [PubMed] [Google Scholar]
- Liu B, Krieger M. Highly purified scavenger receptor class B, type I reconstituted into phosphatidylcholine/cholesterol liposomes mediates high affinity high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2002;277:34125–34135. doi: 10.1074/jbc.M204265200. [DOI] [PubMed] [Google Scholar]
- Liu L, Bortnick AE, Nickel M, Dhanasekaran P, Subbaiah PV, Lund-Katz S, Rothblat GH, Phillips MC. Effects of apolipoprotein A-I on ATP-binding cassette transporter A1-mediated efflux of macrophage phospholipid and cholesterol. J Biol Chem. 2003;278:42976–42984. doi: 10.1074/jbc.M308420200. [DOI] [PubMed] [Google Scholar]
- Liu T, Krieger M, Kan HY, Zannis VI. The effects of mutations in helices 4 and 6 of apo A-I on SR-BI-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted HDL and SR-BI is required for efficient lipid transport. J Biol Chem. 2002;277:21576–21584. doi: 10.1074/jbc.M112103200. [DOI] [PubMed] [Google Scholar]
- Llera-Moya M, Rothblat GH, Connelly MA, Kellner-Weibel G, Sakr SW, Phillips MC, Williams DL. Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J Lipid Res. 1999;40:575–580. [PubMed] [Google Scholar]
- Lu B, Morrow JA, Weisgraber KH. Conformational reorganization of the four-helix bundle of human apolipoprotein E in binding to phospholipid. J Biol Chem. 2000;275:20775–20781. doi: 10.1074/jbc.M003508200. [DOI] [PubMed] [Google Scholar]
- Lu R, Arakawa R, Ito-Osumi C, Iwamoto N, Yokoyama S. ApoA-I facilitates ABCA1 recycle/accumulation to cell surface by inhibiting its intracellular degradation and increases HDL generation. Arterioscler Thromb Vasc Biol. 2008;28:1820–1824. doi: 10.1161/ATVBAHA.108.169482. [DOI] [PubMed] [Google Scholar]
- Lund-Katz S, Liu L, Thuahnai ST, Phillips MC. High density lipoprotein structure. Frontiers in Bioscience. 2003;8:d1044–d1054. doi: 10.2741/1077. [DOI] [PubMed] [Google Scholar]
- Lux SE, Hirz R, Shrager RI, Gotto AM. The influence of lipid on the conformation of human plasma high density apolipoproteins. J Biol Chem. 1972;247:2598–2606. [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Apolipoprotein E: From atherosclerosis to Alzheimer’s disease and beyond. Curr Opin Lipidol. 1999;10:207–217. doi: 10.1097/00041433-199906000-00003. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y, Rall SC. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): Questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–1949. [PubMed] [Google Scholar]
- Mahley RW, Huang Y, Weisgraber KH. Putting cholesterol in its place: apoE and reverse cholesterol transport. J Clin Invest. 2006;116:1226–1229. doi: 10.1172/JCI28632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Rall J. Type III hyperlipoproteinemia (dysbetalipoproteinemia): The role of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 7. McGraw-Hill, Inc; New York: 1995. pp. 1953–1980. [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: Structure determines function – from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50:S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel YL, Ouimet M, Wang MD. Regulation of cholesterol efflux from macrophages. Curr Opin Lipidol. 2008;19:455–461. doi: 10.1097/MOL.0b013e32830f4a1d. [DOI] [PubMed] [Google Scholar]
- Marsche G, Frank S, Raynes JG, Kozarsky KF, Sattler W, Malle E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem J. 2007;402:117–124. doi: 10.1042/BJ20061406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey JB, Gotto AMJ, Pownall HJ. Contribution of α helix formation in human plasma apolipoproteins to their enthalpy of association with phospholipids. J Biol Chem. 1979;254:9359–9361. [PubMed] [Google Scholar]
- Massey JB, Pownall HJ. Cholesterol is a determinant of the structures of discoidal high density lipoproteins formed by the solubilization of phospholipid membranes by apolipoprotein A-I. Biochim Biophys Acta. 2008;1781:245–253. doi: 10.1016/j.bbalip.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson D, Jiang XC, Lagrost L, Tall AR. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J Lipid Res. 2009;50:S201–S206. doi: 10.1194/jlr.R800061-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JA, Hatters DM, Lu B, Hocht P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. J Biol Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- Narayanaswami V, Maiorano JN, Dhanasekaran P, Ryan RO, Phillips MC, Lund-Katz S, Davidson WS. Helix orientation of the functional domains in apolipoprotein E in discoidal high density lipoprotein particles. J Biol Chem. 2004;279:14273–14279. doi: 10.1074/jbc.M313318200. [DOI] [PubMed] [Google Scholar]
- Narayanaswami V, Ryan RO. Molecular basis of exchangeable apolipoprotein function. Biochim Biophys Acta. 2000;1483:15–36. doi: 10.1016/s1388-1981(99)00176-6. [DOI] [PubMed] [Google Scholar]
- Narayanaswami V, Szeto SS, Ryan RO. Lipid association-induced N- and C-terminal domain reorganization in human apolipoprotein E3. J Biol Chem. 2001;276:37853–37860. doi: 10.1074/jbc.M102953200. [DOI] [PubMed] [Google Scholar]
- Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney A, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie EJ, Santamarina-Fojo S, Brewer HBJ. Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S. Molecular mechanism of apolipoprotein E binding to lipoprotein particles. Biochemistry. 2009;48:3025–3032. doi: 10.1021/bi9000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieland TJ, Ehrlich M, Krieger M, Kirchhausen T. Endocytosis is not required for the selective lipid uptake mediated by murine SR-BI. Biochim Biophys Acta. 2005;1734:44–51. doi: 10.1016/j.bbalip.2005.02.007. [DOI] [PubMed] [Google Scholar]
- O’Brien KD, Chait A. Serum amyloid A: The other inflammatory protein. Curr Athero Rep. 2006;8:62–68. doi: 10.1007/s11883-006-0066-0. [DOI] [PubMed] [Google Scholar]
- Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 2003;10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- Oram JF. Tangier Disease and ABCA1. Biochim Biophys Acta. 2000;1529:321–330. doi: 10.1016/s1388-1981(00)00157-8. [DOI] [PubMed] [Google Scholar]
- Oram JF, Heinecke JW. ATP-binding cassette transporter A1: A cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity. Arterioscler Thromb Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- Perugini MA, Schuck P, Howlett GJ. Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J Biol Chem. 2000;275:36758–36765. doi: 10.1074/jbc.M005565200. [DOI] [PubMed] [Google Scholar]
- Peters-Libeu CA, Newhouse Y, Hall SC, Witkowska HE, Weisgraber KH. Apolipoprotein E dipalmitoylphosphatidylcholine particles are ellipsoidal in solution. J Lipid Res. 2007;48:1035–1044. doi: 10.1194/jlr.M600545-JLR200. [DOI] [PubMed] [Google Scholar]
- Peters-Libeu CA, Newhouse Y, Hatters DM, Weisgraber KH. Model of biologically active apolipoprotein E bound to dipalmitoylphosphatidylcholine. J Biol Chem. 2006;281:1073–1079. doi: 10.1074/jbc.M510851200. [DOI] [PubMed] [Google Scholar]
- Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim Biophys Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. J Biol Chem. 1987;262:14352–14360. [PubMed] [Google Scholar]
- Pittman RC, Knecht TP, Rosenbaum MS, Taylor CA., Jr A nonendocytotic mechanism for the selective uptake of high density lipoprotein-associated cholesteryl esters. J Biol Chem. 1987;262:2443–2450. [PubMed] [Google Scholar]
- Pownall H, Pao Q, Hickson D, Sparrow JT, Kusserow SK, Massey JB. Kinetics and mechanism of association of human plasma apolipoproteins with dimyristoylphosphatidylcholine: Effect of protein structure and lipid clusters on reaction rates. Biochemistry. 1981;20:6630–6635. doi: 10.1021/bi00526a017. [DOI] [PubMed] [Google Scholar]
- Pownall HJ. Detergent-mediated phospholipidation of plasma lipoproteins increases HDL cholesterophilicity and cholesterol efflux via SR-BI. Biochemistry. 2006;45:11514–11522. doi: 10.1021/bi0608717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pownall HJ, Ehnholm C. The unique role apolipoprotein A-I in HDL remodeling and metabolism. Curr Opin Lipidol. 2006;17:209–213. doi: 10.1097/01.mol.0000226110.66942.e8. [DOI] [PubMed] [Google Scholar]
- Pownall HJ, Massey JB, Kusserow SK, Gotto AM. Kinetics of lipid-protein interactions: interaction of apolipoprotein A-I from human plasma high density lipoproteins with phosphatidylcholine. Biochemistry. 1978;17:1183–1188. doi: 10.1021/bi00600a008. [DOI] [PubMed] [Google Scholar]
- Pownall HJ, Massey JB, Kusserow SK, Gotto AM., Jr Kinetics of lipid-protein interactions: Effect of cholesterol on the association of human plasma high-density apolipoprotein A-I with L-alpha-dimyristoylphosphatidylcholine. Biochemistry. 1979;18:574–579. doi: 10.1021/bi00571a004. [DOI] [PubMed] [Google Scholar]
- Pownall HJ, Massey JB, Sparrow JT, Gotto AM. Lipid-protein interactions and lipoprotein reassembly. In: Gotto AM, editor. Plasma Lipoproteins. Elsevier Science Publishers B.V; Amsterdam: 1987. pp. 95–127. [Google Scholar]
- Qiu X, Mistry A, Ammirati MJ, Chrunyk BA, Clark RW, Cong Y, Culp JS, Danley DE, Freeman TB, Goeghegan KF, Griffor MC, Hawrylik SJ, Hayward CM, Hensley P, Hoth LR, Karma GA, Lira ME, Lloyd DB, McGrath KM, Stutzman-Engwall KJ, Subashi AK, Subashi TA, Thompson JF, Wang IK, Zhao H, Seddon AP. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat Struct Mol Biol. 2007;14:106–113. doi: 10.1038/nsmb1197. [DOI] [PubMed] [Google Scholar]
- Raussens V, Fisher CA, Goormaghtigh E, Ryan RO, Ruysschaert JM. The low density lipoprotein receptor active conformation of apolipoprotein E. J Biol Chem. 1998;273:25825–25830. doi: 10.1074/jbc.273.40.25825. [DOI] [PubMed] [Google Scholar]
- Reijngoud DJ, Phillips MC. Mechanism of dissociation of human apolipoprotein AI from complexes with dimyristoylphosphatidylcholine as studied by guanidine hydrochloride denaturation. Biochemistry. 1982;21:2969–2976. doi: 10.1021/bi00541a026. [DOI] [PubMed] [Google Scholar]
- Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, Bocharov AV, Vishnyakova TG, Patterson AP, Eggerman TL, Santamarina-Fojo S, Brewer BH. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J Lipid Res. 2003;44:828–836. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- Rigotti A, Trigatti B, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LM, Ray MJ, Shih TW, Hayden E, Reader MM, Brouillette CG. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and -oxidized lipid-free and lipid-bound human apoA-I. Biochemistry. 1997;36:7615–7624. doi: 10.1021/bi962952g. [DOI] [PubMed] [Google Scholar]
- Rodrigueza WV, Thuahnai ST, Temel RE, Lund-Katz S, Phillips MC, Williams DL. Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J Biol Chem. 1999;274:20344–20350. doi: 10.1074/jbc.274.29.20344. [DOI] [PubMed] [Google Scholar]
- Rogers DP, Roberts L, Lebowitz J, Datta G, Anantharamaiah GM, Engler JA, Brouillette CG. The lipid-free structure of apolipoprotein A-I: Effects of amino-terminal deletions. Biochemistry. 1998;37:11714–11725. doi: 10.1021/bi973112k. [DOI] [PubMed] [Google Scholar]
- Roosbeek S, Vanloo B, Duverger N, Caster H, Breyne J, De Beun I, Patel H, Vandekerckhove J, Shoulders C, Rosseneu M, Peelman F. Three arginine residues in apolipoprotein A-I are critical for activation of lecithin: cholesterol acyltransferase. J Lipid Res. 2001;42:31–40. [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Ann Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- Rothblat GH, Bamberger M, Phillips MC. Reverse cholesterol transport. Meth Enzymol. 1986;129:628–644. doi: 10.1016/0076-6879(86)29095-3. [DOI] [PubMed] [Google Scholar]
- Rye KA, Barter PJ. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24:421–428. doi: 10.1161/01.ATV.0000104029.74961.f5. [DOI] [PubMed] [Google Scholar]
- Rye KA, Bursill CA, Lambert G, Tabet F, Barter PJ. The metabolism and anti-atherogenic properties of HDL. J Lipid Res. 2009;50:S195–S200. doi: 10.1194/jlr.R800034-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye KA, Hime NJ, Barter PJ. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J Biol Chem. 1997;272:3953–3960. doi: 10.1074/jbc.272.7.3953. [DOI] [PubMed] [Google Scholar]
- Rye KA, Wee K, Curtiss LK, Bonnet DJ, Barter PJ. Apolipoprotein A-II inhibits high density lipoprotein remodeling and lipid-poor apolipoprotein A-I formation. J Biol Chem. 2008;278:22530–22536. doi: 10.1074/jbc.M213250200. [DOI] [PubMed] [Google Scholar]
- Sahoo D, Darlington YF, Pop D, Williams DL, Connelly MA. Scavenger receptor class B type I (SR-BI) assembles into detergent-sensitive dimers and tetramers. Biochim Biophys Acta. 2007;1771:807–817. doi: 10.1016/j.bbalip.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Saito H, Dhanasekaran P, Baldwin F, Weisgraber K, Lund-Katz S, Phillips MC. Lipid binding-induced conformational change in human apolipoprotein E. J Biol Chem. 2001;276:40949–40954. doi: 10.1074/jbc.M106337200. [DOI] [PubMed] [Google Scholar]
- Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Phillips MC, Lund-Katz S. Effects of polymorphism on the lipid interaction of human apolipoprotein E. J Biol Chem. 2003a;278:40723–40729. doi: 10.1074/jbc.M304814200. [DOI] [PubMed] [Google Scholar]
- Saito H, Dhanasekaran P, Nguyen D, Deridder E, Holvoet P, Lund-Katz S, Phillips MC. Alpha-helix formation is required for high affinity binding of human apolipoprotein A-I to lipids. J Biol Chem. 2004a;279:20974–20981. doi: 10.1074/jbc.M402043200. [DOI] [PubMed] [Google Scholar]
- Saito H, Dhanasekaran P, Nguyen D, Holvoet P, Lund-Katz S, Phillips MC. Domain structure and lipid interaction in human apolipoproteins A-I and E: A general model. J Biol Chem. 2003b;278:23227–23232. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Progress in Lipid Research. 2004b;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Saito H, Miyako Y, Handa T, Miyajima K. Effect of cholesterol on apolipoprotein A-I binding to lipid bilayers and emulsions. J Lipid Res. 1997;38:287–294. [PubMed] [Google Scholar]
- Sakamoto T, Tanaka M, Vedhachalam C, Nickel M, Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H. Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry. 2008;47:2968–2977. doi: 10.1021/bi701923h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamarina-Fojo S, Lambert G, Hoeg JM, Brewer HBJ. Lecithin-cholesterol acyltransferase: role in lipoprotein metabolism, reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol. 2000;11:267–275. doi: 10.1097/00041433-200006000-00007. [DOI] [PubMed] [Google Scholar]
- Scanu A. Binding of human serum high density lipoprotein apoprotein with aqueous dispersions of phospholipids. J Biol Chem. 1967;242:711–719. [PubMed] [Google Scholar]
- Schneeweis LA, Koppaka V, Lund-Katz S, Phillips MC, Axelsen PH. Structural analysis of lipoprotein E particles. Biochemistry. 2005;44:12525–12534. doi: 10.1021/bi050872j. [DOI] [PubMed] [Google Scholar]
- Segall ML, Dhanasekaran P, Baldwin F, Anantharamaiah GM, Weisgraber K, Phillips MC, Lund-Katz S. Influence of apoE domain structure and polymorphism on the kinetics of phospholipid vesicle solubilization. J Lipid Res. 2002;43:1688–1700. doi: 10.1194/jlr.m200157-jlr200. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic alpha-helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, DeLoof H, Harvey SC. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Pownall HJ, Jackson RL, Glenner GG, Pollock PS. Amyloid A: Amphipathic helixes and lipid binding. Biochemistry. 1976;15:3187–3191. doi: 10.1021/bi00660a005. [DOI] [PubMed] [Google Scholar]
- Settasatian N, Duong M, Curtiss LK, Ehnholm C, Jauhianinen M, Huuskonen J, Rye KA. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J Biol Chem. 2001;276:26898–26905. doi: 10.1074/jbc.M010708200. [DOI] [PubMed] [Google Scholar]
- Silva RAG, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence tracking. Biochemistry. 2005;44:2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- Silva RAG, Huang R, Morris J, Fang J, Gracheva EO, Ren G, Kontush A, Jerome WG, Rye KA, Davidson WS. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci USA. 2008;105:12176–12181. doi: 10.1073/pnas.0803626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivashanmugam A, Wang J. A unified scheme for initiation and conformational adaptation of human apolipoprotein E N-terminal domain upon lipoprotein-binding and for receptor-binding activity. J Biol Chem. 2009;284:14657–14666. doi: 10.1074/jbc.M901012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci-Thomas M, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Davidson WS, Lund-Katz S, Phillips MC. Effects of the neutral lipid content of high density lipoprotein on apolipoprotein A-I structure and particle stability. J Biol Chem. 1995;270:26910–26917. doi: 10.1074/jbc.270.45.26910. [DOI] [PubMed] [Google Scholar]
- Stevens FJ. Hypothetical structure of human serum amyloid A protein. Amyloid J Protein Folding Disord. 2004;11:71–80. doi: 10.1080/13506120412331272296. [DOI] [PubMed] [Google Scholar]
- Stonik JA, Remaley AT, Demosky SJ, Neufeld EB, Bocharov A, Brewer HB. Serum amyloid A promotes ABCA1-dependent and ABCA1-independent lipid efflux from cells. Biochem Biophys Res Commun. 2004;321:936–941. doi: 10.1016/j.bbrc.2004.07.052. [DOI] [PubMed] [Google Scholar]
- Tall AR, Small DM, Deckelbaum RJ, Shipley GG. Structure and thermodynamic properties of high density lipoprotein recombinants. J Biol Chem. 1977;252:4701–4711. [PubMed] [Google Scholar]
- Tam SP, Flexman A, Hulme J, Kisilevsky R. Promoting export of macrophage cholesterol: the physiological role of a major acute-phase protein, serum amyloid A 2.1. J Lipid Res. 2002;43:1410–1420. doi: 10.1194/jlr.m100388-jlr200. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Koyama M, Dhanasekaran P, Nguyen D, Nickel M, Lund-Katz S, Saito H, Phillips MC. Influence of tertiary structure domain properties on the functionality of apolipoprotein A-I. Biochemistry. 2008;47:2172–2180. doi: 10.1021/bi702332b. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Bhat S, Sorci-Thomas MG. The use of chemical cross-linking and mass spectrometry to elucidate the tertiary conformation of lipid-bound apolipoprotein A-I. Curr Opin Lipidol. 2006;17:214–220. doi: 10.1097/01.mol.0000226111.05060.f4. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: Implications for its assembly and function. J Lipid Res. 2008;49:1875–1883. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuahnai ST, Lund-Katz S, Anantharamaiah GM, Williams DL, Phillips MC. A quantitative analysis of apolipoprotein binding to scavenger receptor class B, type I (SR-BI): multiple binding sites for lipid-free and lipid-associated apolipoproteins. J Lipid Res. 2003;44:1132–1142. doi: 10.1194/jlr.M200429-JLR200. [DOI] [PubMed] [Google Scholar]
- Thuahnai ST, Lund-Katz S, Dhanasekaran P, Llera-Moya M, Connelly MA, Williams DL, Rothblat GH, Phillips MC. SR-BI-mediated cholesteryl ester selective uptake and efflux of unesterified cholesterol: Influence of HDL size and structure. J Biol Chem. 2004;279:12448–12455. doi: 10.1074/jbc.M311718200. [DOI] [PubMed] [Google Scholar]
- Thuahnai ST, Lund-Katz S, Williams DL, Phillips MC. Scavenger receptor class B, type I-mediated uptake of various lipids into cells. J Biol Chem. 2001;276:43801–43808. doi: 10.1074/jbc.M106695200. [DOI] [PubMed] [Google Scholar]
- Thuren T. Hepatic lipase and HDL metabolism. Curr Opin Lipidol. 2000;11:277–283. doi: 10.1097/00041433-200006000-00008. [DOI] [PubMed] [Google Scholar]
- Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Ziokovic B, Smith JD, LaDu MJ, Rostagno A, Frangione B, Ghiso J. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid β peptides. Biochem J. 2000;348:359–365. [PMC free article] [PubMed] [Google Scholar]
- Tricerri MA, Behling Agree AK, Sanchez SA, Jonas A. Characterization of apolipoprotein A-I structure using a cysteine-specific fluorescence probe. Biochemistry. 2000;39:14682–14691. doi: 10.1021/bi0014251. [DOI] [PubMed] [Google Scholar]
- Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigatti BL, Krieger M, Rigotti A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1732–1738. doi: 10.1161/01.ATV.0000091363.28501.84. [DOI] [PubMed] [Google Scholar]
- van der Westhuijzen DR, de Beer FC, Webb NR. HDL cholesterol transport during inflammation. Curr Opin Lipidol. 2007;18:147–151. doi: 10.1097/MOL.0b013e328051b4fe. [DOI] [PubMed] [Google Scholar]
- van Eck M, Pennings M, Hoekstra M, Out R, Van Berkel TJC. Scavenger receptor BI and ATP-binding cassette transporter A1 in reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol. 2005;16:307–315. doi: 10.1097/01.mol.0000169351.28019.04. [DOI] [PubMed] [Google Scholar]
- Van Tol A. Phospholipid transfer protein. Curr Opin Lipidol. 2002;13:135–139. doi: 10.1097/00041433-200204000-00004. [DOI] [PubMed] [Google Scholar]
- Vanloo B, Morrison J, Fidge N, Lorent G, Lins L, Brasseur R, Ruysschaert JM, Baert J, Rosseneu M. Characterization of the discoidal complexes formed between apoA-I-CNBr fragments and phosphatidylcholine. J Lipid Res. 1991;32:1253–1264. [PubMed] [Google Scholar]
- Vaughan AM, Oram JF. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res. 2006;47:2433–2443. doi: 10.1194/jlr.M600218-JLR200. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007a;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. ABCA1-induced cell surface binding sites for apoA-I. Arterioscler Thromb Vasc Biol. 2007b;27:1603–1609. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, Liu L, Nickel M, Dhanasekaran P, Anantharamaiah GM, Lund-Katz S, Rothblat G, Phillips MC. Influence of apo A-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, Narayanaswami V, Neto N, Forte TM, Phillips MC, Lund-Katz S, Bielicki JK. The C-terminal lipid-binding domain of apolipoprotein E is a highly efficient mediator of ABCA1-dependent cholesterol efflux that promotes the assembly of high-density lipoproteins. Biochemistry. 2007c;46:2583–2593. doi: 10.1021/bi602407r. [DOI] [PubMed] [Google Scholar]
- Vitello LB, Scanu AM. Studies on human serum high density lipoproteins. Self-association of apolipoprotein A-I in aqueous solutions. J Biol Chem. 1976;251:1131–1136. [PubMed] [Google Scholar]
- Wang N, Silver DL, Costet P, Tall AR. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J Biol Chem. 2000;275:33053–33058. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- Wang N, Tall AR. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:1178–1184. doi: 10.1161/01.ATV.0000075912.83860.26. [DOI] [PubMed] [Google Scholar]
- Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007a;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007b;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;31:1503–1511. [PubMed] [Google Scholar]
- Weisgraber KH. Apolipoprotein E: Structure-function relationships. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- Weisgraber KH, Lund-Katz S, Phillips MC. Apolipoprotein E: Structure-function correlations. In: Miller NE, Tall AR, editors. High Density Lipoproteins and Atherosclerosis III. Elsevier Science Publications; 1992. pp. 175–181. [Google Scholar]
- Westerlund JA, Weisgraber KH. Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J Biol Chem. 1993;268:15745–15750. [PubMed] [Google Scholar]
- Williams DL, Llera-Moya M, Thuahnai ST, Lund-Katz S, Connelly MA, Azhar S, Anantharamaiah GM, Phillips MC. Binding and cross-linking studies show that scavenger receptor bi interacts with multiple sites in apolipoprotein A-I and identify the class A amphipathic alpha-helix as a recognition motif. J Biol Chem. 2000;275:18897–18904. doi: 10.1074/jbc.M002411200. [DOI] [PubMed] [Google Scholar]
- Williams KJ, Feig JE, Fisher EA. Cellular and molecular mechanisms for rapid regression of atherosclerosis: from bench top to potentially achievable clinical goal. Curr Opin Lipidol. 2007;18:443–450. doi: 10.1097/MOL.0b013e32823bcb15. [DOI] [PubMed] [Google Scholar]
- Wilson C, Mau T, Weisgraber K, Wardell MR, Mahley RW, Agard DA. Salt bridge relay triggers defective LDL receptor binding by a mutant apolipoprotein. Structure. 1994;2:713–718. doi: 10.1016/s0969-2126(00)00072-1. [DOI] [PubMed] [Google Scholar]
- Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- Wong H, Schotz MC. The lipase gene family. J Lipid Res. 2002;43:993–999. doi: 10.1194/jlr.r200007-jlr200. [DOI] [PubMed] [Google Scholar]
- Yancey PG, Bortnick AE, Kellner-Weibel G, Llera-Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- Yancey PG, Llera-Moya M, Swarnaker S, Monzo P, Klein SM, Connelly MA, Johnson WJ, Williams DL, Rothblat GH. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275:36596–36604. doi: 10.1074/jbc.M006924200. [DOI] [PubMed] [Google Scholar]
- Yokoyama S. Assembly of high-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006;26:20–27. doi: 10.1161/01.ATV.0000195789.39418.e8. [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Kawai Y, Tajima S, Yamamoto A. Behavior of human apolipoprotein E in aqueous solutions and at interfaces. J Biol Chem. 1985;260:16375–16382. [PubMed] [Google Scholar]
- Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Vasudevan S, Sojitrawala R, Zhao W, Cui C, Xu C, Fan D, Newhouse Y, Balestra R, Jerome WG, Weisgraber K, Li Q, Wang J. A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry. 2007;46:10722–10732. doi: 10.1021/bi700672v. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zanotti I, Reilly M, Glick JM, Rothblat GH, Rader DJ. Overexpression of apoA-I promotes reverse cholesterol transport of cholesterol from macrophages to feces. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- Zhu K, Brubaker G, Smith JD. Large disk intermediate precedes formation of apolipoprotein A-I-dimyristoylphosphatidylcholine small disks. Biochemistry. 2007;46:6299–6307. doi: 10.1021/bi700079w. [DOI] [PMC free article] [PubMed] [Google Scholar]