

Abstract
Chronic pain is inadequately managed with currently available classes of analgesic drugs. Recently, peptide antagonists of the α9α10 nicotinic acetylcholine receptor were shown to be analgesic. The present study was conducted to characterize a novel small molecule, non-peptide antagonist at nicotinic receptors. The tetrakis-quaternary ammonium compound ZZ-204G was evaluated for functional activity on cloned nicotinic receptors expressed in Xenopus oocytes. In-vivo efficacy was assessed in rat models of tonic inflammatory pain (formalin test), neuropathic pain (chronic constriction nerve injury), and thermal nociception (tail flick test). ZZ-204G was an antagonist at nicotinic receptors inhibiting the α9α10 subtype with an IC50 of 0.51 (0.35–0.72) nM. Antagonist activity at other nicotinic subtypes (α1β1δε, α2β2, α2β4, α3β2, α3β4, α4β2, α4β4, α6/α3β2β3, α6/α3β4 and α7) was 10–1000-fold lower than at the α9α10 subtype. In competition binding assays, the ki of ZZ-204G at γ-aminobutyric acid(A), serotonin(3), γ-aminobutyric acid(B), κ- and μ-opioid receptors was 1000- to >10,000- fold lower than at α9α10 nicotinic receptors. Parenteral administration of ZZ-204G dose-dependently decreased nociceptive behaviors (paw flinches) in the formalin test and mechanical hyperalgesia in the chronic constriction nerve injury model of neuropathic pain. ZZ-204G was not antinociceptive in the tail flick assay. Results from the rotarod assay indicated that lower doses of ZZ-204G that were analgesic did not alter motor function. In summary, ZZ-204G represents a prototype small molecule antagonist for α9α10 nicotinic receptors and provides a novel molecular scaffold for analgesic agents with the potential to treat chronic inflammatory or neuropathic pain.
Keywords: nicotinic acetylcholine receptors, pain, analgesia, alpha9alpha10
1. Introduction
Chronic pain afflicts approximately 20% of the adult population in developed nations (Brennan 2007). Although numerous analgesic medications are available for treatment, these pharmacotherapies act via a limited number of molecular mechanisms. The primary classes of analgesics include opioids, non-steroidal anti-inflammatory drugs and various adjuvant agents including antidepressants, anticonvulsants, and α2-adrenergic agonists. Altogether, these medications inadequately manage pain, and a belief among many chronic pain patients is that no treatment will alleviate their suffering.
One promising new target is the neuronal nicotinic acetylcholine receptor, which is present in pain signaling pathways, including primary afferents, spinal cord excitatory and inhibitory interneurons, projecting neurons, brain nuclei with descending spinal projection, and dorsal root ganglia (D’Hoedt and Bertrand, 2009; Taly et al., 2009). Nicotinic receptors are penatmeric ligand gated ion channels composed of α and/or β subunit. In mammals, nine α (α1–7, α9 and α10) and four β (β1–4) subunits have been described. Significant efforts have focused on agonists of central α4β2* (* denotes the possible presence of additional subunits) and α7* nicotinic acetylcholine receptors (Vincler, 2005). Activation of either α4β2* or α7* subtypes is associated with analgesic activity in preclinical pain models. Nevertheless, α4β2* and α7 nicotinic acetylcholine receptors are widespread in the CNS and thus, the potential for centrally-mediated side-effects make these agents less desirable analgesic drugs.
α–Conotoxins are small, disulfide rich peptides, isolated from carnivorous marine snails, which selectively target nicotinic receptors (Azam and McIntosh, 2009; Nicke et al., 2004). Recent studies employing α-conotoxins have suggested that the α9α10 nicotinic receptor subtype may be important as a potential target for novel analgesic agents. Interestingly in this case, antagonist rather than agonist activity is associated with analgesia. Transcripts for α9 and α10 subunits have been identified in dorsal root ganglion neurons and a variety of immune cells (Elgoyhen et al., 2001; Gomez-Casati et al., 2005; McIntosh et al., 2009; Weisstaub et al., 2002). Of note, the α9α10 subtype is absent in brain removing the potential for cognitive side effects from blockade of this receptor. To our knowledge, there are no reports of α9 expression in spinal cord.
Peripherally administered α9α10 selective α-conotoxin antagonists produce analgesia in rodent models of neuropathic and tonic inflammatory pain at doses that do not produce motor toxicity (McIntosh et al., 2009; Vincler and McIntosh, 2007; Vincler et al., 2006). In order to characterize non-peptide nicotinic antagonists, we have begun to examine tetrakis-azaaromatic quaternary ammonium salts. Such compounds have previously been shown to be potent inhibitors at α7 nicotinic acetylcholine receptors (Lopez-Hernandez et al., 2009) and to inhibit nicotinic receptor subtypes that mediate nicotine-evoked dopamine release (Zhang et al., 2008). Here, we report that one such analog that has high potency and selectivity for α9α10 nicotinic acetylcholine receptors and has analgesic properties in rat models of pain including chronic constriction nerve injury model of neuropathic pain and the formalin model of tonic inflammatory pain. The current results suggest the feasibility of designing orally active small molecules that exert their therapeutic action via the novel α9α10 target.
2. Materials and methods
2.1. Synthesis of ZZ-204G
ZZ-204G [5,5′,5″, 5‴-(1,2,4,5-benzenetetrayl)tetrakis-[1-(3-phenylpyridinium)-4-pentyne] tetrabromide] was synthesized utilizing the general procedure described by Zhang et al. (2008), by reacting 5,5′,5″, 5‴-(1,2,4,5-benzenetetrayl)tetrakis-[1-bromo-4-pentyne] (0.46 mmol) with 3-phenylpyridine (2 mmol) at 60–70 °C for 18 hours. The resulting mixture was then treated with diethyl ether and the mixture dissolved in water (15 ml). The aqueous solution was extracted extensively with chloroform (5×30ml) to remove unwanted products and starting materials, and the resulting aqueous solution was then lyophilized to afford ZZ-204G in 51% yield.
2.2. Nicotinic acetylcholine receptor expression
The rat nicotinic acetylcholine receptor subunit cDNA clones α2, α3, α4, α7 and β2-β4 were from S. Heinemann (Salk Institute, La Jolla, CA), α9 and α10 were from A.B. Elgoyhen (Universidad de Buenos Aires, Buenos Aires, Argentina), β2 and β3 in the pGEMHE vector were from C. Luetje (University of Miami, Miami, FL). Construction of the α6/α3 chimera has been previously described (McIntosh et al., 2004). Capped cRNA was made using the mMESSAGE mMACHINE T7 in vitro transcription kit (Ambion, Austin, TX) and purified using the Qiagen RNeasy kit (Qiagen, Valencia, CA). The concentration of cRNA was determined by the absorbance at 260 nm. The mouse muscle subunit cDNA clones in the CMV-based pRBG4 vector were provided by S.M. Sine (Mayo Clinic College of Medicine, Rochester, MN). Fifty nl (at least 5–10 ng) of cRNA or 18.3 nl of cDNA (25 ng/μl of each subunit) were injected into each Xenopus oocyte and incubated at 17°C in ND96 (96.0 mM NaCl, 2.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5 mM HEPES, pH 7.5) containing antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, 100 μg/ml amikacin sulfate, 160 μg/ml sulfamethoxazole, and 32 μg/ml trimethoprim). Recordings were made 1–6 days post-injection.
2.3. Voltage Clamp Electrophysiology
Oocytes were voltage-clamped and exposed to acetylcholine and compounds as previously described (Cartier et al., 1996). Nicotine was not used as it has been previously shown to act as an antagonist of the α9α10 nicotinic subtype (Elgoyhen et al., 2001). Briefly, the oocyte chamber consisting of a cylindrical well (~30 μl in volume) was gravity perfused at a rate of ~2 ml/min with ND96 containing 0.01% (wt/vol) BSA and 1 μM atropine to block potential contaminating signal from endogenous muscarinic receptors. For experiments involving α7 and α9α10, atropine was excluded from the perfusion solution because it has been shown to block these receptor subtypes. Oocytes were exposed once a minute to 1 sec pulses of acetylcholine. Acetylcholine concentrations used were 200 μM for α7, 10 μM for α1β1δε and α9α10 and 100 μM for all other subtypes. These concentrations were chosen to approximate EC50 and also ensure that the agonist response returned to baseline after 1 min agonist washout. Test compound was then applied for five min after which Acetylcholine pulses were resumed. The % block was calculated as a % of ND96 control (no compound) response. Concentration-response data were fit to the equation Y = 100/(1 + 10^((LogIC50 − Log[Toxin])*Hill Slope)) by nonlinear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA). Data points in the concentration-response represent the mean ± SEM from at least 3 oocytes.
2.4. Receptor Binding
Radioligand binding assays were performed by the National Institute of Mental Health Psychoactive Drug Screening Program using standard assay protocols (http://pdsp.med.unc.edu/pdspw/binding.php) described briefly below. The specific receptors, respective radioligands and corresponding reference compounds evaluated were as follows: rat -γ-aminobutyric acid(A) receptor, [3H]muscimol [5-(aminomethyl)-isoxazol-3-ol](5 nM), γ-aminobutyric acid; human serotonin(3) receptor- [3H]LY278584 [1-methyl-N-(8-methyl-8-azabicyclo[3.2.1]oct-3-yl)-1H-indazole-3-carboxamide](0.3 nM), zacopride [4-amino-5-chloro-2-methoxy-N-(quinuclidin-3-yl)benzamide]; rat κ-opioid receptor- [3H]U69593 [N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide](0.3 nM), salvinorin A [methyl (2S,4aR,6aR,7R,9S,10aS,10bR)-9-(acetyloxy)-2-(furan-3-yl)-6a,10b-dimethyl-4,10-dioxododecahydro-2H-benzoyl[f]isochromene-7-carboxylate]; human μ-opioid receptor- [3H]DAMGO [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (0.3nM), DAMGO and rat γ-aminobutyric acid(B)(1b,2) receptor- [3H]CGP54626 (2 nM), CGP54626 [[S-(R*,R*)]-[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](cyclohexylmethyl)phosphinic acid]. Crude membrane fractions were from rat brain for γ-aminobutyric acid(A) receptor and from cells expressing recombinant target receptor for the other receptors. ZZ204G [5,5′,5″, 5‴-(1,2,4,5-benzenetetrayl)tetrakis-[1-(3-phenylpyridinium)-4-pentyne] tetrabromide] was diluted to 5x final assay concentration in binding buffer. Then, 50 μl aliquots of buffer (negative control), test compound (triplicate), and reference compound (duplicate) were added in a final volume of 250 μl. Assays were initiated by the addition of crude membrane fractions and the reaction was equilibrated for 1.5 hours at room temperature. Bound radioactivity was isolated by filtration onto 0.3% polyethyleneimine-treated GF/G filter. The IC50 was used to obtain the Ki by applying the Cheng-Prusoff approximation: Ki =IC50/(1 + [ligand]/KD).
2.5. Animals
Male Sprague Dawley rats, approximately 90 days old, weighing about 350 g (Harlan, Indianapolis, IN), were housed individually in transparent cages with a sawdust-covered floor in a humidity- and temperature-controlled AAALAC-accredited facility (a 12 h alternative light/dark cycle) with free access to standard laboratory chow and tap water. Rats were allowed to habituate to the housing facility for at least one week. Next, they were familiarized with the experimental environment/apparatus (three times) before the experiments began. On each day, rats were habituated to the experimental room for at least 30 min prior to testing. All experiments were conducted during the light phase of the cycle (0800-1700). Body weights were determined on the day of experimentation. Surgical procedures were performed under sterile conditions and pentobarbital sodium (40 mg/kg, intraperitoneal ( i.p.) anesthesia. At the end of the experiment, rats were euthanized with a pentobarbital overdose (150 mg/kg i.p.). A cross-over paradigm was used within an experiment (if possible) to minimize the number of rats. All experiments were performed according with a protocol approved by the University of Kentucky Institutional Animal Care and Use Committee and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Publication No 85-23, revised 1983).
2.6. Drug administration
ZZ-204G was dissolved in 0.9% saline and administered intraperitonealy (i.p.,1 ml/kg). Saline vehicle (1 ml/kg) served as a control. Raters were blinded with respect to dose.
2.7. Rodent model of inflammatory pain (the formalin test)
The ability of ZZ-204G to block formalin-induced flinching behavior was assessed using the formalin paw test (Wheeler-Aceto and Cowan, 1991). Briefly, 50 μl of formalin (5%) was injected subcutaneously (s.c). into the dorsal surface of the left hind paw. This procedure typically produces flinching, lifting and licking in a biphasic time course. The early phase (0–10 min) is thought to result from direct stimulation of nociceptors (nociceptive pain) whereas the late phase (20–60 min) involves central sensitization (Tjolsen et al., 1992). Rats were injected with ZZ-204G (3.6, 36, 360 μg/kg, i.p.) 15 min prior to formalin injection (s.c.). This pretreatment time is typically used by us so the antinociceptive activity coincides with the observation period (maximum flinching behavior occurs approximately 45 minutes after formalin injection). Saline preceded formalin in control rats. Incidences of formalin-induced flinching were counted in 5 min intervals for 60 min. Each rat received only one dose of ZZ-204G.
2.8. Chronic Constriction Nerve Injury Rodent Model of Neuropathic Pain
A well-established chronic constriction nerve injury rodent model of neuropathic pain (Bennett and Xie, 1988) was utilized to determine the antihyperalgesic effect of ZZ-204G. Chronic constriction injury results in enhanced sensitivity to noxious stimuli (hyperalgesia) that develops typically within 7 days and lasts, in our hands, approximately 28 days after surgery in the injured paw. Briefly, rats were subjected to a sciatic nerve ligation (left paw) and sham surgery (right paw) as follows. Proximal to the sciatic trifurcation, the nerve (approximately 7 mm) was freed from adhering tissue and four loose ligatures were tied around the nerve (1 mm apart) using 4.0 chromic catgut, barely constricting the diameter of the nerve. In sham surgery, the nerve was exposed but was not ligated. The incision was closed in layers with silk thread 3.0. There are no significant changes in the sham-operated paw.
The presence of mechanical hyperalgesia was determined by the paw pressure test (32 g/s; cut-off 300 g) (Randall and Selitto, 1957) utilizing the Basile Analgesimeter (UGO Basile, Italy). Vocalization was used as the end point. Assessments were carried out prior to surgery (pre-chronic constriction injury baseline) and on post-surgery days 7, 9, 11 and 14, when abnormal pain behavior was at a stable maximum. Typically, vocalization threshold values were equal to 200 g and 100 g prior to and after chronic constriction injury, respectively, in control rats. Each rat received four doses of ZZ-204 (3.6, 36, 360, 3600 μg/kg, randomized order of administration). Control rats were treated with saline. Responsiveness to mechanical noxious stimuli (vocalization threshold) was determined prior to (twice, 15 min apart) and at several time points (15–240 min) after injection. The nerve-injured (chronic constriction injury) and sham-operated paws were tested in each rat.
2.9. Rodent model of thermal acute nociception (the tail flick test)
The antinociceptive effect of ZZ-204G was determined by the responsiveness to radiant heat in the tail-flick test (D’Amour and Smith, 1941). Tail flick latency (s) was measured by recording the time from the onset of heat stimulus to the tail to withdrawal of the tail from the heat source using a standard tail-flick apparatus (ITTA Life Science, Woodland Hills, CA). Heat intensity was adjusted to yield average baseline tail flick latency equal to 2–3 s. A cut-off at 10 s was used to prevent tissue damage. Each rat was injected with three doses of ZZ-204G (3.6, 36, 360 μg/kg) and saline (randomized order of administration at weekly intervals). Responsiveness (tail flick latency) was measured prior to (twice, 15 min apart) and at several time points after administration (15–240 min).
2.10. Motor coordination (the rotarod test)
The motor effect of ZZ-204G was determined using the rotarod performance test (Watzman et al., 1964). Intact rats were trained to run on the Rat Rota Rod (Ugo Basile, Comeno, Italy) at a constant speed (10 rev/ min) for 180 s on two consequent days. Thereafter, the rats were treated with ZZ-204G (3.6, 36, 360, 1800, 3600 μg/kg; randomized administration of doses at weekly intervals) and latency to fall off the rotarod was assessed prior to and at several time points (15–360 min) after injection. Cut-off time was 180 s.
2.11. Data and statistics
Data were normalized for baseline values for each rat at each time point. The areas under the curves (AUC) were calculated for baseline-normalized data (the trapezoidal rule). Percent maximum possible effect was calculated as follows: formalin test: (flinches saline – flinches drug/flinches saline)*100; chronic constriction injury paw-pressure test: [(vocalization threshold-baseline post-surgery)/(baseline pre-surgery – baseline post-surgery)]*100; tail-flick test: [(tail flick latency – baseline)/(10 – baseline)]*100; and rotarod test: (latency to fall/180)*100. Dose-response curves were generated for percent maximum possible effect (at peak time) as a function of log dose. A dose of drug causing 50% maximum possible effect (ED50) and 95% confidence limits (95% CL) were calculated using the method of Tallarida and Murray (1987). The data were analyzed using linear regression, one-way analysis of variance (ANOVA), repeated measures (RM) one-way ANOVA, post-hoc Dunnett’s test. Statistical analysis was performed using SigmaPlot for Windows version 11.0 (Systat Software, Inc.). The level of significance was P< 0.05. All data are presented as mean ± standard error of the mean
3. Results
3.1 Activity at nicotinic acetylcholine receptor subtypes and off target receptors
To assess the functional effects of ZZ-204G (structure illustrated in Fig. 1) the compound was applied to Xenopus oocytes heterologously expressing either neuronal or muscle nicotinic acetylcholine receptors. ZZ-204G potently blocked α9α10 nicotinic acetylcholine receptors, with 10 nM compound blocking 93.9 ± 1.4% inhibition of the response (n=6 oocytes). Block was reversed upon washout of ZZ-204G. By comparison, ZZ-204G (100 nM) blocked only ~ 5% of the acetylcholine-induced currents from α4β2 nicotinic acetylcholine receptors (Fig 2). α9α10 nicotinic acetylcholine receptors are highly permeable to Ca++. A substantial portion of the acetylcholine response in oocytes is due to secondary activation of Ca++ dependent Cl− currents (Elgoyhen et al., 2001). Therefore, ZZ-204G was also evaluated using oocytes in which Ca++ in the buffer was replaced with equimolar Ba++; ZZ-204G (100 nM) abolished the current under these conditions (n=3, data not shown) indicating block of α9α10 nicotinic acetylcholine receptors.
Figure 1. Structure of ZZ-204G.
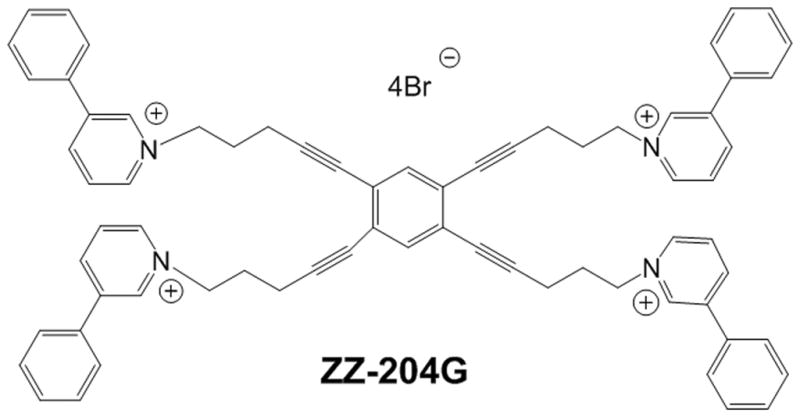
Figure 2. ZZ-204G selectively blocks α9α10 vs. α4β2 nicotinic acetylcholine receptors.

ZZ-204G was applied to oocytes heterologously expressing the indicated nicotinic acetylcholine receptors. The response to a 1 sec application of acetylcholine was measured as described in Methods. Traces from individual oocytes are shown. Note that 10 nM ZZ-204G blocks ~ 95% of the acetylcholine induced response on the α9α10 nicotinic acetylcholine receptors whereas a 10-fold higher concentration blocked < 5% of α4β2 nicotinic acetylcholine receptors.
Concentration response analysis indicated an IC50 of 0.51 nM with a 95% confidence interval (CI) of 0.35–0.72 nM for α9α10 nicotinic acetylcholine receptors. Activity at other subtypes is shown in Fig. 3 and Table 1. In contrast to other small molecule nicotinic acetylcholine receptor ligands, ZZ-204G has highest potency for the α9α10 subtype. The next highest potency was for homomeric nicotinic acetylcholine receptors composed of α7, the subunit evolutionarily closest to α9 and α10. Activity was 190–7500-fold lower at all other Nicotinic acetylcholine receptor subtypes compared to α9α10. No agonist activity was noted at any nicotinic acetylcholine receptor subtype evaluated (data not shown).
Figure 3. Concentration response of ZZ-204G on nicotinic acetylcholine receptors.

ZZ-204G was applied to oocytes expressing the indicated subtype of nicotinic acetylcholine receptor as described in Methods. Each data point represents the average of 3–7 oocytes. Error bars indicated SEM. IC50 values and Hill slopes are shown in Table 1.
Table 1.
Activity of ZZ-204G on nicotinic acetylcholine receptor subtypes
| Subtype | IC50 (nM)a | Hill Slopea |
|---|---|---|
| α9α10 | 0.51 (0.35–0.72) | 0.84 (0.59–1.1) |
| α1β1δε | 120 (98–145) | 1.61 (1.2–2.1) |
| α2β2 | 2330 (1940–2810) | 1.25 (0.98–1.5) |
| α2β4 | 2900 (2340–3600) | 1.36 (0.98–1.7) |
| α3β2 | 750 (353–1590) | 1.11 (0.28–1.9) |
| α3β4 | 792 (641–977) | 1.56 (1.1–2.0) |
| α4β2 | 3830 (2880–5090) | 1.05 (0.73–1.4) |
| α4β4 | 1730 (1320–2260) | 1.6 (0.99–2.2) |
| α6/α3β2β3 | 98.2 (55.6–173) | 0.44 (0.33–0.56) |
| α6/α3β4 | 6140 (4440–8500) | 1.2 (0.73–1.8) |
| α7 | 8.8 (5.0–15.7) | 1.03 (0.48–1.6) |
Numbers in parentheses are 95% confidence intervals
Competition binding assays were performed to assess activity at other receptor subtypes. Nicotinic acetylcholine receptors are structurally related to serotonin type 3 and γ-aminobutyric acid(A) receptors. The Ki values for ZZ-204G displacing [3H]LY27854 and [3H]muscimol were 2.2 μM for 5HT3 and > 10 μM for γ-aminobutyric acid(A) receptors respectively. Activities at κ- and μ-opioid receptors were assessed with [3H]U69593 and [3H]DAMGO; ZZ-204G had Ki values of 783 nM and 659 nM respectively. Thus, for all of these receptors the Ki values were three orders of magnitude higher than the IC50 for α9α10 nicotinic acetylcholine receptors.
3.2. ZZ-204G inhibits formalin-induced flinching behavior in a rodent model of inflammatory pain (the formalin test)
Formalin injection (s.c.) into the paw caused a biphasic flinching behavior in control (saline) rats. The effects of ZZ-204G on the first phase (0–10 min) and the second phase (20–60 min) were investigated. ZZ-204G attenuated flinching behavior during both phases of the formalin test (Fig. 5). A dose related effect was found during the second phase (F3,23 = 12; P < 0.001). Dose-effect relationship only approached significance (F3,23 = 3.05, P = 0.053, ANOVA) during the first phase. The ED50 (95% CI) values were equal to 2.12 (1.24 – 5.65) μg/kg (the second phase) and 16.2 (2.9–90.3) μg/kg (the first phase).
Figure 5. Effect of ZZ-204G on formalin-induced flinching behavior.

The paw formalin test was performed as described in Methods. [A] Flinches versus time (min); Area under the curve (AUC) versus dose (μg/kg): [B] phase 1 (0–10 min) and [C] phase 2 (20–60 min). Mean ± standard error of the mean (n = 6 rats/dose). * Significantly different from saline (P<0.05; Dunnett’s test).
3.3. ZZ-204G alleviates mechanical hyperalgesia in a chronic constriction nerve injury rodent model of neuropathic pain
Chronic constriction injury of the sciatic nerve resulted in the development of hyperalgesia, i.e. enhanced responsiveness to mechanical noxious stimuli, as evidenced by decrease in vocalization threshold of 111.9±7.6, 99.4±5.2, 106.2±6.9 and 128.1±8.0 g on post-chronic constriction injury days 7, 9, 11 and 14, respectively) compared to the pre-chronic constriction injury baseline value (vocalization threshold 228.7±3.3 g). Sham surgery did not enhance pain sensitivity (vocalization threshold, 235.0±1.7, 215.5±3.3, 222.6±2.3, 232.5±4.8 and 218.7±3.7 g, respectively on days 0, 7, 9, 11 and 14).
In a dose-related fashion, ZZ-204G attenuated the mechanical hyperalgesia induced by chronic constriction injury (Fig. 6, F4,35 = 7.3, P<0.01). The ED50 (95% CL) value was 96.2 (35.7–255.3) μg/kg. The maximum possible effect (81.9±7.6%) was achieved within 60 min and was maintained for 240 min after administration of the high dose (3600 μg/kg) of ZZ-204G. Of note, this dose of ZZ-204G produced no effect in the sham-operated condition; vocalization threshold, 235.0±1.7 g and 250±9.69 g before and after (at peak time) drug administration, respectively. The antihyperalgesic effects produced by lower doses of ZZ-204G declined during the time of testing. There was no cumulative effect with repeated dosing (weekly intervals); AUC0–120min = 932.8±672.1, 1845.3±1008.0, 3126.6±840.9 and 6946.9±1632.2 for 3.6, 36, 360 and 3600 μg/kg, respectively.
Figure 6. The antihyperalgesic effect of ZZ-204G.

chronic constriction injury nerve injury model of neuropathy and the paw pressure test were performed as described in Methods. [A] Vocalization threshold (VT, g) versus time (min); [B] Percentage maximum possible effect (% MPE) versus dose (μg/kg). Mean ± standard error of the mean (n = 8 and 4 rats for ZZ-204G and saline, respectively). * Significantly different from saline (P<0.05; Dunnett’s test).
3.4. Activity in a rodent model of thermal nociception (tail-flick assay)
The effect of ZZ-204G did not significantly differ from that of saline control in the tail-flick assay (Fig 7). The antinociceptive effect of ZZ-204G was clearly less pronounced (maximum possible effect was 8.4±4.6%, 6.2±1.75% and 15.6±2.4% for 3.6, 36 and 360 μg/kg, respectively) compared to its actions against mechanical hyperalgesia and formalin-induced flinching behavior.
Figure 7. The antinociceptive effect of ZZ-204G.

The tail-flick test was conducted as described in Methods. Tail-flick latency (s) versus time after injection (min). Mean ± standard error of the mean (n = 6 rats).
3.5 Effect ZZ-204G on motor performance (rotarod)
ZZ-204G produced a dose-related motor incoordination evident by impairment of rotarod performance (F5,35 = 7.07, P<0.001) in intact rats (Fig. 8). The ED50 (95% CI) value was 1884 (942–3767) μg/kg. Of note, the ED50 for this undesirable motor effect was substantially (20–800-fold) higher than the ED50s for the analgesic effects in rodent models of inflammatory and neuropathic pain (Fig. 9).
Figure 8. Effect of ZZ-204G on motor coordination.

The rotarod performance test was conducted as described in Methods. [A] Time to fall (s) versus time after injection (min); [B] Area under the curve (AUC 0–240min) versus dose (μg/kg). Mean ± standard error of the mean (n = 6 rats/dose). * Significantly different from saline (P<0.05; Dunnett’s test).
Fig. 9. Comparison between ZZ-2004-produced analgesia (formalin and chronic constriction injury models) and toxicity (motor incoordination).

Maximum possible effect (% MPE) versus log dose (μg/kg). Mean ± standard error of the mean (number of rats are indicated in the legends of figures 5, 6 and 8).
4. Discussion
The current results show that the tetrakis-quaternary ammonium compound ZZ-204G is a potent antagonist of nicotinic acetylcholine receptors. To our knowledge this is the first report of a small molecule nicotinic ligand that shows selectivity for the α9α10 nicotinic acetylcholine receptor subtype. Extensive prior work on nicotine analogs and related compounds has focused on the development of analgesic agonists (Arneric et al., 2007; Buccafusco, 2004; D’Hoedt and Bertrand, 2009). Given the wide variety of nicotinic acetylcholine receptor subtypes, efforts have been aimed at elucidating the specific subtypes of nicotinic acetylcholine receptors associated with analgesia. The α4β2* subtype and to a lesser extent, the α7 subtype have been implicated in the analgesic effects of nicotinic agonists (Arneric et al., 2007). In contrast, the α3β4* (peripheral “ganglionic” subtype) has been associated with undesirable side effects. Efforts to produce α4β2- or α7- targeted compounds have led to more selective agents with varying degrees of efficacy (Gao et al., 2010) (Rowley et al., 2010).
α9α10 nicotinic acetylcholine receptors were first associated with analgesia upon the discovery that the venom derived peptides α–conotoxin RgIA and α-conotoxin Vc1.1 are potent, selective antagonists of α9α10 nicotinic acetylcholine receptors (Vincler and McIntosh, 2007; Vincler et al., 2006). Parenteral administration of either of these compounds produces antinociception in rodent models of pain. The present study demonstrates that non-peptide small molecules also act as potent and selective α9α10 antagonists that possess analgesic activity. The tetrakis analogs were developed as a sub-library of novel subtype-selective antagonists at nicotinic acetylcholine receptors that mediate nicotine-evoked dopamine release (Zhang et al., 2008) and are structurally related to the tris-quaternary ammonium analogs, which were demonstrated to be potent inhibitors at these nicotinic acetylcholine receptors (Zheng et al., 2007). The tetrakis compound ZZ-204G exhibited poor affinity for α4β2* nicotinic acetylcholine receptors in binding assays (Zhang et al., 2008) and was also a weak antagonist at nicotinic acetylcholine receptors mediating nicotine-evoked dopamine release. In the present study, ZZ-204G was found to block α9α10 nicotinic acetylcholine receptors with subnanomolar potency, and also potently blocks α7 nicotinic acetylcholine receptors (0.51 and 8.8 nM respectively). Agonists of α7 nicotinic acetylcholine receptors are analgesic and this effect is reversed by an antagonist of α7 nicotinic acetylcholine receptors (Medhurst et al., 2008). Thus, the α7 nicotinic acetylcholine receptor antagonist activity of ZZ-204G is unlikely related to the analgesic mechanism. Indeed it is possible that block of α7 may mask some of the analgesic efficacy of ZZ-204G.
The effect of ZZ-204G was evaluated in several rodent models of pain thought to mimic some of the aspects of clinical pain in humans. The current results demonstrate that ZZ-204G lacked activity in the tail flick test suggesting a limited role for antagonism of the α9α10 nicotinic acetylcholine receptor in acute nociceptive pain. In contrast, the efficacy of ZZ-204G was evident in both the chronic constriction nerve injury model of neuropathic pain and formalin model of persistent inflammatory pain. Chronic constriction of the sciatic nerve produces an inflammatory response at the site of injury. This inflammatory response is thought to contribute to the development of neuropathic pain following peripheral nerve injury (Moalem and Tracey, 2006). In contrast, the early phase of the formalin test is thought to result from direct activation of peripheral nociceptors, while the later phase is dependent on further activation of peripheral nociceptors by inflammatory mediators with resultant central sensitization in the dorsal horn of the spinal cord (Tjolsen et al., 1992). Importantly, the antihyperalgesic and antinociceptive effects of ZZ-204G were evident in both the chronic constriction nerve injury and formalin rodent pain models, revealing the ability to alleviate neuropathic pain and persistent inflammatory pain, respectively. Inhibition of formalin-induced flinching behavior was evident in both early (acute) and late (tonic) phases of the formalin test; however, the effect in the later phase was dose-related and occurred at lower doses. The more prominent effect of ZZ-204G in the second phase suggests that this small molecule is more effective in the inflammatory phase, where there is prolonged activation of nociceptors resulting in the barrage of afferent information to the dorsal horn producing central sensitization. Thus, ZZ-204G may be effective in persistent pain states involving inflammatory chemical mediator induced central sensitization occurring as a result of an injury to nerve or tissue. ZZ-204G showed greater potency (~50-fold) against formalin-evoked inflammatory pain than neuropathic pain due to chronic constriction injury. However, caution is indicated in making direct comparisons between different models of pain as responses are related to both intensity and modality of stimuli.
The separation in the dose of ZZ-204G producing motor side-effects from that producing antihyperalgesic or antinociceptive effect (~20- to 800-fold) is a desirable quality for a potential clinically useful analgesic agent. The α4β2 nicotinic agonist S(−) nornicotine was recently characterized using similar paradigms. In contrast to ZZ-204G, the ED50 values (mg/kg, i.p.) were only 3–9-fold higher for motor impairment vs. analgesia, 13.6 (9.2–19.9), rotarod; 5.2 (3.1–8.5), chronic constriction injury; and 1.5 (0.6–3.6), formalin model (Holtman et al., 2010b). Full evaluation of the potential toxicity of ZZ-204G is warranted. We note that the highest antihyperalgesic dose used in chronic constriction injury rats also produced significant motor effect in naïve rats (3,600 ug/kg). Encouraging data are noted in that another α9α10 antagonist, α-conotoxin Vc1.1 did not produce any adverse effects in preclinical and clinical Phase I studies, reviewed in (Vincler and McIntosh, 2007).
Interestingly, earlier studies with the α9α10 nicotinic acetylcholine receptor antagonists α-conotoxin Vc1.1 and RgIA (small peptides) (Ellison et al., 2008; Sandall et al., 2003) also showed antinociceptive effects in the chronic constriction nerve injury model (Livett et al., 2006; Satkunanathan et al., 2005; Vincler and McIntosh, 2007; Vincler et al., 2006). Although the specific mechanism has not been fully elucidated, antagonism of α9α10 nicotinic acetylcholine receptor has been shown to reduce the number of immune cells, both macrophages and lymphocytes, including those positive for choline acetyltransferase, at the site of constriction induced nerve injury (Vincler et al., 2006). This reduction would be expected to modulate the inflammatory response at the nerve injury site and therefore inhibit or reduce the development of neuropathic pain. ZZ-204G may also act by a similar mechanism, that is, through antagonism of α9α10 nicotinic acetylcholine receptors at the site of nerve injury to produce its analgesic effects.
An alternative antinociceptive mechanism has been proposed for α-conotoxins, that is, stimulation of γ-aminobutyric acid(B) receptors in dorsal root ganglia resulting in inhibition of N-type calcium channels (Callaghan et al., 2008; Klimis et al., 2011; Nevin et al., 2007). Yet, when the α-conotoxins were tested on cloned γ-aminobutyric acid(B) receptors the conotoxins failed to displace the γ-aminobutyric acid(B) ligand [3H]CGP54626. In addition, examination of the functional effects of the conotoxins on γ-aminobutyric acid(B) receptors failed to show agonist activity (McIntosh et al., 2009). Thus, the analgesic mechanism of action of these peptides remains debated. The present study shows that an entirely different class of α9α10 antagonist, a tetrakis- ammonium compound, has potent analgesic activity. The low affinity of ZZ-204G at the γ-aminobutyric acid(B) receptor demonstrated in the current study is not supportive of a likely action of ZZ-204G at γ-aminobutyric acid(B) receptors. In addition, there is a preliminary report of two other small molecule α9α10 nicotinic acetylcholine receptor antagonists that are effective in rodent models of pain (Holtman et al., 2010a). These also are inactive at γ-aminobutyric acid(B) receptors (NIMH psychotherapeutic drug discovery program, unpublished data). ZZ-204G does have submicromolar affinity for κ- and μ-opioid receptors. It is not known whether ZZ-204G acts as an agonist or antagonist of opioid receptors. However, opioid agonists are active in the radiant heat tail flick assay, whereas ZZ-204G was not active at tested doses. Thus, the antinociceptive and antihyperalgesic effects of ZZ-204G in the formalin and chronic constriction injury tests is unlikely the result of activity at opioid receptors. In addition, the potency of ZZ-204G at κ- and μ-opioid receptors is 1000-fold less than that at α9α10 nicotinic acetylcholine receptors.
In summary, ZZ-204G is a potent, first in class, small molecule antagonist of α9α10 nicotinic acetylcholine receptors. Based on its activity on α9α10 nicotinic acetylcholine receptors, we tested ZZ-204G in standard animal models of pain. The potent pain reducing activity and relatively low motor side-effect profile provide further incentive to investigate α9α10 antagonists as novel analgesics.
Figure 4. Concentration response of ZZ-204G on non-nicotinic acetylcholine receptors.

Compound was tested in quadruplicate in competition binding assays as described in methods. Response in the presence of reference compound and ZZ-204G is shown. A, 5HT3 receptor. B, κ-opioid receptor. C, μ-opioid receptor.
Table 2.
Activity of ZZ-204G in receptor binding assays
| Receptor | Ki (nM)a | Hill Slopea |
|---|---|---|
| 5HT3 | 2230 (1595–3117) | 1.05 (0.73–1.37) |
| GABAA | >10,000 | -- |
| GABAB | >10,000 | -- |
| κ-opioid | 783 (560–1095) | 0.95 (0.67–1.2) |
| μ-opioid | 659 (560–776) | 1.1 (0.94–1.3) |
Numbers in parentheses are 95% confidence intervals
Acknowledgments
This work was supported by U19 DA017548, NIH MH53631, GM48677 and a seed grant from the University of Utah Research Foundation to JMM. Radioligand binding studies were generously performed by the NIMH Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C. The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arneric SP, Holladay M, Williams M. Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem Pharmacol. 2007;74:1092–1101. doi: 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Azam L, McIntosh JM. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol Sin. 2009;30:771–783. doi: 10.1038/aps.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ. Neuronal nicotinic receptor subtypes: defining therapeutic targets. Mol Interv. 2004;4:285–295. doi: 10.1124/mi.4.5.8. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ. Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J Neurosci. 2008;28:10943–51109. doi: 10.1523/JNEUROSCI.3594-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- D’Amour FE, Smith DL. A Method For Determining Loss of Pain Sensation. J Pharmacol Exp Therap. 1941;72:74–79. [Google Scholar]
- D’Hoedt D, Bertrand D. Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin Ther Targets. 2009;13:395–411. doi: 10.1517/14728220902841045. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J. alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A. 2001;98:3501–3506. doi: 10.1073/pnas.051622798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison M, Feng ZP, Park AJ, Zhang X, Olivera BM, McIntosh JM, Norton RS. Alpha-RgIA, a novel conotoxin that blocks the alpha9alpha10 nAChR: structure and identification of key receptor-binding residues. J Mol Biol. 2008;377:1216–1227. doi: 10.1016/j.jmb.2008.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Hierl M, Clarkin K, Juan T, Nguyen H, Valk M, Deng H, Guo W, Lehto SG, Matson D, McDermott JS, Knop J, Gaida K, Cao L, Waldon D, Albrecht BK, Boezio AA, Copeland KW, Harmange JC, Springer SK, Malmberg AB, McDonough SI. Pharmacological effects of nonselective and subtype-selective nicotinic acetylcholine receptor agonists in animal models of persistent pain. Pain. 2010;149:33–49. doi: 10.1016/j.pain.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Gomez-Casati ME, Fuchs PA, Elgoyhen AB, Katz E. Biophysical and pharmacological characterization of nicotinic cholinergic receptors in rat cochlear inner hair cells. J Physiol. 2005;566:103–118. doi: 10.1113/jphysiol.2005.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman JR, Dwoskin LP, Wala E, Crooks PA, McIntosh JM. Novel small molecule alpha9alpha10 nicotinic receptor antagonists for pain management. American Pain Society. J of Pain. 2010a:S33. [Google Scholar]
- Holtman JR, Jr, Crooks PA, Johnson-Hardy JK, Wala EP. The analgesic and toxic effects of nornicotine enantiomers alone and in interaction with morphine in rodent models of acute and persistent pain. Pharmacol Biochem Behav. 2010b;94:352–362. doi: 10.1016/j.pbb.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW, Mozar CA, Christie MJ. A novel mechanism of inhibition of high-voltage activated calcium channels by alpha-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain. 2011;152:259–266. doi: 10.1016/j.pain.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Livett BG, Sandall DW, Keays D, Down J, Gayler KR, Satkunanathan N, Khalil Z. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon. 2006;48:810–829. doi: 10.1016/j.toxicon.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Lopez-Hernandez GY, Thinschmidt JS, Zheng G, Zhang Z, Crooks PA, Dwoskin LP, Papke RL. Selective inhibition of acetylcholine-evoked responses of alpha7 neuronal nicotinic acetylcholine receptors by novel tris- and tetrakis-azaaromatic quaternary ammonium antagonists. Mol Pharmacol. 2009;76:652–666. doi: 10.1124/mol.109.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol. 2009;78:693–702. doi: 10.1016/j.bcp.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- Medhurst SJ, Hatcher JP, Hille CJ, Bingham S, Clayton NM, Billinton A, Chessell IP. Activation of the alpha7-nicotinic acetylcholine receptor reverses complete freund adjuvant-induced mechanical hyperalgesia in the rat via a central site of action. J Pain. 2008;9:580–587. doi: 10.1016/j.jpain.2008.01.336. [DOI] [PubMed] [Google Scholar]
- Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51:240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Nevin ST, Clark RJ, Klimis H, Christie MJ, Craik DJ, Adams DJ. Are alpha9alpha10 nicotinic acetylcholine receptors a pain target for alpha-conotoxins? Mol Pharmacol. 2007;72:1406–1410. doi: 10.1124/mol.107.040568. [DOI] [PubMed] [Google Scholar]
- Nicke A, Wonnacott S, Lewis RJ. Alpha-conotoxins as tools for the elucidation of structure and function of neuronal nicotinic acetylcholine receptor subtypes. Eur J Biochem. 2004;271:2305–2319. doi: 10.1111/j.1432-1033.2004.04145.x. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–419. [PubMed] [Google Scholar]
- Rowley TJ, McKinstry A, Greenidge E, Smith W, Flood P. Antinociceptive and anti-inflammatory effects of choline in a mouse model of postoperative pain. Br J Anaesth. 2010;105:201–207. doi: 10.1093/bja/aeq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandall DW, Satkunanathan N, Keays DA, Polidano MA, Liping X, Pham V, Down JG, Khalil Z, Livett BG, Gayler KR. A novel alpha-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry. 2003;42:6904–6911. doi: 10.1021/bi034043e. [DOI] [PubMed] [Google Scholar]
- Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005;1059:149–158. doi: 10.1016/j.brainres.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Vincler M. Neuronal nicotinic receptors as targets for novel analgesics. Expert Opin Investig Drugs. 2005;14:1191–1198. doi: 10.1517/13543784.14.10.1191. [DOI] [PubMed] [Google Scholar]
- Vincler M, McIntosh JM. Targeting the alpha9alpha10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin Ther Targets. 2007;11:891–897. doi: 10.1517/14728222.11.7.891. [DOI] [PubMed] [Google Scholar]
- Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc Natl Acad Sci U S A. 2006;103:17880–17884. doi: 10.1073/pnas.0608715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watzman N, Barry H, 3rd, Buckley JP, Kinnard WJ., Jr Semiautomatic System for Timing Rotarod Performance. J Pharm Sci. 1964;53:1429–1430. doi: 10.1002/jps.2600531142. [DOI] [PubMed] [Google Scholar]
- Weisstaub N, Vetter DE, Elgoyhen AB, Katz E. The α9α10 nicotinic acetylcholine receptor is permeable to and is modulated by divalent cations. Hear Res. 2002;167:122–135. doi: 10.1016/s0378-5955(02)00380-5. [DOI] [PubMed] [Google Scholar]
- Wheeler-Aceto H, Cowan A. Standardization of the rat paw formalin test for the evaluation of analgesics. Psychopharmacology (Berl) 1991;104:35–44. doi: 10.1007/BF02244551. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zheng G, Pivavarchyk M, Deaciuc AG, Dwoskin LP, Crooks PA. Tetrakis-azaaromatic quaternary ammonium salts: novel subtype-selective antagonists at neuronal nicotinic receptors that mediate nicotine-evoked dopamine release. Bioorg Med Chem Lett. 2008;18:5753–5757. doi: 10.1016/j.bmcl.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Sumithran SP, Deaciuc AG, Dwoskin LP, Crooks PA. Tris-azaaromatic quaternary ammonium salts: Novel templates as antagonists at nicotinic receptors mediating nicotine-evoked dopamine release. Bioorg Med Chem Lett. 2007;17:6701–6706. doi: 10.1016/j.bmcl.2007.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]