

Abstract
Chronic granulomatous disease (CGD) is a primary immunodeficiency defined by mutations in the NADPH oxidase complex leading to reduced superoxide production, increased susceptibility to infection, chronic inflammation, and recurring abscess and granuloma formation. Here, we found that CGD mice were hyperresponsive to abscess-inducing T-cell-dependent carbohydrate antigens (glycoantigens) due to a ten-fold increase in NO production within APCs, which is known to be necessary for glycoantigen presentation on MHC class II. CGD mice exhibited increased Th1 pro-inflammatory T-cell responses in vitro and in vivo, characterized by more severe abscess pathology. This phenotype was also seen in WT animals following adoptive transfer of neutrophil-depleted APCs from CGD animals, demonstrating that this phenotype was independent of neutrophil and T-cell defects. Finally, pharmacological attenuation of NO production to WT levels in vivo reduced abscess incidence and severity in CGD without overt increases in inflammation or the ability to clear infection, suggesting a potential new treatment option for early stage CGD-associated infections.
Keywords: Antigen presentation/processing, Immunodeficiencies, Inflammation
Introduction
Oxidative killing of microbes by phagocytes represents a leading edge of the innate immune response. The released microbial contents following killing also serve as a pool of potential antigens for the adaptive immune response within the endosomal class II major histocompatibility complex (MHCII) pathway. Oxidation is achieved through the production of both reactive oxygen species ROS and reactive nitrogen species (RNS) that attack surface molecules and lyse pathogens. The primary source of reactive nitrogen species is NO synthesized by the inducible nitric oxide synthase (iNOS) enzyme [1], which is transcriptionally activated via the NF-κB signaling cascade upon recognition of microbial “molecular patterns” at the cell surface [2]. The NADPH oxidase complex is responsible for the production of superoxide, which fuels the synthesis of hydrogen peroxide and hypochlorous acid through the serial enzymatic actions of superoxide dismutase and myeloperoxidase respectively [3, 4].
Chronic granulomatous disease (CGD) is characterized by any of a number of deleterious mutations within the NADPH oxidase complex [5-7]. While the reduction of superoxide production varies in severity depending on the mutation, patients with CGD show heightened susceptibility to bacterial and fungal infections, have increased incidence of abscess and granuloma formation, and suffer from chronic inflammation [7-9], all of which highlight the central role for oxidation in controlling infectious disease. Although CGD is classified as a primary immunodeficiency, the increases in abscess and granuloma formation as well as the chronic unresolved inflammation represent hyperresponsiveness to infection and microbial products [8-10]. The granulomas are often sterile and form in response to unregulated and widespread inflammation [7-11]. In contrast, abscess formation is a T-cell-dependent adaptive pathway [12] that normally serves to quarantine the offending pathogen. Despite the role in reducing dissemination of the pathogen throughout the body, abscesses reduce the efficacy of antibiotics due to isolation of the bacteria from the blood stream and they require surgical drainage, collectively increasing risk of secondary infections [8].
CGD patients are susceptible to abscess formation induced by microbes carrying antigenic capsular carbohydrates, including the fungus Aspergillus sp., the Gram-positive Staphylococcus aureus, and other catalase-positive organisms [7, 13]. Bacteroides fragilis, also catalase-positive, is the most common anaerobic bacteria isolate from clinical abscess samples [14], and both S. aureus and B. fragilis carry zwitterionic glycoantigens (GlyAgs) that have been shown to induce abscess formation in animal models through the activation of pro-inflammatory CD4+ T lymphocytes [15, 16]. The canonical member of the GlyAg family is polysaccharide A (PSA) from the capsule of B. fragilis. PSA is comprised of a tetrasaccharide repeating unit with both positively and negatively charged groups [17] that facilitate its ability to be presented by MHCII molecules [18]. GlyAgs are endocytosed by professional APCs and trigger the production of NO [19], which is responsible for the oxidative cleavage of the antigen to low molecular weight fragments for MHCII-mediated presentation [20, 21]. This NO-dependent oxidative processing and presentation mechanism is essential for GlyAg-specific T-cell recognition and activation. Animals lacking the iNOS gene fail to form abscesses in response to GlyAg challenge [20].
With NO-mediated oxidation at the root of GlyAg-induced abscess formation, we sought to understand the nature of the hyperresponsiveness in CGD. Using the gp91phox-deficient animal model of CGD, we discovered that the loss of a functional NADPH oxidase results in a ten-fold increase in sensitivity against GlyAg challenge, with CGD abscesses being consistently larger compared with WT C57BL/6 (WT) controls. Ex vivo experiments further reveal an earlier and more robust T-cell activation response against GlyAg that correlated with increased NO and iNOS protein production in CGD animals and increased GlyAg processing in CGD APCs. Remarkably, CGD hyperresponsiveness was transferrable to WT animals through adoptive transfer of neutrophil-depleted CGD APCs, demonstrating that increased abscess formation was a result of aberrant APC function and the resulting downstream T-cell activation, rather than changes in neutrophil or T-cell activity resulting from changes in ROS production. Perhaps most significantly, we discovered that attenuation of iNOS activity with 1400W (N-(3-(aminomethyl)benzyl)acetamidine, 2HCl) effectively and safely reduced the incidence and severity of abscesses in CGD. These findings reveal that the abscess hyperresponsiveness in CGD is mediated at least in part through greater sensitivity to GlyAg via an increase in NO-dependent T-cell activation and that treatment with 1400W could represent a novel approach to improving infection outcomes for CGD patients.
Results
Dysregulated oxidation leads to excessive GlyAg-mediated abscesses
GlyAg-mediated abscess formation in rodent models of sepsis is dependent upon MHCII presentation [20, 22, 23] and CD4+ T-cell activation [16, 23-26], while being exquisitely sensitive to NO production in responding APCs [19-21, 23]. Given the dependence upon oxidation, we measured the impact of the CGD mutation on GlyAg-specific responses. CGD and WT mice were challenged i.p. with either 200 μg GlyAg containing undiluted sterile cecal contents (SCC) (dilution = 1), SCC alone, or dilutions of each inoculum. On day 7, the number of mice with at least one abscess was scored (Fig. 1A). CGD animals were ten-fold more sensitive to GlyAg challenge compared with WT control animals (C1/2 = four-fold dilution for WT; 40 for CGD). The SCC-only controls showed no abscesses in WT and limited abscesses in several CGD animals, but only at high SCC concentrations. CGD abscesses were consistently larger than in WT animals with equivalent challenge (Fig. 1B) and flow cytometry of collagenase D-released abscess cells indicated that a majority of cells were Gr-1+neutrophils, F4/80+ macrophages, and CD11c+ DCs (Fig. 1C). The total number of cells within a WT abscess was 5.3 × 106 while the CGD abscess yielded 3.06 × 107 cells, a 5.7-fold increase that correlates well with the increased abscess size. Finally, H&E staining of abscess sections showed the difference in overall size and revealed distinct areas of increased neutrophilic infiltrate in the CGD abscess (Fig. 1D). These data establish that the CGD mutation results in extreme sensitivity to abscess formation in response to GlyAg/SCC exposure characterized by more severe pathology and either increased neutrophil infiltrate or defective clearance (e.g. efferocytosis) following the initial insult.
Figure 1.

CGD mice are hyper-responsive to GlyAg challenge. (A) WT and CGD mice were challenged i.p. with 200 μg GlyAg with undiluted sterile cecal contents (SCC, dilution = 1), SCC alone (red), or serial dilutions of the inoculum (e.g. dilution of 4 = 50 μg GlyAg and 1:4 diluted SCC), then scored for abscesses (n = 8 per dilution). CGD animals (right) were much more sensitive to GlyAg+SCC challenge compared with WT (left). (B) Abscesses were isolated and weighed 7 days post GlyAg challenge at a dilution of 4 (individual weight data points shown). (C) After collagenase digestion, the cellular contents of abscesses were analyzed by flow cytometry (n = 4; blue = Gr-1+, green = CD11c+, red = F4/80+) to quantify total cell numbers. (D) H&E-stained sections of CGD abscesses were analyzed to determine any differences in abscess infiltration and composition (representative sections from four independent abscesses shown). All error bars represent mean±SEM and p values were determined using an unpaired Student’s t-test with a two-tailed p value.
CGD mice exhibit increased NO production in response to GlyAg
To discern the hyperresponsiveness mechanism, mice were challenged with 100 μg GlyAg and 1:4 diluted SCC for analysis of cellular infiltration (Fig. 2). At the times indicated, a peritoneal lavage was performed. Recovered cells were analyzed by flow cytometry while lavage supernatants were tested for nitrate and nitrite levels as markers of NO synthesis. Unchallenged CGD animals showed elevated baseline NO levels compared with WT (p<0.03); however, this difference increased dramatically over the first 24-h period upon challenge (Fig. 2A), demonstrating the hyperresponsiveness to the GlyAg+SCC stimulation despite the modestly increased baseline. Remarkably, total cellular influx into the peritoneum was not significantly different at most time points (Fig. 2B) and no consistent proportional differences in neutrophil, macrophage, or CD4+ T-cell populations were seen between WT and CGD (Fig. 2C and D), although modest differences in neutrophils were seen at 24 h (Fig. 2D). These data suggest that the proportional increase in neutrophils visible by H&E within the abscess (Fig. 1C and D) was mostly likely due to defects in neutrophil clearance rather than increased peritoneal infiltration, which is consistent with previous reports [27-29]. More importantly, these findings suggest that the > 10-fold increase in NO detected in the peritoneal lavage (Fig. 2A) was not due to increased cell numbers, but was more likely the result of changes in per-cell production of NO.
Figure 2.

CGD animals have increased NO but similar cellular influx in response to GlyAg challenge. (A) WT and CGD mice were challenged with GlyAg and SCC; then, peritoneal lavage with PBS was performed at the indicated time points (n = 3 independent animals per time point, per genotype on all Fig. 2 data). The resulting supernatants were tested for nitrates and nitrites using the Griess assay as a measure of NO production. (B–D) WT and CGD mice were challenged with GlyAg and SCC; then, peritoneal lavage with PBS was performed at the indicated time points and the cellular influx was characterized using flow cytometry and antibodies specific for neutrophils (α-GR-1), macrophages (α-F4/80), and CD4+ T cells (α-CD4). (B) The total responding cellular influx, (C) percent of each cell type, and (D) relative number (% multiplied by total cell count) of each cell type at the various time points are shown to compare WT and CGD animal responses. All data points represent n = 3 mice, all error bars represent mean±SEM, and comparisons were performed using an unpaired Student’s t-test with a two-tailed p value. All data points are not significantly different (p>0.05), unless indicated.
CGD cells up-regulate iNOS in response to GlyAg
iNOS expression was examined in isolated WT and CGD cells to establish the source of increased NO levels. Lavage cells were collected from GlyAg challenged mice for mRNA isolation and detection of the iNOS transcript using RT-PCR. In vivo challenge induced CGD cells to transcribe iNOS mRNA to a remarkably greater extent compared with WT cells at 24 h (Fig. 3A). These bulk lavage cells were also separated via magnetic beads into neutrophil and macrophage populations and used to isolate mRNA to determine the cell-type specific response with quantitative PCR (Fig. 3B). The data reveal that the individual CGD cells up-regulate the transcription of the iNOS gene (NOS2) beyond WT cells in both neutrophil and macrophages upon challenge.
Figure 3.
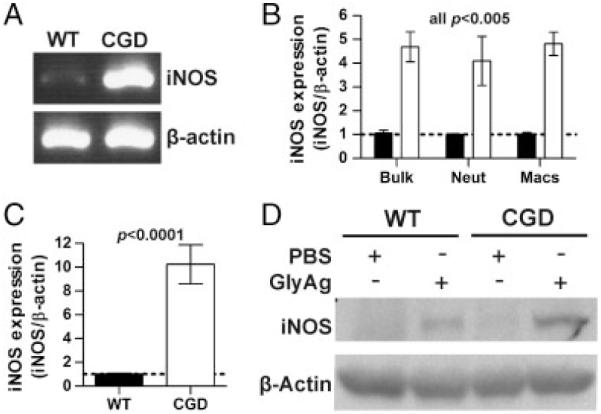
iNOS is induced to a greater extent in CGD cells compared with WT cells. (A) WT and CGD animals were challenged with GlyAg and SCC for 24 h. After peritoneal lavage, mRNA was isolated from the harvested cells and subjected to PCR using primers specific for iNOS gene transcript, using β-actin as a housekeeping control. The resulting PCR products were run on an agarose gel and the data shown are representative of n = 4 experiments. (B) Using quantitative PCR, we examined the upregulation of iNOS transcript in RNA isolated from bulk lavage cells (Bulk) or purified neutrophils (Neut) and macrophages (Macs) from CGD and WT animals (normalized to WT; n = 9). The data were first normalized to β-actin; then, WT levels were set to unity. (C) BMDCs were stimulated with PBS or GlyAg in vitro for 24 h and then evaluated for iNOS expression as before (normalized to WT; n = 9). (D) Western blots of BMDC lysates following stimulation were performed to compare the iNOS protein levels in WT and CGD BMDCs stimulated with GlyAg. Data shown are representative of n = 3 independent experiments. All error bars represent mean±SEM.
The response of bone marrow-derived dendritic cells (BMDCs) from unchallenged WT and CGD mice to GlyAg alone was also tested. At 24 h, mRNA and cell extracts were isolated and analyzed by qPCR and Western blot respectively. We found that iNOS transcription was increased by nearly ten-fold over WT in response to GlyAg (Fig. 3C) and this difference was readily apparent at the protein level (Fig. 3D). These data demonstrate that GlyAg-stimulated CGD cells up-regulate the iNOS gene to a significantly greater extent than WT cells in neutrophils, macrophages, and BMDCs, and this difference accounts for the increased NO produced in the peritoneal cavity upon challenge (Fig. 2A).
Excessive NO in CGD cells leads to increased GlyAg processing and T-cell activation
Given that GlyAg-induced abscess formation is dependent on NO-dependent processing, presentation on MHCII, and subsequent CD4+ T-cell activation [20], we examined the CGD effect on the amount of GlyAg processing. CGD and WT APCs were incubated for 48 h with radiolabeled GlyAg, then intracellular GlyAg was analyzed for changes in molecular mass as a measure of processing. Greater amounts of the MHCII-presentable low molecular weight form of GlyAg were found in CGD cells compared with WT (Fig. 4A, arrow), demonstrating that increases in NO correlates with greater processed GlyAg available for MHCII presentation.
Figure 4.

CGD APCs process more GlyAg and activate T cells earlier to produce more pro-inflammatory cytokine than WT T cells. (A) The ability of WT or CGD APCs to process GlyAg was assessed in vitro by incubating APCs with radiolabeled GlyAg for 48 h and then comparing the molecular mass of intracellular GlyAg to an unprocessed GlyAg control. Low molecular weight products are found in the late elution volumes (19–20 mL; arrow). CPM, counts per minute. Data shown are representative of n = 3 independent experiments. (B) In vitro T-cell activation assays with co-cultures of WT or CGD APCs and CD4+ T cells stimulated with GlyAg. At the times indicated, supernatants were removed and analyzed for IFN-γ production by ELISA as a marker of T-cell activation (n = 3 independent experiments per day, per genotype). (C) As a control, a similar T-cell activation assay was set up using iNOS−/− (NO deficient) APCs and CD4+ T cells and IFN-γ production was measured on day 3 (n = 3 independent experiments per day, per genotype). (D) The number of in vivo GlyAg-specific T cells were analyzed at 24 h post challenge in WT and CGD animals using the early activation marker CD69 and flow cytometry. Data shown are representative of n = 3 independent experiments. (E) Confocal microscopy of CD4+ T cells (green) in abscesses was performed to determine the relative number of T cells within the walls of each abscess (representative image of n = 8 shown). (F) Splenocytes from either WT or CGD animals depleted of both T cells and neutrophils were adoptively transferred into WT recipient mice (eight animals per group) to determine whether the CGD phenotype could be transferred to the WT animals using APCs alone. These WT mice were then challenged with a seven-fold dilution of GlyAg and SCC inoculum, which normally generates an abscess in only 10% of the WT animals (see Fig. 1A). The number of positive animals is given above each bar. Error bars represent SEM; scale bar represents 20 μm for confocal images; comparisons were performed using an unpaired Student’s t-test with a two-tailed p value.
Next, to determine if the increased NO production and antigen processing seen in CGD mice would lead to aberrant T-cell activation, syngeneic APCs and CD4+ T cells were cultured and stimulated with GlyAg and analyzed for IFN-γ by ELISA. We found that the CGD T cells responded earlier and more robustly than WT T cells, with strong IFN-γ production by day 3 in CGD assays (Fig. 4B). The relationship between NO production and T-cell response was further demonstrated by comparing the T-cell responses from WT, CGD, and iNOS−/− animals at day 3. IFN-γ production was modest for WT, heightened for CGD, and reduced for iNOS−/− cells (Fig. 4C), showing a direct correlation between NO concentration and T-cell response amplitude.
To differentiate between greater individual cell responses and a greater number of cells responding, we challenged WT and CGD animals with GlyAg and compared the number of CD4+ T cells expressing CD69, an early activation marker (Fig. 4D). At 24 h, the number of CD4+CD69+ cells without GlyAg challenge was indistinguishable between WT and CGD animals (12.3 and 11.4% respectively), while in vivo stimulation with GlyAg yielded ~4% increases in CD69+ T cells in both backgrounds (Fig. 4D). Since responding CD4+ T cells have been previously localized to the abscess wall following GlyAg challenge [24], we also performed immunohistochemistry on abscess cryosections. Confocal microscopy failed to reveal significant differences in the CD4+ T-cell composition of abscess walls (Fig. 4E). These data indicate that the overall number of responding T cells is constant between WT and CGD, but that the inflammatory signal amplitude (i.e. IFN-γ) is increased within individual T cells in proportion to the CGD-associated increase in NO production within APCs.
To directly test whether CGD APCs drive increased T-cell-dependent abscess formation in CGD, splenocytes were harvested from WT and CGD animals and depleted of both neutrophils and T cells. The remaining cells (B cells, macrophages, and DCs; data not shown) were adoptively transferred into WT animals and then each animal was challenged with a seven-fold dilution of the GlyAg and SCC inoculum, which generated an abscess in 0–10% of WT animals (see Fig. 1A). We found that when WT APCs were transferred into the WT animals, 1 out of 8 mice developed an abscess, as before. In contrast, 75% of the WT animals receiving CGD APCs developed an abscess (Fig. 4F). These findings demonstrate that CGD APCs are sufficient to transfer the CGD phenotype characterized by increased GlyAg-induced abscess formation.
Attenuation of NO reduces GlyAg-induced abscess formation
Based on our findings, attenuation of NO production in the first 24 h post challenge should reduce T-cell activation and abscess incidence in CGD. We therefore performed in vitro T-cell activation experiments with CGD cells with and without the specific iNOS inhibitor 1400W. We found that 1400W reduced the amount of IFN-γ produced by up to 50% as compared with mock-treated cultures (Fig. 5A).
Figure 5.

Attenuation of NO reduces GlyAg-induced abscess formation. (A) T-cell activation assays were set up as before using CGD APCs and T cells with or without an irreversible iNOS inhibitor, 1400W, and compared with WT cells to determine the ability to reduce the CGD-mediated changes in T-cell response via reduction in NO production (n = 3 independent experiments per day, per treatment). (B) To test the effectiveness of 1400W in vivo, WT and CGD mice were challenged with GlyAg and SCC; then, peritoneal lavage was performed at 24 h (n = 4 independent experiments per genotype). An additional group of CGD mice were treated with 0.5 mg 1400W at 0 and 6 h post challenge (n = 4). The lavage fluid was then assayed for NO production as before. (C and D) To determine the relative safety of NO inhibition in CGD mice, animals were challenged with live B. fragilis with (left) or without (right) 1400W (n = 4 per group; shown individually) and monitored closely for weight and activity levels over 10 days. No overt differences in activity level were seen and no bacteria were detectable in any non-intestinal tissue (data not shown). (E) To verify that no differences in inflammatory mediators accompany 1400W treatment, WT and CGD macrophages were stimulated in vitro with GlyAg±1400W for 24 h; then, culture supernatants were tested for IL-1β production. p>0.05 for all WT versus CGD values. (F) CGD animals were challenged with GlyAg±0.5 mg 1400W at 0, 6, and 24 h post challenge (n = 14 per group). Seven days after challenge, mice were scored for abscess formation and size (in mm) to determine the effectiveness of iNOS inhibition in reducing abscess incidence and severity. (G) Representative photos of CGD abscesses in the presence and absence of 1400W, illustrating the effect of 1400W in abscess size. For all panels, error bars represent mean±SEM and comparisons were performed using an unpaired Student’s t-test with a two-tailed p value.
Next, WT and CGD animals were challenged with a four-fold dilution of the standard inoculum and compared with another group of CGD animals also treated 0 and 6 h post challenge with 0.5 mg 1400W. Twenty-four hours later, peritoneal lavage fluid was collected and analyzed for NO production. We found a large increase in NO production in CGD animals over WT (Fig. 5B), reflecting increased iNOS expression (Fig. 3). In addition, 1400W did not eliminate NO production, but reduced NO levels to that seen in WT animals (Fig. 5B).
Reducing NO production to WT levels in already immunocompromised CGD mice could result in the inability to clear bacterial challenge, thus we examined bacterial clearance in CGD animals treated with 1400W. Mice were challenged with 106 live B. fragilis, a leading cause of peritonitis associated with intestinal leakage [30, 31], and were treated with 0.5 mg 1400W or PBS vehicle at 0, 6, and 24 h post inoculation. All mice maintained body weight (Fig. 5C and D) and no overt change in activity levels was seen over a 10-day period. On day 8, one mouse in each group was sacrificed and blood agar plates were streaked with tissue samples of blood, liver, spleen, and peritoneal lavage and incubated under anaerobic conditions for 48 h. No bacterial growth was detected from any tissue sample from the PBS or 1400W treated mice (not shown), indicating that 1400W had no deleterious effect on the ability to clear B. fragilis.
To address the possibility that increased inflammation could result from changes in oxidant concentration following 1400W treatment, BM-derived macrophages (BMMs) from WT and CGD animals were stimulated in vitro with LPS or GlyAg in the presence and absence of 1400W for 24 h. The culture supernatants were analyzed for the inflammatory mediator IL-1β (Fig. 5E), and we found that while both GlyAg and LPS stimulated IL-1β production, the response in WT and CGD cells were indistinguishable, even with 1400W present.
Finally, we tested the efficacy of 1400W in reducing abscess incidence in CGD mice. Using the four-fold dilution challenge (50 μg GlyAg and 1:4 SCC), we found that 1400W treatment significantly reduced the number of CGD animals that developed abscesses from 93 to 57% (Fig. 5F). Moreover, the abscesses found in 1400W-treated CGD animals were also significantly reduced in clinical score as judged by size (1.9 mm average diameter) compared with those found in CGD animals without 1400W (3.6 mm average diameter; Fig. 5F and G). These data show that modulation of iNOS activity via 1400W decreases NO production in vivo compared with that seen in WT animals, resulting in the reduced incidence and severity of GlyAg-mediated abscess formation in CGD.
Discussion
We show that the gp91phox mutation in CGD results in the upregulation of NO production, leading to increased T-cell-mediated abscess formation in response to GlyAg. We further demonstrate that inhibition of iNOS in vivo with 1400W decreases abscess incidence and severity in CGD without increasing risk of bacterial sepsis, raising the possibility of iNOS inhibition as a clinical approach for CGD patients.
CGD is characterized by recurring abscess and granuloma formation [7-9]. While granulomas are usually sterile and result from chronic inflammation [7, 11], abscesses tend to form in response to microbial stimuli [13]. For example, S. aureus, a GlyAg-expressing pathogen [16], is commonly associated with liver and brain abscesses [8, 11, 13, 32]. Although abscesses are an important response to contain microbes and prevent sepsis, once formed, they preclude antibiotic effectiveness and require surgical drainage [7, 8]. As a result, attenuation of abscess formation could provide a significant reduction in infection morbidity and possibly even mortality through improving antibiotic efficacy and reducing surgical intervention.
CGD has traditionally been viewed as a neutrophil-mediated disease since neutrophils are early responders to infection and produce high bactericidal oxidant concentrations. In addition, apoptosis of responding neutrophils is known to be abnormal through multiple mechanisms including deficient surface expression of phosphatidylserine (PS) [27, 28, 33], or diminished production of the apoptosis-inducers TGFβ and prostaglandin D2 [34]. However, an emphasis on the involvement of other cell populations (e.g. macrophages, DCs, and even T cells) in CGD has more recently challenged the neutrophil-centered model. For example, phosphatidylserine-mediated efferocytosis of apoptotic cells by macrophages is reduced in CGD, although normal levels of efferocytosis can be restored with IL-4 [27]. Changes in protein antigen processing and T-cell activation have also been reported in CGD [35], while studies using human cells have reported increased pro-inflammatory and decreased anti-inflammatory mediators when compared with healthy controls [34, 36-38].
We focused upon a recently described family of GlyAgs expressed by commensal and pathogenic bacteria (e.g. S. aureus, S. pneumoniae, and B. fragilis) that have been shown to induce abscess formation via CD4+ T-cell activation [12, 16, 20, 23, 39]. Lack of intact αβ T-cell receptor expression or blockade of costimulatory pathways in mice translates into a failure to develop abscesses in response to GlyAg [24]. GlyAgs require processing via NO-dependent oxidation [20, 21, 23] and presentation on MHCII molecules [16, 20, 23], providing an unexpected link to oxidative disorders. Our results reveal that CGD mice showed a dramatically increased immune response against GlyAgs, resulting in more frequent and severe abscesses. This differential response was mediated by APCs rather than neutrophils as might be expected and appears to be a result of increased NO and more efficient GlyAg processing. Likewise, the CGD phenotype was transferrable to WT animals via APC transfer, which indicates that the difference in T-cell activation is due to changes in the APC and not the responding T cells. Although we cannot completely rule out direct NO effects on responding T cells, it is clear that NO is required for processing [20, 23] and that CGD APCs are better GlyAg processors than their WT counterparts.
The NADPH oxidase complex is also known to maintain a neutral pH environment within endo/lysosomes [35], and thus changes impact acid-dependent protein antigen processing. In fact, CGD favors vesicular acidification and increased conventional antigen proteolysis [35]. In sharp contrast, GlyAg processing is dependent upon a neutral pH and acidification stops GlyAg processing in cells [40]. As a result, one might expect the CGD cells to process GlyAg less than the WT counterparts due to increased acidification, yet we observed the opposite. With the role of NO firmly established within this pathway [20, 23] and together with the ability to ameliorate the CGD effect by iNOS inhibition and the effectiveness of APC transfer into WT animals, we conclude that CGD results in GlyAg hyperresponsiveness because of increased GlyAg processing by resident APCs via increased NO levels, resulting in greater T-cell activation and downstream sequelae.
Another unexpected observation was that the level of IL-1β, used as a crude measure of inflammation, was not altered in CGD cells. While this may seem counterintuitive, recent evidence in humans has indicated that asymptomatic CGD patients do not make more IL-1β in response to a number of stimuli compared with healthy controls [41]. Thus, our findings are consistent with the human CGD condition.
Our data further suggest that the production of ROS and NO is linked. Since transcriptional regulation of iNOS is altered, this linkage is most likely at the level of the signaling pathways and ultimately NFκB associated. It remains unclear whether this effect is mediated by the ROS molecules themselves, the changes in vesicular pH, or another mechanism; however, our data are supported by findings in which the anti-inflammatory regulator Nrf2 was found to be defective in CGD [42]. This raises the possibility that increased iNOS transcription in CGD upon GlyAg stimulation could be a result of an inability to shut down the initial GlyAg-mediated TLR2-dependent signal [19] to activate iNOS synthesis in the first place. The difference between WT and CGD responses to an actual antigen like PSA from B. fragilis provides an ideal model system to explore the relationship between the control of ROS and NO production.
Taken together, our findings suggested that NO in macrophages, but not neutrophils, is the primary mediator of hyperresponsiveness to GlyAg in CGD. Our adoptive transfer experimental data further suggest that the loss of ROS in the T-cell population, which has been linked to a switch between T effector and T regulatory cells [43], does not explain the enhanced GlyAg response. These interpretations were confirmed in vivo using iNOS inhibition which completely prevented abscess formation in 6 of 14 animals while significantly reducing the abscess severity in the remaining mice. Since 1400W did not appear to increase the risk of bacterial sepsis, this strategy may represent a new pathway of treatment for CGD patients, although far more stringent testing with more invasive organisms would be needed to confirm these initial findings.
In contrast to the CGD T-cell studies in which non-specific anti-CD3/anti-CD28 stimulation of T cells was used [44, 45], our findings suggest a novel pathway responsible for CGD-associated recurring abscess formation that is centered upon professional APCs, increased GlyAg processing, and antigen-mediated T-cell activation. This pathway can be specifically targeted through inhibition of iNOS activity in vivo, resulting in attenuation of CGD-associated immune pathology arising from bacterial infection. This approach could significantly improve treatment outcomes for CGD patients through increasing antibiotic efficacy and reducing the need for surgical drainage of abscesses.
Materials and methods
Mice
WT (C57BL/6J, stock 000664) and X-linked gp91phox-deficient CGD (B6. 129S6-Cybbtm1Din/J, stock 002365) breeders were purchased from Jackson Labs and colonies were housed at CWRU Animal Resource Center. Experiments were performed in accordance with the guidelines of the National Institutes of Health (NIH) and protocols approved by the Institutional Animal Care and Use Committee. All experimental mice were at least 12 wk old.
Primary cells
Neutrophils and macrophages were harvested by peritoneal lavage at various time points after antigenic challenge and were purified by magnetic beads using anti-F4/80-Biotin (Biolegend), anti-Ly6G-Biotin, anti-Biotin microbeads, and magnetic columns (Miltenyi Biotec). BMDCs were obtained by culturing BM cells in RPMI 1640 with 15 ng/mL GM-CSF (Invitrogen) for 11–13 days. BM macrophages were derived via culture with LADMAC for 7 days.
Antibodies and general reagents
For flow cytometry: FITC-anti-mouse CD4, FITC-anti-mouse Gr-1, FITC-anti-mouse F4/80, FITC-anti-mouse I-Ab, FITC-mouse IgG2a isotype, FITC-rat IgG2b isotype (all from Biolegend); FITC-anti-mouse CD3, PE-anti-mouse CD69, FITC-Hamster IgG isotype, PE-Hamster IgG isotype, PE-rat IgG1 isotype (all from eBioscience). For Western blotting: mouse anti-iNOS/Nos2 (BD Biosciences), mouse anti-actin (Santa Cruz Biotechnology). iNOS inhibitor 1400W was purchased from Cayman Chemical.
Primers and PCR
Primers for iNOS and β-actin for PCR were purchased from Integrated DNA Technologies: iNOS sense: 5′-GTC CTA CAC CAC ACC AAA-3′, iNOS anti-sense: 5′-CAA TCT CTG CCT ATC CGT CTC-3′ (product size, 197 bps); β-actin sense: 5′-TGA GAG GGA AAT CGT GCG TGA C-3′, β-actin anti-sense: 5′-GAA CCG GTT GCC AAT AGT G-3′ (product size, 154 bps). Isolated RNA was standardized, converted to cDNA via First Strand cDNA synthesis (Invitrogen), and then rt-PCR was performed with SuperScript III (Invitrogen) and MultiGene II thermocycler (Labnet International). Quantitative PCR was done using SYBR®GreenER™ (Invitrogen) and iCycler (Bio-Rad Laboratories). Data were analyzed using the Pfaffl Method.
GlyAg purification and SCC preparation
GlyAg from the capsule of B. fragilis was purified as described previously [46]. Briefly, B. fragilis was anaerobically grown for 24 h, harvested, and extracted with phenol. The soluble phenol sample was extracted with diethyl ether and then digested with DNase and RNase, followed by Pronase. The resulting mixture of LPS and capsule was separated on a Sephacryl S-300 column in 3% deoxycholate.
SCC were made by harvesting cecal contents, diluting with enough PBS to make it easy to transfer via pipette, and then sterilization in an autoclave. The SCC was stored in aliquots at −80°C until use. All experiments in the present study were performed with the same batch of SCC to ensure dilution consistency.
Nitrate and nitrite detection
Lavage supernatants were tested for nitrate/nitrite concentrations using Nitrate Reductase kit and Griess Reagent (Caymen Chemical) according to the manufacturer’s protocol. Color change was quantified on a Victor 3V multilabel plate reader.
Flow cytometry
To measure cellular influx, mice were injected with 100 μg GlyAg and 1:4 SCC and at various time points, peritoneal lavage was performed with 1 mL of sterile PBS. The collected lavage samples were counted, divided, and stained for CD4, Gr-1, F4/80, or the appropriate isotype controls. The relative cell number was determined for each by multiplying the percent of positive stained cells by the total cell number. To determine T-cell phenotype, cells were collected from spleens of unchallenged WT and CGD mice or from peritoneal lavage samples taken 24 h after challenge with 100 μg GlyAg and 1:4 SCC. The bulk cells were stained for CD4, CD69, or isotype controls and analyzed. Cells were gated on CD4. All experiments were performed using C6 Flow Cytometer (Accuri).
Abscess experiments
For abscess induction, mice were injected with a challenge inoculum (200 μL i.p.) consisting of GlyAg and SCC at various dilutions. At day 7, mice were euthanized and scored for abscess formation (≥1 abscess = positive). Abscesses were removed and weighed and the diameter was measured. Some abscesses were sectioned and stained with H&E, or cryosectioned for confocal microscopy. Abscess digestion was done for 2 h using 2 mg/mL collagenase D at 37°C. The resulting cell suspensions were stained with antibodies and analyzed via flow cytometry. For 1400W administration, CGD mice were treated challenged with 50 μg GlyAg and 1:4 SCC and 100 μL of either PBS or 0.5 mg 1400W in PBS. Additional injections of either PBS or 1400W were administered at 6 and 24 h post challenge.
Western blot
Performed as described [47]. Briefly, NP-40 cellular extracts were boiled in standard SDS-PAGE loading buffer containing 1% SDS and loaded onto a 10% polyacrylamide gel. Protein was transferred to a nitrocellulose membrane and blotted with anti-NOS2 monoclonal antibody. Bands were visualized with a HRP-conjugated secondary antibody and ECL (GE Healthcare) according to the manufacturer’s protocol.
Processing assays
Intracellular processing was assessed by incubating splenocytes with 50 μg/mL [3H]GlyAg (PSA) for 48 h. Processed radioactive GlyAg was isolated as previously described [20, 23] and analyzed for molecular mass on a SuperDex 75 column in PBS using an Akta® Purifier10 HPLC system (GE Healthcare Biosciences) to measure cleavage compared with the input, unprocessed GlyAg.
T-cell assays
APCs and CD4+ T cells were purified from WT, CGD, or iNOS−/− splenocytes using microbeads for CD90.2 (for T-cell-depleted APCs) or CD4 (CD4+ T-cell purification) and magnetic columns (Miltenyi Biotec, Auburn, CA, USA). 1.5 × 105 APCs and 2.5 × 105 T cells were added to wells of 96-well plates in triplicates and treated with 100 μg/mL GlyAg in PBS or PBS alone. At various time points, supernatant was removed and analyzed for IFN-γ production via ELISA (eBioscience). Additional experiments were set up as described above but wells were also treated with 0.1 mM 1400W or PBS.
Adoptive transfer
5 × 106 WT or CGD splenic APCs (T cell and neutrophil depleted by anti-CD90.2 or anti-Ly6G microbeads respectively; Miltenyi Biotec) were transferred i.p. into WT animals which were then challenged with 50 μg GlyAg and 1:7 SCC. After 7 days, mice were scored for abscess formation.
IL-1β production
9 × 104 WT or CGD BM-derived macrophages were plated in triplicates in 96-well plates, then stimulated with 100 ng/mL LPS (Sigma), 100 μg/mL GlyAg±100 μM 1400W for 24 h. Cells were treated with 5 mM ATP (Sigma) 45 min prior to collection of supernatant and IL-1β was detected via ELISA (Biolegend).
Statistical analysis
Data are expressed as mean±standard error of the mean (SEM). Graphs were generated using GraphPad Prism v. 4 graphing software and data analyzed using unpaired t-test with a Student’s two-tailed p value using GraphPad Instat3.
Acknowledgements
The authors thank Dr. Derek Abbott and Jill Marinis for help with the Western blots and H&E staining of abscess sections. The authors also thank Nile Chang and Dr. Alex Huang for assistance with cryosections and for use of Imaris image analysis software and Dr. Lakshmi Ramachandra for providing LADMAC-derived macrophages. This work was funded by grants to B. A. Cobb (NIH, AI062707 and NIH, OD004225 and CGD Research Trust, Grant # J4G/06/01).
Abbreviations
- 1400W
(N-(3-aminomethyl) benzylacetamidine, 2HCl)
- BMDC
bone marrow-derived dendritic cells
- CGD
chronic granulomatous disease
- GlyAg
glycoantigen
- iNOS
inducible nitric oxide synthase
- MHCII
MHC class II
- PSA
polysaccharide A from B.fragilis
- SCC
sterilized cecal contents
Footnotes
Conflict of interest: The work described herein is the subject of a provisional patent application (#61332896) filed with the United States Patent and Trademark Office governing the use of 1400W in abscess prevention in CGD and other patients at risk for abscess formation.
References
- 1.Stuehr DJ. Mammalian nitric oxide synthases. Biochim. Biophys. Acta. 1999;1411:217–230. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 2.Bogdan C. Nitric oxide and the immune response. Nat. Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 3.Segal AW. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int. J. Biochem. Cell Biol. 2008;40:604–618. doi: 10.1016/j.biocel.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shatwell KP, Segal AW. NADPH oxidase. Int. J. Biochem. Cell Biol. 1996;28:1191–1195. doi: 10.1016/s1357-2725(96)00084-2. [DOI] [PubMed] [Google Scholar]
- 5.Ellson CD, Davidson K, Ferguson GJ, O’Connor R, Stephens LR, Hawkins PT. Neutrophils from p40phox−/− mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J. Exp. Med. 2006;203:1927–1937. doi: 10.1084/jem.20052069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, Marchal CC, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Seger RA. Modern management of chronic granulomatous disease. Br. J. Haematol. 2008;140:255–266. doi: 10.1111/j.1365-2141.2007.06880.x. [DOI] [PubMed] [Google Scholar]
- 9.Rosenzweig SD. Inflammatory manifestations in chronic granulomatous disease (CGD) J. Clin. Immunol. 2008;28:S67–S72. doi: 10.1007/s10875-007-9160-5. [DOI] [PubMed] [Google Scholar]
- 10.Dinauer MC. Chronic granulomatous disease and other disorders of phagocyte function. Hematology (Am. Soc. Hematol. Educ. Program.) 2005:89–95. doi: 10.1182/asheducation-2005.1.89. [DOI] [PubMed] [Google Scholar]
- 11.Schappi MG, Jaquet V, Belli DC, Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin. Immunopathol. 2008;30:255–271. doi: 10.1007/s00281-008-0119-2. [DOI] [PubMed] [Google Scholar]
- 12.Tzianabos AO, Kasper DL. Role of T cells in abscess formation. Curr. Opin. Microbiol. 2002;5:92–96. doi: 10.1016/s1369-5274(02)00292-8. [DOI] [PubMed] [Google Scholar]
- 13.Huang A, Abbasakoor F, Vaizey CJ. Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis. 2006;8:637–644. doi: 10.1111/j.1463-1318.2006.01030.x. [DOI] [PubMed] [Google Scholar]
- 14.Polk BF, Kasper DL. Bacteroides fragilis subspecies in clinical isolates. Ann. Intern. Med. 1977;86:569–571. doi: 10.7326/0003-4819-86-5-569. [DOI] [PubMed] [Google Scholar]
- 15.Tzianabos AO, Onderdonk AB, Rosner B, Cisneros RL, Kasper DL. Structural features of polysaccharides that induce intra-abdominal abscesses. Science. 1993;262:416–419. doi: 10.1126/science.8211161. [DOI] [PubMed] [Google Scholar]
- 16.Tzianabos AO, Wang JY, Lee JC. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc. Natl. Acad. Sci. USA. 2001;98:9365–9370. doi: 10.1073/pnas.161175598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumann H, Tzianabos AO, Brisson JR, Kasper DL, Jennings HJ. Structural elucidation of two capsular polysaccharides from one strain of Bacteroides fragilis using high-resolution NMR spectroscopy. Biochemistry. 1992;31:4081–4089. doi: 10.1021/bi00131a026. [DOI] [PubMed] [Google Scholar]
- 18.Kreisman LS, Friedman JH, Neaga A, Cobb BA. Structure and function relations with a T-cell-activating polysaccharide antigen using circular dichroism. Glycobiology. 2007;17:46–55. doi: 10.1093/glycob/cwl056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, McLoughlin RM, Cobb BA, Charrel-Dennis M, Zaleski KJ, Golenbock D, Tzianabos AO, et al. A bacterial carbohydrate links innate and adaptive responses through Toll-like receptor 2. J. Exp. Med. 2006;203:2853–2863. doi: 10.1084/jem.20062008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cobb BA, Wang Q, Tzianabos AO, Kasper DL. Polysaccharide processing and presentation by the MHCII pathway. Cell. 2004;117:677–687. doi: 10.016/j.cell.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan J, Avci FY, Kasper DL. Microbial carbohydrate depolymerization by antigen-presenting cells: deamination prior to presentation by the MHCII pathway. Proc. Natl. Acad. Sci. USA. 2008;105:5183–5188. doi: 10.1073/pnas.0800974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cobb BA, Kasper DL. Characteristics of carbohydrate antigen binding to the presentation protein HLA-DR. Glycobiology. 2008;18:707–718. doi: 10.1093/glycob/cwn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velez CD, Lewis CJ, Kasper DL, Cobb BA. Type I Streptococcus pneumoniae carbohydrate utilizes a nitric oxide and MHC II-dependent pathway for antigen presentation. Immunology. 2009;127:73–82. doi: 10.1111/j.1365-2567.2008.02924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, Tzianabos AO. CD4+T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J. Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- 25.Kalka-Moll WM, Tzianabos AO, Wang Y, Carey VJ, Finberg RW, Onderdonk AB, Kasper DL. Effect of molecular size on the ability of zwitterionic polysaccharides to stimulate cellular immunity. J. Immunol. 2000;164:719–724. doi: 10.4049/jimmunol.164.2.719. [DOI] [PubMed] [Google Scholar]
- 26.Tzianabos AO, Russell PR, Onderdonk AB, Gibson FC, III, Cywes C, Chan M, Finberg RW, et al. IL-2 mediates protection against abscess formation in an experimental model of sepsis. J. Immunol. 1999;163:893–897. [PubMed] [Google Scholar]
- 27.Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, Henson PM, et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood. 2009;113:2047–2055. doi: 10.1182/blood-2008-05-160564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hampton MB, Vissers MC, Keenan JI, Winterbourn CC. Oxidant-mediated phosphatidylserine exposure and macrophage uptake of activated neutrophils: possible impairment in chronic granulomatous disease. J. Leukoc. Biol. 2002;71:775–781. [PubMed] [Google Scholar]
- 29.Arroyo A, Modriansky M, Serinkan FB, Bello RI, Matsura T, Jiang J, Tyurin VA, et al. NADPH oxidase-dependent oxidation and externalization of phosphatidylserine during apoptosis in Me2SO-differentiated HL-60 cells. Role in phagocytic clearance. J. Biol. Chem. 2002;277:49965–49975. doi: 10.1074/jbc.M204513200. [DOI] [PubMed] [Google Scholar]
- 30.Kasper DL, Onderdonk AB. Infection with Bacteroides fragilis: pathogenesis and immunoprophylaxis in an animal model. Scand. J. Infect. Dis. Suppl. 1982;31:28–33. [PubMed] [Google Scholar]
- 31.Bartlett JG, Onderdonk AB, Louie T, Kasper DL, Gorbach SL. A review. Lessons from an animal model of intra-abdominal sepsis. Arch. Surg. 1978;113:853–857. doi: 10.1001/archsurg.1978.01370190075013. [DOI] [PubMed] [Google Scholar]
- 32.Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect. Immun. 2005;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanford AN, Suriano AR, Herche D, Dietzmann K, Sullivan KE. Abnormal apoptosis in chronic granulomatous disease and autoantibody production characteristic of lupus. Rheumatology (Oxford) 2006;45:178–181. doi: 10.1093/rheumatology/kei135. [DOI] [PubMed] [Google Scholar]
- 34.Brown JR, Goldblatt D, Buddle J, Morton L, Thrasher AJ. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD) J. Leukoc. Biol. 2003;73:591–599. doi: 10.1189/jlb.1202599. [DOI] [PubMed] [Google Scholar]
- 35.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 36.Lekstrom-Himes JA, Kuhns DB, Alvord WG, Gallin JI. Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J. Immunol. 2005;174:411–417. doi: 10.4049/jimmunol.174.1.411. [DOI] [PubMed] [Google Scholar]
- 37.Tsuji S, Taniuchi S, Hasui M, Yamamoto A, Kobayashi Y. Increased nitric oxide production by neutrophils from patients with chronic granulomatous disease on trimethoprim-sulfamethoxazole. Nitric Oxide. 2002;7:283–288. doi: 10.1016/s1089-8603(02)00110-6. [DOI] [PubMed] [Google Scholar]
- 38.Bylund J, MacDonald KL, Brown KL, Mydel P, Collins LV, Hancock RE, Speert DP. Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-kappa B. Eur. J. Immunol. 2007;37:1087–1096. doi: 10.1002/eji.200636651. [DOI] [PubMed] [Google Scholar]
- 39.Cobb BA, Kasper DL. Zwitterionic capsular polysaccharides: the new MHCII-dependent antigens. Cell Microbiol. 2005;7:1398–1403. doi: 10.1111/j.1462-5822.2005.00591.x. [DOI] [PubMed] [Google Scholar]
- 40.Lewis CJ, Cobb BA. Carbohydrate oxidation acidifies endosomes, regulating antigen processing and TLR9 signaling. J. Immunol. 2010;184:3789–3800. doi: 10.4049/jimmunol.0903168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–1573. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, et al. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Efimova O, Szankasi P, Kelley TW. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+effector T cells. PLoS One. 2011;6:e16013. doi: 10.1371/journal.pone.0016013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, Ottenhoff TH, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA. 2010;107:17686–17691. doi: 10.1073/pnas.1012016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Veen RC, Dietlin TA, Hofman FM, Pen L, Segal BH, Holland SM. Superoxide prevents nitric oxide-mediated suppression of helper T lymphocytes: decreased autoimmune encephalomyelitis in nicotinamide adenine dinucleotide phosphate oxidase knockout mice. J. Immunol. 2000;164:5177–5183. doi: 10.4049/jimmunol.164.10.5177. [DOI] [PubMed] [Google Scholar]
- 46.Tzianabos AO, Pantosti A, Baumann H, Brisson JR, Jennings HJ, Kasper DL. The capsular polysaccharide of Bacteroides fragilis comprises two ionically linked polysaccharides. J. Biol. Chem. 1992;267:18230–18235. [PubMed] [Google Scholar]
- 47.Harlow E, Lane D. Immunoblotting. Using Antibodies, A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1999. pp. 269–309. [Google Scholar]