

Abstract
Cytokine antibody arrays were used to establish the profiles of cytokine release from THP-1 monocytes exposed to different amphotericin B (AMB) drug delivery systems. Fungizone (FZ) and Amphotec (ABCD) caused the release of significantly more inflammatory molecules and the release of inflammatory molecules at higher levels than either AmBisome (L-AMB) or Abelcet (ABLC) after 6 h of treatment. Specifically, tumor necrosis factor alpha (TNF-α), interleukin-8 (IL-8), GRO-(αβγ), monocyte chemoattractant protein-1 (MCP-1), RANTES, IL-10, and IL-6 were detected and semiquantified with a chemiluminscence imaging system. TNF-α, IL-8, and MCP-1 were the most predominant; however, little if any TNF-α was present in ABLC- or L-AMB-treated cultures. The TNF- α and IL-8 levels determined by quantitative enzyme-linked immunosorbent assay correlated with the relative cytokine levels measured by using the antibody arrays. Although the viabilities of THP-l monocytes in all AMB-treated cultures were similar by trypan blue exclusion, the amount of lactic dehydrogenase released was significantly larger in FZ- and ABCD-treated cultures than in L-AMB- and ABLC-treated cultures, indicating more membrane perturbations with those formulations. Membrane cation channel formation was also measured in model cholesterol-containing large unilamellar vesicles to directly assess the ion channel formation ability of the system. Only FZ and ABCD induced significant ion currents at concentrations less than 1.5 × 10−5 M. These results may help provide rationales for the immediate cytokine-mediated side effects observed with FZ and ABCD and the reduced side effects observed with L-AMB and ABLC.
Amphotericin B (AMB) is a polyene antifungal antibiotic and for a number of years has been the “gold standard” for the treatment of systemic fungal infections. Three new lipid-based formulations, Abelcet (ABLC), Ambisome (L-AMB), and Amphotec (ABCD), have been developed in an attempt to obtain improved efficacies, therapeutic indices, and tolerabilities compared to those of the traditional AMB formulation, Fungizone (FZ). In clinical trials, improvements in renal tolerability were shown for all three lipid formulations; however, the levels of acute toxicity, including fever, chills, hypotension, and nausea, vary with each formulation (8, 23). L-AMB has been shown to promote the least adverse effects, while immediate reactions to ABCD are often as frequent and severe as those to FZ (23). Clinical toxicity has been associated with increases in plasma tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) levels during infusion of the drug (1), while in vitro studies with the human THP-1 monocytic cell line have further identified and characterized a number of proinflammatory cytokines that are induced and released following exposure to FZ (5, 19, 20). Because the clinical effects of the four AMB formulations are different, we hypothesized that each AMB formulation would produce a different cytokine profile. Using recently developed antibody arrays (13, 27) and the THP-1 monocytic cell model, we show that the four commercially available AMB delivery systems fall into two groups, with FZ and ABCD in one group and L-AMB and ABLC in the other group, with respect to cytokine release profiles.
MATERIALS AND METHODS
Cell culture.
The THP-1 (monocytic leukemic) cell line was grown in a 5% CO2 incubator at 37°C to a saturation density of 1 × 106 cells/ml in RPMI medium containing 10% endotoxin-free fetal bovine serum, glutamine, 100 U of penicillin per ml, 100 μg of streptomycin per ml, 1 μg of gentamicin per ml, and 2 × 10−5 M β-mercaptoethanol, as described previously (24). The cells were then split and placed into 96-well plates or 35-mm dishes for treatment with the AMB formulations.
AMB formulations.
L-AMB (Gilead Sciences, Inc.), ABLC (The Liposome Company, Inc.), and ABCD (InterMune Inc.) were compared with traditional FZ (Pharmacia & Upjohn Co.). Each pyrogen-free sterile powder formulation was weighed under aseptic conditions in a hood. One milliliter of sterile filtered nanopure water was added to obtain an approximate concentration of 5 mg of AMB equivalent per ml, according to the formulations of the manufacturers and the storage conditions. The actual molar concentration of AMB was then determined by dissolving an aliquot of each preparation in methanol and using an extinction coefficient (ɛ406) of 150,000 liters/mol cm.
Incubation of THP-1 monocytes with AMB formulations.
For AMB treatment, the cells were seeded either into the wells of 96-well round-bottom plates at a density of 100,000 cells/200 μl or in 35-mm dishes at a density of 2.9 × 106 cells in 5.8 ml of medium. Each AMB formulation was added to THP-1 cell cultures at a final concentration per well (96 well plate) or dish (35 mm) of 2 or 5 μg of AMB equivalent per ml. Escherichia coli O55:B5 lipopolysaccharide (LPS) was added to the other wells as a positive control, and untreated cells (cultured in complete medium only) were used as negative controls. THP-1 cells were exposed for 6 h. Supernatants were removed (150 μl from each well of the 96-well plates and 3 ml from each 35-mm dish) and stored at −20°C until use.
Measurement of cytokine and chemokine release using antibody arrays.
Human Cytokine Array I (Ray Biotech Inc., Norcross, Ga.) consisted of 23 different cytokine and chemokine antibodies spotted in duplicate onto a membrane (27). The membranes were blocked with 10% bovine serum albumin in Tris-buffered saline according to the instructions of the manufacturer. One and a half milliliters of conditioned medium was added to each membrane in separate wells of a six-well plate. The membranes were shaken at 110 rpm at room temperature for an hour and a half. All washes were done in new six-well plates, according to the instructions of the manufacturer. Two milliliters of a 1:500 dilution of biotin-conjugated antibodies was added to each membrane, and the mixture was incubated on a shaker for an hour and a half at room temperature. Following the wash, the membranes were incubated with a 1:40,000 dilution of strepavidin-conjugated peroxidase for 1 h at room temperature, according to the instructions of the manufacturer. Following a thorough wash, the membranes were exposed to a peroxidase substrate (ChemGlow West; AlphaInnotech Corp., San Leandro, Calif.) for 5 min in the dark before imaging. Two or six individual membranes were placed side by side in a plastic protective folder and sealed. Imaging was done with a UVP AutoChemi imaging system within 30 min of exposure to the substrate. Exposure times ranged from 1 to 10 min. Chemiluminescence was quantified with LabWorks imaging and analysis software. Horseradish peroxidase (HRP)-conjugated antibody served as a positive substrate control at six spots and was also used to identify membrane orientation. For each spot the net density gray level was determined by subtracting the background gray levels from the total raw density gray levels. The relative fold difference in cytokine amount was determined in reference to the amount present on the control culture membrane on the basis of the following: average treated culture cytokine spot gray levels/average control culture cytokine spot gray levels.
ELISA.
IL-8 was quantified by using the Flexia antibody pair system (BioSource Intl., Camarillo, Calif.). Basically, the enzyme-linked immunosorbent assays (ELISAs) were optimized by using 96-well plates (MaxiSorp; Nalge Nunc International Corp., Naperville, IL) coated with 100 μl of strepavidin (Sigma, St. Louis, Mo.) at 5 μg/ml in water overnight at 37°C or until all water was evaporated. All plates were stored desiccated at 4°C until use. Biotinylated F(ab′)2 coating antibody (5 μg/ml) was added to each well, and the plate was incubated at 37°C for 2 h. All wells were blocked and washed according to the instructions of the manufacturer. Standards or samples (100 μl) were added to the wells, and the plates were incubated at 37°C for 1 h. HRP-conjugated antibody (10 μg/ml) was added for an additional hour at 37°C. The plates were then washed thoroughly according to the instructions of the manufacturer. One hundred microliters of tetramethlybenzidine substrate (Roche, Indianapolis, Ind.) was added to each well, and the plates were held for 30 min in the dark. The reaction was stopped by the addition of 100 μl of 2 N H2SO4 to each well. The absorbance was read on an automated plate reader (Tecan U.S. Inc.) at 450 nm (reference wavelength, 650 nm). Values (in picograms per milliliter) were calculated from a standard curve. Samples were diluted up to 1:20, depending on the concentration.
SearchLight proteome arrays.
Some samples from the 96-well plates were sent directly to Pierce Boston Technology Center (Woburn, Mass.) for quantification of TNF-α. Each sample (120 μl) was frozen in dry ice and shipped overnight (Federal Express). The ELISA for quantification of TNF-α generates a chemiluminescent signal that is imaged with a camera with a charge-coupled device (CCD). The values for the test samples were compared to those on standard curves, and exact values were determined.
Measurement of proliferation and cell viability.
THP-1 monocyte proliferation and viability were assayed by both trypan blue exclusion and a lactic dehydrogenase (LDH) assay (Roche). In the LDH assay, THP-1 monocytes were plated at a concentration of 100,000 cells per 200 μl in round-bottom 96-well plates. The cells were exposed to 2 or 5 μg of each AMB formulation per ml or 25 μg of LPS per ml for 6 h or to 1% Triton X-100 for 1 h. The cells were spun down in the plate, and 100 μl was removed from each well and placed in a flat-bottom plate. A total of 100 μl of the LDH reagent was added to each well, and the plates were incubated in the dark at room temperature for 10 min. The reaction was stopped with 50 μl of 1 N HCl. The absorbance at 490 nm was read in a plate reader. The reference wavelength was 650 nm.
Membrane ion current detection by fluorescence.
Experiments for determination of ion channel activity were carried out as described previously (11, 22). Briefly, large unilamellar egg phosphatidylcholine vesicles containing 2 mM pyranine, 3 mM FCCP [carbonyl cyanide p-(trifluoromethoxy)phenyl hydrazone], and 10 mol% cholesterol were prepared by manual extrusion. The lipids were dispersed in a 15 mM K2HPO4-2 mM pyranine buffer with 200 mM KCl at pH 7.20. The lipid dispersions were then frozen-thawed and extruded 10 times through 100-nm-pore-size Nuclepore filters with a manual extruder (Avanti Polar Lipids, Alabaster, Ala.) to make large unilamellar vesicles (LUVs) of ca. 100 nm in diameter. The external pyranine solution was removed and replaced by a 15 mM K2HPO4 osmotically balanced (with sucrose) external buffer by gel filtration through a Sephadex G-25 column, thus forming a [K+] gradient of 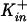 /
/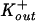 across the membrane of ∼6.7. FCCP (3 × 10−6 M; Sigma-Aldrich) was added to pyranine-containing vesicles to allow the free equilibration of H+ ions. The pyranine molecules entrapped in the target LUVs can then be used in a sensitive fluorescence assay for detection of the resulting interior vesicular pH change. The fluorescence intensity of the pyranine is linear between about pH 7.6 and pH 6.8, and so the fluorescence intensity may be converted directly to the internal pH. Hence, these AMB-induced ion currents can be observed as a dynamic change in pH, and the activity of AMB on the membrane can be reported as the change in pH (ΔpH) per second. The lipid-complex drug delivery systems, particularly ABCD, often exhibited a lag period as a result of the kinetics of vehicle transfer and ion channel assembly before ion channel currents resulted. As such, after our standard 200-s runs we chose to report the activities of these systems as final pH rather than ΔpH per second. Valinomycin, an ideal K+-selective ionophore, was used as a control. In these experiments, the AMB in the various delivery forms (prepared according to the instructions of the manufacturers) was introduced from an external buffer solution at two times the final AMB concentration at 37°C. The experiment was initiated by use of a stopped-flow pulse by mixing pyranine-containing vesicles (∼3 mg of total lipid per ml) and drug containing buffer at 1:1 by using a microvolume stopped-flow reaction analyzer (SX.18MV; Applied Photophysics Ltd., Leatherhead, United Kingdom). The data typically reflect the averages of three separate traces.
across the membrane of ∼6.7. FCCP (3 × 10−6 M; Sigma-Aldrich) was added to pyranine-containing vesicles to allow the free equilibration of H+ ions. The pyranine molecules entrapped in the target LUVs can then be used in a sensitive fluorescence assay for detection of the resulting interior vesicular pH change. The fluorescence intensity of the pyranine is linear between about pH 7.6 and pH 6.8, and so the fluorescence intensity may be converted directly to the internal pH. Hence, these AMB-induced ion currents can be observed as a dynamic change in pH, and the activity of AMB on the membrane can be reported as the change in pH (ΔpH) per second. The lipid-complex drug delivery systems, particularly ABCD, often exhibited a lag period as a result of the kinetics of vehicle transfer and ion channel assembly before ion channel currents resulted. As such, after our standard 200-s runs we chose to report the activities of these systems as final pH rather than ΔpH per second. Valinomycin, an ideal K+-selective ionophore, was used as a control. In these experiments, the AMB in the various delivery forms (prepared according to the instructions of the manufacturers) was introduced from an external buffer solution at two times the final AMB concentration at 37°C. The experiment was initiated by use of a stopped-flow pulse by mixing pyranine-containing vesicles (∼3 mg of total lipid per ml) and drug containing buffer at 1:1 by using a microvolume stopped-flow reaction analyzer (SX.18MV; Applied Photophysics Ltd., Leatherhead, United Kingdom). The data typically reflect the averages of three separate traces.
Statistical analysis.
Analysis was performed by two-tailed Student's t test for comparison between two groups. A P value <0.05 was considered statistically significant. The correlation between two groups was quantified by use of the linear correlation coefficient.
RESULTS
Cytokine antibody array analysis.
Seven cytokines were detected and semiquantified on membrane arrays containing 23 different cytokine antibodies (Fig. 1A to C; Fig. 2A to C). The supernatants of nontreated THP-1 control cultures contained detectable levels of IL-8, monocyte chemoattractant protein-1 (MCP-1), RANTES, and GRO-(αβγ) (GRO) after 6 h of incubation. IL-8 and MCP-1 were the most predominant, with RANTES and GRO being present at relatively lower levels. The presence of these cytokines was not due to the fetal bovine serum in the RPMI medium, since incubation with the medium alone did not result in reactivity with any of the antibody spots (data not shown). These four cytokines were induced to higher levels by some but not all of the AMB formulations (2 μg/ml) or LPS (Table 1). Three other cytokines (TNF-α, IL-6, IL-10) not detected in control cultures were induced in THP-1 monocytes exposed to 25 μg of E. coli LPS per ml and some of the AMB formulations. The cytokines released in relatively large amounts were IL-8, MCP-1, and TNF-α. IL-6, IL-10, GRO, and RANTES were present at relatively smaller amounts. Overall, exposure of THP-1 monocytes to 2 μg of each of the four AMB formulations per ml for 6 h resulted in unique cytokine profiles (Fig. 1). The relative amount (based on gray levels or brightness values) of each cytokine spot present was converted into the fold difference between the negative control culture and the treated culture (Table 1). Values were obtained by using the area density tool in LabWorks software, as described in Materials and Methods. Although cultures exposed to both FZ and ABCD had the highest total cumulative levels of cytokines and the highest levels of each cytokine at 6 h of incubation, the levels were lower than those for the positive control (LPS). ABCD in fact caused the release of slightly more inflammatory cytokines than FZ. On the other hand, ABLC and L-AMB treatment caused little if any increase in the levels of any of the seven cytokines, and for the most part the cytokine profiles for these cultures were similar to those for control cultures (Table 1). After 24 h of exposure the grouping of the four AMB formulations in terms of cytokine release by array analysis was similar to that after 6 h (data not shown).
FIG. 1.

Detection of cytokines on membrane antibody arrays by chemiluminscence. THP-1 monocytes were exposed to 2 μg of the AMB formulations (FZ, ABCD, L-AMB, ABLC) per ml for 6 h or to 25 μg of LPS per ml (control). (A) Image obtained with a camera with a CCD after an exposure of 90 s using a UVP AutoChemi imaging system. Each cytokine is represented by duplicate spots in the following locations: A1 and A2, HRP-conjugated antibody (positive control); A3 and A4, IL-1α; A5 and A6, IL-13; A7 and A8, TGF- 1; B1 and B2, HRP-conjugated antibody (positive control); B3 and B4, IL-2; B5 and B6, IL-15; B7 and B8, TNF-α; C1 and C2, bovine serum albumin (negative control); C3 and C4, IL-3; C5 and C6, IFN-γ; C7 and C8, TNF-α; D1 and D2, Tris-buffered saline (negative control); D3 and D4, IL-5; D5 and D6, MCP-1; D7 and D8, blank; E1 and E2, granulocyte colony-stimulating factor; E2 and E3, IL-6; E5 and E6, MCP-2; E7 and E8, blank; F1 and F2, granulocyte-macrophage colony-stimulating factor; F3 and F4, IL-7; F5 and F6, MCP-3; F7 and F8, blank; G1 and G2, GRO; G3 and G4, IL-8; G5 and G6, gamma interferon-induced monokine; G7 and G8, blank; H1 and H2, GRO-α; H3 and H4, IL-10; H5 and H6, RANTES; H7 and H8, HRP-conjugated antibody (positive control). (B and C) Average net light intensity for each pair of cytokine spots detected on the basis of gray-scale levels using LabWorks software.
FIG. 2.

Cytokines from L-AMB- and FZ-treated cultures detected on antibody arrays. THP-1 monocytes were exposed to 5 μg of either L-AMB or FZ per ml for 6 h. (A) Image obtained with a camera with a CCD after an exposure of 8 min with a UVP AutoChemi imaging system. The spots are as described in the legend to Fig. 1. (B and C) Average net light intensity of each pair of cytokine spots detected on the basis of gray-scale levels using LabWorks software.
TABLE 1.
Cytokine release profiles in THP-1 monocyte cultures treated with different AMB formulationsa
| Cytokineb | Array locationc | Relative levels in control cultured | Fold increase from levels in control culture at 6 he
|
Relative levels after induction | ||||
|---|---|---|---|---|---|---|---|---|
| LPS | FZ | ABCD | ABLC | L-AMB | ||||
| IL-1α | A3-4 | − | − | − | − | − | − | − |
| IL-2 | B3-4 | − | − | − | − | − | − | − |
| IL-3 | C3-4 | − | − | − | − | − | − | − |
| IL-5 | D3-4 | − | − | − | − | − | − | − |
| IL-6 | E3-4 | − | ∼112 | ∼15 | ∼21 | NC | NC | L |
| IL-7 | F3-4 | − | − | − | − | − | − | − |
| IL-8 | G3-4 | H | 7 | 4 | 41 | NC | NC | H |
| IL-10 | H3-4 | − | ∼3 | NC | ∼3 | NC | NC | L |
| IL-13 | A5-6 | − | − | − | − | − | − | − |
| IL-15 | B5-6 | − | − | − | − | − | − | − |
| GCSF | E1-2 | − | − | − | − | − | − | − |
| GM-CSF | F1-2 | − | − | − | − | − | − | − |
| GRO-(αβγ) | G1-2 | L | 15 | 2 | 5 | NC | NC | L |
| GRO-α | H1-2 | − | − | − | − | − | − | − |
| IFN-γ | C5-6 | − | − | − | − | − | − | − |
| MCP-1 | D5-6 | H | 3.6 | 2.3 | 2.5 | NC | NC | H |
| MCP-2 | E5-6 | − | − | − | − | − | − | − |
| MCP-3 | F5-6 | − | − | − | − | − | − | − |
| MIG | G5-6 | − | − | − | − | − | − | − |
| RANTES | H5-6 | L | ∼3.5 | ∼2 | ∼2.3 | NC | NC | L |
| TGF-β1 | A7-8 | − | − | − | − | − | − | − |
| TNF-α | B7-8 | − | ∼162 | ∼25 | ∼36 | NC | NC | H |
| TNF-β | C7-8 | − | − | − | − | − | − | − |
The cultures with the different formulations contained AMB at 2 μg/ml and LPS (positive control) at 25 μg/ml.
G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, gamma interferon; MIG, gamma interferon-induced monokine; TGF-β1, transforming growth factor β1.
See Fig. 1 for the locations of the duplicate spots in the matrix.
Relative levels: −, undetectable; H, high; L, low.
When the control level was essentially zero, the “∼” symbol was used to indicate an approximation; the values indicate the fold increase in induction and up-regulation. NC, no change (less than or equal to fold difference from the level in the control culture).
Comparison of effects of FZ and L-AMB at higher dosages.
Since the cytokine expression profiles of cells treated with 2 μg of L-AMB per ml were the same as those of untreated control cultures, we increased the level of L-AMB to 5 μg/ml for 6 h to see if the higher dose of L-AMB would change the levels of expression of any of these cytokines. We also increased the FZ concentration to 5 μg/ml and compared the differences in cytokine profiles between the two cultures (Fig. 2). The relative levels of cytokines in the FZ-treated cultures compared to those in the L-AMB-treated cultures at 6 h are shown in Table 2. In contrast to the lower L-AMB dose of 2 μg/ml, the higher L-AMB dose of 5 μg/ml induced a small amount of TNF-α. In the FZ-treated cultures all seven cytokines except RANTES were present at larger amounts compared to the amounts present in the L-AMB-treated cultures. For example, FZ-treated cultures had 110- and 14-fold more TNF-α and IL-8, respectively, than the L-AMB-treated cultures (Table 2).
TABLE 2.
Cytokine release profiles of FZ- and L-AMB-treated THP-1 monocyte culturesa
| Cytokine | Array locationb | Relative level in L-AMB cultures at 6 hc | Fold increase in level in FZ-treated cultures at 6 hd |
|---|---|---|---|
| IL-6 | E3-4d | − | ∼28 |
| IL-8 | G3-4 | H | 13 |
| IL-10 | H3-4 | − | ∼2 |
| GRO | G1-2 | VL | 8.9 |
| MCP-1 | D5-6 | H | 3.8 |
| RANTES | H5-6 | L | NC |
| TNF-α | B7-8 | VL | 102 |
THP-1 cells were cultured for 6 h with either L-AMB or FZ (5 μg/ml).
See Fig. 2 for the locations of the duplicate spots in the matrix.
Relative levels: −, undetectable; H, high; L, low; VL, very low.
When the cytokine level in L-AMB cultures was essentially zero, the “∼” symbol was used to indicate an approximation; NC, no change (less than or equal to twofold difference).
Confirmation of cytokine levels.
In order to check the reliability of the relative differences observed among AMB-treated cultures, we quantified TNF-α and IL-8, two cytokines that showed large fold differences in levels among cultures. The results for these two cytokines were independently confirmed by two similar quantitative procedures, as shown in Table 3. First, the actual IL-8 levels in the cell supernatants used in the antibody arrays were determined by a conventional ELISA, as described in Material and Methods. The actual IL-8 levels (in picograms per milliliter) showed the same pattern seen in the arrays with LPS-treated cultures containing larger IL-8 amounts than in FZ- and ABCD-treated cultures. ABLC- and L-AMB-treated cultures contained amounts of IL-8 equivalent to those found in the control cultures (data not shown). Second, we generated samples from drug-treated THP-1 monocyte cultures set up in microtiter plates instead of 35-mm culture dishes (see Materials and Methods). Table 3 shows that the levels of IL-8 and TNF-α (in picograms per milliliter) correlated well with the relative fold difference seen in the antibody array experiments. The linear correlation coefficients (r values) were 0.97 and 0.99 for IL-8 and TNF-α, respectively.
TABLE 3.
Comparison of cytokine levels obtained by antibody array analysis and ELISAa
| Culture treatment | TNF-α levels in:
|
IL-8 levels in:
|
||
|---|---|---|---|---|
| 35-mm dish (fold increaseb) | Microtiter wells (concn [pg/ml])c | 35-mm dish (fold increase) | Microtiter wells (concn [pg/ml])d | |
| None | — | 68 ± 4 | — | 100 ± 12 |
| LPS | ∼162 | 7,779 ± 593 | 7 | 4,013 ± 238 |
| FZ | ∼25 | 426 ± 23 | 4 | 1,143 ± 72 |
| ABCD | ∼36 | 838 ± 236 | 4.1 | 1,612 ± 37 |
| ABLC | NC | 61 ± 13 | NC | 136 ± 13 |
| L-AMB | NC | 99 ± 29 | NC | 78 ± 15 |
For the assays with 35-mm dishes, 2.9 × 106 THP-1 monocytes were seeded into a 35-mm dish in 5.8 ml of RPMI complete medium and incubated with formulations containing AMB at 2 μg/ml. For the assays with microtiter plates, 1 × 105 THP-1 monocytes were seeded into individual round-bottom wells of a 96-well plate in 200 μl of RPMI complete medium and incubated with 2 μg of AMB per ml.
Fold increase in net light intensity in treated culture compared to that in untreated control culture. —, not applicable; ∼, an approximation when the cytokine level in the control culture was zero.
Quantified by the Pierce Biotechnology Searchlite multiplex sample testing service. The values are averages ± standard errors of the means.
Quantified by ELISA (see Material and Methods). The values are averages ± standard errors of the means. NC, no change (less than or equal to a two difference fold from the level in the control culture).
Cell viability and proliferation.
The viabilities of the treated THP-1 monocytes did not differ from those of the control monocytes under any experimental condition when viabilities were assayed by trypan blue exclusion. Greater than 95% of the cells in all cultures were viable (data not shown). However, by use of the more sensitive LDH assay, which measures the amount of LDH enzyme released into the supernatant, we found that significantly (P < 0.05) more LDH was released as a result of membrane perturbation in FZ- and ABCD-treated cultures than in ABLC- and L-AMB-treated cultures when AMB was used at both 2 and 5 μg/ml (Table 4). There was no apparent difference in cell numbers (by counting by trypan blue exclusion) at 6 h, suggesting that none of the drugs had a proliferative effect on the cells.
TABLE 4.
Percent LDH release from AMB-treated THP-1 monocytes at 6 h
| Dose (μg/ml) | % LDH releasea
|
|||||
|---|---|---|---|---|---|---|
| Control | Treated cells
|
|||||
| LPS | FZ | ABCD | ABLC | L-AMB | ||
| None | 10.8 ± 1.1 | — | — | — | — | — |
| 2 | — | — | 20.3 ± 3b | 25.6 ± 2.3b | 11.7 ± 1.5 | 10.8 ± 1.5 |
| 5 | — | — | 35.8 ± 3.3b | 30.6 ± 1.4b | 11.0 ± 1.0 | 10.4 ± 0.2 |
| 25 | — | 78.5 ± 6.7b | — | — | — | — |
The percent LDH release was calculated as the (average optical density at 490 nm for the control or treated culture supernatant/optical density at 490 nm for total LDH release) × 100. Total LDH release was obtained by treatment with Triton X-100 for 1 h. Each value represents the average ± standard error of the mean for four wells. —, not done or not applicable
Significantly different (P < 0.05) from the value for the control.
FZ and ABCD cause ion channel activity.
Figure 3 shows the intrinsic activities of the AMB pharmaceutical preparations at a therapeutically reasonable concentration of 5 μM AMB. Only FZ- and ABCD-treated cultures showed significantly more K+ selective ion channel activity in this time frame and at this concentration. This trend also held for increasing concentrations of the drug delivery systems and against ergosterol-containing membranes, although the ergosterol-containing membranes were far more sensitive (unpublished data). The method used measures only electrogenic ion channel activity, so AMB activity could lead to changes in cell membrane potential, depending on the specific ionic conditions.
FIG. 3.

Final pH values (after 200 s) for 10 mol% cholesterol-containing LUVs after exposure to various concentrations of drug delivery vehicles. Micromolar concentrations of AMB are reported. Only ABCD and FZ exhibited significant channel-inducing activities in this time frame, especially at concentrations below 15 μM. The results of control experiments with valinomycin and each AMB carrier (data not shown) indicated a strongly cation-selective channel with minimal Cl− permeation. Dynamically, ABCD exhibited a lag time for induction of channel formation of 30 to 60 s, after which significant activity was observed. A 0.1 μM valinomycin control served as an ideal K+-selective carrier, and equilibrium activity was reached within 60 s with a final pH of 6.79. Typically, three final pH values were averaged for each point. Diamonds, FZ; triangles, ABCD; circles, ABLC; filled squares, L-AMB; open square, valinomycin.
DISCUSSION
The antifungal drug of choice for the treatment of fungal infections is AMB because of its known property of complexing with fungal membrane sterols. The most common formulation, FZ, is quite toxic in some patients and is also known to activate the innate immune system (26). This may not necessarily be all bad, since activation of the innate immune system and perhaps some acquired defense mechanisms are interesting therapeutic strategies that can be used to supplement the direct antifungal activity of the drug. However, side effects likely caused in part by cytokine activation, especially nephrotoxicity, have been severe enough that new AMB delivery systems that offer fewer complications have been developed.
The immunomodulating activity of AMB on THP-1 cells in vitro has primarily been studied with FZ or heat-treated FZ. The drug causes the release of important proinflammatory molecules from THP-1 cells and the activation of various other genes (5, 19, 20). The goal of the present study was to directly compare the immunomodulating activities of all four commercially available AMB formulations in the THP-1 monocyte model. We established limited cytokine release profiles for each formulation and found that the four AMB formulations fell into two groups with respect to the number and the amount of cytokines induced in cultures of THP-1 monocytes. Cells exposed to FZ and ABCD for 6 h produced the highest levels of proinflammatory cytokines. On the other hand, the cytokine profiles of ABLC- or L-AMB-treated cultures were similar to those of untreated control cultures. Exposure to both FZ and ABCD was associated with the induction and release of high levels of TNF-α. TNF-α is believed to contribute to the infusion-related adverse drug reactions (1, 17), and on the basis of the results of earlier in vitro studies and in vivo clinical studies, the release of TNF-α from monocytes is associated with exposure to FZ (1, 17, 26). Other cytokines released from THP-1 monocytes include IL-8, MCP-1, GRO, RANTES, IL-6, and IL-10. Fairly high levels of IL-8 and MCP-1 were detected in untreated control cultures of THP-1 cells. Constitutive expression of these two cytokines in monocytes has been reported before (2, 7, 19, 21) and may represent occurrences during the differentiation stage of the THP-1 monocyte (12). Significantly more IL-8 and MCP-1 was induced and released from FZ- and ABCD-treated cultures than from L-AMB- and ABLC-treated cultures. In fact, the levels of IL-8 and MCP-1 in L-AMB- and ABLC-treated cultures were less than those in control cultures. Our cytokine profiles correlated with clinical observations that show that the rates of occurrence of fever and chills are generally lower after L-AMB and ABLC treatment than after FZ treatment (23). Likewise, both FZ and ABCD therapy have been associated with infusion-related side effects, such as fever and chills, with similar severities and frequencies (23). L-AMB has been reported to be the safest and best-tolerated formulation of the three formulations presently available (8).
The trade-off between clinical safety or a lack of adverse effects and efficacy remains unknown, in part due to the complexity of the inflammatory response. The net effect of these interactions between proinflammatory cytokines, such as TNF-α, IL-1, IL-6, IL-8, and GRO, and anti-inflammatory cytokines, such as IL-1ra, IL-6, and IL-10, determines the nature or degree of response over time. Thus, immunomodulatory AMB formulations that recruit the innate immune system response and perhaps the acquired immune response could be beneficial or detrimental to the host, depending on which cytokines are induced. In our study, ABCD was the only drug that also induced IL-6 and IL-10, both of which known to have anti-inflammatory properties (6, 25). In another study, FZ was found to induce inhibitors of IL-1 (17). The picture is far from complete, and what this means in terms of activation or inhibition of the innate immune response is unknown. Neutrophils and macrophages constitute the main mechanism of host defense against disseminated yeast infections (26). In this study, the important neutrophil-activating and -recruiting cytokines TNF-α, IL-8, and GRO were also upregulated by FZ and ABCD but not by ABLC or L-AMB. Therefore, ABLC or L-AMB treatment may be more appropriate in fungus-infected patients with persistently febrile neutropenia because TNF-α would not be induced and increase the fever response. In fact, in a study by Prentice et al. (15) there was a statistically higher success rate for resolution of fever with L-AMB treatment than with FZ treatment. Although the use of AMB derivatives has also been evaluated as a therapeutic approach to human immunodeficiency virus infection, the use of AMB formulations that induce TNF-α could increase the rate of human immunodeficiency virus replication, as reported previously (4). However, in the nonimmunocompromised patient, as long as nephrotoxicity can be tolerated, a systemic fungal infection might be better treated with FZ or ABCD because the recruitment of neutrophils and monocytes may be enhanced by the expression of proinflammatory cytokines.
The mechanism of AMB activation of macrophages is thought to involve the drug's ion channel activity. Stimulation of Ca2+ influx is a possible mechanism and has been observed in human monocytes (18). However, it is unclear whether Ca2+ is directly transported since the AMB-induced channels in cholesterol-containing model membranes are not appreciably Ca2+ permeant (10, 16). Alternatively, monovalent cation permeation could lead to membrane polarization or depolarization, leading to a secondary release of Ca2+ from internal stores or from stimulated outer membrane Ca2+ channels. In either case, if ion transport is key to macrophage stimulation, there should be a correlation between the intrinsic ion channel activities of AMB drug delivery systems and their stimulatory activities. Jensen and coworkers (14) have compared the longer-term activities of the four AMB formulations against red blood cells and Candida cells by K+ release assays. Their results agree well with ours for model systems with FZ and ABCD, which are generally more active against mammalian red blood cells; L-AMB is the least active. Admittedly, the serum and cell-free ion channel studies described in this report are not the equivalent of in vivo studies; nonetheless, the apparent correlation of intrinsic ion channel-forming ability in model and in vitro cellular systems with cytokine stimulation suggests that channel activity may be a key factor and should be further studied.
This study also demonstrates the usefulness and limitations of chemiluminescence-based membrane antibody arrays for the analysis of more than one protein at a time. This new assay of the array family combines the advantages of the specificity of the ELISA and the sensitivity of chemiluminescence analysis (13, 27). Furthermore, if the arrays are combined in a high-throughput microspot format, fairly high-density arrays should be possible. We used 12-bit imaging with a camera with a CCD to rapidly detect and quantify signals for 23 different cytokines and determine the relative levels and the fold differences in levels among the different treatment regimens. The relative levels correlated quite well with the actual levels (in picograms per milliliter) of selected cytokines by more quantitative assays. However, the assumption that there is a linear response between the chemiluminescent signal and quantity may not always hold, as suggested by Budowle et al. (3). This nonlinearity seemed to occur as IL-8 reached higher levels. Another minor concern occurred when the control chemiluminescent signal was essentially zero. When a signal was detected in a treated culture, the fold increase from the signal for the control represented only an approximation and could be quite large.
Despite some limitations, use of the relatively inexpensive membrane antibody arrays used in the present study for determination of the relative levels of a large number of cytokines simultaneously and as a confirmation of differential gene expression should gain in popularity. Such confirmation is important, since experimental evidence shows that a disparity between relative mRNA levels and the levels of the corresponding proteins can exist (9). Therefore, in the future the use of antibody and gene arrays in combination should help us understand the cellular responses to AMB formulations.
Acknowledgments
This work was supported by a grant from the National Science Foundation (MCB-9603582 to S.C.H. and L.W.T.).
REFERENCES
- 1.Arning, M., K. O. Kliche, A. H. Heer-Sonderhoff, and A. Wehmeier. 1995. Infusion-related toxicity of three different amphotericin B formulations and its relation to cytokine plasma levels. Mycoses 38:459-465. [DOI] [PubMed] [Google Scholar]
- 2.Baggiolini, M., B. Dewald, and B. Moser. 1994. Interleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Adv. Immunol. 55:97-179. [PubMed] [Google Scholar]
- 3.Budowle, B., W. R. Hudlow, S. B. Lee, and L. Klevan. 2001. Using a CCD camera imaging system as a recording device to quantify human DNA by slot blot hybridization. BioTechniques 30:680-685. [DOI] [PubMed] [Google Scholar]
- 4.Clayette, P. M., M. Martin, V. Beringue, N. Dereudde-Bosquet, K. Adjou, M. Seman, and D. Dormont. 2000. Effects of MS-8209, an amphotericin B derivative, on tumor necrosis factor alpha synthesis and human immunodeficiency virus replication in macrophages. Antimicrob. Agents Chemother. 44:405-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cleary, J. D., P. D. Rogers, and S. W. Chapman. 2001. Differential transcription factor expression in human mononuclear cells in response to amphotericin B: identification with complementary DNA microarray technology. Pharmacotherapy 21:1046-1054. [DOI] [PubMed] [Google Scholar]
- 6.Conti, P., D. Kempuraj, K. Kandere, M. D. Gioacchino, R. C. Barbacane, M. L. Castellani, M. Felaco, W. Boucher, R. Letourneau, and T. C. Theoharides. 2003. IL-10, an inflammatory/inhibitory cytokine, but not always. Immunol. Lett. 86:123-129. [DOI] [PubMed] [Google Scholar]
- 7.Cushing, S. D., and A. M. Fogelman. 1992. Monocytes may amplify their recruitment into inflammatory lesions by inducing monocyte chemotactic protein. Arterioscler. Thromb. 12:78-82. [DOI] [PubMed] [Google Scholar]
- 8.Dupont, B. 2002. Overview of the lipid formulations of amphotericin B. J. Antimicrob. Chemother. 49:31-36. [DOI] [PubMed] [Google Scholar]
- 9.Gygi, S. P., Y. Rochon, B. R. Franza, and R. Aeversold. 1999. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19:1720-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartsel, S. C., S. K. Benz, R. P. Peterson, and B. S. Whyte. 1991. Potassium selective amphotericin B channels are predominant in vesicles regardless of sidedness. Biochemistry 30:77-82. [DOI] [PubMed] [Google Scholar]
- 11.Hartsel, S. C., E. A. Bauer, E. H. Kwong, and K. M. Wasan. 2001. The effect of serum albumin on amphotericin B aggregate structure and activity. Pharm. Res. 18:1305-1309. [DOI] [PubMed] [Google Scholar]
- 12.Hemmi, H., and T. R. Breitman. 1985. Induction of functional differentiation of a human monocytic leukemia cell line (THP-1) by retinoic acid and cholera toxin. Jpn. J. Cancer Res. 76:345-351. [PubMed] [Google Scholar]
- 13.Huang, R. 2001. Detection of multiple proteins in an antibody-based protein microarray system. J. Immunol. Methods 255:1-13. [DOI] [PubMed] [Google Scholar]
- 14.Jensen, G. M., C. R. Skenes, T. H. Bunch, C. A. Weissman, N. Amirghahari, A. Satorius, K. L. Moynihan, and C. G. S. Eley. 1999. Determination of the relative toxicity of amphotericin B formulations: a red blood cell potassium release assay. Drug Delivery 6:81-88. [Google Scholar]
- 15.Prentice, H. G., I. M. Hann, R. Herbrecht, M. Aoun, S. Kvaloy, D. Catovsky, C. R. Pinkerton, S. A. Schey, F. Jacobs, A. Oakhill, R. F. Stevens, P. J. Darbyshire, and B. E. Gibson. 1997. A randomized comparison of liposomal versus conventional amphotericin B for the treatment of pyrexia of unknown origin in neutropenic patients. Br. J. Haematol. 98:711-718. [DOI] [PubMed] [Google Scholar]
- 16.Ramos, H., A. Attias de Murciano, B. E. Cohen, and J. Bolard. 1989. The polyene antibiotic amphotericin B acts as a Ca2+ ionophore in sterol-containing liposomes. Biochim. Biophys. Acta 982:303-306. [DOI] [PubMed] [Google Scholar]
- 17.Rogers, P. D., J. K. Jenkins, S. W. Chapman, K. Ndebele, B. A. Chapman, and J. D. Cleary. 1998. 1998. Amphotericin B activation of human genes encoding for cytokines. J. Infect. Dis. 178:1726-1733. [DOI] [PubMed] [Google Scholar]
- 18.Rogers, P. D., R. E. Kramer, S. W. Chapman, and J. D. Cleary. 1999. Amphotericin B-induced interleukin-1β expression in human monocytic cells is calcium and calmodulin dependent. J. Infect. Dis. 180:1259-1266. [DOI] [PubMed] [Google Scholar]
- 19.Rogers, P. D., J. K. Stiles, S. W. Chapman, and J. D. Cleary. 2000. Amphotericin B induces expression of genes encoding chemokines and cell adhesion molecules in the human monocytic cell line THP-1. J. Infect. Dis. 182:1280-1283. [DOI] [PubMed] [Google Scholar]
- 20.Rogers, P. D., K. S. Barker, V. Herring, and M. Jacob. 2003. Heat-induced superaggregation of amphotericin B attenuates its ability to induce cytokine and chemokine production in the human monocytic cell line THP-1. J. Antimicrob. Chemother. 51:405-408. [DOI] [PubMed] [Google Scholar]
- 21.Rollins, B. J., P. Stier, T. Ernst, and G. W. Wong. 1989. The human homolog of the JE gene encodes a monocyte secretory protein. Mol. Cell. Biol. 9:4687-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruckwardt, T., A. Scott, J. Scott, P. Mikulecky, and S. C. Hartsel. 1998. Lipid and stress dependence of amphotericin B ion selective channels in sterol-free membranes. Biochim. Biophys. Acta 1372:283-288. [DOI] [PubMed] [Google Scholar]
- 23.Tiphine, M., V. Letscher-Bru, and R. Herbrecht. 1999. Amphotericin B and its new formulations: pharmacologic characteristics, clinical efficacy, and tolerability. Transplant. Infect. Dis. 1:273-283. [DOI] [PubMed] [Google Scholar]
- 24.Turtinen, L. W., A. Assimacopoulos, and A. T. Haase. 1989. Increased monokines in cytomegalovirus infected myelomonocytic cell cultures. Microb. Pathog. 7:135-145. [DOI] [PubMed] [Google Scholar]
- 25.Xing, Z., J. Gauldie, G. Cox, H. Baumann, M. Jordana, X. Lei, and M. K. Achong. 1998. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Investig. 101:311-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi, H., S. Abe, and Y. Tokuda. 1993. Immunomodulating activity of antifungal drugs. Ann. N. Y. Acad. Sci. 685:447-457. [DOI] [PubMed] [Google Scholar]
- 27.Ying, L., R. Huang, X. Cao, S.-M. Wang, Q. Shi, and R.-P. Huang. 2003. Detection of multiple cytokines by protein arrays from cell lysate and tissue lysate. Clin. Chem. Lab. Med. 41:139-145. [DOI] [PubMed] [Google Scholar]